
G Protein-Coupled Receptors in Cancer


Abstract
:1. Introduction
2. Biasing towards Specific G-Proteins in Cancer
3. G Protein-Coupled Receptor (GPCR) and Oncogenicity
4. Gep Oncogenes
5. Lysophosphatidic Acid (LPA) Receptors in Tumor Biology
6. Chemokine Receptors
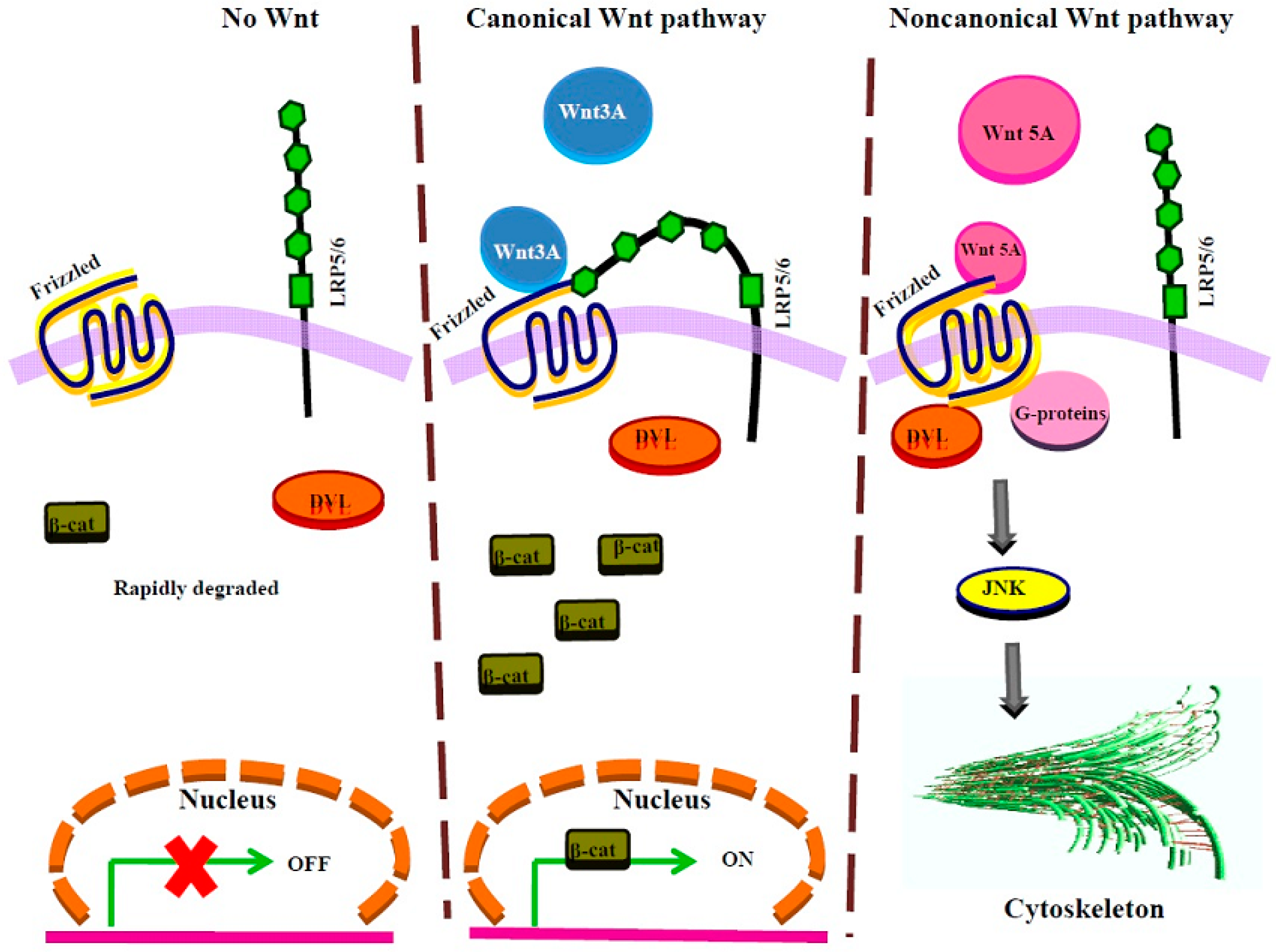
7. Wnt Signaling
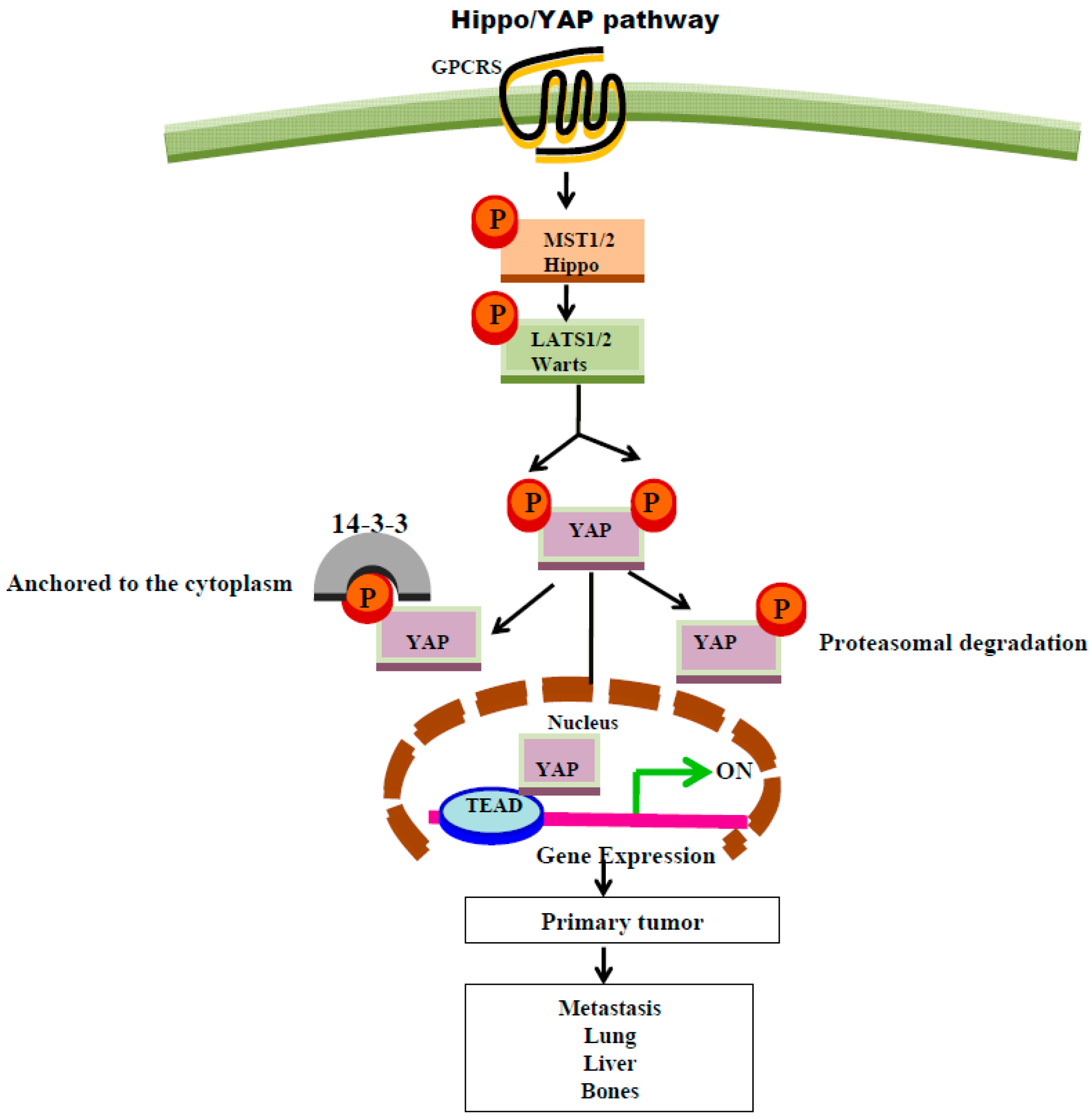
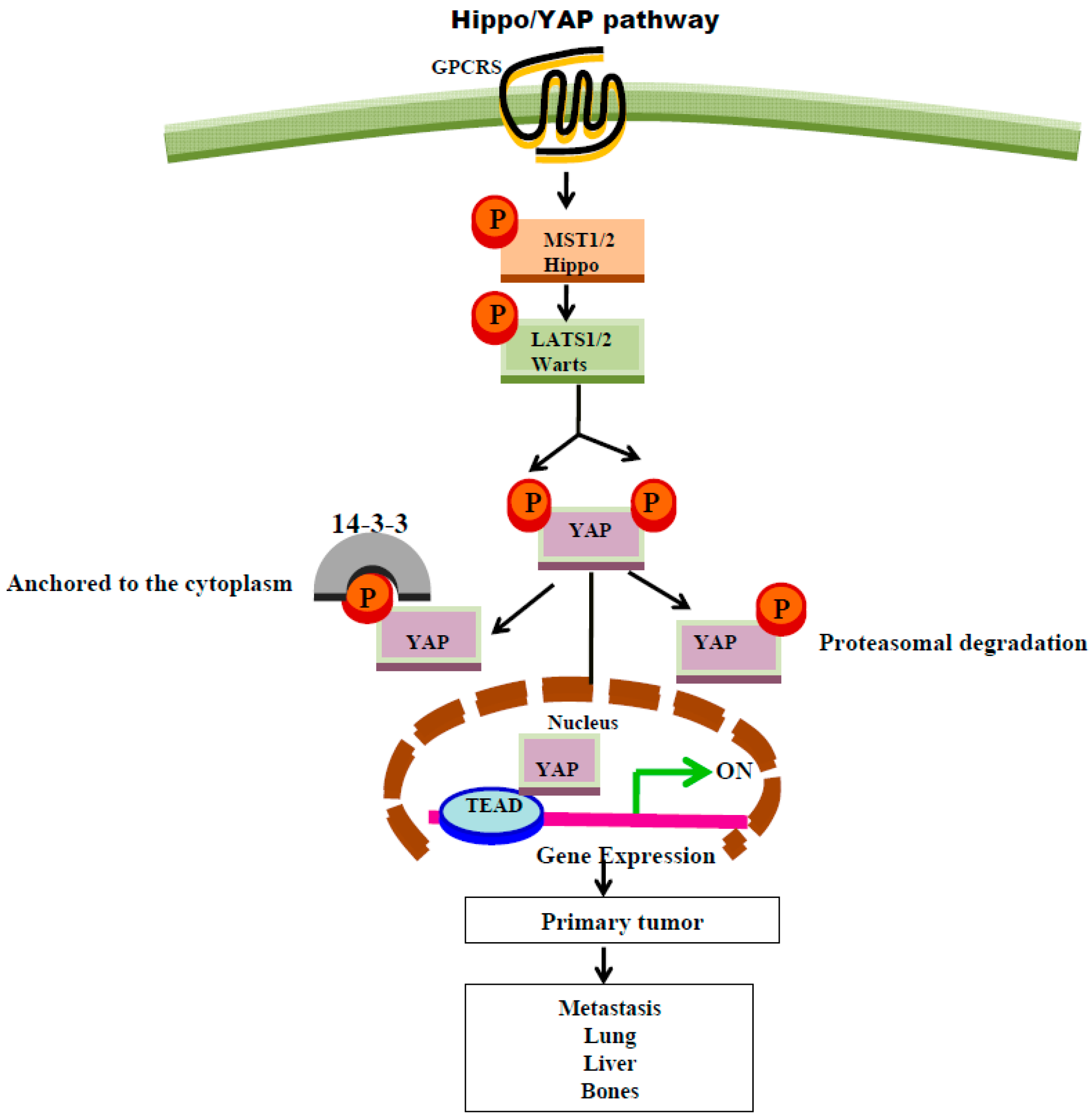
8. GPCR Regulation of Hippo Signaling Pathway
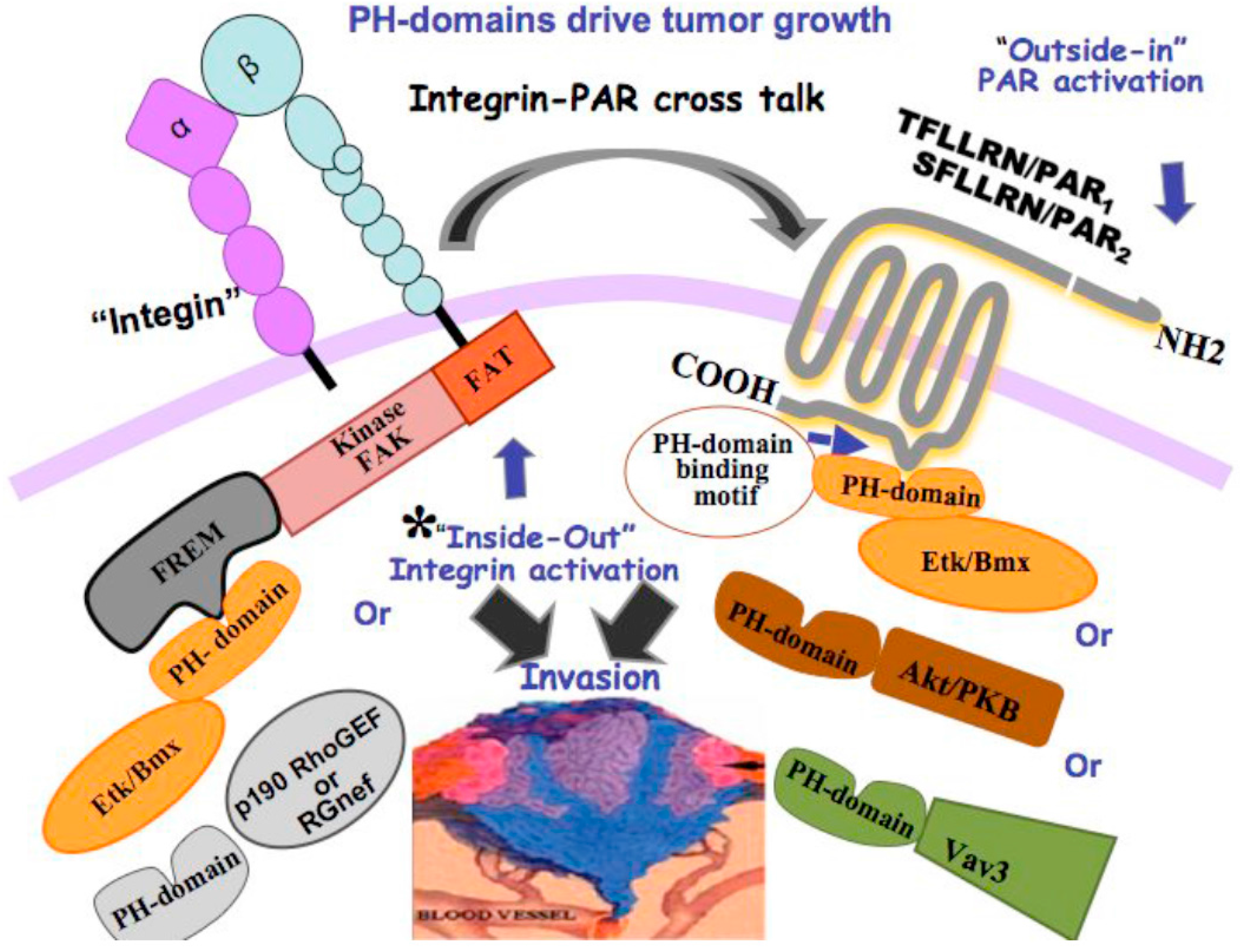
9. Protease-Activated Receptors, PARs, and Cancer
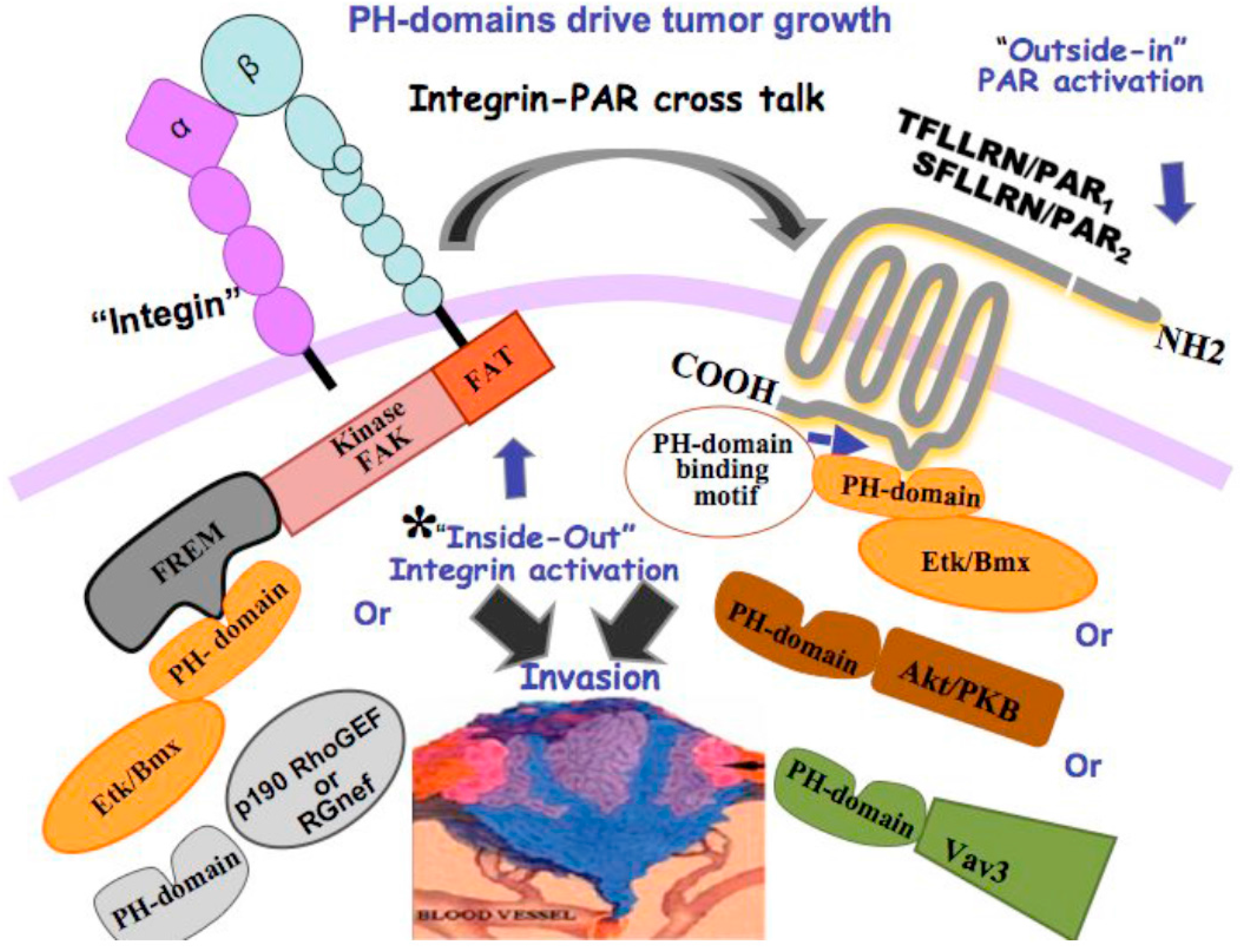
10. Novel Signaling of PARs Endowing Critical PH-Domain Binding Motifs
11. Concluding Remarks and Future Directions
Acknowledgments
Conflicts of Interest
References
- Wisler, J.W.; Xiao, K.; Thomsen, A.R.; Lefkowitz, R.J. Recent developments in biased agonism. Curr. Opin. Cell Biol. 2014, 27, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. The potential for selective pharmacological therapies through biased receptor signaling. BMC Pharmacol. Toxicol. 2012, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, M.D.; Mihara, K.; Polley, D.; Suen, J.Y.; Han, A.; Fairlie, D.P.; Ramachandran, R. Biased signalling and proteinase-activated receptors (PARs): Targeting inflammatory disease. Br. J. Pharmacol. 2014, 171, 1180–1194. [Google Scholar] [CrossRef] [PubMed]
- Feigin, M.E. Harnessing the genome for characterization of G-protein coupled receptors in cancer pathogenesis. FEBS J. 2013, 280, 4729–4738. [Google Scholar] [CrossRef] [PubMed]
- Whalen, E.J.; Rajagopal, S.; Lefkowitz, R.J. Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol. Med. 2011, 17, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Cianfrocca, R.; Tocci, P.; Semprucci, E.; Spinella, F.; di Castro, V.; Bagnato, A.; Rosanò, L. β-Arrestin 1 is required for endothelin-1-induced NF-κB activation in ovarian cancer cells. Life Sci. 2014, 118, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Rosanò, L.; Cianfrocca, R.; Tocci, P.; Spinella, F.; di Castro, V.; Spadaro, F.; Salvati, E.; Biroccio, A.M.; Natali, P.G.; Bagnato, A. β-arrestin-1 is a nuclear transcriptional regulator of endothelin-1-induced β-catenin signaling. Oncogene 2013, 32, 5066–5077. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drugdiscovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer. 2007, 7, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Bradley, L.; Ambdukar, I.; Gutkind, J.S. A mutant α subunit of G12 potentiates the eicosanoid pathway and is highly oncogenic in NIH 3T3 cells. Proc. Natl. Acad. Sci. USA 1993, 90, 6741–6745. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wu, D.; Simon, M.I. The transforming activity of activated G α 12. FEBS Lett. 1993, 330, 319–322. [Google Scholar] [CrossRef]
- Voyno-Yasenetskaya, T.A.; Pace, A.M.; Bourne, H.R. Mutant α subunits of G12 and G13 proteins induce neoplastic transformation of Rat-1 fibroblasts. Oncogene 1994, 9, 2559–2565. [Google Scholar] [PubMed]
- Dhanasekaran, N.; Prasad, M.V.; Wadsworth, S.J.; Dermott, J.M.; van Rossum, G. Protein kinase C-dependent and -independent activation of Na+/H+ exchanger by Gα12 class of G proteins. J. Biol. Chem. 1994, 269, 11802–11806. [Google Scholar] [PubMed]
- Hooley, R.; Yu, C.Y.; Symons, M.; Barber, D.L. Gα13 stimulates Na+-H+ exchange through distinct Cdc42-dependent and RhoA-dependent pathways. J. Biol. Chem. 1996, 271, 6152–6158. [Google Scholar] [PubMed]
- Offermanns, S.; Mancino, V.; Revel, J.P.; Simon, M.I. Vascular system defects and impaired cell chemokinesis as a result of Gα13 deficiency. Science 1997, 275, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Tigyi, G. Aiming drug discovery at lysophosphatidic acid targets. Br. J. Pharmacol. 2010, 161, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Contos, J.J.; Ishii, I.; Chun, J. Lysophosphatidic acid receptors. Mol. Pharmacol. 2000, 58, 1188–1196. [Google Scholar] [PubMed]
- Willier, S.; Butt, E.; Grunewald, T.G. Lysophosphatidic acid (LPA) signalling in cell migration and cancer invasion: A focused review and analysis of LPA receptor gene expression on the basis of more than 1700 cancer microarrays. Biol. Cell 2013, 105, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Kim, M.; Choi, Y.; Lee, C.K.; Ka, H. Analysis of lysophosphatidic acid (LPA) receptor and LPA-induced endometrial prostaglandin-endoperoxide synthase 2 expression in the porcine uterus. Endocrinology 2008, 149, 6166–6167. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, X.; Chen, W.; Liu, J.; Wang, X. The lysophosphatidic acid (LPA) receptors their expression and significance in epithelial ovarian neoplasms. Gynecol. Oncol. 2007, 104, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Rosanò, L.; Spinella, F.; Bagnato, A. Endothelin 1 in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer. 2013, 13, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular signatures of G-protein-coupled receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Bodor, E.T.; Offermanns, S. Nicotinic acid: An old drug with a promising future. Br. J. Pharmacol. 2008, 153, S68–S75. [Google Scholar] [CrossRef] [PubMed]
- Soto, A.G.; Smith, T.H.; Chen, B.; Bhattacharya, S.; Cordova, I.C.; Kenakin, T.; Vaidehi, N.; Trejo, J. N-linked glycosylation of protease-activated receptor-1 at extracellular loop 2 regulates G-protein signaling bias. Proc. Natl. Acad. Sci. USA 2015, 112, E3600–E3608. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. Functional selectivity and biased receptor signaling. J. Pharmacol. Exp. Ther. 2011, 336, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Gesty-Palmer, D.; Chen, M.; Reiter, E.; Ahn, S.; Nelson, C.D.; Wang, S.; Eckhardt, A.E.; Cowan, C.L.; Spurney, R.F.; Luttrell, L.M.; et al. Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J. Biol. Chem. 2006, 281, 10856–10864. [Google Scholar] [CrossRef] [PubMed]
- Bryja, V.; Gradl, D.; Schambony, A.; Arenas, E.; Schulte, G. Β-arrestin is a necessary component of Wnt/β-catenin signaling in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 6690–6695. [Google Scholar] [CrossRef] [PubMed]
- Rosanò, L.; Cianfrocca, R.; Masi, S.; Spinella, F.; di Castro, V.; Biroccio, A.; Salvati, E.; Nicotra, M.R.; Natali, P.G.; Bagnato, A. Β-arrestin links endothelin A receptor to β-catenin signaling to induce ovarian cancer cell invasion and metastasis. Proc. Natl. Acad. Sci. USA 2009, 106, 2806–2811. [Google Scholar] [CrossRef] [PubMed]
- Young, D.; Waitches, G.; Birchmeier, C.; Fasano, O.; Wigler, M. Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell 1986, 45, 711–719. [Google Scholar] [CrossRef]
- Davenport, A.P.; Alexander, S.P.; Sharman, J.L.; Pawson, A.J.; Benson, H.E.; Monaghan, A.E.; Liew, W.C.; Mpamhanga, C.P.; Bonner, T.I.; Neubig, R.R.; et al. International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: Recommendations for new pairings with cognate ligands. Pharmacol. Rev. 2013, 65, 967–986. [Google Scholar] [CrossRef] [PubMed]
- Zohn, I.E.; Klinger, M.; Karp, X.; Kirk, H.; Symons, M.; Chrzanowska-Wodnicka, M.; Der, C.J.; Kay, R.J. G2A is an oncogenic G protein-coupled receptor. Oncogene 2000, 19, 3866–3877. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Fluckiger, A.C.; Nisitani, S.; Wahl, M.I.; Le, L.Q.; Hunter, C.A.; Fernal, A.A.; Le Beau, M.M.; Witte, O.N. A DNA damage and stress inducible G protein-coupled receptor blocks cells in G2/M. Proc. Natl. Acad. Sci. USA 1998, 95, 12334–12339. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.B.; Mahon, G.M.; Klinger, M.B.; Kay, R.J.; Symons, M.; Der, C.J.; Whitehead, I.P. The thrombin receptor, PAR-1, causes transformation by activation of Rho-mediated signaling pathways. Oncogene 2001, 20, 1953–1963. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, I.; Kirk, H.; Kay, R. Expression cloning of oncogenes by retroviral transfer of cDNA libraries. Mol. Cell. Biol. 1995, 15, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Even-Ram, S.; Uziely, B.; Cohen, P.; Grisaru-Granovsky, S.; Maoz, M.; Ginzburg, Y.; Reich, R.; Vlodavsky, I.; Bar-Shavit, R. Thrombin receptor overexpression in malignant and physiological invasion processes. Nat. Med. 1998, 4, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Voyno-Yasenetskaya, T.; Gutkind, J.S. Potent transforming activity of the G13α subunit defines a novel family of oncogenes. Biochem. Biophys. Res. Commun. 1994, 201, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Kawanabe, Y.; Okamoto, Y.; Nozaki, K.; Hashimoto, N.; Miwa, S.; Masaki, T. Molecular mechanism for endothelin-1-induced stress-fiber formation: Analysis of G proteins using a mutant endothelin(A) receptor. Mol. Pharmacol. 2002, 61, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.; Casey, P.J.; Meigs, E. Biologic functions of the G12 subfamily of heterotrimeric G proteins: Growth, migration, and metastasis. Biochemistry 2007, 46, 6677–6687. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Yang, J.W.; Cho, I.J.; Kim, W.D.; Cho, M.K.; Lee, C.H.; Kim, S.G. The Gep oncogenes, Gα and Gα, upregulate the transforming growth factor-β1 gene. Oncogene 2009, 28, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.I.; Strathmann, M.P.; Gautam, N. Diversity of G proteins in signal transduction. Science 1991, 252, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Hepler, J.R.; Gilman, A.G. G proteins. Trends Biochem. Sci. 1992, 17, 383–395. [Google Scholar] [CrossRef]
- Conklin, B.R.; Bourne, H.R. Structural elements of G α subunits that interact with Gβγ, receptors, and effectors. Cell 1993, 73, 631–641. [Google Scholar] [CrossRef]
- Chan, A.M.; Fleming, T.P.; McGovern, E.S.; Chedid, M.; Miki, T.; Aaronson, S.A. Expression cDNA cloning of a transforming gene encoding the wild-type Gα12 gene product. Mol. Cell. Biol. 1993, 13, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, N.; Dermott, J.M. Signaling by the G12 class of G proteins. Cell Signal. 1996, 8, 235–245. [Google Scholar] [CrossRef]
- Goldsmith, Z.G.; Dhanasekaran, D.N. G protein regulation of MAPK networks. Oncogene 2007, 26, 3122–3142. [Google Scholar] [CrossRef] [PubMed]
- Kuner, R.; Swiercz, J.M.; Zywietz, A.; Tappe, A.; Offermanns, S. Characterization of the expression of PDZ-RhoGEF, LARG and G(α)12/G(α)13 proteins in the murine nervous system. Eur. J. Neurosci. 2002, 16, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Kurose, H. Gα12 and Gα13 as key regulatory mediator in signal transduction. Life Sci. 2003, 74, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Onohara, N.; Maruyama, Y.; Tanabe, S.; Kobayashi, H.; Fukutomi, M.; Nagamatsu, Y.; Nishihara, N.; Inoue, R.; Sumimoto, H.; et al. Gα12/13-mediated production of reactive oxygen species is critical for angiotensin receptor-induced NFAT activation in cardiac fibroblasts. J. Biol. Chem. 2005, 280, 23041–23047. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.N.; Shore, S.K.; Dhanasekaran, N. Neoplastic transformation by the Gep oncogene, Gα12, involves signaling by STAT3. Oncogene 2006, 25, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Ishii, S.; Shimizu, T. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J. Biol. Chem. 2003, 278, 25600–25606. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Rivera, R.; Gardell, S.; Dubin, A.E.; Chun, J. GPR92 as a new G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J. Biol. Chem. 2006, 281, 23589–23597. [Google Scholar] [CrossRef] [PubMed]
- Tabata, K.; Baba, K.; Shiraishi, A.; Ito, M.; Fujita, N. The orphan GPCR GPR87 was deorphanized and shown to be a lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2007, 363, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Mills, G.B.; Moolenaar, W.H. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhong, W.W.; Srivastava, N.; Slavin, A.; Yang, J.; Hoey, T.; An, S. G protein-coupled lysophosphatidic acid receptors stimulate proliferation of colon cancer cells through the β-catenin pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 6027–6032. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Towers, L.N.; O’Connor, K.L. LPA2 (EDG4) mediates Rho-dependent chemotaxis with lower efficacy than LPA1 (EDG2) in breast carcinoma cells. Am. J. Physiol. Cell Physiol. 2007, 292, C1927–C1933. [Google Scholar] [CrossRef] [PubMed]
- Bian, D.; Mahanivong, C.; Yu, J.; Frisch, S.M.; Pan, Z.K.; Ye, R.D.; Huang, S. The G12/13-Rho a signaling pathway contributes to efficient lysophosphatidic acid-stimulated cell migration. Oncogene 2006, 25, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Aoki, J.; Taira, A.; Takanezawa, Y.; Kishi, Y.; Hama, K.; Kishimoto, T.; Mizuno, K.; Saku, K.; Taguchi, R.; Arai, H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J. Biol. Chem. 2002, 277, 48737–48744. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Burkhardt, A.M.; Homey, B. Homeostatic chemokine receptors and organ-specific metastasis. Nat. Rev. Immunol. 2011, 11, 597–606. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Salanga, C.L.; Handel, T.M.; Allen, S.J. Chemokines and cancer: Migration, intracellular signalling and intercellular communication in the microenvironment. Biochem. J. 2008, 409, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Kipps, T.J. CXCR4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Staller, P.; Sulitkova, J.; Lisztwan, J.; Moch, H.; Oakeley, E.J.; Krek, W. Chemokine receptor CXCR4 down regulated by von Hippel-Lindau tumour suppressor pVHL. Nature 2003, 425, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Tan, W.; Dillenburg-Pilla, P.; Armando, S.; Amornphimoltham, P.; Simaan, M.; Weigert, R.; Molinolo, A.A.; Bouvier, M.; Gutkind, J.S. A synthetic biology approach reveals a CXCR4-G13-Rho signaling axis driving transendothelial migration of metastatic breast cancer cells. Sci. Signal. 2011, 4, ra60. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/β-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. Wnt signaling in disease and in development. Cell Res. 2005, 15, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Korinek, V.; Barker, N.; Morin, P.J.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of β-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [PubMed]
- Van Amerongen, R. Alternative Wnt pathways and receptors. Cold Spring Harb. Perspect. Biol. 2012, 4, a007914. [Google Scholar] [CrossRef] [PubMed]
- Katanaev, V.L.; Ponzielli, R.; Sémériva, M.; Tomlinson, A. Trimeric G protein-dependent frizzled signaling in Drosophila. Cell 2005, 120, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; DeCostanzo, A.J.; Liu, X.; Wang, Hy.; Hallagan, S.; Moon, R.T.; Malbon, C.C. G protein signaling from activated rat frizzled-1 to the β catenin-Lef-Tcf pathway. Science 2001, 292, 1718–1722. [Google Scholar] [CrossRef] [PubMed]
- Slusarski, D.C.; Corces, V.G.; Moon, R.T. Interaction of Wnt and a Frizzled homologue triggers G-protein-linked phosphatidylinositol signalling. Nature 1997, 390, 410–413. [Google Scholar] [PubMed]
- Major, M.B.; Roberts, B.S.; Berndt, J.D.; Marine, S.; Anastas, J.; Chung, N.; Ferrer, M.; Yi, X.; Stoick-Cooper, C.L.; von Haller, P.D.; et al. New regulators of Wnt/β-catenin signaling revealed by integrative molecular screening. Sci. Signal. 2008, 1, ra12. [Google Scholar] [CrossRef] [PubMed]
- Regard, J.B.; Cherman, N.; Palmer, D.; Kuznetsov, S.A.; Celi, F.S.; Guettier, J.M.; Chen, M.; Bhattacharyya, N.; Wess, J.; Coughlin, S.R.; et al. Wnt/B-catenin signaling is differentially regulated by Ga proteins and contributes to fibrous dysplasia. Proc. Natl. Acad. Sci. USA 2011, 108, 20101–20106. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.; Yang, J.; DeRan, M.; Wu, C.; Su, A.I.; Bonamy, G.M.; Liu, J.; Peters, E.C.; Wu, X. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem. Biol. 2012, 19, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.S.; Yu, F.X.; Gong, R.; Brown, J.H.; and Guan, K.L. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs). Genes Dev. 2012, 26, 2138–2143. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Luo, J.; Mo, J.S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhang, Y.; Park, H.W.; Jewell, J.L.; Chen, Q.; Deng, Y.; Pan, D.; Taylor, S.S.; Lai, Z.C.; Guan, K.L. Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev. 2013, 27, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Camargo, F.D. The Hippo signaling pathway and stem cell biology. Trends Cell Biol. 2012, 22, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.L. The Hippo-YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Sudol, M.; Bork, P.; Einbond, A.; Kastury, K.; Druck, T.; Negrini, M.; Huebner, K.; Lehman, D. Characterization of the mammalian YAP (Yes-associated protein) gene and its role in defining a novel protein module, the WW domain. J. Biol. Chem. 1995, 270, 14733–14741. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.X.; Alexander, C.M.; et al. Alternative Wnt signaling activates YAP/TAZ. Cell 2015, 162, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluh, J.; Nijhawan, D.; Cox, G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Puente, X.S.; Gutiérrez-Fernández, A.; Ordóñez, G.R.; Hillier, L.W.; López-Otín, C. Comparative genomic analysis of human and chimpanzee proteases. Genomics 2005, 86, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.M. Proteolytic enzymes initiating cell division and escape from contact inhibition of growth. Nature 1970, 227, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.B.; Buchanan, J.M. Mitogenic activity of blood components. I. Thrombin and prothrombin. Proc. Natl. Acad. Sci. USA 1975, 72, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Carney, D.H.; Cunningham, D.D. Initiation of check cell division by trypsin action at the cell surface. Nature 1977, 268, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Carney, D.H.; Cunningham, D.D. Cell surface action of thrombin is sufficient to initiate division of chick cells. Cell 1978, 14, 811–823. [Google Scholar] [CrossRef]
- Rasmussen, U.B.; Vouret-Craviari, V.; Jallat, S.; Schlesinger, Y.; Pagès, G.; Pavirani, A.; Lecocq, J.P.; Pouysségur, J.; van Obberghen-Schilling, E. cDNA cloning and expression of a hamster α-thrombin receptor coupled to Ca2+ mobilization. FEBS Lett. 1991, 288, 123–128. [Google Scholar] [CrossRef]
- Vu, T.K.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068. [Google Scholar] [CrossRef]
- Coughlin, S.R. Protease-activated receptors start a family. Proc. Natl. Acad. Sci. USA 1994, 91, 9200–9202. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, H.; Connolly, A.J.; Zeng, D.; Kahn, M.L.; Zheng, Y.W.; Timmons, C.; Tram, T.; Coughlin, S.R. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature 1997, 386, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M.L.; Zheng, Y.W.; Huang, W.; Bigornia, V.; Zeng, D.; Moff, S.; Farese, R.V.J.; Tam, C.; Coughlin, S.R. A dual thrombin receptor system for platelet activation. Nature 1998, 394, 690–694. [Google Scholar] [PubMed]
- Xu, W.F.; Andersen, H.; Whitmore, T.E.; Presnell, S.R.; Yee, D.P.; Ching, A.; Gilbert, T.; Davie, E.W.; Foster, D.C. Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad. Sci. USA 1998, 95, 6642–6646. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, S.; Emilsson, K.; Wahlestedt, C.; Sundelin, J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 9208–9210. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R. How the protease thrombin talks to cells. Proc. Natl. Acad. Sci. USA 1999, 96, 11023–11027. [Google Scholar] [CrossRef] [PubMed]
- Oldham, W.M.; Hamm, H.E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2008, 9, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Kancharla, A.; Maoz, M.; Jaber, M.; Agranovich, D.; Peretz, T.; Grisaru-Granovsky, S.; Uziely, B.; Bar-Shavit, R. PH motifs in PAR1&2 endow breast cancer growth. Nat. Commun. 2015, 6, 8853–8865. [Google Scholar] [PubMed]
- Konishi, H.; Kuroda, S.; Kikkawa, U. The pleckstrin homology domain of RAC protein kinase associates with the regulatory domain of protein kinase C zeta. Biochem. Biophys. Res. Commun. 1994, 205, 1770–1775. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Kawakami, Y.; Kawakami, T. The pleckstrin homology domain of Bruton tyrosine kinase interacts with protein kinase C. Proc. Natl. Acad. Sci. USA 1994, 91, 9175–9179. [Google Scholar] [CrossRef] [PubMed]
- Even-Ram, S.C.; Maoz, M.; Pokroy, E.; Reich, R.; Katz, B.Z.; Gutwein, P.; Altevogt, P.; Bar-Shavit, R. Tumor cell invasion is promoted by activation of protease activated receptor-1 in cooperation with the α vβ 5 integrin. J. Biol. Chem. 2001, 276, 10952–10962. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.J.; Park, S.Y.; Cho, K.H.; Sohn, J.S.; Lee, J.; Kim, Y.K.; Kang, J.; Park, C.G.; Han, J.W.; Lee, H.Y. The Rho/ROCK pathway for lysophosphatidic acid-induced proteolytic enzyme expression and ovarian cancer cell invasion. Oncogene 2012, 31, 4279–4289. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Kim, N.H.; Ji, L.; Kim, S.H.; Lee, J.; Rhee, H.J. Lysophosphatidic acid activates β-catenin/T cell factor signaling, which contributes to the suppression of apoptosis in H19–7 cells. Mol. Med. Rep. 2013, 8, 1729–1733. [Google Scholar] [PubMed]
- Burkhalter, R.J.; Westfall, S.D.; Liu, Y.; Stack, M.S. Lysophosphatidic acid initiates epithelial to mesenchymal transition and induces β-catenin-mediated transcription in epithelial ovarian carcinoma. J. Biol. Chem. 2015, 290, 22143–22154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bialkowska, A.; Rusovici, R.; Chanchevalap, S.; Shim, H.; Katz, J.P.; Yang, V.W.; Yun, C.C. Lysophosphatidic acid facilitates proliferation of colon cancer cells via induction of Krüppel-like factor 5. J. Biol. Chem. 2007, 282, 15541–15549. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Park, H.W.; Qin, B.; Chen, Q.; Meng, Z.; Plouffe, S.W.; Taniguchi, K.; Yu, F.X.; Karin, M.; Pan, D.; et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 2015, 29, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.J.; Katz, V.; Salah, Z.; Maoz, M.; Cohen, I.; Uziely, B.; Turm, H.; Grisaru-Granovsky, S.; Suzuki, H.; Bar-Shavit, R. Mammary gland tissue targeted overexpression of human protease-activated receptor 1 reveals a novel link to β-catenin stabilization. Cancer Res. 2006, 66, 5224–5233. [Google Scholar] [CrossRef] [PubMed]
- Turm, H.; Maoz, M.; Katz, V.; Yin, Y.J.; Offermanns, S.; Bar-Shavit, R. Protease-activated receptor-1 (PAR1) acts via a novel Gα13-dishevelled axis to stabilize β-catenin levels. J. Biol. Chem. 2010, 285, 15137–15148. [Google Scholar] [CrossRef] [PubMed]
- Romero, G.; Sneddon, W.B.; Yang, Y.; Wheeler, D.; Blair, H.C.; Friedman, P.A. Parathyroid hormone receptor directly interacts with dishevelled to regulate β-Catenin signaling and osteoclastogenesis. J. Biol. Chem. 2010, 285, 14756–14763. [Google Scholar] [CrossRef] [PubMed]
- Malbon, C.C. Frizzleds: New members of the superfamily of G-protein-coupled receptors. Front. Biosci. 2004, 9, 1048–1058. [Google Scholar] [CrossRef] [PubMed]
- Cojoc, M.; Peitzsch, C.; Trautmann, F.; Polishchuk, L.; Telegeev, G.D.; Dubrovska, A. Emerging targets in cancer management: Role of the CXCL12/CXCR4 axis. Onco. Targets Ther. 2013, 6, 1347–1361. [Google Scholar] [PubMed]
- Rosanò, L.; Bagnato, A. Endothelin therapeutics in cancer: Where are we? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R469–R475. [Google Scholar] [CrossRef] [PubMed]
- Dannenberg, A.J.; Subbaramaiah, K. Targeting cyclooxygenase-2 in human neoplasia: Rationale and promise. Cancer Cell 2003, 4, 431–436. [Google Scholar] [CrossRef]
- Hull, M.A.; Ko, S.C.; Hawcroft, G. Prostaglandin EP receptors: Targets for treatment and prevention of colorectal cancer? Mol. Cancer Ther. 2004, 3, 1031–1039. [Google Scholar] [PubMed]
- O’Callaghan, G.; Houston, A. Prostaglandin E2 and the EP receptors in malignancy: Possible therapeutic targets? Br. J. Pharmacol. 2015, 172, 5239–5250. [Google Scholar] [CrossRef] [PubMed]
- Taub, J.S.; Guo, R.L.; Leeb-Lundberg, M.F.; Madden, J.F.; Daaka, Y. Bradykinin receptor subtype 1 expression and function in prostate cancer. Cancer Res. 2003, 63, 2037–2041. [Google Scholar] [PubMed]
- Liu, Y.; An, S.; Ward, R.; Yang, Y.; Guo, X.X.; Li, W.; Xu, T.R. G protein-coupled receptors as promising cancer targets. Cancer Lett. 2016, 376, 226–239. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Receptor | Ligand | Pathways |
|---|---|---|
| Lysophosphatidic acid Receptors (LPA1-6) | Lysophosphatidic acid | Rho-dependent pathway [37,106] |
| β-Catenin stabilization [107,108] | ||
| Kruppel-like factor 5 [109] | ||
| Protease activated receptors (PAR1&2) LPA | Thrombin, Trypsin, respectively, or TFLLRN (G12/13, PAR1) or SLIGKV (G12/13, PAR2) [79] Lysophosphatidic acid (Gαq) [80] | Hippo/YAP pathways via activation of Gα12/13-coupled receptors or Gαq. Inhibition of Hippo pathway (via the inhibition of Lats1/2 kinases ) results in activation of YAP co-transcriptional activity [110] |
| Frizzled (Fz) PAR1 Parathyroid receptor1 (PTHR1) | Wnt 3A (canonical pathway) | Canonical Wnt signaling stabilization of β-catenin [66,114] and its transcription activity |
| Thrombin or TFLLRN [111,112] | ||
| PTH [113] | ||
| Chemokine receptor (CXCR4) | CXCL12, SDF-1 | PI3K, Akt, Src PIP2, IP3, Ras, Raf, ERK1/2, PLC, JNK [115] |
| PAR1 and PAR2 | Thrombin or TFLLRN (PAR1) Trypsin or SLIGKV (PAR2) | PH domain signal partners such as Etk/Bmx or Akt [102] Gα12/13, Rho [24] |
| Endothelin receptors (ETAR and ETBR) | endothelin-1-3 (ET-1, ET-2, ET-3) | C-Src/cross talk with EGFR |
| β-arrestin -1or-2 PDZRhoGEF and Rho A, C | ||
| β-catenin stabilization [7,21,116] | ||
| Prostaglandin receptors (PE2, PE4) | PGE2 | Cyclooxygenase (COX-2)pathway P13K (coupling to Gs) [117,118,119] |
| Bradykinin Receptor Type 1 and 2 (B1R, B2R) | Kinins | Gαq and Cross talk with EGFR Ras, Raf, ERK |
| Sphingosine1-phosphate receptor1 (S1PR1) | S1P | Ras-ERK, PI3K-Akt-Rac, Rho, STAT3 (coupling to Gαi) [120,121] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G Protein-Coupled Receptors in Cancer. Int. J. Mol. Sci. 2016, 17, 1320. https://doi.org/10.3390/ijms17081320
Bar-Shavit R, Maoz M, Kancharla A, Nag JK, Agranovich D, Grisaru-Granovsky S, Uziely B. G Protein-Coupled Receptors in Cancer. International Journal of Molecular Sciences. 2016; 17(8):1320. https://doi.org/10.3390/ijms17081320
Chicago/Turabian StyleBar-Shavit, Rachel, Myriam Maoz, Arun Kancharla, Jeetendra Kumar Nag, Daniel Agranovich, Sorina Grisaru-Granovsky, and Beatrice Uziely. 2016. "G Protein-Coupled Receptors in Cancer" International Journal of Molecular Sciences 17, no. 8: 1320. https://doi.org/10.3390/ijms17081320
APA StyleBar-Shavit, R., Maoz, M., Kancharla, A., Nag, J. K., Agranovich, D., Grisaru-Granovsky, S., & Uziely, B. (2016). G Protein-Coupled Receptors in Cancer. International Journal of Molecular Sciences, 17(8), 1320. https://doi.org/10.3390/ijms17081320