1. Introduction
The Centers for Disease Control and Prevention continues to address multistate foodborne outbreaks that have impacted the health of the nation over the last 10 years [
1]. One area of concern is the need for reduction of
Salmonella as a foodborne pathogen. Despite control efforts that cost over a half a billion dollars annually, foodborne illnesses due to
Salmonella continue to impact the consumer. Poultry are commonly identified as a major source of
Salmonella. Asymptomatic carrier states are poorly understood. “Normal infections” include infection of chicks through an oral route and is characterized by a translocation through the intestinal epithelial cells followed with a splenic infection [
2]. While asymptomatic carriers can be infected by
Salmonella Enteritidis (SE) and
Salmonella Typhmurium (ST), these bacteria can survive in the gastrointestinal tract of birds for months without showing clinical signs [
3]. These
Salmonella carriers have an infected gastrointestinal tract without showing clinical signs while excreting high concentrations of
Salmonella into the environment [
3,
4,
5,
6,
7,
8,
9,
10]. These healthy carriers can be a risk to affect other birds by horizontal transmission or affect newly hatched chicks.
Despite the importance of
Salmonella as a human pathogen, relatively little is known about the host immune response or virulence mechanisms of persistent asymptomatic infections in the avian intestine. The most fundamental question to answer is how do these organisms manage to escape clearance for so long in the presence of the host immune response? Upon infection with
Salmonella, an up-regulation of the innate inflammatory response is generated and is characterized by pro-inflammatory cytokines and granulocyte (heterophils in chickens) influx within hours [
11,
12,
13,
14]. Yet, this intestinal inflammatory response is somehow dampened facilitating pathogen survival and persistent infection [
15] for up to 10 weeks or more [
3,
16]. One must keep in mind that this dampened inflammatory response in chickens may be a host-developed mechanism to minimize immune-mediated damage to the intestine at a time when the gut microbiome is being established (disease tolerance). Disease tolerance has recently been described as a “distinct host defense strategy” [
17,
18,
19,
20,
21,
22]. Thus, a diminished immune response provides conceivable advantages to both the host and bacterium during a persistent infection in chickens. The mechanisms involved in this down-regulation of the mucosal immune response are currently unknown. However, one can speculate that mucosal disease tolerance is required to establish a persistent infection. During host tolerance, defined as coping with a pathogenic encounter without a consequent reduction in health [
17,
18,
19,
20,
21,
22], the host’s strategy is to avoid a harmful excessive inflammatory response [
23,
24]. However, this strategy may enable pathogen persistence, such as that observed with
Salmonella infections of poultry [
15,
16].
We, and others, have recently demonstrated the development of a Th2, anti-inflammatory response in the cecum of chickens that begins at least four days after an initial infection with
Salmonella and continues for weeks [
11,
25,
26,
27,
28]. Moreover, we have noted a significant increase in CD4
+CD25
+ (T regulatory) cells in the cecum that corresponds to this shift from a pro-inflammatory to an anti-inflammatory environment [
29]. T regulatory cells (Tregs) have been linked to play crucial roles sustaining a balance between the host immune response and immunological tolerance in many infections in mammals [
30,
31,
32,
33,
34]. Further, a role for Tregs during a persistent
Salmonella infection was recently described using a mouse model of persistence [
35]. More recently, we also found alterations in the tissue phenotype of the cecum of the
Salmonella-infected animals that is distinguished by metabolic signatures indicative of metabolic reprogramming with a shift from anabolic to catabolic reactions [
28]. It is during this phase that we speculate that
Salmonella takes advantage of a reduction of host response to infection to begin to establish a persistent cecal colonization [
28].
In this study, our hypothesis was that Salmonella enterica serovar Enteritidis (S. Enteritidis) induces a disease tolerance host defense mechanism in chickens that allows the bacteria to colonize persistently the cecum of poultry. To test the hypothesis, we analyzed a time-course of chicken-specific kinomic immune changes and interferon-γ (IFN-γ) mRNA transcription in avian cecal tissue during a persistent infection by S. Enteritidis. Using these techniques, we were able to identify specific phosphorylation based immune post-translational signaling changes during a chronic Salmonella colonization in chickens that provide confirmation for the transition from an early mucosal pro-inflammatory response to the development of a disease tolerant mucosal response.
3. Discussion
Relatively little is known about how and why
Salmonella enterica persist in the avian intestine, specifically the interactions between the virulence mechanisms and host immune response. The persistent colonization of the gut, the carrier state, is established, and the
Salmonella is able to stay in the ceca for months without triggering clinical signs of infection [
4,
5,
7,
8]. A persistent, chronic, subclinical
Salmonella infection of the intestinal tract is important to continued bacterial propagation and the contamination of poultry as it is nearly impossible to detect and isolate infected birds [
41]. We, and others, have speculated that the bacterium is involved in redirecting, or subverting, the host response toward disease tolerance [
11,
26,
27,
28,
42]. The present study was designed to address the question of immune tolerance induction during a persistent paratyphoid
Salmonella infection in chickens.
Host responses to infectious agents are often regulated through phosphorylation. However, proteomic mechanisms of
Salmonella acute infection biology and host responses to the bacteria have been investigated only in murine models [
43,
44,
45,
46,
47]. Until recently, studies in poultry have been limited to the genomic responses of the host to infection (reviews in [
11,
48,
49]). Our recent development of chicken-specific peptide arrays for kinome analysis of host phosphorylation-based cellular signaling responses provided us with the opportunity to develop a more detailed understanding of the chicken host-pathogen interactions with
Salmonella [
50,
51].
Based on the findings here, our kinomic analysis demonstrate a phenotypic change in the avian cecum as it orchestrates the dynamics of immune signaling pathways, cytokine secretion, transcription factor expression, and the launch of a different immune microenvironment during the establishment of a persistent
Salmonella infection and a return to intestinal homeostasis. Four days post-infection (pi)
Salmonella induces an immune transition from an acute pro-inflammatory response to an established infection and a dampened or eliminated innate response [
52]. By 4 days pi, we have described a substantial down-regulation of the expression pro-inflammatory cytokines that coincides with the up-regulation of the expression of anti-inflammatory cytokines [
27,
28]. Further, by day four pi a dramatic increase in Tregs (CD4
+CD25
+) in the cecum and remains elevated through 14 days pi [
29]. This coordinated production of pro- versus anti-inflammatory responses is a central mechanism of an effective early inflammatory response and later return to tissue immune homeostasis. Finally, we used a kinomics approach to uncover the mechanisms used by
S. Enteritidis to impact the avian inflammatory responses and determine host signaling events altered by the bacteria to create the conditions for a persistent infection. Our results identified multiple changes to the host kinome during the establishment of a persistent
Salmonella infection in the avian cecum. This immune analysis that compared the immune responses between the
S. Enteritidis-infected avian cecum and non-infected cecum provides novel information on host cellular signaling cascades that are altered during the establishment of
Salmonella persistence (
Table 5 and
Table 6) [
27,
28]. Additionally, the relative lack of differential phosphorylation events found in the signaling pathways between the infected and non-infected ceca 7–14 days pi indicate that a level of immune homeostasis had been achieved and that the
Salmonella were no longer being recognized as infectious agents and were now part of the commensal population. Further experiments are underway to further characterize and contrast this homeostasis to that of the non-infected controls.
Here we have further described a series of phosphorylation-mediated changes in the ceca of chickens during the development of a persistent Salmonella infection. The most significant differences in host immune kinase activities in infected animals occurred within four days pi. These changes were localized to select pathways, specifically the T cell receptor and the JAK-STAT signaling pathways, which were altered by the persistent colonization of the cecum by S. Enteritidis.
Stimulation of the T cell receptor results in the activation of the TCR signal transduction pathway. This pathway activates the transcription factors nuclear factor κB (NF-κB), nuclear factor of activated T-cells (NFAT), and activator protein 1 (AP-1), that induce expression of cytokine genes [
53]. The results of this study clearly point to changes in the activity of all three of the central transcription factors, specifically at 4 days pi (
Table 4). First, we found no significant effect on the phosphorylation of the AP-1 transcription factors between the
S. Enteritidis-infected and non-infected tissues. The AP-1 pathway is dependent on activated of mitogen-activated protein kinases (MAPKs), such as extracellular signal–regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38, which promote the synthesis, phosphorylation, and activation of the Fos and Jun proteins that together comprise the AP-1 transcription factor [
53]. However, we found a significant dephosphorylation of MAPKs, including MEK1, ERK1, MAP3K8, and p38 involved in the T cell receptor signaling cascade; thus, pointing to the lack of involvement of AP-1-induced genes during a persistent
S. Enteritidis infection in the chicken (
Table 4). Second, NFAT (phosphorylated) and NF-κB (dephosphorylated) were significantly differentially phosphorylated in the ceca of
S. Enteritidis-infected chickens (
Table 4). The central question is whether there is a common thread that could account for this differential response of these transcription factors. This thread appears to be phospholipase C-γ1 (PLCG1) that was significantly dephosphorylated in the
S. Enteritidis-infected cecal tissue at 4 days pi when compared to the non-infected control cecal tissue. Activation of both NF-κB and NFAT requires the activity of PLC-γ1, which generates the second messengers diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP
3). DAG leads to activation of protein kinase C θ (PKCθ), which, in turn, activates the inhibitor of κB (IκB) kinase (IKK) complex, resulting in the phosphorylation and degradation of IκBα and the translocation of the NF-κB p50:p65 heterodimer to the nucleus [
54]. IP
3 induces an increase in the concentration of cytoplasmic calcium (Ca
2+) and activation of the Ca
2+-dependent phosphatase calcineurin, which results in the rapid activation of NFAT, which is followed by its translocation to the nucleus [
55]. To our knowledge, our report is the first to implicate
Salmonella targeting of PLCG1 to manipulate the NF-κB and NFAT pathways to inhibit pro-inflammatory responses. What bacterial factors may be involved in dephosphorylating PLCG1 are unknown at this time and will be the focus of future experiments.
NFAT proteins, a family of transcription factors, are critical to the transcription of cytokine genes and other genes that are critical for the control of inflammation and regulation of the immune response [
55,
56]. Further, NFAT must ultimately bind to additional transcription factors, such as AP-1 to form transcriptional complexes that regulate gene expression that are inducibly transcribed by immune-system cells [
55,
57]. NFAT functions to regulate the interaction of the innate immune cells with acquired immunity and to promote anti-inflammatory programs (reviewed by [
58]). Thus, the increased phosphorylation of NFAT peptides would suggest the initiation of anti-inflammatory signals. NF-κB is a transcription factor whose activity is triggered in response to infectious agents and pro-inflammatory cytokines via the IκB kinase (IKK) complex and plays a key role in regulating the pro-inflammatory response [
59,
60]. Therefore, dephosphorylation of both IKK and NF-κB would result in a down-regulation of pro-inflammatory cytokines as we observed in the present experiments. As a result, the T cell receptor signaling pathway analysis data provide evidence that the establishment of a persistent infection by
S. Enteritidis in the avian cecum appears to be partially due to the targeting of signaling cascades that inhibit the transcription of pro-inflammatory responses and induce the beginning of a transition from T
H1/T
H17 cells to the development of Tregs [
61,
62,
63,
64].
Further pathway analysis of the kinome data indicated differential phosphorylation of the JAK-STAT pathway, a signaling cascade that provides a direct mechanism to translate an extracellular signal into a transcriptional response, in
S. Enteritidis-infected cecal tissue. The JAK-STAT system consists of three main components: (1) a receptor; (2) Janus kinase (JAK); and (3) Signal Transducers and Activator of Transcription (STAT) [
65]. Based on the results from them present experiments (
Table 6), the IFN-α, IL-2, IL-4, and IL-10 receptors were phosphorylated; whereas, IFN-γ, IL-7, and IL-6 cytokine family (gp130) receptors were dephosphorylated. IFN-γ is characteristic of a Th1 response whereas IL-4 is a signature cytokine of Th2 responses. IL-7 is involved in early T cell development and IL-6 is a pro-inflammatory cytokine involved in stimulating an immune response during infection. IL-2 is normally produced by T cells during an immune response and involved in growth, proliferation, and differentiation of T cells to become “effector” T cells [
66,
67]. When combined with the cytokine expression observed previously [
27,
28], the down-regulation of IFN-γ mRNA transcription shown here (
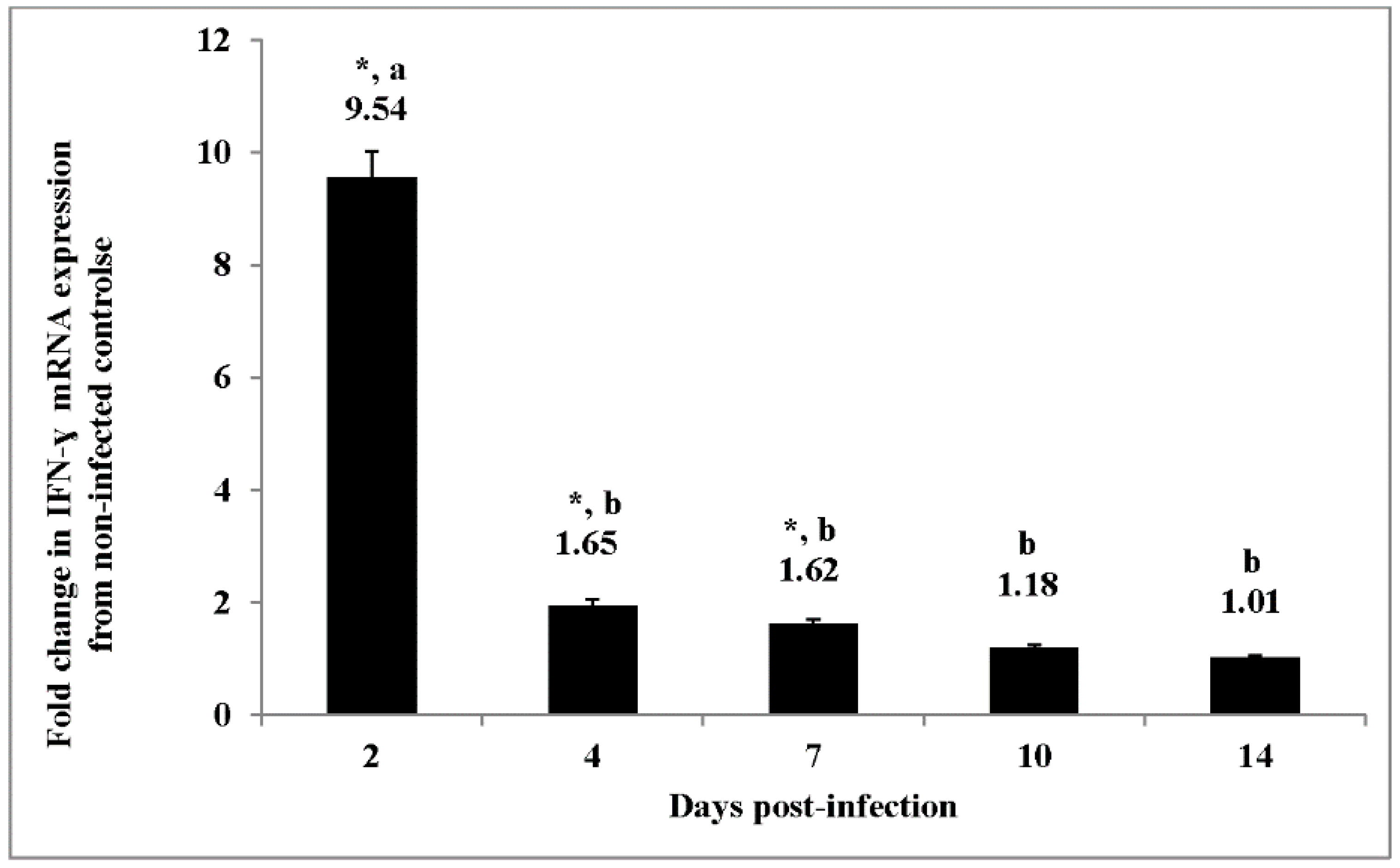
Figure 1) provides a clear pattern of down-regulation of the pro-inflammatory cytokines (IL-6, IL-1β, IFN-γ) and an up-regulation of anti-inflammatory cytokines, IL-10 and TGF-β4 [
27,
28]. We speculate there is a profound immune transition from an active inflammatory response where the immune system was working to reduce the number of bacteria to an environment of homeostasis where the immune response is allowing for a persistent state of infection in the
S. Enteritidis-infected cecal tissue.
Cytokine receptor proteins lack enzymatic activity, thus are dependent upon JAKs to initiate signaling upon binding of their ligands. The JAK family has four members: JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2) [
68]. TYK2 is the only JAK family member that was activated (phosphorylated) in the ceca from the
S. Enteritidis-infected chickens (
Table 6). Although primarily involved in IL-12 and type I-IFN signaling, TYK2 is activated by IL-10 [
69].
Most importantly, based on these experiments, the development of a persistent cecal infection by
S. Enteritidis triggers a dephosphorylation of both JAK2 and JAK3 proteins (
Table 6). JAK2 is an essential tyrosine kinase for modulating the immune response and whose activation contributes to the severe inflammatory response in sepsis [
70,
71]. Inhibition of JAK2 prevents NF-κB activation; thus “rescuing” mice from polymicrobial sepsis [
72]. Therefore, we can conclude that the dephosphorylation of both JAK2 and NF-κB found via our kinomic analysis is indicative of a negative regulation of a pro-inflammatory response; in this case brought about by the establishment of a persistent
Salmonella infection. Further experiments are required to confirm this hypothesis.
JAK3 is predominantly expressed in hematopoietic lineage such as NK cells, T cells and B cells and intestinal epithelial cells [
73,
74,
75]. JAK3 is the only JAK family member involved in all phases of T cell biology: development, proliferation, and differentiation [
76,
77,
78]. For T cell differentiation, JAK3, along with IL-4, steer Th2 cell differentiation [
78], but inhibition of JAK3 generates the induction of Tregs [
79,
80]. Therefore, the dephosphorylation of JAK2 and JAK3 found in the present studies would result in a change in the functional immune phenotype of the cecal environment that benefits the establishment of a tolerant mucosal immune response against the bacterial colonization. Although these studies cannot confirm what provoked this dephosphorylation of JAK2 and JAK3, we speculate that the mechanism is a specific action of the
Salmonella organism as it begins to establish its long term colonization. These results are the first to infer that
Salmonella have evolved a time-dependent strategy that blocks responsiveness of the JAK proteins that down-regulates the host response to infection.
STAT4 is a decisive factor in host resistance to a variety of viral, bacterial, and protozoan pathogens while serving as the central regulator of IFN-γ production during inflammation [
81]. Intestinal IFN-γ mRNA expression levels are a prevailing indicator of a reduced immune response associated with persistence of
Salmonella in the chicken gastrointestinal tract [
42]. Furthermore, the ratio between STAT1 and STAT4 are crucial for IFN-γ production during viral and
Salmonella infections [
82,
83]. Herein, we found a reduced IFN-γ mRNA expression during the establishment of the persistent
Salmonella infection (
Figure 1) and an increased phosphorylation of STAT1 and dephosphorylation of STAT4 (
Table 6). Our results are in agreement with two recent studies where
N-ethyl-
N-nitrosourea-induced mutations of mice resulted in increased STAT1 phosphorylation, suppressed STAT4 expression, and altered IFN-γ production that led to the increased susceptibility of the animals to
S. Typhimurium infection [
83,
84]. IFN-γ has been shown to play a fundamental role in the resolution of intestinal Salmonella infection [
13,
42,
85]. Further, our observation of a dramatic decrease in IFN-γ mRNA expression at day four p.i. is in agreement with previously reported results by other laboratories [
13,
26].
4. Materials and Methods
4.1. Experimental Animals
Experiments were conducted according to the regulations established by the United States Department of Agriculture Animal Care and Use Committee. Broiler chickens used in this study were obtained from a commercial breeder and were all of the same genetic background and were not vaccinated at any time. Chicks were placed in floor pens containing wood shavings, provided supplemental heat, water, and a balanced, unmedicated corn and soybean meal-based chick starter diet ad libitum that met or exceeded the levels of critical nutrients recommended by the National Research Council [
86].
Salmonella was not detected in the feed or from the paper tray liners.
4.2. S. Enteritidis Challenge
A poultry isolate of Salmonella enterica serovar Enteritidis (S. Enteritidis; (ID 9711771, part 24)) was obtained from the National Veterinary Services Laboratory (Ames, IA, USA), and was selected for resistance to nalidixic acid and novobiocin and maintained in tryptic soy broth (Difco Laboratories, Sparks, MD, USA) containing antibiotics (20 µg/mL nalidixic acid and 25 µg/mL novobiocin; Sigma Chemical Co.; St. Louis, MO, USA). A stock culture was prepared in sterile PBS and adjusted to a concentration of 1 × 109 colony forming units (CFU/mL). The viable cell concentration of the challenge dose for each experiment was determined by colony counts on XLT4 agar base plates with XLT4 supplement (Difco) and nalidixic acid and novobiocin (XLT-NN).
4.3. Experimental Design
One-day-old broiler chickens were randomly distributed into two experimental groups: non-infected control and infected. Each group contained 200 birds fed a balanced, unmedicated corn and soybean meal-based diet. Four days post-hatch, all chickens were orally challenged with 1 mL of either 5 × 106 CFU/mL S. Enteritidis or mock challenged with 1 mL sterile PBS. Four, 7, 10, and 14 days after challenge, 50 chickens from each group were killed by cervical dislocation, cecal contents were analyzed for S. Enteritidis colonization, 10 of these chickens were used for: (a) cecal tonsils for quantitative real-time PCR (qRT-PCR); and (b) cecal tissue was flash frozen in liquid nitrogen and stored for use in the peptide and antibody arrays.
All experiments were conducted three times. Therefore, the ceca from a total of 30 chickens for each of the 2 groups (10 chickens each in 3 experiments) were used to prepare the mRNA for the qRT-PCR IFN-γ assay described below. RNA from each bird (n = 10) was isolated and assayed separately and not pooled. Each RNA sample was replicated 3 times for IFN-γ expression per experiment.
4.4. Sample Collection for Peptide and Antibody Arrays
At 4, 7, 10, and 14 days post infection, both ceca were removed from each of 10 birds from each group (non-infected and infected) and immediately flash frozen in liquid nitrogen to preserve kinase enzymatic activity. Samples were taken from liquid nitrogen and transferred to a −80 °C freezer until further experimental procedures were conducted.
4.5. Kinome Array
At each of the time points and under each condition (infected and uninfected), 4 cecal samples from 4 different animals were taken from storage for analysis (32 samples total). Infected birds were selected based on a consistent high level of
S. Enteritidis colonization. Cecal tissue samples were weighed to obtain a consistent 40 mg sample for the array protocol. Samples were homogenized by a hand-held Qiagen TissueRuptor (Valencia, CA, USA) in 100 μL of lysis buffer (20 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM Ethylene glycol tetraacetic acid (EGTA), 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM Na
3VO
4, 1 mM NaF, 1 μg/mL leupeptin, 1 g/mL aprotinin and 1 mM Phenylmethylsulphonyl fluoride (all products from Sigma Aldrich (St. Louis, MO, USA), unless indicated). Following homogenization, the peptide array protocol was carried out as per Jalal et al. [
87], with alterations described in Arsenault et al. [
51,
88].
4.6. Antibody Array
The antibody array assay kit was procured from Full Moon BioSystems (Sunnyvale, CA, USA). This technique was used as an alternative to procuring phosphospecific antibodies individually and performing several western blot assays. The protocol was carried out as per manufacturer’s instructions (Antibody Array User’s Guide Rev 11.3) with the following alteration to the homogenization step: instead of using the bead and vortex homogenization indicated in the kit, the hand-held Qiagen Tissue Ruptor was used.
4.7. Data Analysis: Kinome and Antibody Arrays
Data normalization and PCA analysis was performed for both the peptide and antibody microarrays as per Li et al. [
89] using the PIIKA2 online platform (
http://saphire.usask.ca/saphire/piika/index.html). Briefly, the array data were analyzed by subtracting the background intensity from the foreground intensity, variance stabilization normalization was conducted to bring all of the arrays onto the same scale, and then
t-test, clustering and pathway analysis were performed. This consistent analysis method facilitated a more direct comparison between the two distinct array datasets and allowed for a statistically robust analysis of the phosphorylation events being measured. Geneontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed by uploading the statistically significant peptide lists to the Search Tool for the Retrieval of Interacting Genes (STRING) [
36].
4.8. Sample Collection for Bacterial Contents
The ceca from each chicken was removed aseptically, and the contents (0.25 g) were serially diluted to 1:100, 1:1000, or 1:10,000 and spread onto XLT-NN plates. The plates were incubated at 37 °C for 24 h, and the number of NN-resistant S. Enteritidis cells per gram of cecal contents was determined. The data from each experimental group were pooled from three separate trials for statistical analysis.
4.9. Sample Collection for mRNA
Chickens from each experimental group were euthanized at 4, 7, 10, and 14 days post-infection. A 25-mg piece of tissue was removed from the cecal tonsils. The tissue was washed in PBS and placed in a 2-mL microcentrifuge tube with 1 mL of RNAlater (Qiagen, Inc., Valencia, CA, USA) and stored at −20 °C until processed.
4.10. RNA Isolation
Tissues (50 mg) were removed from RNAlater and transferred to pre-filled 2 mL tubes containing Triple-Pure™ 1.5 mm zirconium beads. RLT lysis buffer (600 μL) from the RNeasy mini kit (Qiagen, Valencia, CA, USA) was added and the tissue was homogenized for 1–2 min at 4000 rpm in a Bead Bug microtube homogenizer (Benchmark Scientific, Inc., Edison, NJ, USA). Total RNA was extracted from the homogenized lysates according to the manufacturer’s instructions, eluted with 50 μL RNase-free water, and stored at −80 °C until qRT-PCR analyses performed. RNA was quantified and the quality was evaluated using a spectrophotometer (NanoDrop Products, Wilmington, DE, USA). The data from these three repeated experiments were pooled for presentation and statistical analysis. Total RNA (300 ng) from each sample was prepared.
4.11. Quantitative Real-Time PCR
The primer and probe sets for IFN-γ and 28S rRNA were designed using the Primer Express software program (Applied Biosystems, Foster City, CA, USA). IFN-γ mRNA expression was quantitated using a well-described method. Primers and probes for IFN-γ and 28S rRNA-specific amplification have been described [
25,
47] and are provided in
Table 7. The qRT-PCR was performed using the TaqMan fast universal PCR master mix and one-step RT-PCR master mix reagents [
27,
28] (Applied Biosystems). Amplification and detection of specific products were performed using the Applied Biosystems 7500 Fast real-time PCR system with the following cycle profile: one cycle of 48 °C for 30 min and 95 °C for 20 s and 40 cycles of 95 °C for 3 s and 60 °C for 30 s. Quantification was based on the increased fluorescence detected by the 7500 Fast sequence detection system due to hydrolysis of the target-specific probes by the 5 = nuclease activity of the r
Tth DNA polymerase during PCR amplification. Normalization was carried out against 28S rRNA, which was used as a housekeeping gene. To correct for differences in RNA levels between samples within the experiment, the correction factor for each sample was calculated by dividing the mean threshold cycle (
Ct) value for 28S rRNA-specific product for each sample by the overall mean
Ct value for the 28S rRNA-specific product from all samples. The corrected cytokine mean was calculated as follow: (average of each replicate × cytokine slope)/(28S slope × 28S correction factor). Fold changes in mRNA levels were calculated from mean 40
Ct values by the formula 2
(40 Ct infected group − 40 Ct in non-infected control).
4.12. Statistical Analysis: mRNA Expression
The mean and standard error of the mean were calculated and differences between groups were determined by analysis of variance. Significant differences were further separated using Duncan’s multiple range test [
27]. Fold changes in RNA levels were calculated from mean 40
Ct values using formula 2
(40 Ct infected group − 40 Ct in non−infected control). A
p value of ≤0.05 was considered statistically significant.





{kind=link}