
Trichostatin A Enhances the Apoptotic Potential of Palladium Nanoparticles in Human Cervical Cancer Cells

Abstract
:1. Introduction
2. Results and Discussion
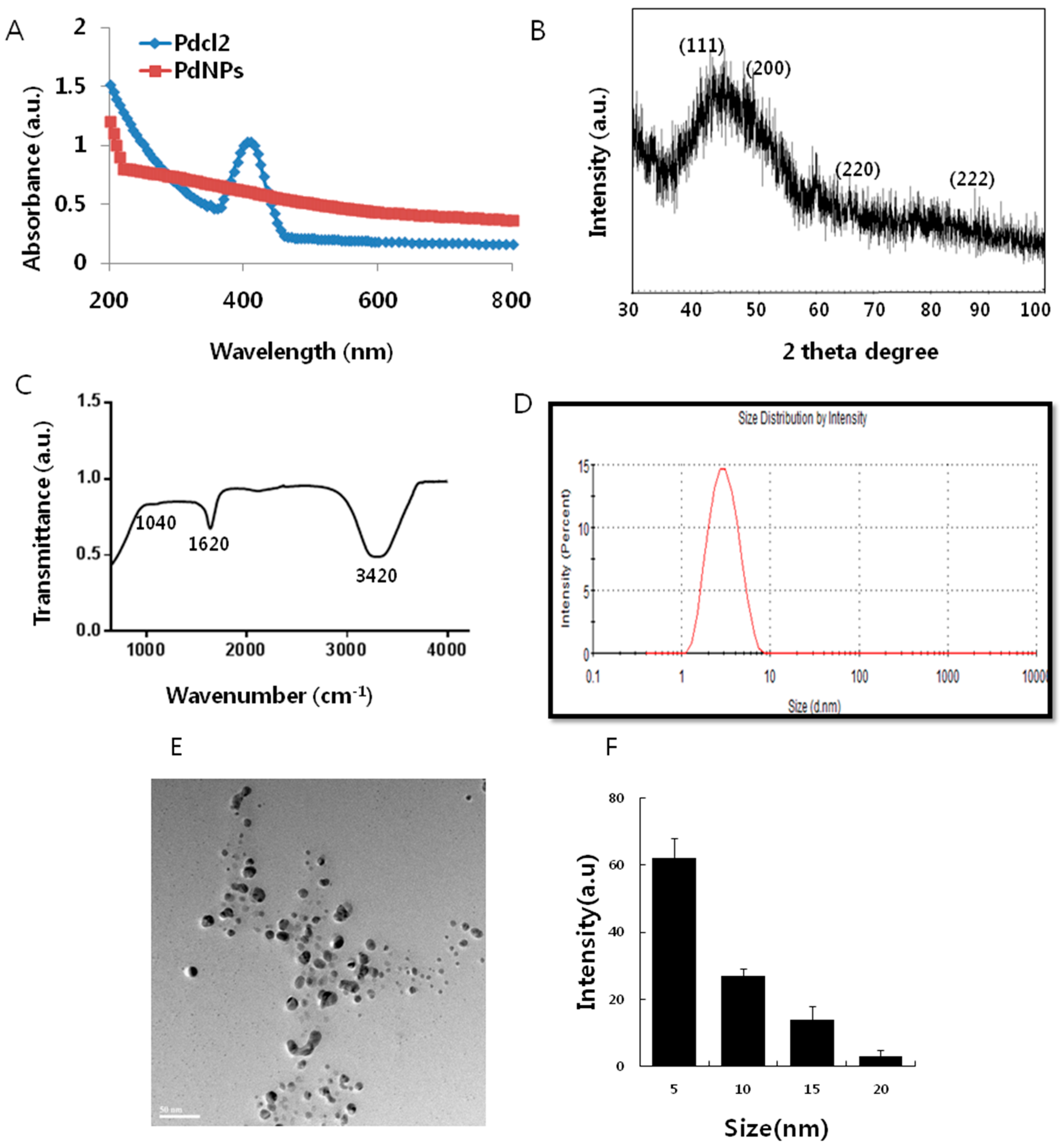
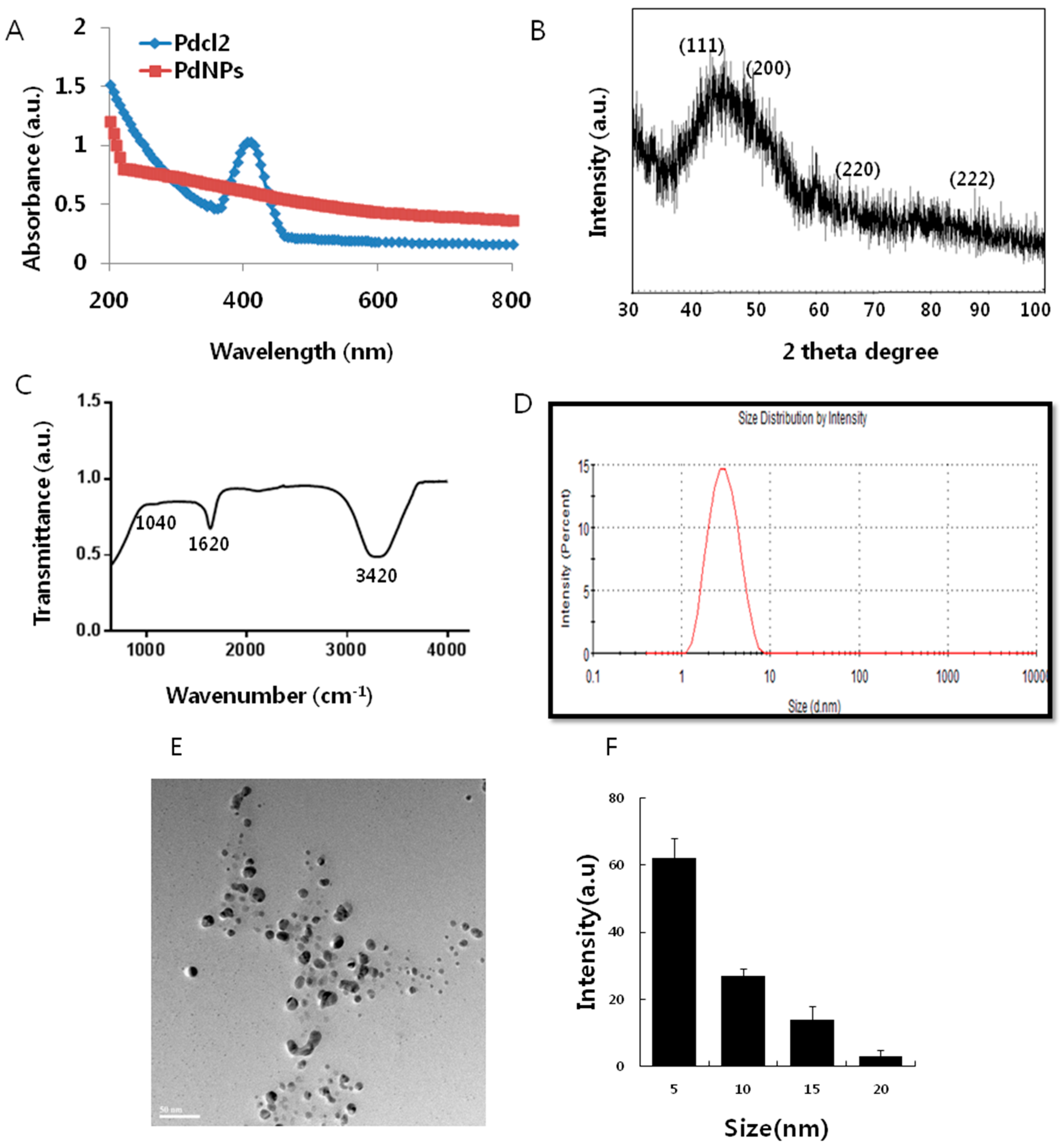
2.1. Synthesis and Characterization of Palladium Nanoparticles (PdNPs)
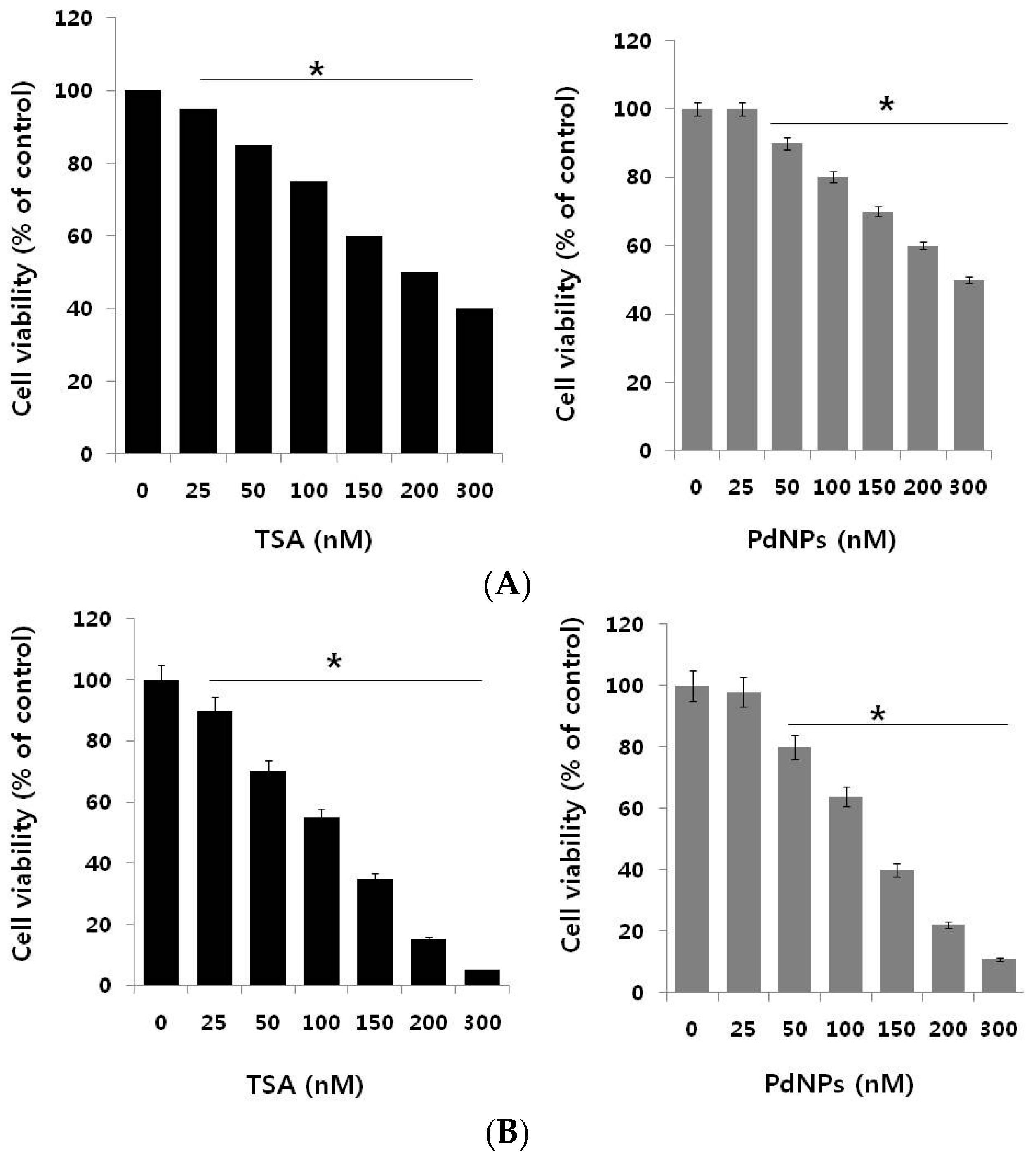
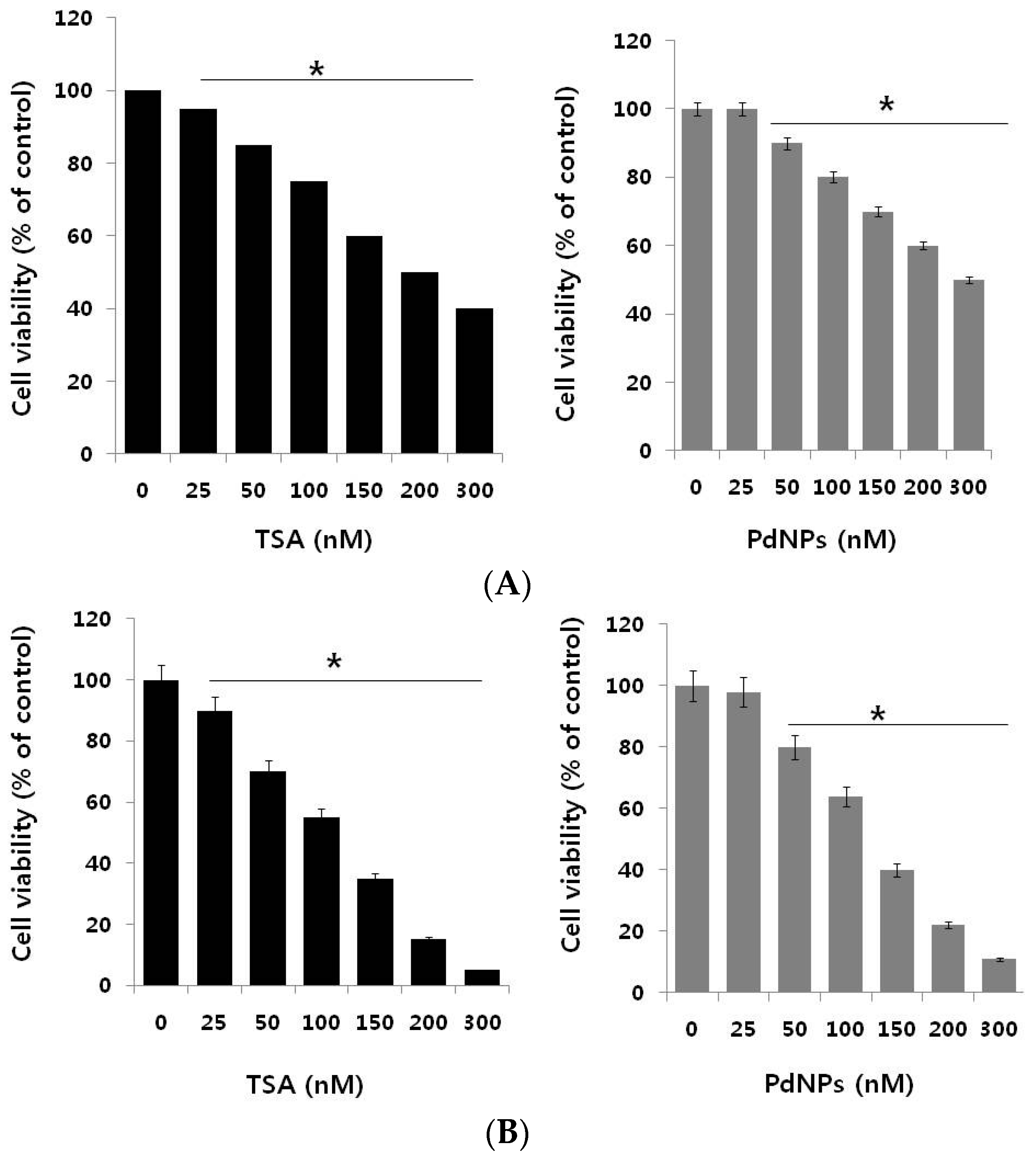
2.2. Trichostatin A (TSA) and PdNPs Inhibit Breast Cancer and HeLa Cell Viability
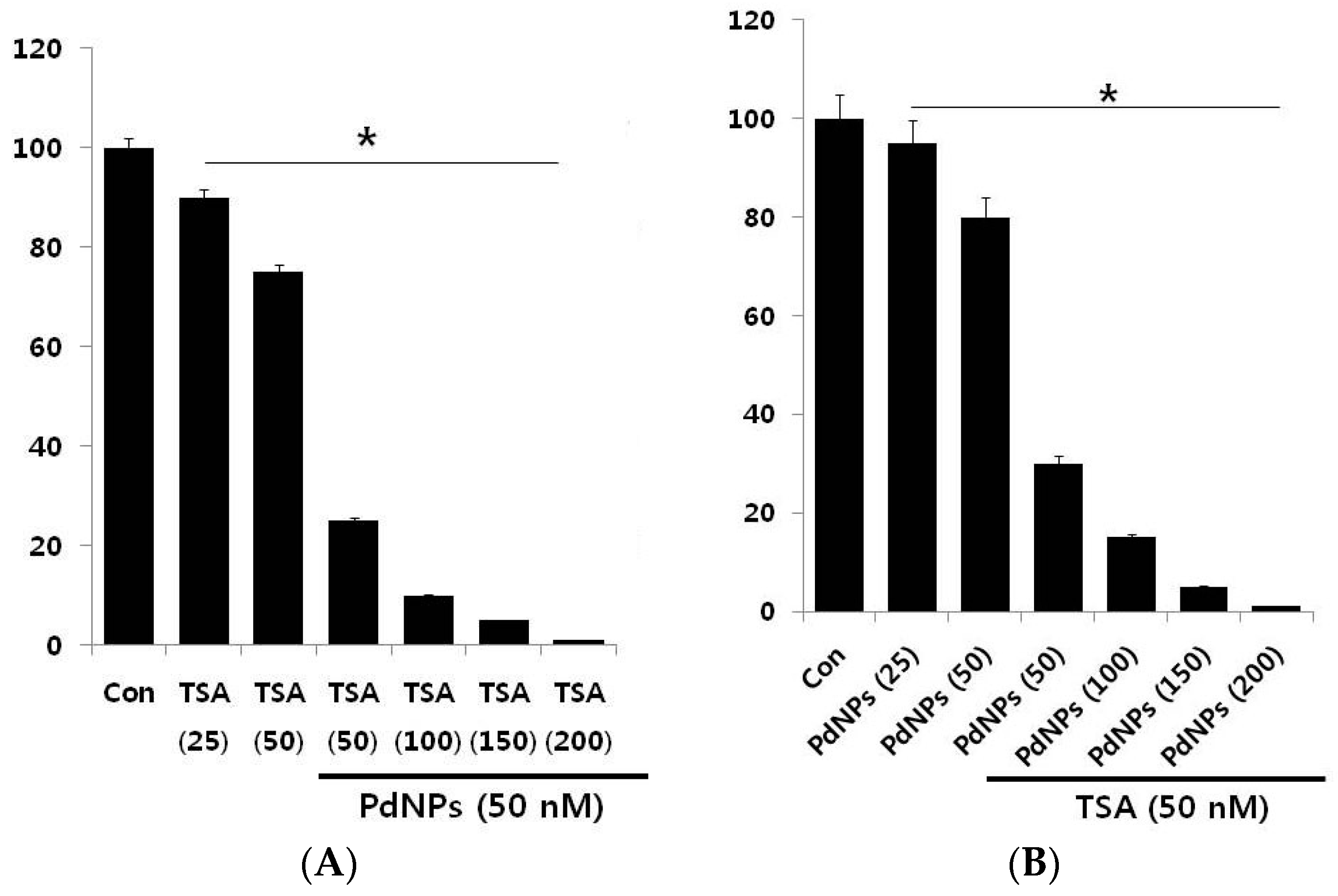
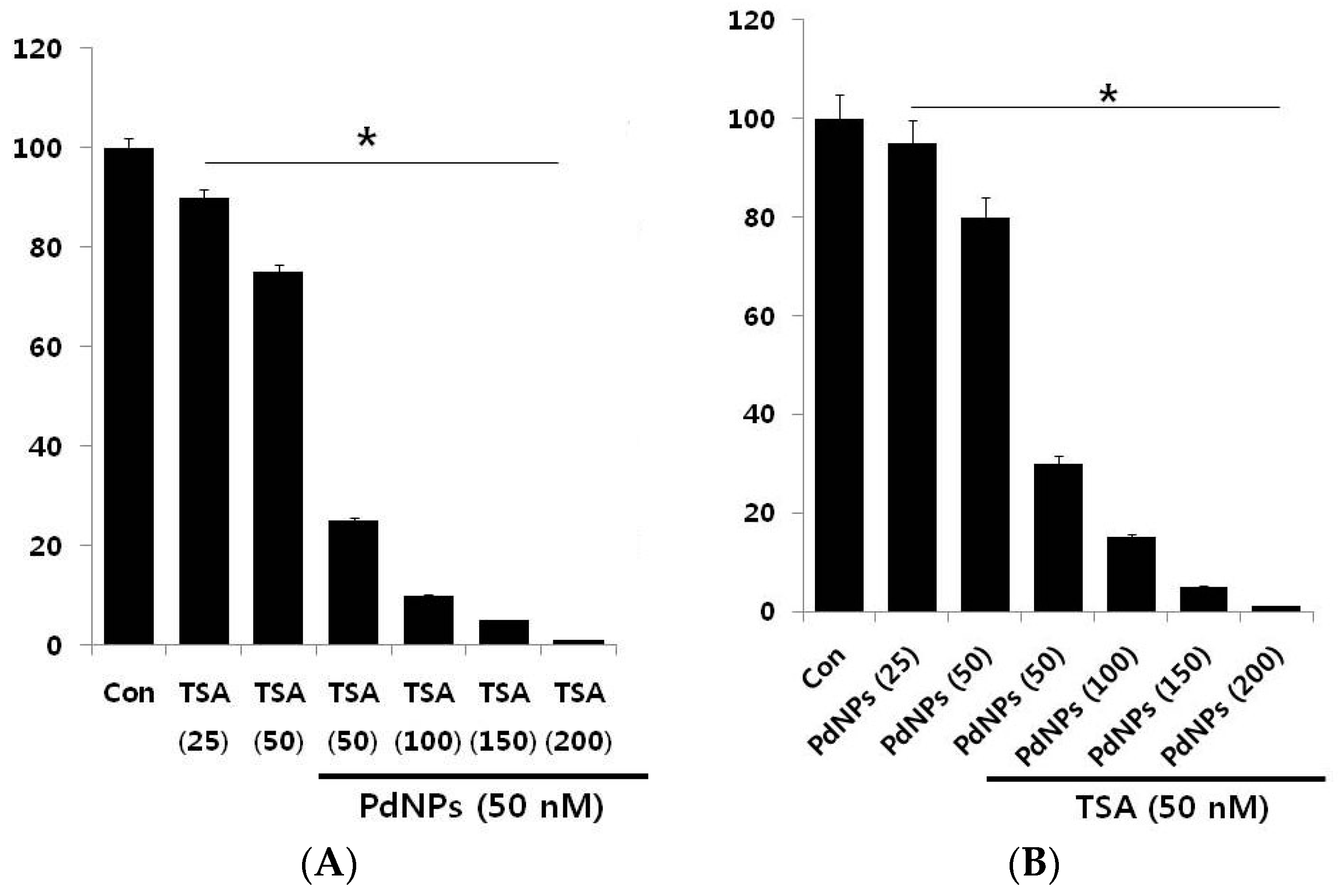
2.3. A Combination of TSA and PdNPs Dose-Dependently Inhibits HeLa Cell Viability
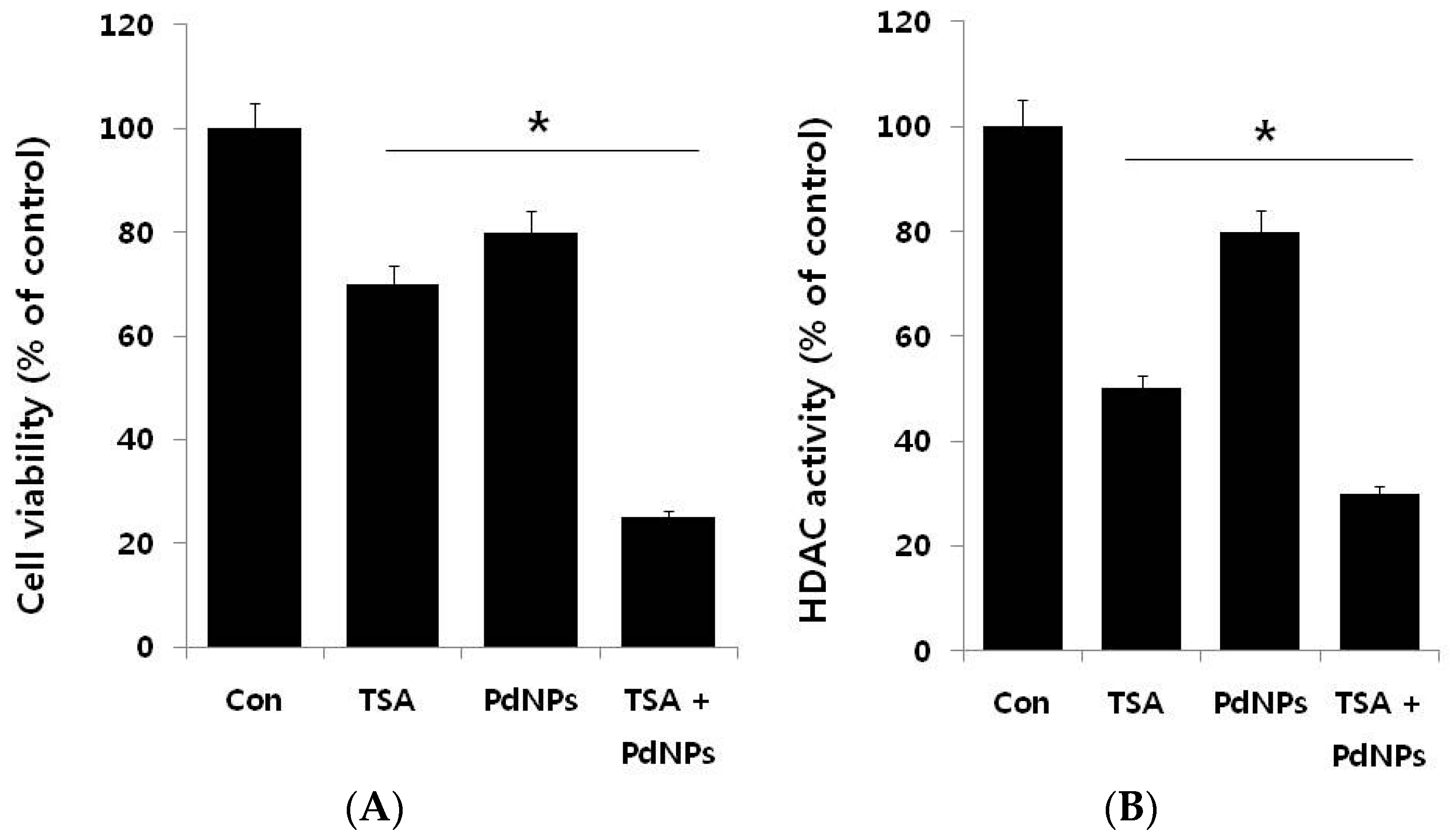
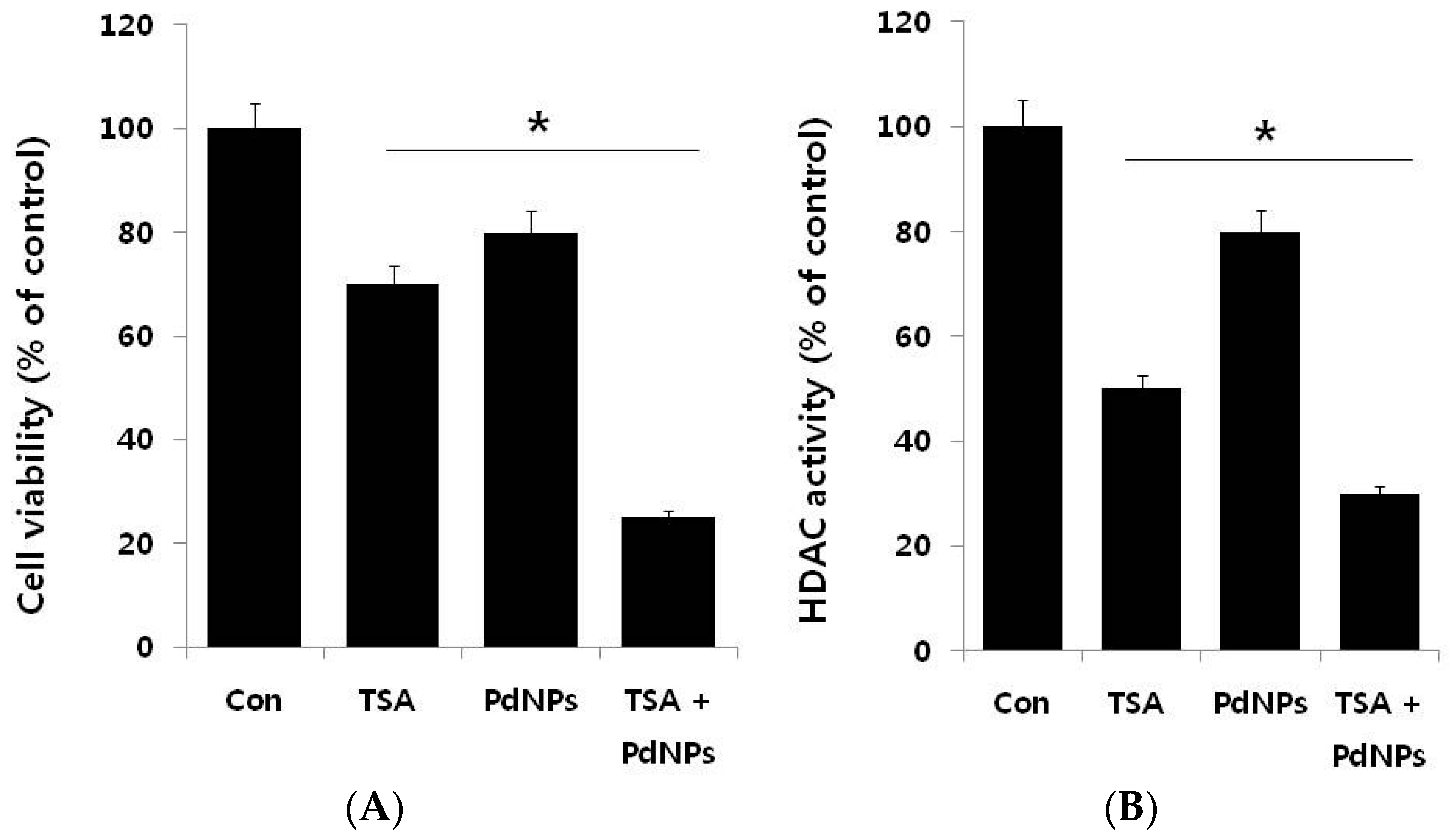
2.4. The Combination of TSA and PdNPs Inhibits Cell Viability and Histone Deacetylase (HDAC) Activity
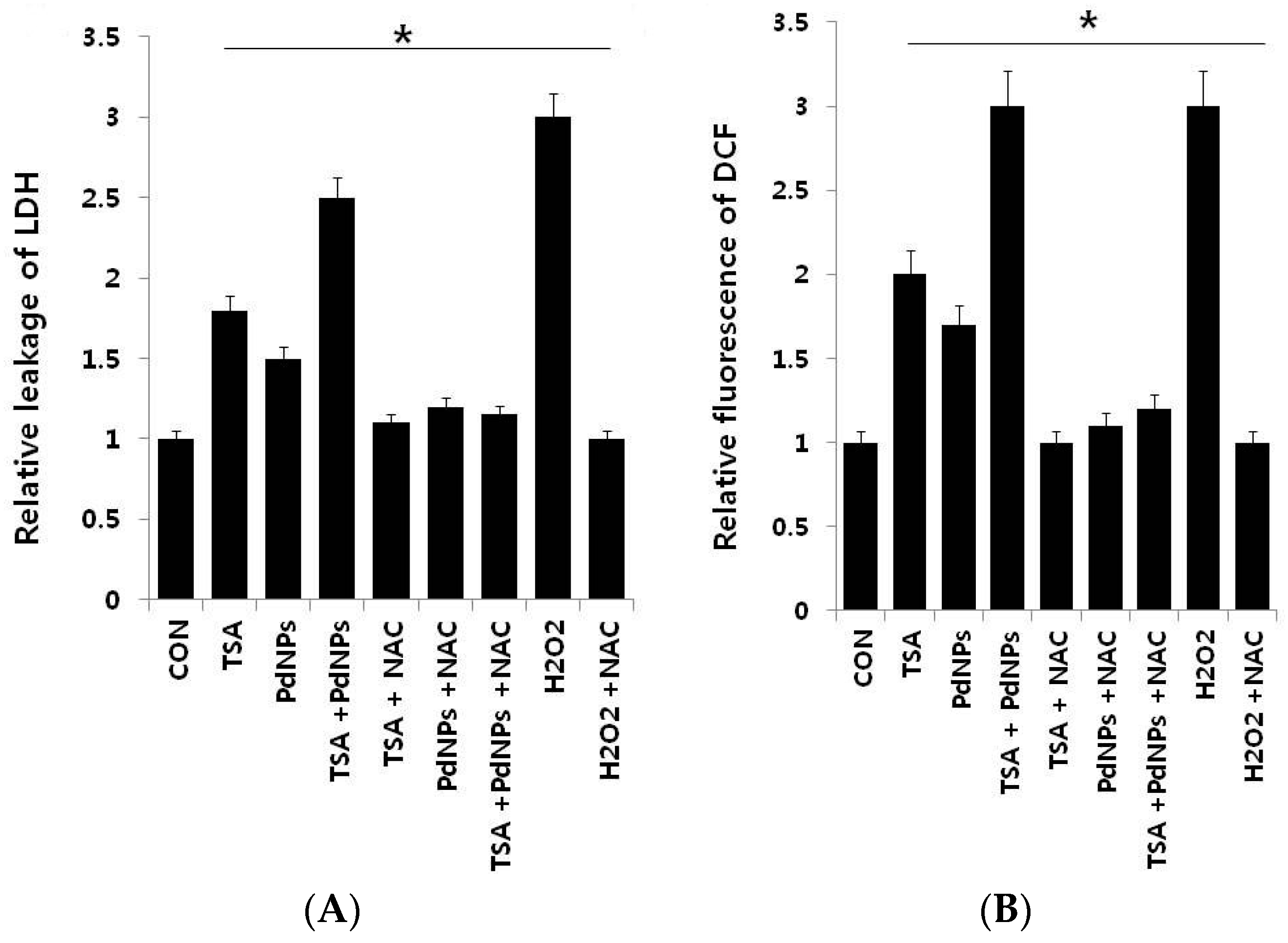
2.5. Combination of TSA and PdNPs Enhances Cytotoxicity
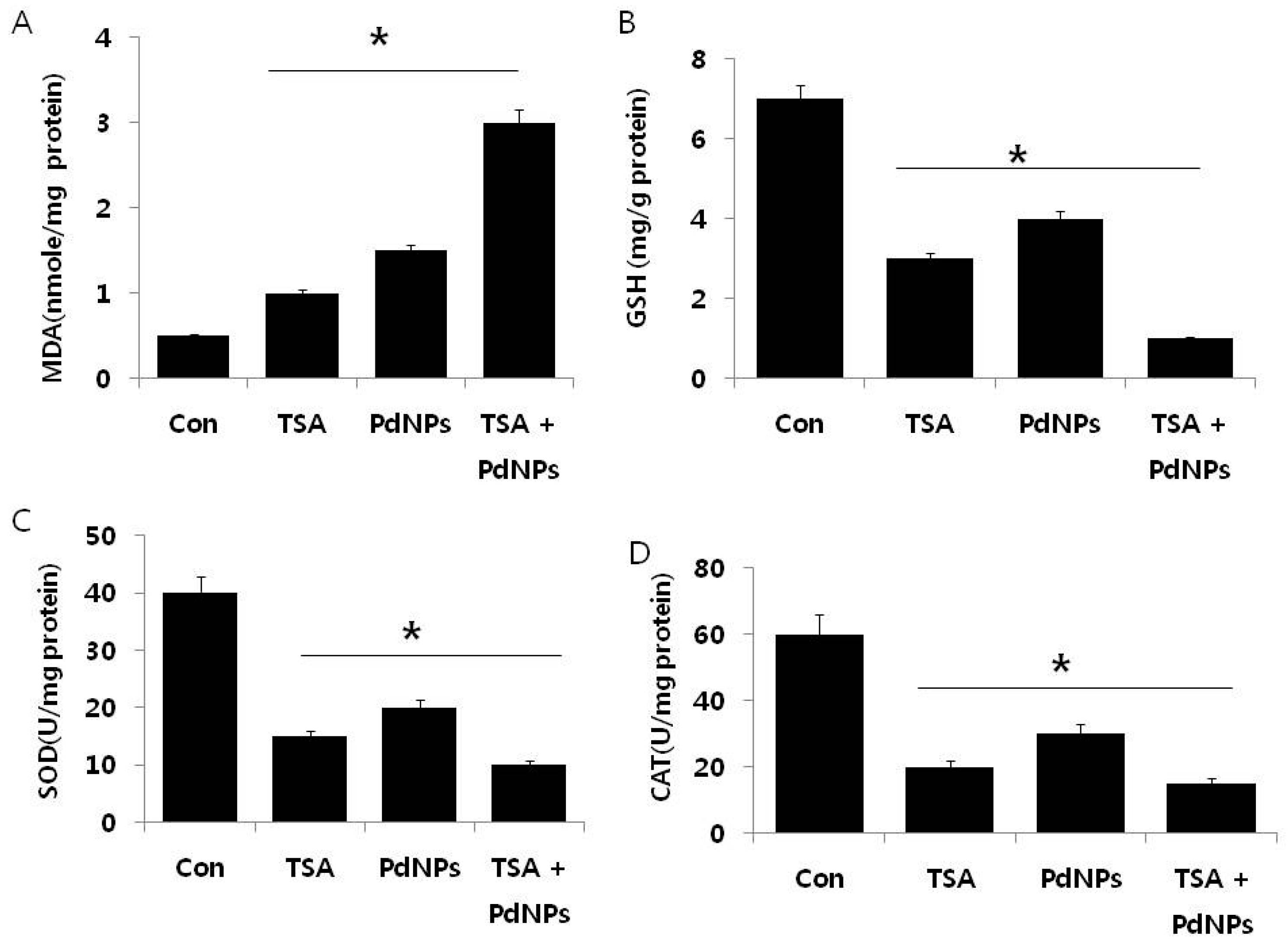
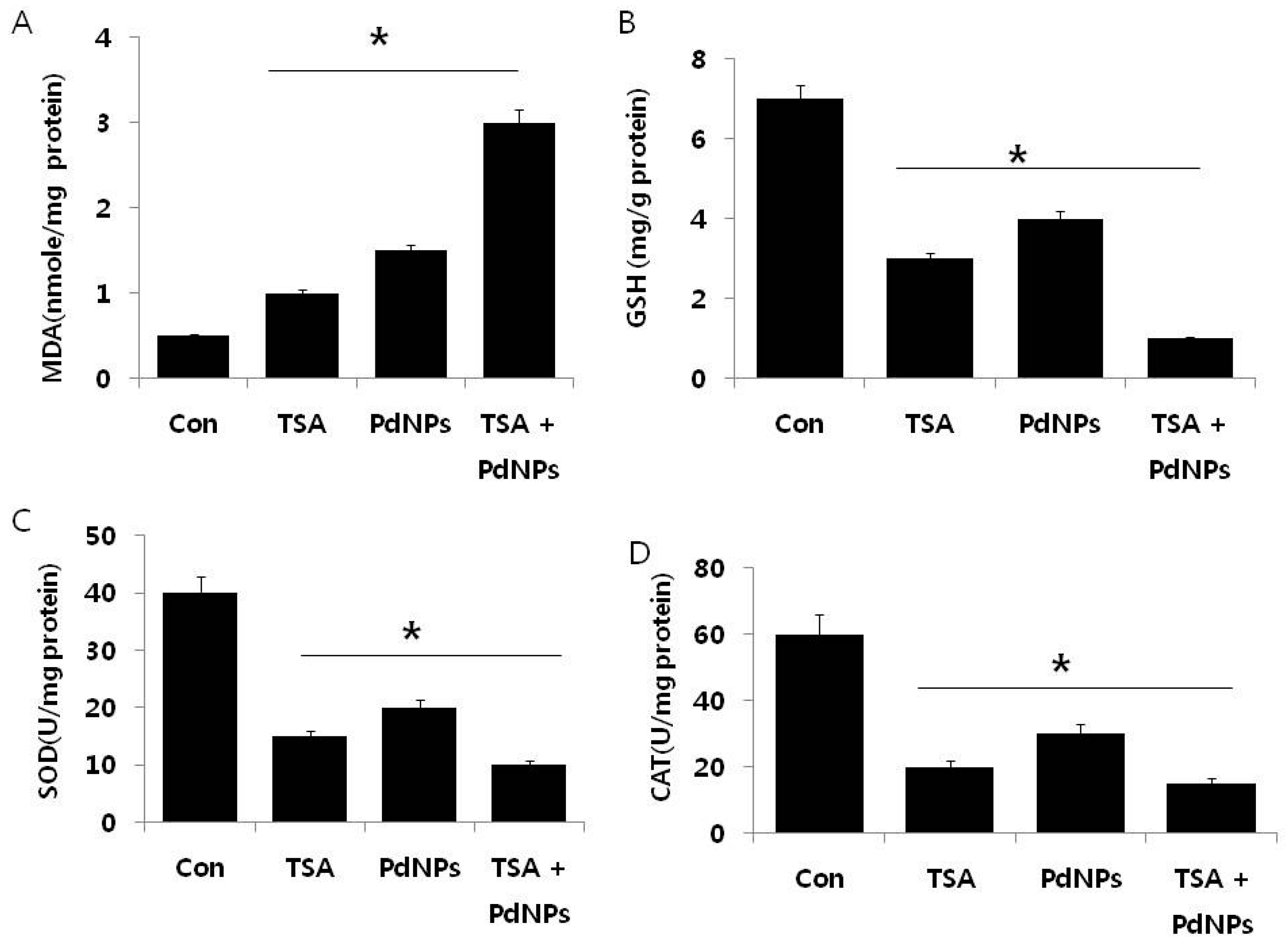
2.6. Effect of TSA and PdNPs on Oxidative Stress Markers
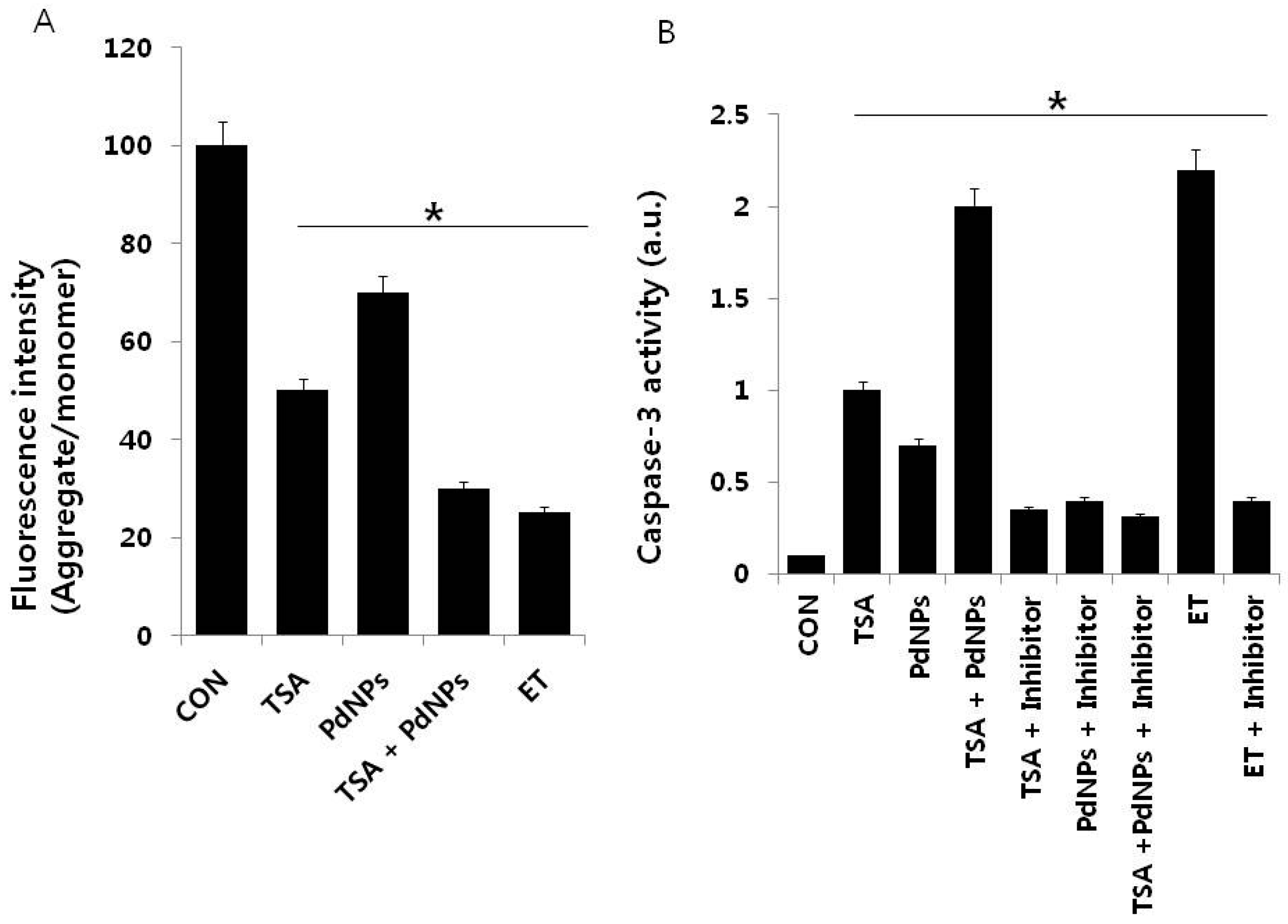
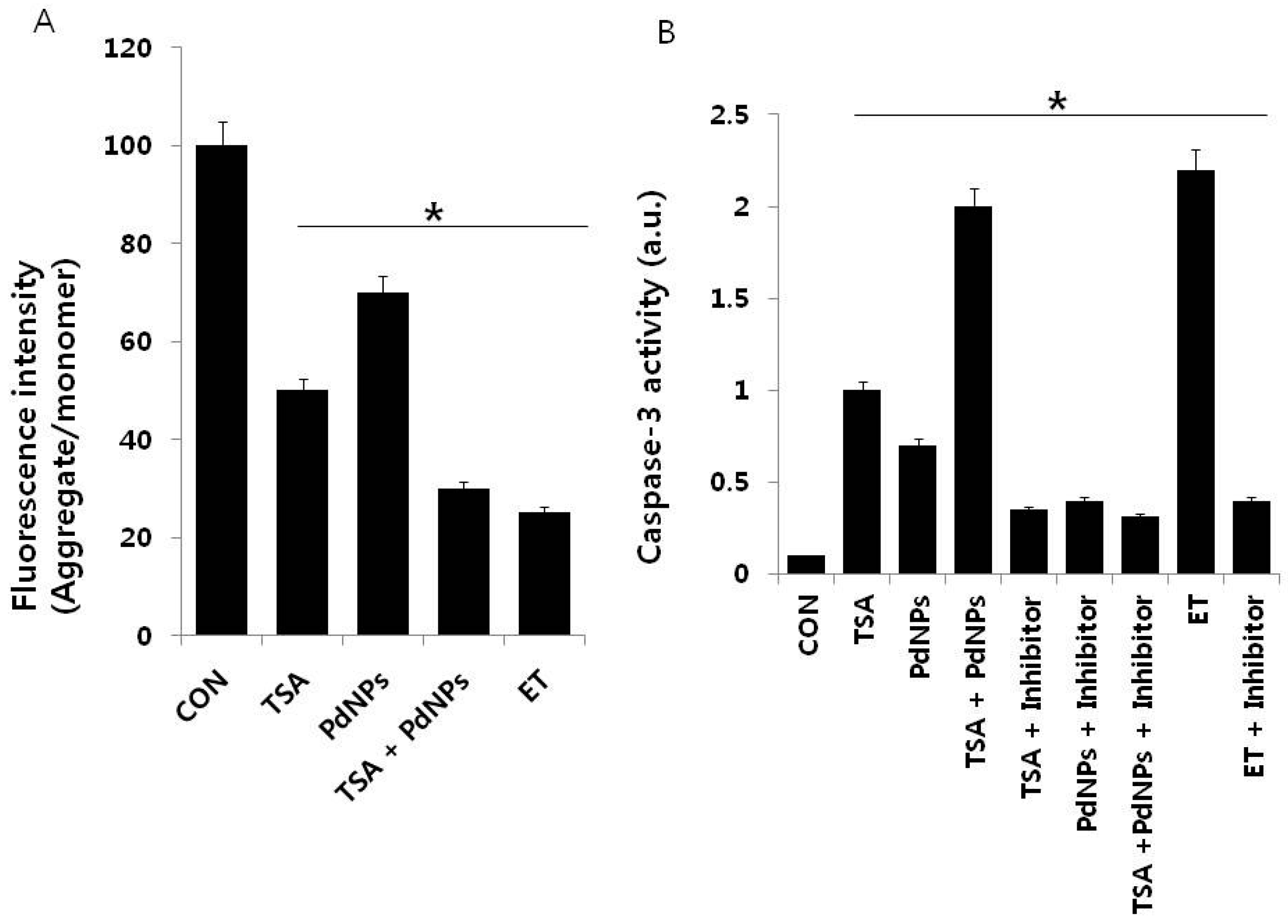
2.7. Combination of TSA and PdNPs Disrupts Membrane Potential (MMP) and Enhances Caspase-3 Activity
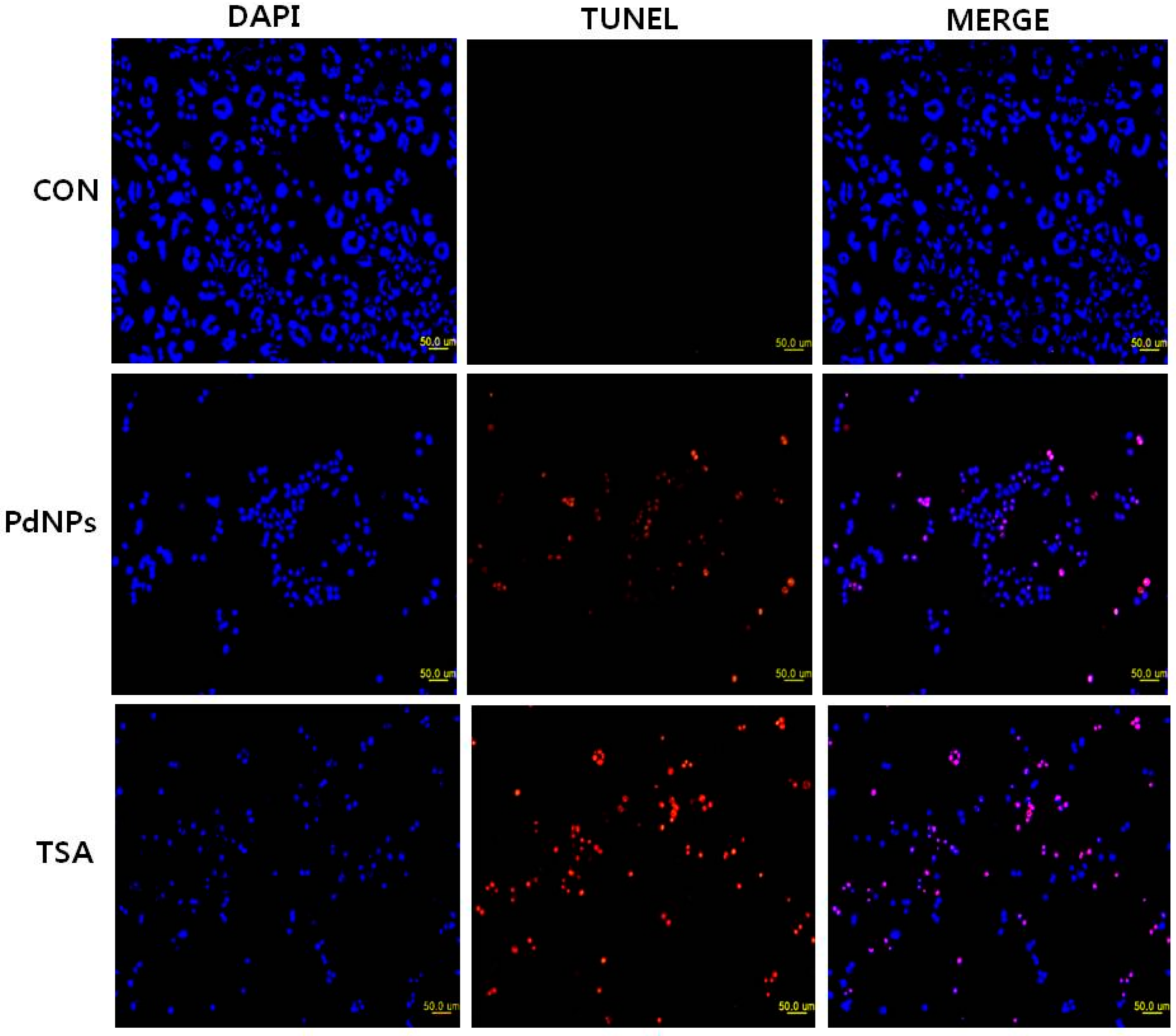
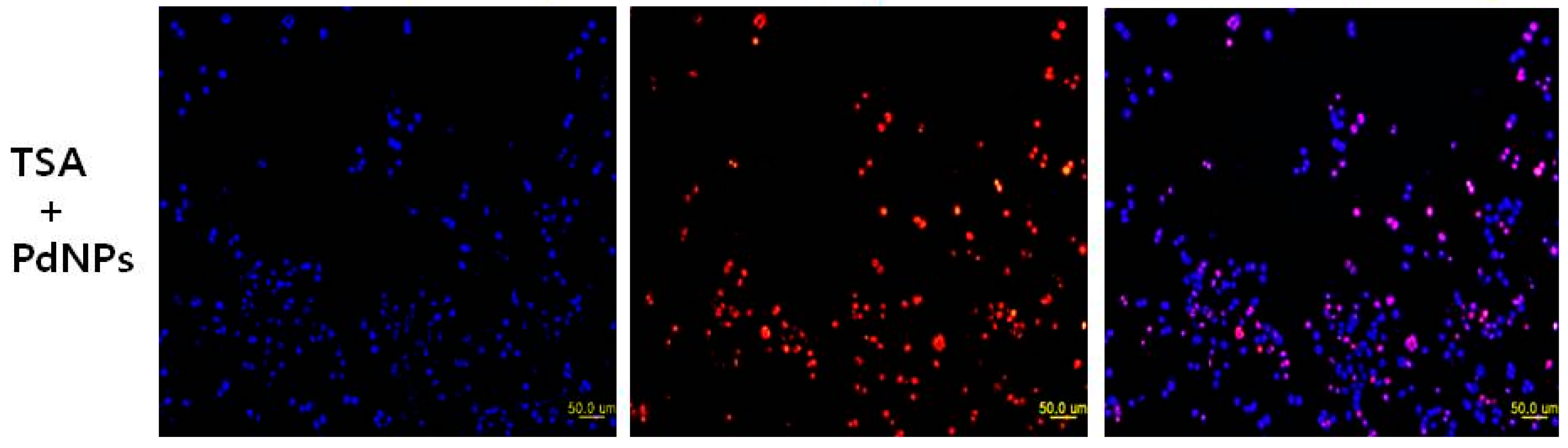
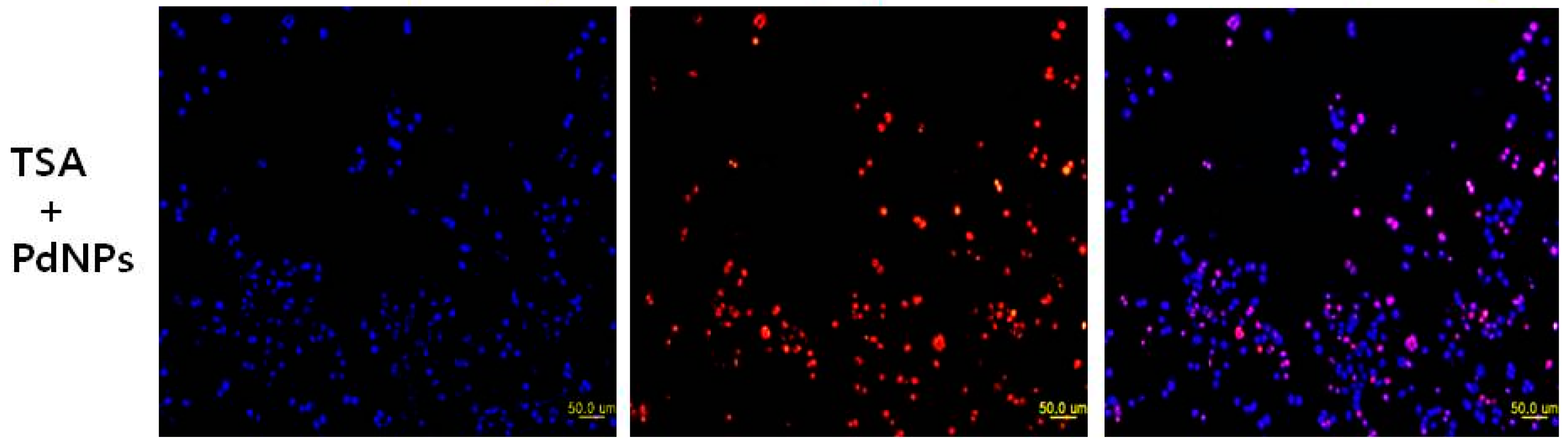
2.8. Combination of TSA and PdNPs Enhances Apoptosis
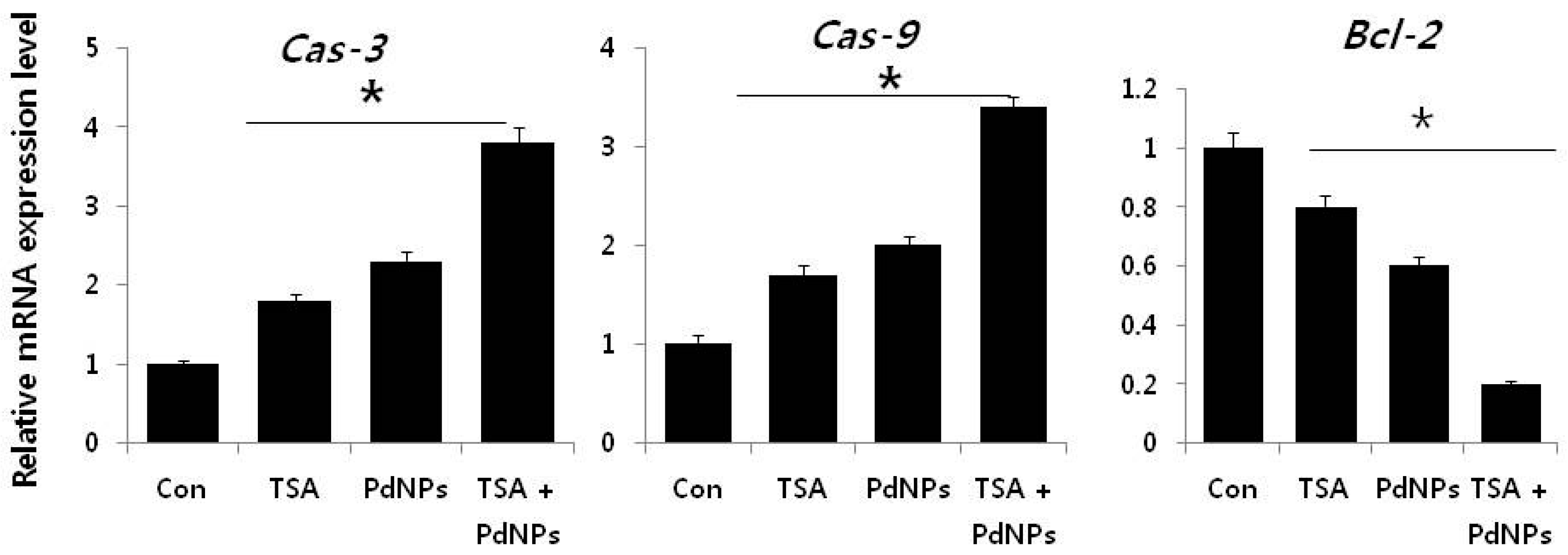
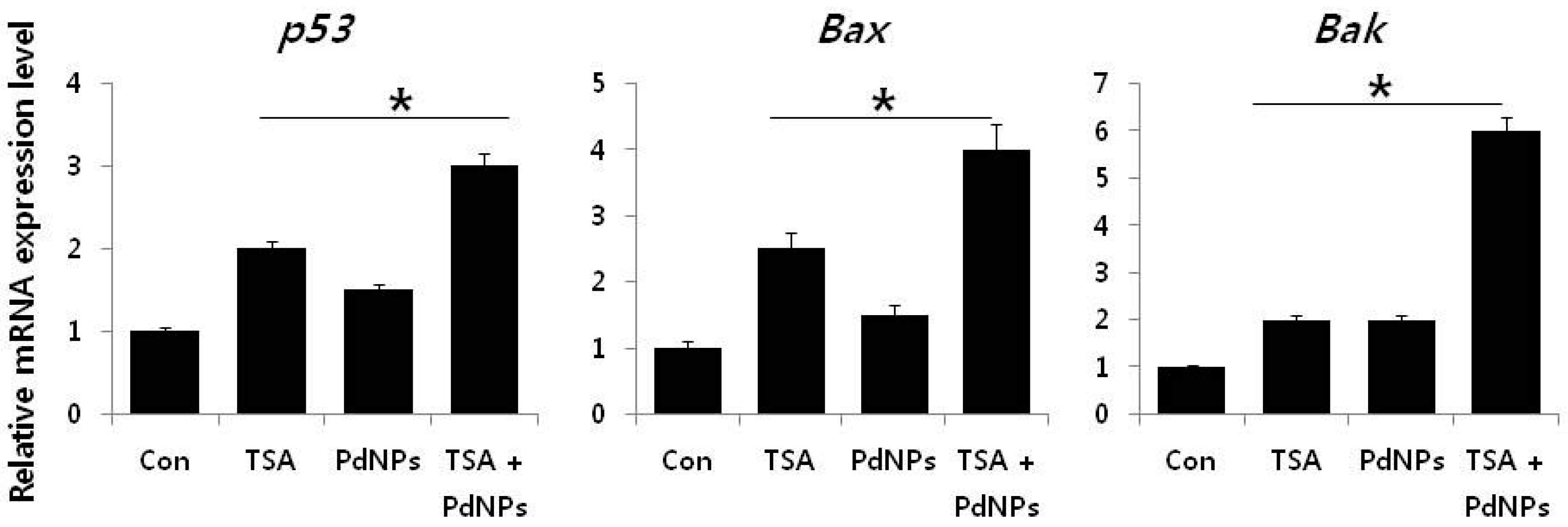
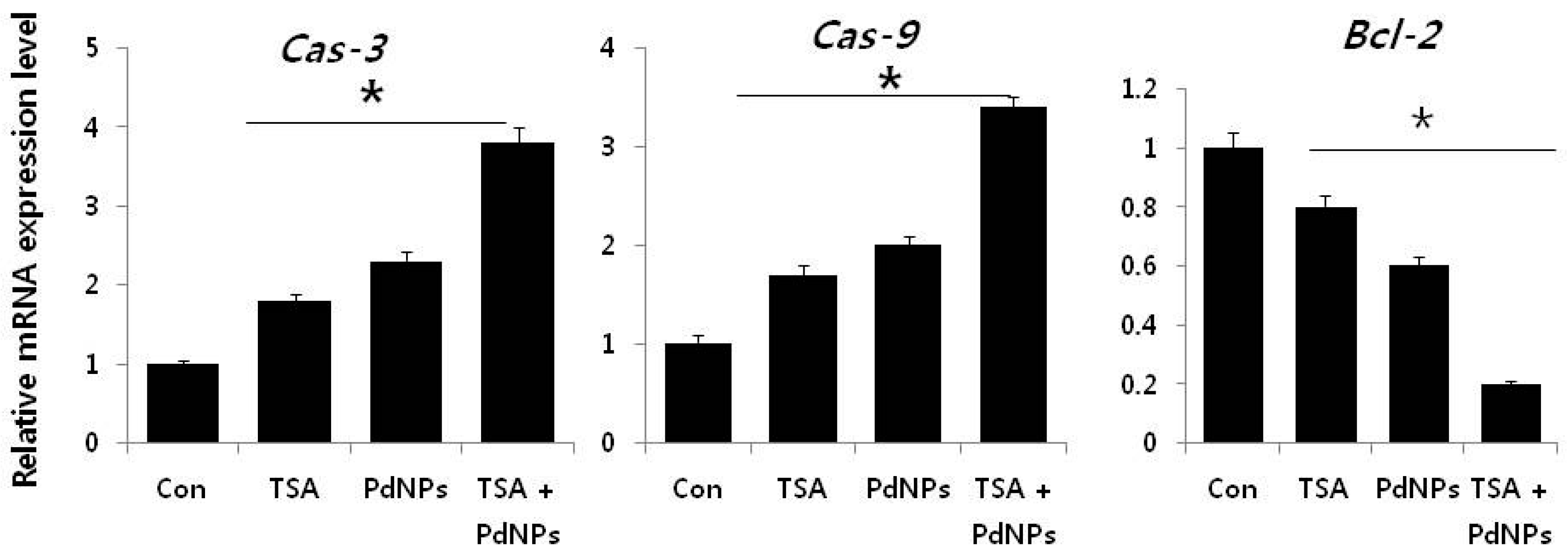
2.9. Combination of TSA and PdNPs Upregulates Apoptotic Genes
3. Materials and Methods
3.1. Materials
3.2. Synthesis of PdNPs
3.3. Characterization of PdNPs
3.4. Cell Culture
3.5. Cell Viability Assay
3.6. Histone Deacetylase Activity
3.7. Cytotoxicity Assay
3.8. Measurement of Oxidative Stress Markers
3.9. Measurement of Mitochondrial Membrane Potential
3.10. Measurement of Caspase-3 Activity and TUNEL Assay
3.11. Extraction and Amplification of mRNA
3.12. Statistical Analyses
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cervical Cancer Statistics. World Cancer Research Fund International GLOBOCAN Cancer Fact Sheets: Cervical Cancer—Iarc. Available online: http://www.globocan.iarc.fr (accessed on 6 July 2016).
- Chemotherapy for Cervical Cancer—American Cancer Society. Available online: http://www.cancer.org (accessed on 6 July 2016).
- Anton, M.; Horký, M.; Kuchtícková, S.; Vojtĕsek, B.; Bláha, O. Immunohistochemical detection of acetylation and phosphorylation of histone H3 in cervical smears. Ceska Gynekol. 2004, 69, 3–6. [Google Scholar] [PubMed]
- Dueñas-González, A.; Lizano, M.; Candelaria, M.; Cetina, L.; Arce, C.; Cervera, E. Epigenetics of cervical cancer. An overview and therapeutic perspectives. Mol. Cancer 2005, 25, 38. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Fields, C.R.; Su, N.; Pan, Y.X.; Robertson, K.D. Pharmacologic inhibition of epigenetic modifications, coupled with gene expression profiling, reveals novel targets of aberrant DNA methylation and histone deacetylation in lung cancer. Oncogene 2007, 26, 2621–2634. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Eller, M.S.; Elm, C.; Larocca, C.A.; Ryu, B.; Panova, I.P.; Dancy, B.M.; Bowers, E.M.; Meyers, D.; Lareau, L.; et al. Selective inhibition of p300 HAT blocks cell cycle progression, induces cellular senescence, and inhibits the DNA damage response in melanoma cells. J. Investig. Dermatol. 2013, 133, 2444–2452. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.H.; Allis, C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 1998, 20, 615–626. [Google Scholar] [CrossRef]
- Cress, W.D.; Seto, E. Histone deacetylases, transcriptional control, and cancer. J. Cell. Physiol. 2000, 184, 1–16. [Google Scholar] [CrossRef]
- Gray, S.G.; Ekström, T.J. The human histone deacetylase family. Exp. Cell Res. 2001, 262, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.A.; Kim, I.H.; Kim, H.J.; Chie, E.K.; Kim, J.S. HDAC inhibitor-mediated radiosensitization in human carcinoma cells: A general phenomenon? J. Radiat. Res. 2010, 51, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Koul, N.; Joseph, C.; Dixit, D.; Ghosh, S.; Sen, E. HDAC inhibitor, scriptaid, induces glioma cell apoptosis through JNK activation and inhibits telomerase activity. J. Cell. Mol. Med. 2010, 14, 2151–2161. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Mandal, M.; Mazumdar, A.; Kumar, R. Dynamic chromatin remodeling on the HER2 promoter in human breast cancer cells. FEBS Lett. 2001, 507, 88–94. [Google Scholar] [CrossRef]
- Ranganathan, P.; Rangnekar, V.M. Exploiting the TSA connections to overcome apoptosis-resistance. Cancer Biol. Ther. 2005, 4, 391–392. [Google Scholar] [CrossRef] [PubMed]
- Hajji, N.; Wallenborg, K.; Vlachos, P.; Nyman, U.; Hermanson, O.; Joseph, B. Combinatorial action of the HDAC inhibitor trichostatin A and etoposide induces caspase-mediated AIF-dependent apoptotic cell death in non-small cell lung carcinoma cells. Oncogene 2008, 27, 3134–3144. [Google Scholar] [CrossRef] [PubMed]
- Platta, C.S.; Greenblatt, D.Y.; Kunnimalaiyaan, M.; Chen, H. The HDAC inhibitor trichostatin A inhibits growth of small cell lung cancer cells. J. Surg. Res. 2007, 142, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Q.; Zhang, R.; Chao, C.; Zhang, J.F.; Zhang, Y.Q. Histone deacetylase inhibitor trichostatin A induced caspase-independent apoptosis in human gastric cancer cell. Chin. Med. J. 2007, 120, 2112–2118. [Google Scholar] [PubMed]
- Sonnemann, J.; Hüls, I.; Sigler, M.; Palani, C.D.; Hong, I.T.T.; Völker, U.; Kroemer, H.K.; Beck, J.F. Histone deacetylase inhibitors and aspirin interact synergistically to induce cell death in ovarian cancer cells. Oncol. Rep. 2008, 20, 219–224. [Google Scholar] [PubMed]
- Gurunathan, S.; Raman, J.; Abd Malek, S.N.; John, P.A.; Vikineswary, S. Green synthesis of silver nanoparticles using Ganoderma neo-japonicum Imazeki: A potential cytotoxic agent against breast cancer cells. Int. J. Nanomed. 2013, 8, 4399–4413. [Google Scholar]
- Gurunathan, S.; Han, J.W.; Park, J.H.; Kim, E.; Choi, Y.J.; Kwon, D.N.; Kim, J.H. Reduced graphene oxide-silver nanoparticle nanocomposite: A potential anticancer nanotherapy. Int. J. Nanomed. 2015, 10, 6257–6276. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, S.; Park, J.H.; Han, J.W.; Kim, J.H. Comparative assessment of the apoptotic potential of silver nanoparticles synthesized by Bacillus tequilensis and Calocybe indica in MDA-MB-231 human breast cancer cells: Targeting p53 for anticancer therapy. Int. J. Nanomed. 2015, 10, 4203–4222. [Google Scholar] [CrossRef] [PubMed]
- Rosi, N.L.; Mirkin, C.A. Nanostructures in biodiagnostics. Chem. Rev. 2005, 105, 1547–1562. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R.; El-Sayed, M.A. FTIR study of the mode of binding of the reactants on the Pd nanoparticle surface during the catalysis of the Suzuki reaction. J. Phys. Chem. B 2005, 109, 4357–4360. [Google Scholar] [CrossRef] [PubMed]
- Ariga, K.; Li, J.; Fei, J.; Ji, Q.; Hill, J.P. Nanoarchitectonics for dynamic functional materials from atomic-/molecular-level manipulation to macroscopic action. Adv. Mater. 2016, 28, 1251–1286. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, W.; Minami, K.; Shrestha, L.K.; Ji, Q.M.; Hill, J.P.; Ariga, K. Bioactive nanocarbon assemblies: Nanoarchitectonics and applications. Nanotoday 2014, 9, 378–394. [Google Scholar] [CrossRef]
- Elhusseiny, A.F.; Hassan, H.H. Antimicrobial and antitumor activity of platinum and palladium complexes of novel spherical aramides nanoparticles containing flexibilizing linkages: Structure-property relationship. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 103, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Tang, S.; Liu, P.; Fang, X.; Gong, J.; Zheng, N. Pd nanosheet-covered hollow mesoporous silica nanoparticles as a platform for the chemo-photothermal treatment of cancer cells. Small 2012, 8, 3816–3822. [Google Scholar] [CrossRef] [PubMed]
- Balbin, A.; Gaballo, F.; Ceballos-Torres, J.; Prashar, S.; Fajardo, M.; Kaluderović, G.N.; Gómez-Ruiz, S. Dual application of Pd nanoparticles supported on mesoporous silica SBA-15 and MSU-2: Supported catalysts for C-C coupling reactions and cytotoxic agents against human cancer cell lines. RSC Adv. 2014, 4, 54775–54787. [Google Scholar] [CrossRef]
- Boscolo, P.; Bellante, V.; Leopold, K.; Maier, M.; di Giampaolo, L.; Antonucci, A.; Iavicoli, I.; Tobia, L.; Paoletti, A.; Montalti, M.; et al. Effects of palladium nanoparticles on the cytokine release from peripheral blood mononuclear cells of non-atopic women. J. Biol. Regul. Homeost. Agents 2010, 24, 207–214. [Google Scholar] [PubMed]
- Fontana, L.; Leso, V.; Marinaccio, A.; Cenacchi, G.; Papa, V.; Leopold, K.; Schindl, R.; Bocca, B.; Alimonti, A.; Iavicoli, I. The effects of palladium nanoparticles on the renal function of female Wistar rats. Nanotoxicology 2015, 9, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, S.; Kim, E.; Han, J.W.; Park, J.H.; Kim, J.H. Green chemistry approach for synthesis of effective anticancer palladium nanoparticles. Molecules 2015, 20, 22476–22498. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhao, C.; Wang, L. Molecular-targeted agents combination therapy for cancer: Developments and potentials. Int. J. Cancer 2014, 134, 1257–1269. [Google Scholar] [CrossRef] [PubMed]
- Klement, G.; Baruchel, S.; Rak, J.; Man, S.; Clark, K.; Hicklin, D.J.; Bohlen, P.; Kerbel, R.S. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J. Clin. Investig. 2000, 105, R15–R24. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kang, J.H. Quercetin and trichostatin A cooperatively kill human leukemia cells. Pharmazie 2005, 60, 856–860. [Google Scholar] [PubMed]
- Jang, E.R.; Kim, Y.J.; Myung, S.C.; Kim, W.; Lee, C.S. Different effect of protein kinase B/Akt and extracellular signal-regulated kinase inhibition on trichostatin A-induced apoptosis in epithelial ovarian carcinoma cell lines. Mol. Cell. Biochem. 2011, 353, 1–11. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Franklin, R.A.; Abrams, S.L.; Chappell, W.H.; Wong, E.W.; Lehmann, B.D.; Terrian, D.M.; Basecke, J.; Stivala, F.; et al. Targeting the RAF/MEK/ERK, PI3K/AKT and p53 pathways in hematopoietic drug resistance. Adv. Enzym. Regul. 2007, 47, 64–103. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Graham, K.; Olson, M.F. The RAS signalling pathway as a target in cancer therapy. Recent Results Cancer Res. 2007, 172, 125–153. [Google Scholar] [PubMed]
- Adjei, A.A. RAS signaling pathway proteins as therapeutic targets. Curr. Pharm. Des. 2001, 7, 1581–1594. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Y.; Liu, Y.; Wu, G.S. The role of mitogen-activated protein kinase phosphatase-1 in oxidative damage-induced cell death. Cancer Res. 2006, 66, 4888–4894. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Koeffler, H.P.; Yokoyama, A. Blockade of mTOR signaling potentiates the ability of histone deacetylase inhibitor to induce growth arrest and differentiation of acute myelogenous leukemia cells. Leukemia 2008, 22, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Zhang, Q.; Li, Q.; Song, H. The biosynthesis of palladium nanoparticles by antioxidants in Gardenia jasminoides Ellis: Long lifetime nanocatalysts for p-nitrotoluene hydrogenation. Nanotechnology 2009, 20, 385601. [Google Scholar] [CrossRef] [PubMed]
- Sheny, D.S.; Philip, D.; Mathew, J. Rapid green synthesis of palladium nanoparticles using the dried leaf of Anacardium occidentale. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 91, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Khan, M.; Kuniyil, M.; Adil, S.F.; Al-Warthan, A.; Alkhathlan, H.Z.; Tremel, W.; Tahir, M.N.; Siddiqui, M.R. Biogenic synthesis of palladium nanoparticles using Pulicaria glutinosa extract and their catalytic activity towards the Suzuki coupling reaction. Dalton Trans. 2014, 43, 9026–9031. [Google Scholar] [CrossRef] [PubMed]
- Nadagouda, M.N.; Varma, R.S. Green synthesis of silver and palladium nanoparticles at room temperature using coffee and tea extract. Green Chem. 2008, 10, 859–862. [Google Scholar] [CrossRef]
- Vigushin, D.M.; Ali, S.; Pace, P.E.; Mirsaidi, N.; Ito, K.; Adcock, I.; Coombes, R.C. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin. Cancer Res. 2001, 7, 971–976. [Google Scholar] [PubMed]
- Wu, P.; Meng, L.; Wang, H.; Zhou, J.; Xu, G.; Wang, S.; Xi, L.; Chen, G.; Wang, B.; Zhu, T.; et al. Role of hTERT in apoptosis of cervical cancer induced by histone deacetylase inhibitor. Biochem. Biophys. Res. Commun. 2005, 335, 36–44. [Google Scholar] [CrossRef] [PubMed]
- You, B.R.; Park, W.H. Trichostatin A induces apoptotic cell death of HeLa cells in a Bcl-2 and oxidative stress-dependent manner. Int. J. Oncol. 2013, 42, 359–366. [Google Scholar] [PubMed]
- Chan, S.T.; Yang, N.C.; Huang, C.S.; Liao, J.W.; Yeh, S.L. Quercetin enhances the antitumor activity of trichostatin A through upregulation of p53 protein expression in vitro and in vivo. PLoS ONE 2013, 8, e54255. [Google Scholar] [CrossRef] [PubMed]
- Sack, M.; Alili, L.; Karaman, E.; Das, S.; Gupta, A.; Seal, S.; Brenneisen, P. Combination of conventional chemotherapeutics with redox-active cerium oxide nanoparticles—A novel aspect in cancer therapy. Mol. Cancer Ther. 2014, 13, 1740–1749. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.S.; Yoon, Y.H.; Choi, J.A.; Lee, S.J.; Koh, J.Y. Induction of autophagy and cell death by tamoxifen in ultured retinal pigment epithelial and photoreceptor cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5344–5353. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, S.; Han, J.W.; Dayem, A.A.; Eppakayala, V.; Kim, J.H. Oxidative stress-mediated antibacterial activity of graphene oxide and reduced graphene oxide in Pseudomonas aeruginosa. Int. J. Nanomed. 2012, 7, 5901–5914. [Google Scholar] [CrossRef] [PubMed]
- Niki, T.; Pekny, M.; Hellemans, K.; Bleser, P.D.; Berg, K.V.; Vaeyens, F.; Quartier, E.; Schuit, F.; Geerts, A. Class VI intermediate filament protein nestin is induced during activation of rat hepatic stellate cells. Hepatology 1999, 29, 520–527. [Google Scholar] [CrossRef] [PubMed]
- De la Vega, L.; Grishina, I.; Moreno, R.; Krüger, M.; Braun, T.; Schmitz, M.L. A redox-regulated SUMO/acetylation switch of HIPK2 controls the survival threshold to oxidative stress. Mol. Cell 2012, 46, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Rosato, R.R.; Almenara, J.A.; Dai, Y.; Grant, S. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol. Cancer Ther. 2003, 2, 1273–1284. [Google Scholar] [PubMed]
- Xu, Z.; Meng, X.; Cai, Y.; Koury, M.J.; Brandt, S.J. Recruitment of the SWI/SNF protein Brg1 by a multiprotein complex effects transcriptional repression in murine erythroid progenitors. Biochem. J. 2006, 399, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Ungerstedt, J.S.; Sowa, Y.; Xu, W.S.; Shao, Y.; Dokmanovic, M.; Perez, G.; Ngo, L.; Holmgren, A.; Jiang, X.; Marks, P.A. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.F.; Zhang, Y.J.; Tian, X.Q.; Chen, Y.X.; Fang, J.Y. Inhibition of mTOR signalling potentiates the effects of trichostatin A in human gastric cancer cell lines by promoting histone acetylation. Cell Biol. Int. 2014, 38, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Z. Increased oxidative stress as a selective anticancer therapy. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Boonstra, J.; Post, J.A. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 2004, 337, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S.; Okamoto, K.; Yodoi, J.; Hiai, H. Persistent oxidative stress in cancer. FEBS Lett. 1995, 358, 1–3. [Google Scholar] [CrossRef]
- Oberley, T.D.; Oberley, L.W. Antioxidant enzyme levels in cancer. Histol. Histopathol. 1997, 12, 525–535. [Google Scholar] [PubMed]
- Saydam, N.; Kirb, A.; Demir, O.; Hazan, E.; Oto, O.; Saydam, O.; Guner, G. Determination of glutathione, glutathione reductase, glutathione peroxidase and glutathione S-transferase levels in human lung cancer tissues. Cancer Lett. 1997, 119, 13–19. [Google Scholar] [CrossRef]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Fath, M.A.; Ahmad, I.M.; Smith, C.J.; Spence, J.; Spitz, D.R. Enhancement of carboplatin-mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. J. Am. Assoc. Cancer Res. 2011, 17, 6206–6217. [Google Scholar] [CrossRef] [PubMed]
- Brechbuhl, H.M.; Kachadourian, R.; Min, E.; Chan, D.; Day, B.J. Chrysin enhances doxorubicin-induced cytotoxicity in human lung epithelial cancer cell lines: The role of glutathione. Toxicol. Appl. Pharmacol. 2012, 258, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Feng, L.; Oldham, E.A.; Keating, M.J.; Plunkett, W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature 2000, 407, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Colburn, N.H.; Oberley, L.W. Depression of catalase gene expression after immortalization and transformation of mouse liver cells. Carcinogenesis 1993, 14, 1505–1510. [Google Scholar] [CrossRef] [PubMed]
- Zohre, S.; Kazem, N.K.; Abolfazl, A.; Mohammad, R.Y.; Aliakbar, M.; Effat, A.; Zahra, D.; Hassan, D.; Nosratollah, Z. Trichostatin A-induced apoptosis is mediated by Kruppel-like factor 4 in ovarian and lung cancer. Asian. Pac. J. Cancer Prev. 2014, 15, 6581–6586. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Zhivotovsky, B.; Orrenius, S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ. 2007, 14, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tan, J.; Zhuang, L.; Jiang, X.; Liu, E.T.; Yu, Q. Inhibitors of histonedeacetylases target the Rb-E2F1 pathway for apoptosis induction throughactivation of proapoptotic protein Bim. Proc. Natl. Acad. Sci. USA 2005, 102, 16090–16095. [Google Scholar] [CrossRef] [PubMed]
- Martinou, J.C.; Youle, R.J. Mitochondria in apoptosis: Bcl-2 family members and mtochondrial dynamics. Dev. Cell 2011, 21, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Heckman, C.A.; Boxer, L.M. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol. Cell. Biol. 2005, 25, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Han, M.H.; Park, C.; Kwon, T.K.; Kim, G.Y.; Kim, W.J.; Hong, S.H.; Yoo, Y.H.; Choi, Y.H. The histone deacetylase inhibitor trichostatin a Sensitizes human renal carcinoma cells to TRAIL-Induced Apoptosis through down-regulation of c-FLIPL. Biomol. Ther. 2015, 23, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Srinivasula, S.M.; Ashwell, J.D. IAPs: What’s in a name? Mol. Cell 2008, 30, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Fortson, W.S.; Kayarthodi, S.; Fujimura, Y.; Xu, H.L.; Matthews, R.; Grizzle, W.E.; Rao, V.N.; Bhat, G.K.; Reddy, E.S.P. Histone deacetylase inhibitors, valproic acid and trichostatin-A induce apoptosis and affect acetylation status of p53 in ERG-positive prostate cancer cells. Int. J. Oncol. 2011, 39, 111–119. [Google Scholar] [PubMed]
- Lee, E.; Furukubo, T.; Miyabe, T.; Yamauchi, A.; Kariya, K. Involvement of histone hyperacetylation in triggering DNA fragmentation of rat thymocytes undergoing apoptosis. FEBS Lett. 1996, 395, 183–187. [Google Scholar] [CrossRef]
- Yee, S.B.; Kim, M.S.; Baek, S.J.; Kim, G.C.; Yoo, K.S.; Yoo, Y.H.; Park, B.S. Trichostatin A induces apoptosis of p815 mastocytoma cells in histone acetylation- and mitochondria-dependent fashion. Int. J. Oncol. 2004, 25, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Tóth, K.F.; Knoch, T.A.; Wachsmuth, M.; Frank-Stöhr, M.; Stöhr, M.; Bacher, C.P.; Müller, G.; Rippe, K. Trichostatin A-induced histone acetylation causes decondensation of interphase chromatin. J. Cell Sci. 2004, 117, 4277–4287. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Gao, Z.; Marks, P.A.; Jiang, X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl. Acad. Sci. USA 2004, 101, 18030–18035. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Hong, J.; Zee, O.; Ohuchi, K. Possible mechanism of action of the histone deacetylase inhibitors for the induction of differentiation of HL-60 clone 15 cells into eosinophils. Br. J. Pharmacol. 2004, 142, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Kankaanranta, H.; Janka-Junttila, M.; Ilmarinen-Salo, P.; Ito, K.; Jalonen, U.; Ito, M.; Adcock, I.M.; Moilanen, E.; Zhang, X.Z. Histone deacetylase inhibitors induce apoptosis in humaneosinophils and neutrophils. J. Inflamm. 2010, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, P.; Donadelli, M.; Costanzo, C.; Moore, P.S.; Palmieri, M.; Scarpa, A. Trichostatin A enhances the response of chemotherapeutic agents in inhibiting pancreatic cancer cell proliferation. Virchows Arch. 2006, 448, 797–804. [Google Scholar]
- Reed, J. Bcl-2 family proteins. Oncogene 1998, 17, 3225–3236. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Luo, R.Z.; Peng, H.; Huang, M.; Nishmoto, A.; Hunt, K.K.; Helin, K.; Liao, W.S.L.; Yu, Y. E2F–HDAC complexes negatively regulate the tumor suppressor gene ARHI in breast cancer. Oncogene 2006, 25, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Van Den Heuvel, A.P.; Prabhu, V.V.; Zhang, S. Targeting tumor suppressor p53 for cancer therapy: Strategies, challenges and opportunities. Curr. Drug Targets 2014, 15, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hu, C.P.; Gu, Q.H.; Li, Y.P.; Song, M. Trichostatin A sensitizes cisplatin-resistant A549 cells to apoptosis by up-regulating death-associated protein kinase. Acta Pharmacol. Sin. 2010, 31, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Modulation of TRAIL-induced apoptosis by HDAC inhibitors. Curr. Cancer Drug Targets 2008, 8, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Rikiishi, H. Autophagic and apoptotic effects of HDAC inhibitors on cancercells. BioMed Res. Int. 2011, 4, 830260. [Google Scholar]
- Yang, X.M.; Shi, Z.L.; Zhang, N.; Ou, Z.; Fu, S.; Hu, X.H.; Shen, Z.Z. Suberoyl bis-hydroxamic acid enhances cytotoxicity induced by proteasome inhibitors in breast cancer cells. Cancer Cell Int. 2014, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Han, K.S.; Li, N.; Raven, P.A.; Fazli, L.; Ettinger, S.; Hong, S.J.; Gleave, M.E.; So, A.I. Targeting integrin-linked kinase suppresses invasion and metastasis through downregulation of epithelial-to-mesenchymal transition in renal cell carcinoma. Mol. Cancer Ther. 2015, 14, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Anh, T.D.; Ahn, M.Y.; Kim, S.A.; Yoon, J.H.; Ahn, S.G. The histone deacetylase inhibitor, Trichostatin A, induces G2/M phase arrest and apoptosis in YD-10B oral squamous carcinoma cells. Oncol. Rep. 2011, 27, 455–460. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial Number | Gene | Direction | Primers(5′–3′) |
|---|---|---|---|
| 1 | Bax | F | GAG AGG TCT TTT TCC GAG TGG |
| R | GGA GGA AGT CCA ATG TCC AG | ||
| 2 | P53 | F | AGG AAA TTT GCG TGT GGA GTA T |
| R | TCC GTC CCA GTA GAT TAC CAC T | ||
| 3 | Bak | F | CTC AGA GTT CCA GAC CAT GTT G |
| R | CAT GCT GGT AGA CGT GTA GGG | ||
| 4 | CAS3 | F | CAT ACT CCA CAG CAC CTG GTT A |
| R | ACT CAA ATT CTG TTG CCA CCT T | ||
| 5 | CAS9 | F | ACT TTC CCA GGT TTT GTT TCC T |
| R | GAA ATT AAA GCA ACC AGG CAT C | ||
| 6 | Bcl2 | F | CTG AGT ACC TGA ACC GGC A |
| R | GAG AAA TCA AAC AGA GGC CG |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.-F.; Yan, Q.; Shen, W.; Gurunathan, S. Trichostatin A Enhances the Apoptotic Potential of Palladium Nanoparticles in Human Cervical Cancer Cells. Int. J. Mol. Sci. 2016, 17, 1354. https://doi.org/10.3390/ijms17081354
Zhang X-F, Yan Q, Shen W, Gurunathan S. Trichostatin A Enhances the Apoptotic Potential of Palladium Nanoparticles in Human Cervical Cancer Cells. International Journal of Molecular Sciences. 2016; 17(8):1354. https://doi.org/10.3390/ijms17081354
Chicago/Turabian StyleZhang, Xi-Feng, Qi Yan, Wei Shen, and Sangiliyandi Gurunathan. 2016. "Trichostatin A Enhances the Apoptotic Potential of Palladium Nanoparticles in Human Cervical Cancer Cells" International Journal of Molecular Sciences 17, no. 8: 1354. https://doi.org/10.3390/ijms17081354
APA StyleZhang, X.-F., Yan, Q., Shen, W., & Gurunathan, S. (2016). Trichostatin A Enhances the Apoptotic Potential of Palladium Nanoparticles in Human Cervical Cancer Cells. International Journal of Molecular Sciences, 17(8), 1354. https://doi.org/10.3390/ijms17081354