
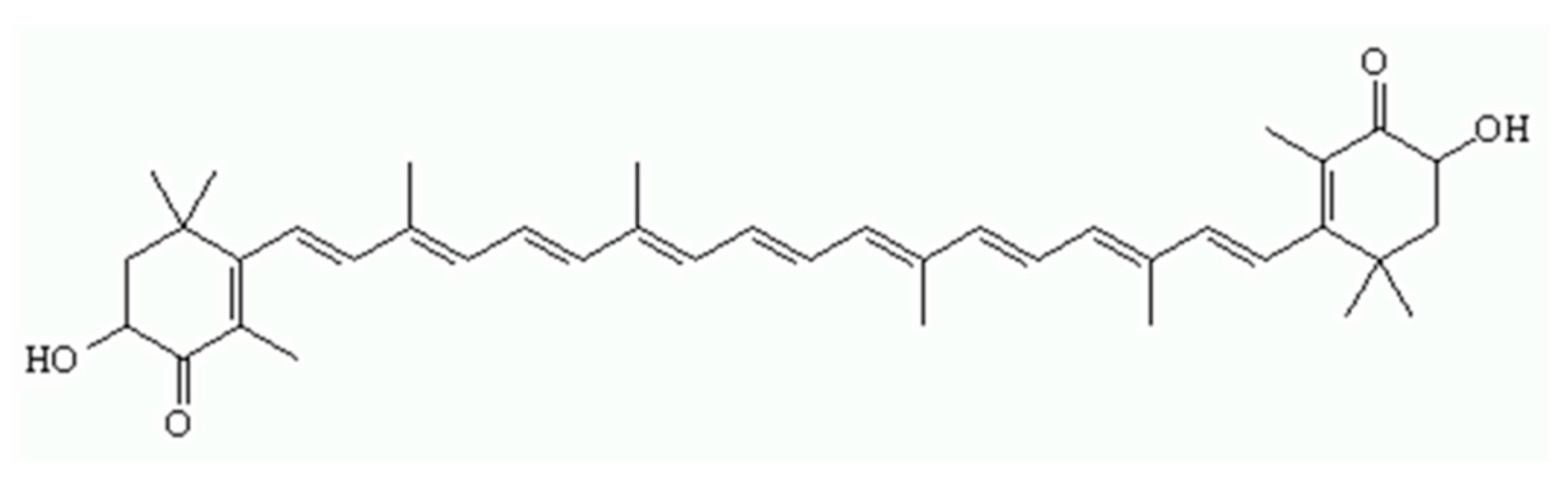
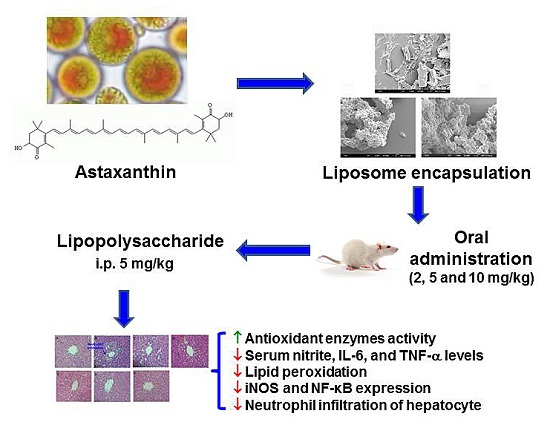
Improved Hepatoprotective Effect of Liposome-Encapsulated Astaxanthin in Lipopolysaccharide-Induced Acute Hepatotoxicity


Abstract
:
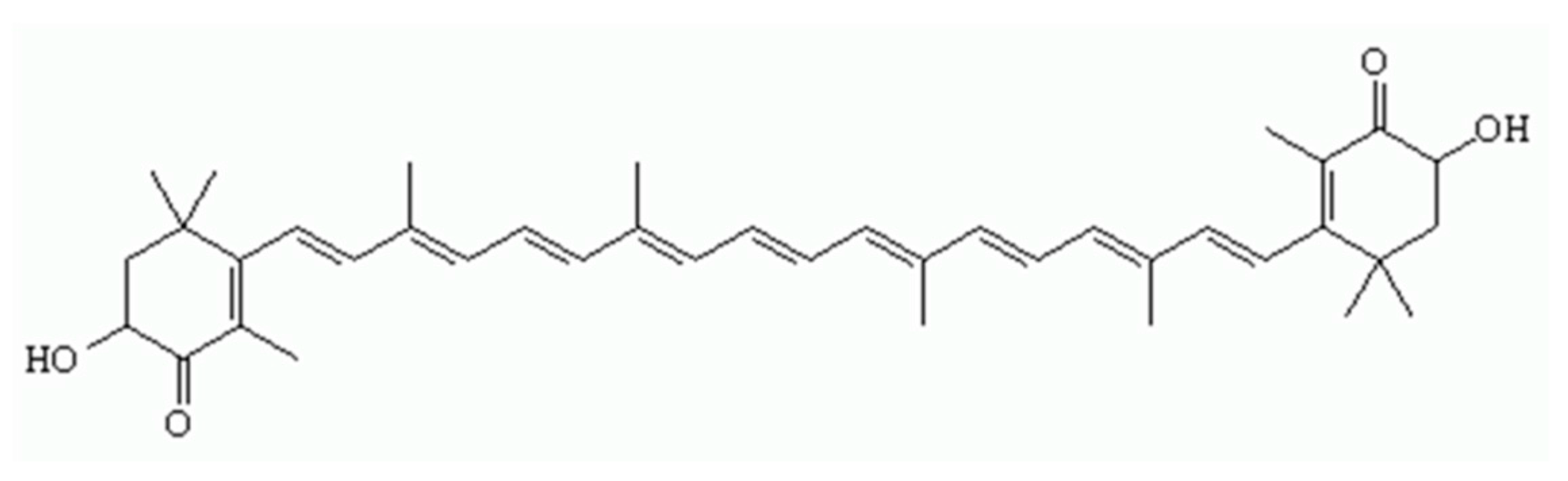
1. Introduction
2. Results and Discussion
2.1. Change in Body and Organ Weights
2.2. Field Emission Scanning Electron Microscope (FE-SEM) Image of the Liposomal Nanocapsules
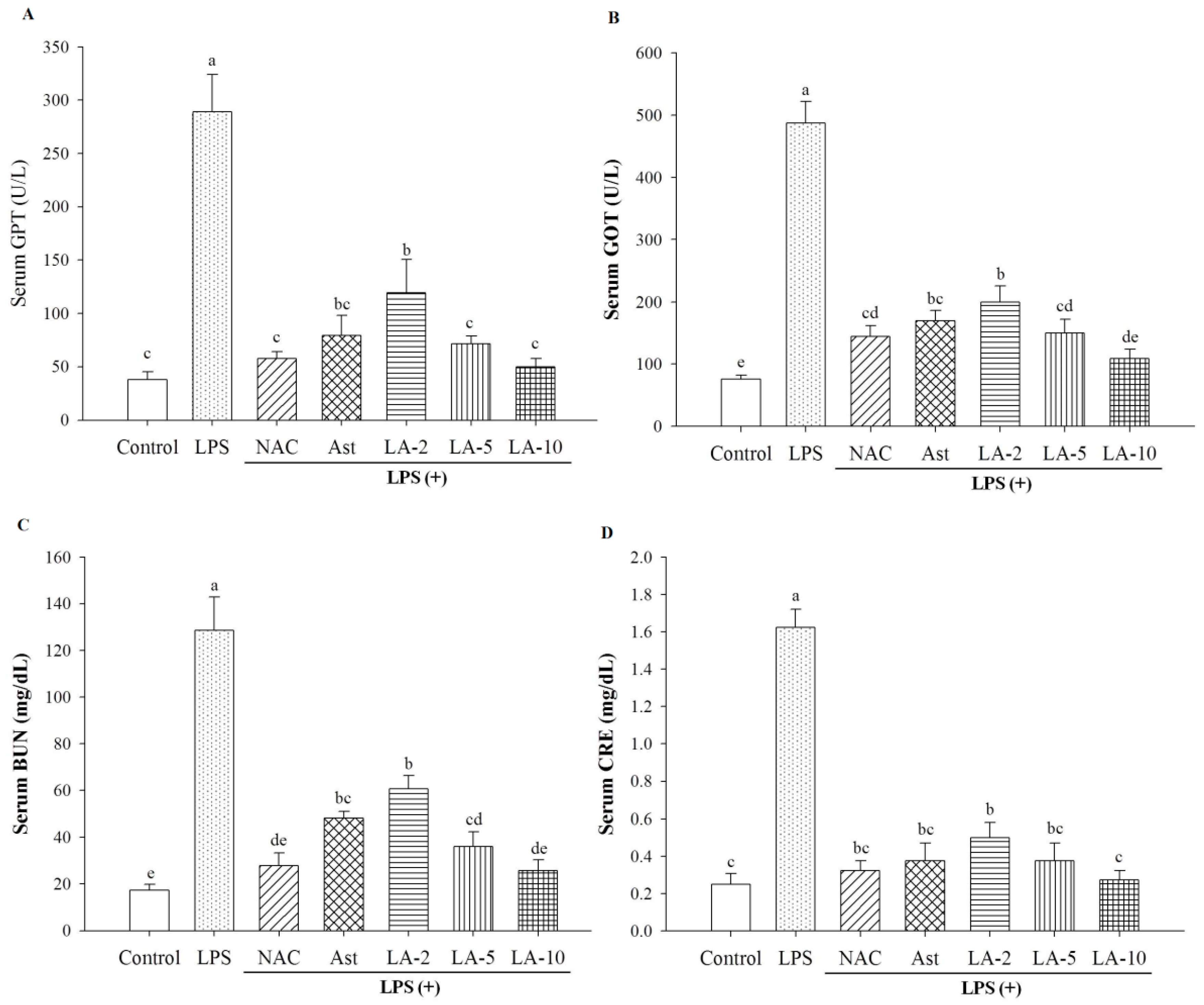
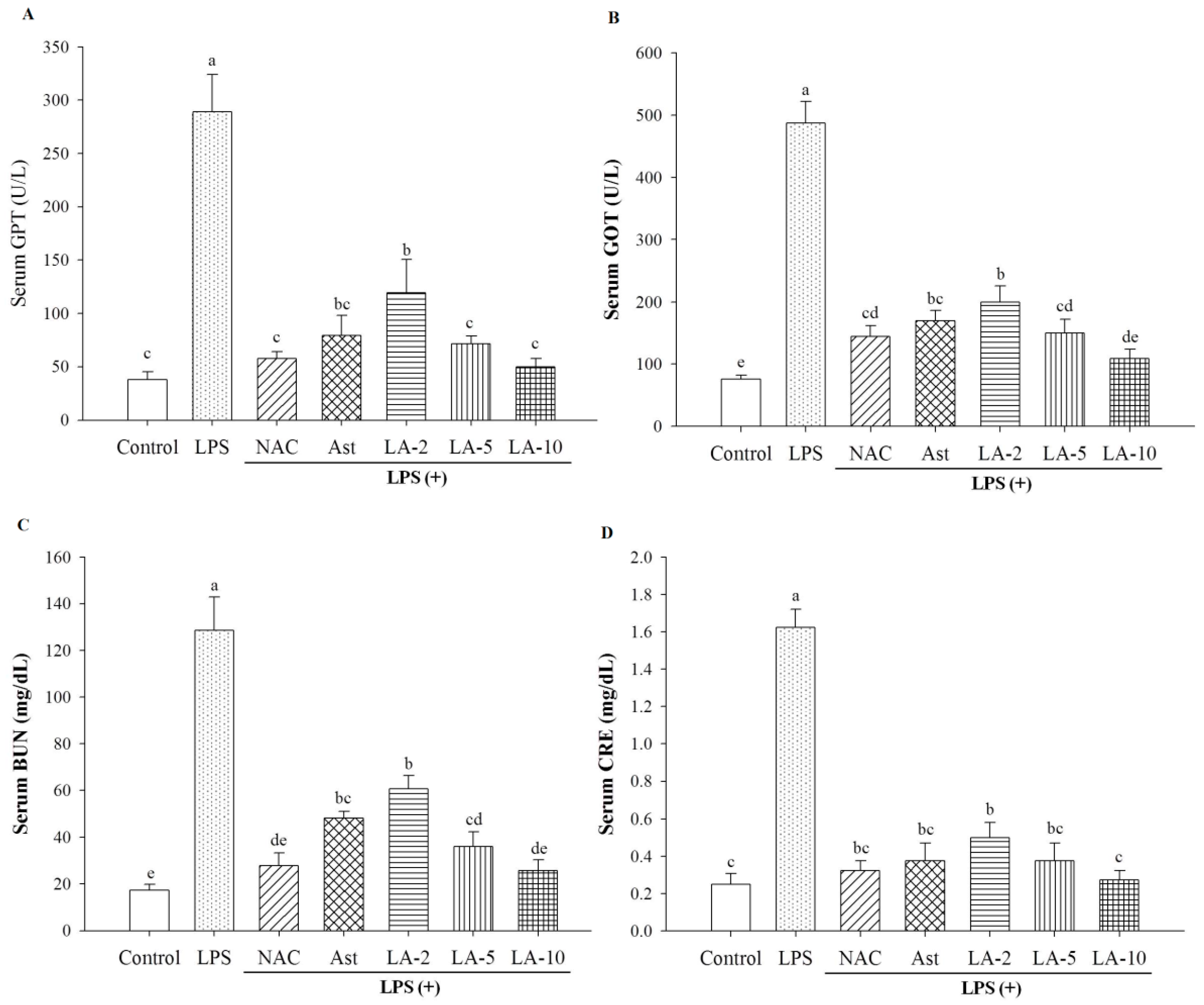
2.3. Level of Parameters Clinically Most Related to Liver Function
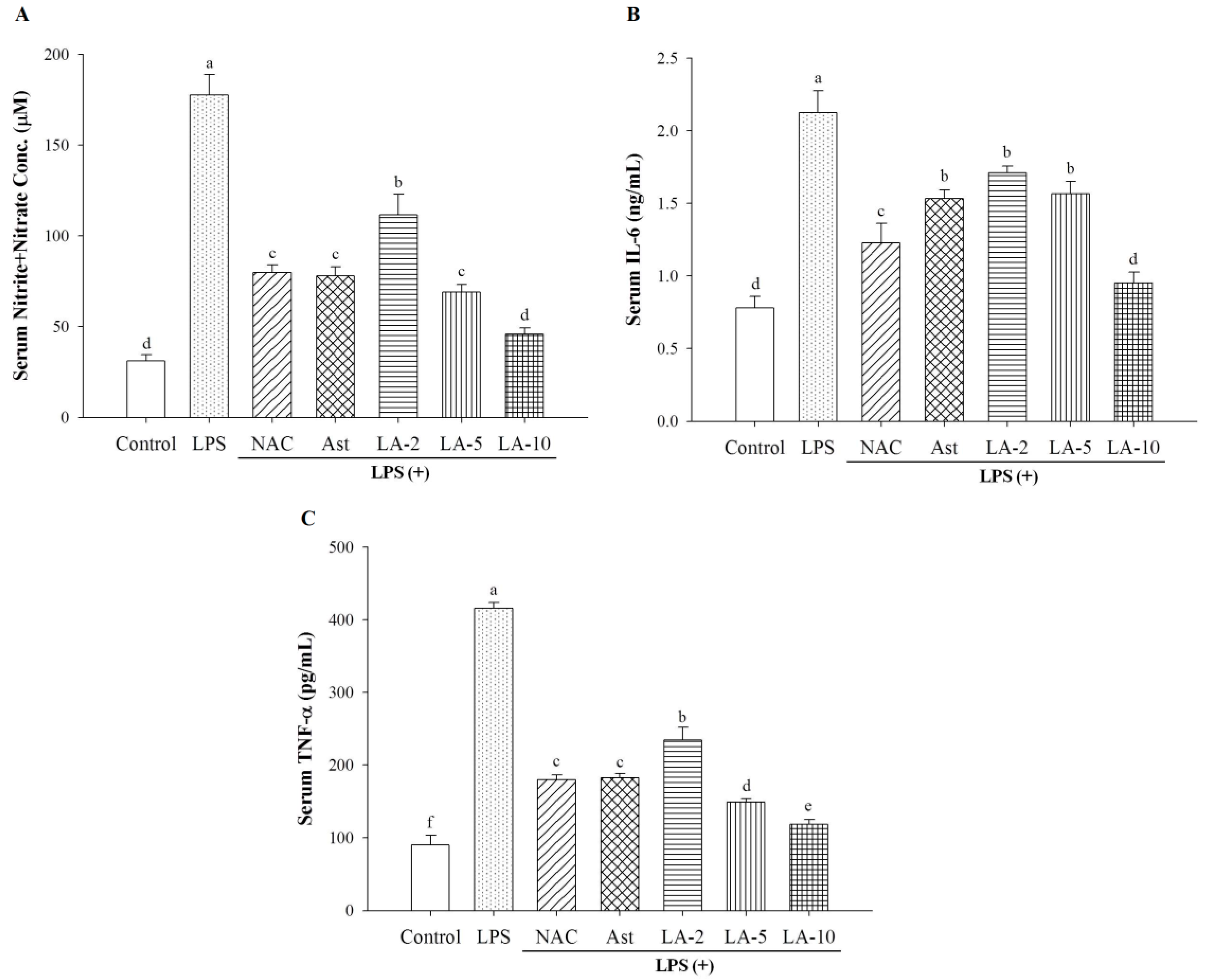
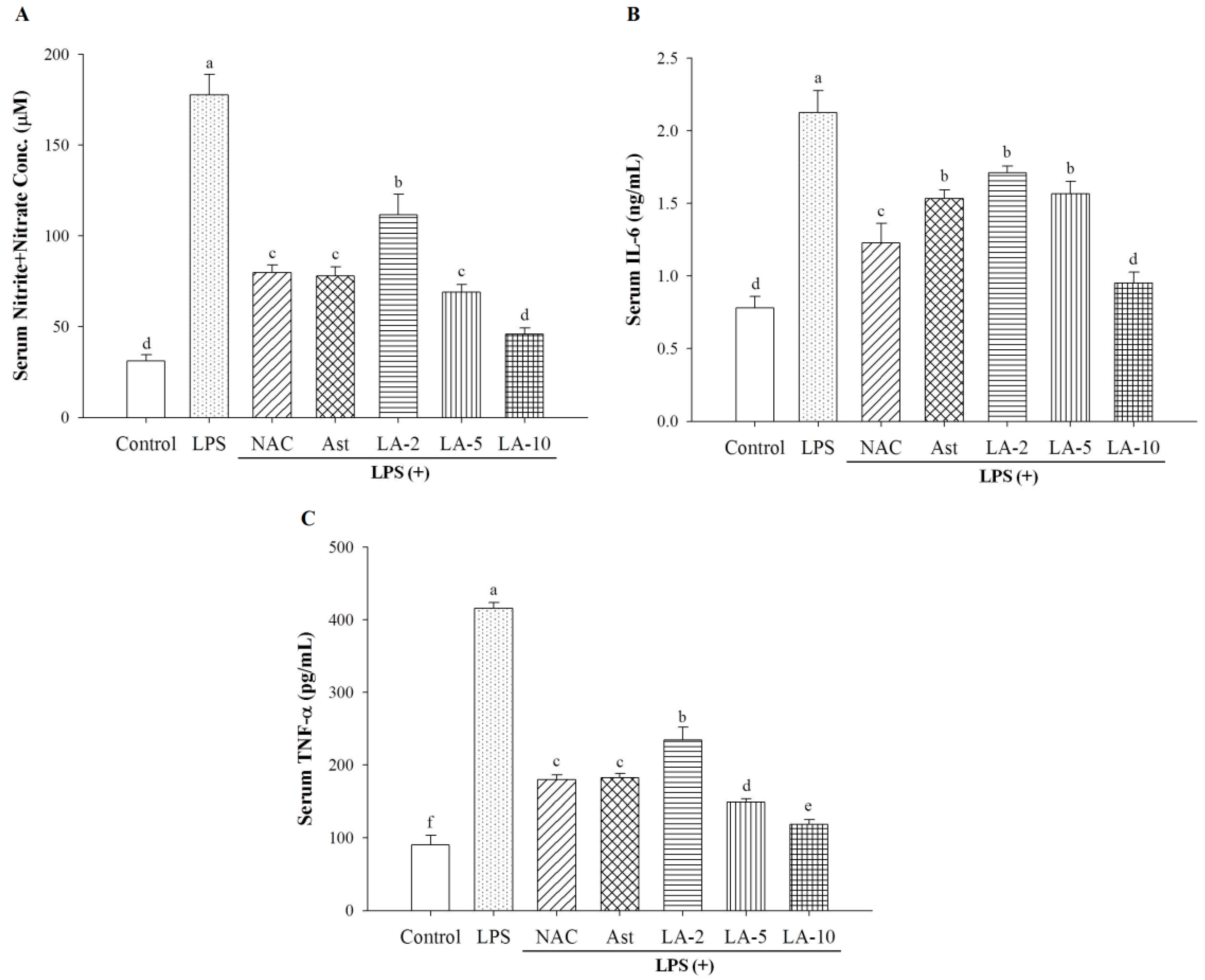
2.4. Changes of Serum Nitrite, IL-6, and TNF-α
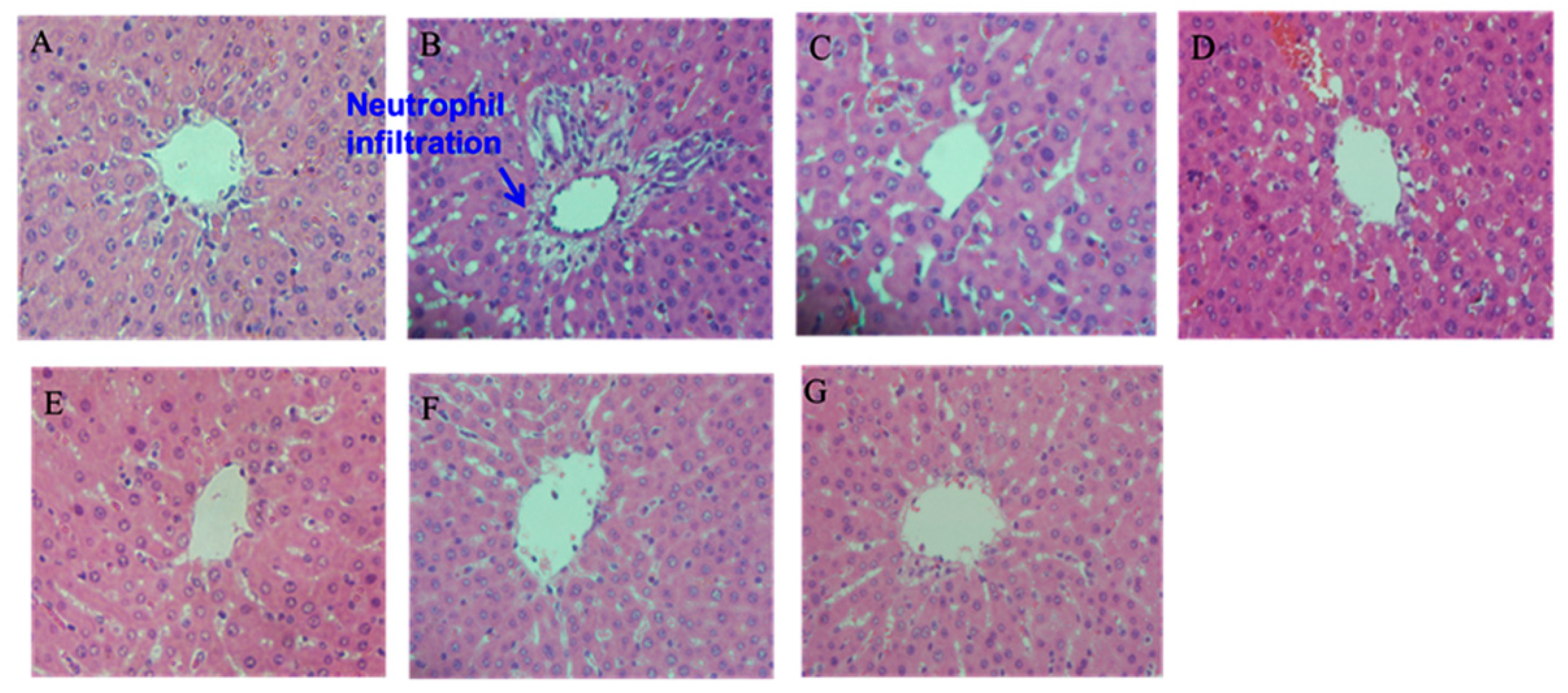
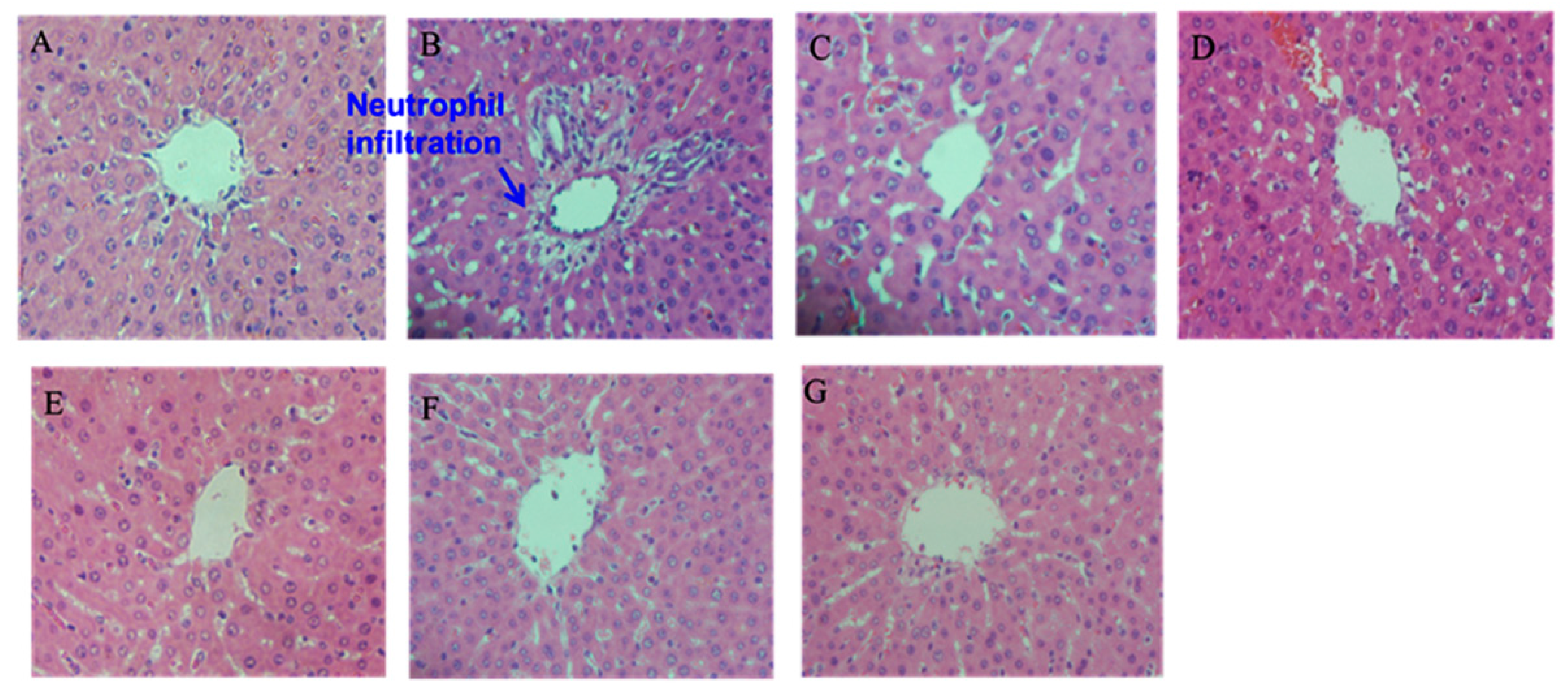
2.5. Histopathological Examination
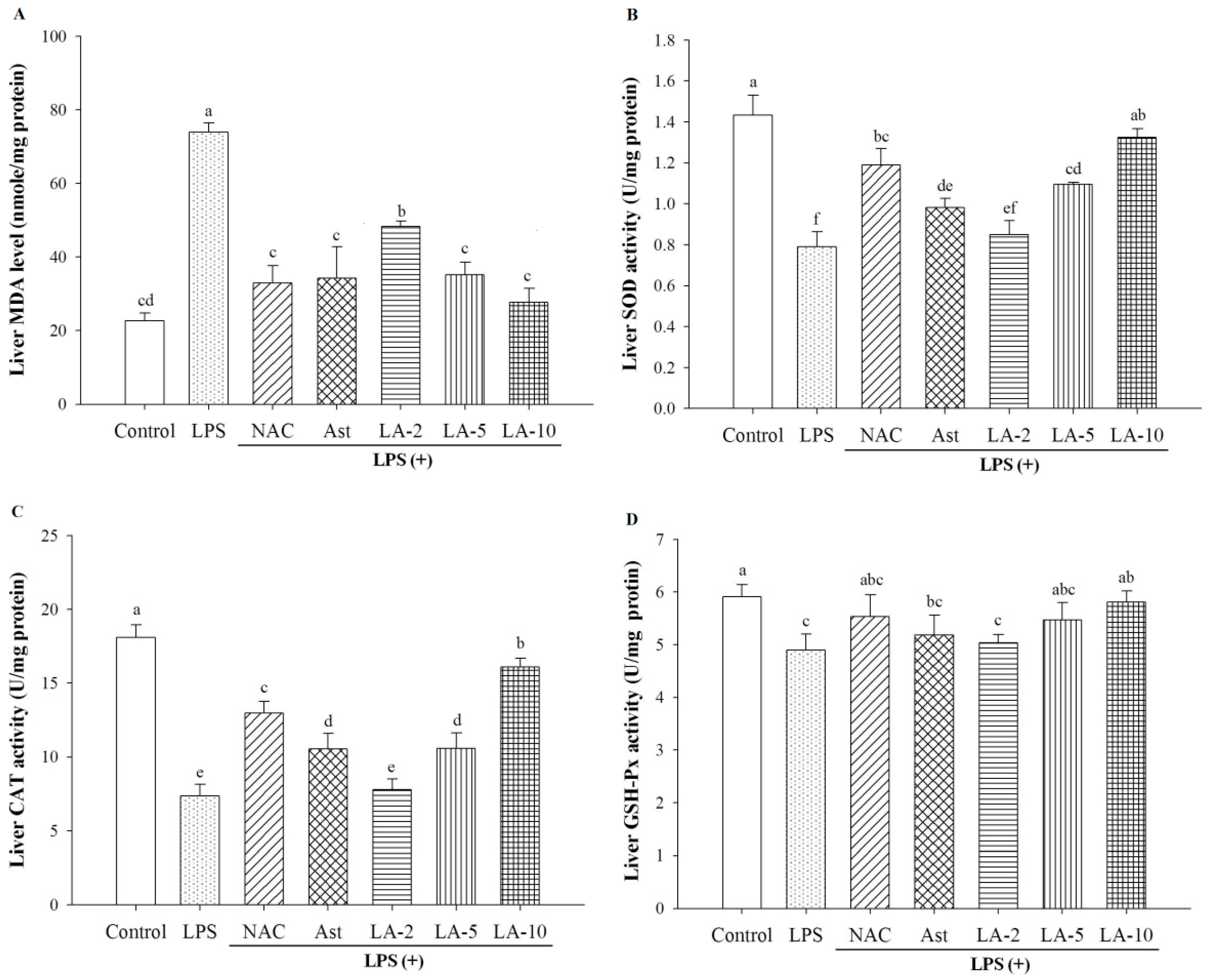
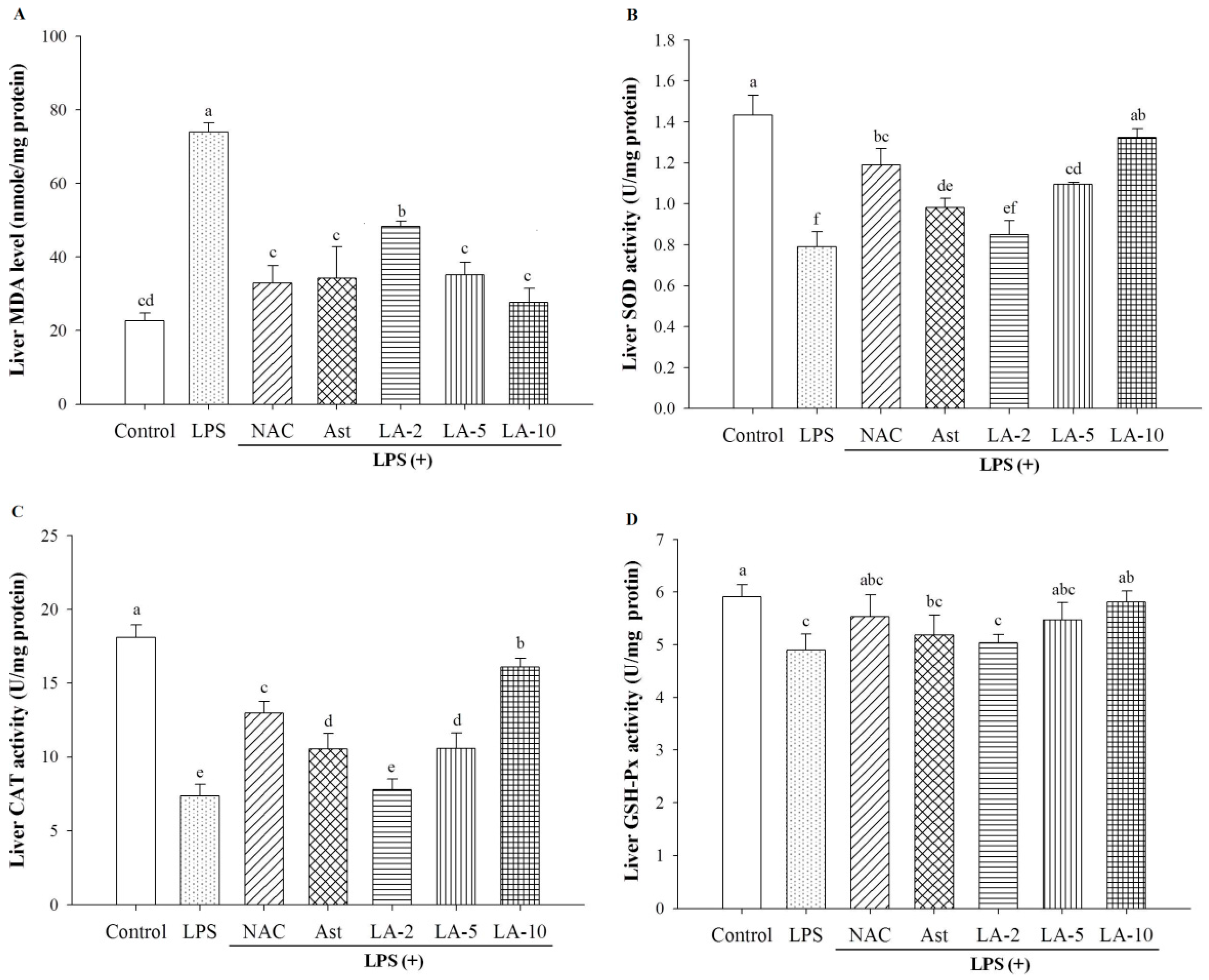
2.6. Oxidative Stress and Antioxidative Enzymes
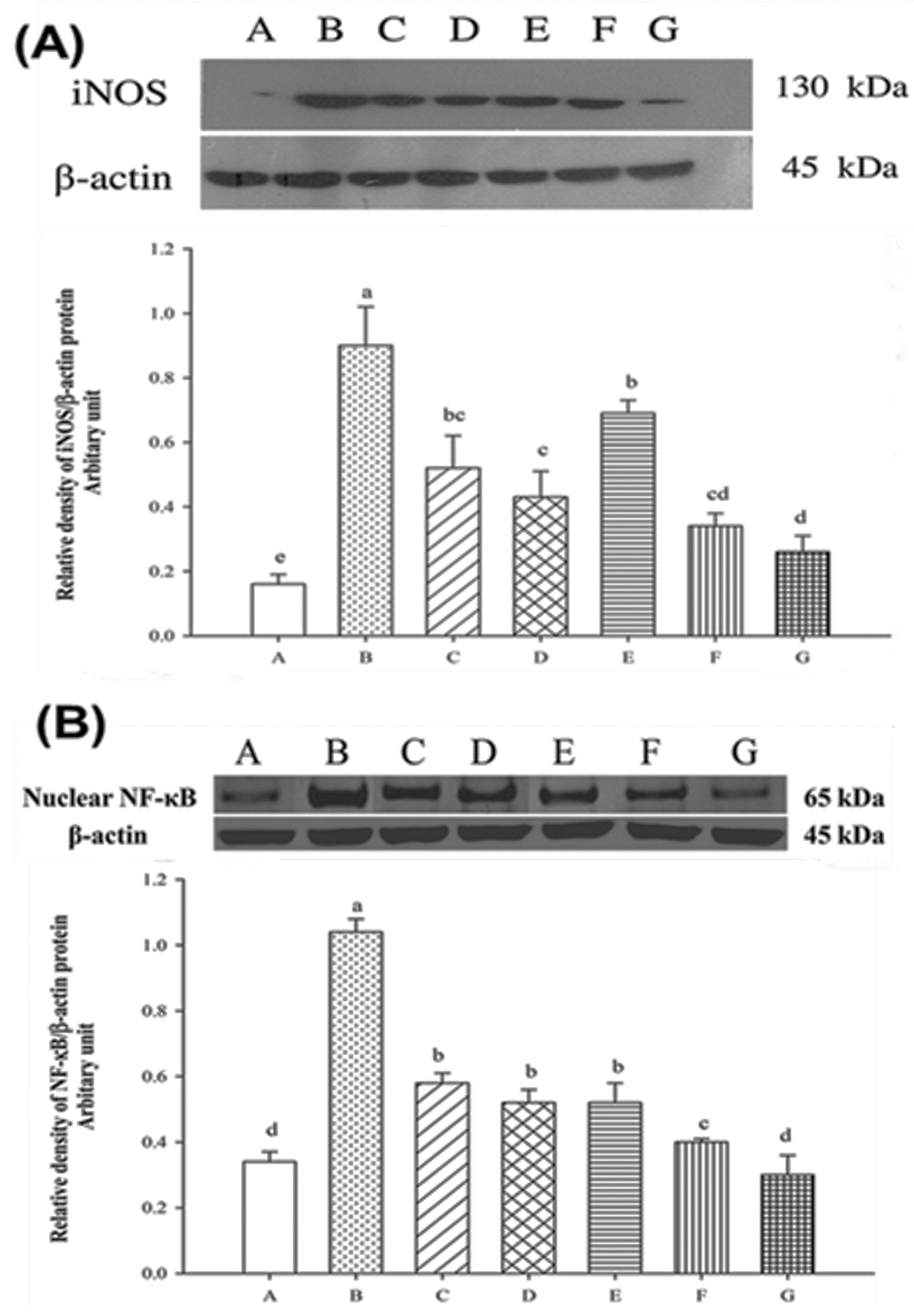
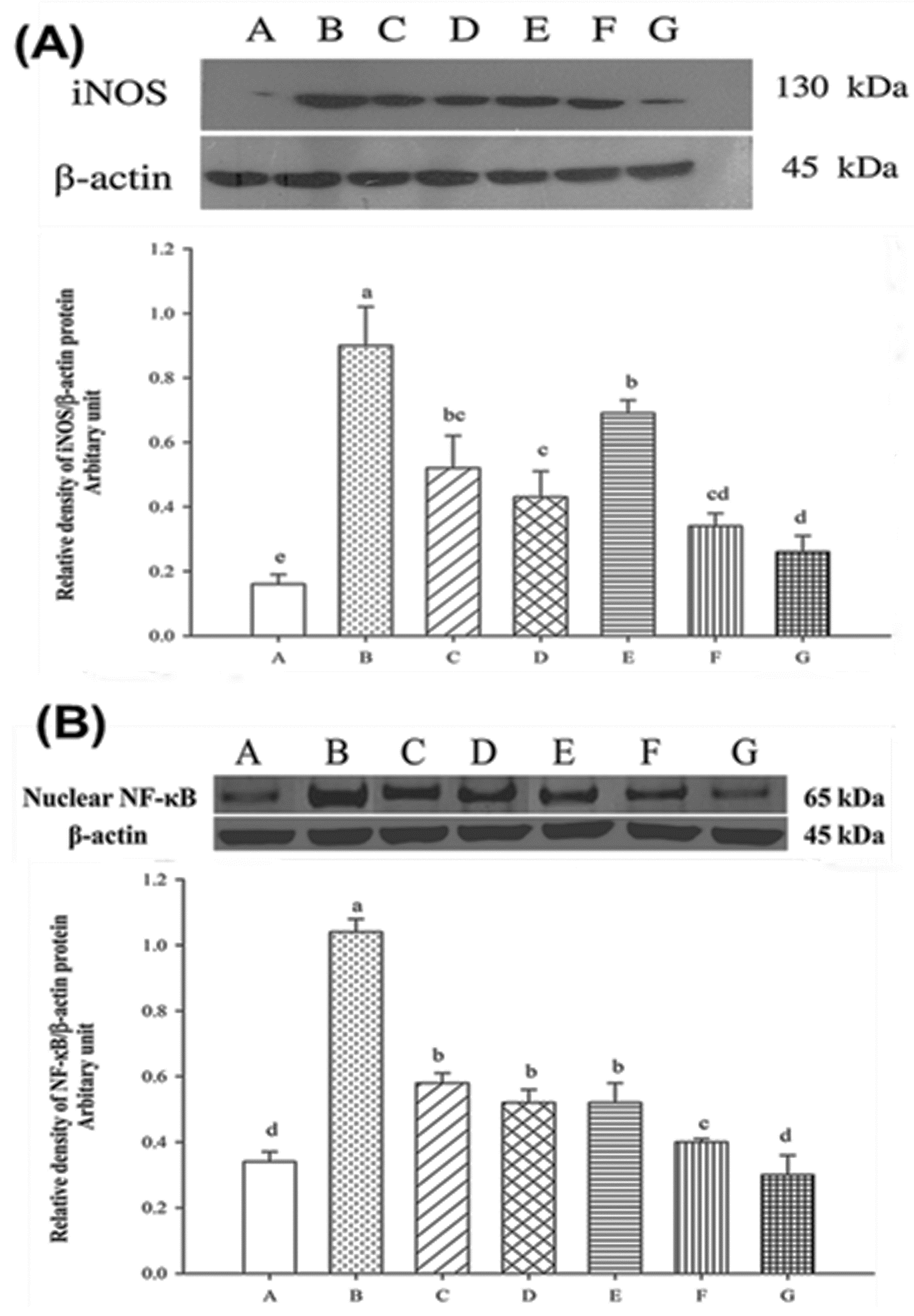
2.7. Expression of Nuclear iNOS and NF-κB
3. Materials and Methods
3.1. Chemicals
3.2. Methods
3.2.1. Preparation of Liposome-Encapsulated Astaxanthin
3.2.2. High-Performance Liquid Chromatographic (HPLC) Analysis for Astaxanthin Content in the Liposome-Encapsulated Particles
3.2.3. FE-SEM Imaging of Liposome-Encapsulated Astaxanthin
3.2.4. Animals and Treatments
3.2.5. Assay for Serum GOT, GPT, CRE and BUN
3.2.6. Assay for Serum Nitrite Level
3.2.7. Assay for Serum IL-6 and TNF-α Levels
3.2.8. Hematoxylin-Eosin Staining (H & E Staining)
3.2.9. Analysis of the Hepatic Malondialdehyde Level
3.2.10. Protein Extraction and Quantification
3.2.11. Assay for Hepatic Antioxidant Enzymes
3.2.12. Extraction of the Cytosolic Proteins from Hepatic Tissues
3.2.13. Extraction of Nuclear Proteins Extraction
3.2.14. SDS-PAGE and Protein Transfer
3.2.15. Antibody-Antigen Binding
3.3. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications—A review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Dose, J.; Matsugo, S.; Yokokawa, H.; Koshida, Y.; Okazaki, S.; Seidel, U.; Eggersdorfer, M.; Rimbach, G.; Esatbeyoglu, T. Free radical scavenging and cellular antioxidant properties of astaxanthin. Int. J. Mol. Sci. 2016, 17, 103. [Google Scholar] [CrossRef] [PubMed]
- Naguib, Y.M. Antioxidant activities of astaxanthin and related carotenoids. J. Agric. Food Chem. 2000, 48, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.R.; Sarada, R.; Ravishankar, G.A. Stabilization of astaxanthine in edible oils and its use as an antioxidant. J. Sci. Food Agric. 2007, 87, 957–965. [Google Scholar] [CrossRef]
- Yuan, J.P.; Peng, J.; Yin, K.; Wang, J.H. Potential health-promoting effects of astaxanthin: A high-value carotenoid mostly from microalgae. Mol. Nutr. Food Res. 2011, 55, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Tsuji, S.; Satoh, A.; Ishikura, M.; Shirasawa, T.; Shimizu, T. Protective effects of astaxanthine on 6-hydroxydopamine-induced apoptosis in human neuroblastoma SH-SY5Y cells. J. Neurochem. 2008, 107, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Kistler, A.; Liechti, H.; Pichard, L.; Wolz, E.; Oesterhelt, G.; Hayes, A.; Maurel, P. Metabolism and CYP-inducer properties of astaxanthine in man and primary human hepatocytes. Arch. Toxicol. 2002, 75, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Jyonouchi, H.; Sun, S.; Iijima, K.; Gross, M.D. Antitumor activity of astaxanthin and its mode of action. Nutr. Cancer 2000, 36, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Franceschelli, S.; Pesce, M.; Ferrone, A.; de Lutiis, M.A.; Patruno, A.; Grilli, A.; Felaco, M.; Speranza, L. Astaxanthin treatment confers protection against oxidative stress in U937 cells stimulated with lipopolysaccharide reducing O2− production. PLoS ONE 2014, 9, e88359. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Chen, K.; Lu, J.; Cheng, P.; Xu, L.; Dai, W.; Wang, F.; He, L.; Zhang, Y.; Chengfen, W.; et al. Protective effect of astaxanthin on liver fibrosis through modulation of TGF-β1 expression and autophagy. Mediat. Inflamm. 2014, 2014, 954502. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xia, Y.; Liu, T.; Wang, J.; Dai, W.; Wang, F.; Zheng, Y.; Chen, K.; Li, S.; Abudumijiti, H.; et al. Protective effects of astaxanthin on ConA-induced autoimmune hepatitis by the JNK/p-JNK pathway-mediated inhibition of autophagy and apoptosis. PLoS ONE 2015, 10, e0120440. [Google Scholar] [CrossRef] [PubMed]
- Elpek, G.Ö. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J. Gastroenterol. 2014, 20, 7260–7276. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Pham, T.X.; Wegner, C.J.; Kim, B.; Ku, C.S.; Park, Y.K.; Lee, J.Y. Astaxanthin lowers plasma TAG concentrations and increases hepatic antioxidant gene expression in diet-induced obesity mice. Br. J. Nutr. 2014, 112, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Amar, E.C.; Kiron, V.; Satoh, S.; Watanabe, T. Enhancement of innate immunity in rainbow trout (Oncorhynchus mykiss Walbaum) associated with dietary intake of carotenoids from natural products. Fish Shellfish Immunol. 2004, 16, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.H.; Chang, C.H.; Peng, R.Y.; Chyau, C.C. Improved membrane transport of astaxanthine by liposomal encapsulation. Eur. J. Pharm. Biopharm. 2010, 75, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Taga, T.; Kishimoto, T. Interleukin-6 in biology and medicine. Adv. Immunol. 1993, 54, 1–78. [Google Scholar] [PubMed]
- Gao, B. Hepatoprotective and anti-inflammatory cytokines in alcoholic liver disease. J. Gastroenterol. Hepatol. 2012, 27, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Damas, P.; Ledoux, D.; Nys, M.; Vrindts, Y.; de Groote, D.; Franchimont, P.; Lamy, M. Cytokine serum levels during severe sepsis in human: IL-6 as a marker of severity. Ann. Surg. 1992, 15, 362–365. [Google Scholar] [CrossRef]
- Osawa, Y.; Hoshi, M.; Yasuda, I.; Saibara, T.; Moriwaki, H.; Kozawa, O. Tumor necrosis factor-α promotes cholestasis-induced liver fibrosis in the mouse through tissue inhibitor of metalloproteinase-1 production in hepatic stellate cells. PLoS ONE 2013, 8, e65251. [Google Scholar] [CrossRef] [PubMed]
- Chastre, A.; Be’langer, M.; Beauchesne, E.; Nguyen, B.N.; Desjardins, P.; Butterworth, R.F. Inflammatory cascades driven by tumor necrosis factor-α play a major role in the progression of acute liver failure and its neurological complications. PLoS ONE 2012, 7, e49670. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, M.C.; Tyagi, C.; Tomar, L.K.; Kumar, P.; Choonara, Y.E.; Pillay, V. Nutraceutical-based therapeutics and formulation strategies augmenting their efficiency to complement modern medicine: An overview. J. Funct. Foods 2014, 6, 82–99. [Google Scholar] [CrossRef]
- Lorenz, R.T.; Cysewski, G.R. Commercial potential for Haematococcus microalgae as a natural source of astaxanthine. Trends Biotechnol. 2000, 18, 160–167. [Google Scholar] [CrossRef]
- Green, S.J.; Scheller, L.F.; Marletta, M.A.; Seguin, M.C.; Klotz, F.W.; Slayter, M.; Nelson, B.J.; Nacy, C.A. Nitric oxide: Cytokine-regulation of nitric oxide in host resistance to intracellular pathogens. Immunol. Lett. 1994, 43, 87–94. [Google Scholar] [CrossRef]
- Mungrue, I.N.; Husain, M.; Stewart, D.J. The role of NOS in heart failure: Lessons from murine genetic models. Heart Fail. Rev. 2002, 7, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim. Biophys. Acta 2001, 1504, 46–57. [Google Scholar] [CrossRef]
- Mateo, J.; Garcia-Lecea, M.; Cadenas, S.; Hernandez, C.; Moncada, S. Regulation of hypoxia-inducible factor-1α by nitric oxide through mitochondria-dependent and -independent pathways. Biochem. J. 2003, 376, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Higgs, E.A. The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S193–S201. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Charles, I.G.; Moncada, S. Nitric oxide: Orchestrating hypoxia regulation through mitochondrial respiration and the endoplasmic reticulum stress response. Cell Res. 2005, 15, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Zelickson, B.R.; Benavides, G.A.; Johnson, M.S.; Chacko, B.K.; Venkatraman, A.; Landar, A.; Betancourt, A.M.; Bailey, S.M.; Darley-Usmar, V.M. Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes. Biochim. Biophys. Acta 2011, 1807, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Chen, C.; Hou, J.; Zhao, B.; Xin, W.; Mori, A. The antioxidant EPC-K1 attenuates NO-induced mitochondrial dysfunction, lipid peroxidation and apoptosis in cerebellar granule cells. Toxicology 1999, 134, 117–126. [Google Scholar] [CrossRef]
- Stewart, V.C.; Heales, S.J. Nitric oxide-induced mitochondrial dysfunction: Implications for neurodegeneration. Free Radic. Biol. Med. 2003, 34, 287–303. [Google Scholar] [CrossRef]
- Wei, Y.H.; Lu, C.Y.; Lee, H.C.; Pang, C.Y.; Ma, Y.S. Oxidative damage and mutation to mitochondrial DNA and age dependent decline of mitochondrial respiratory function. Ann. N. Y. Acad. Sci. 1998, 854, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M.; Pérez-Gómez, C.; Núñez de Castro, I. Antioxidant enzymes and human diseases. Clin. Biochem. 1999, 32, 595–603. [Google Scholar] [CrossRef]
- Breimer, L.H. Molecular mechanisms of oxygen radical carcinogenesis and mutagenesis: The role of DNA base damage. Mol. Carcinog. 1990, 3, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Pajović, S.B.; Saičić, Z.S.; Pejić, S.; Kasapović, J.; Stojiljković, V.; Kanazir, D.T. Antioxidative biomarkers and cancerogenesis. Jugosl. Med. Biohem. 2006, 25, 397–402. [Google Scholar] [CrossRef]
- Miranda, K.M.; Espey, M.G.; Wink, D.A. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide 2001, 1, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Buege, J.A.; Aust, S.D. Microsomal lipid peroxidation. Methods Enzymol. 1978, 52, 302–310. [Google Scholar] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microquantities of protein utilizing the principle of protein-dye binding. Anal. Chem. 1976, 72, 248–254. [Google Scholar]
- Marklund, S.; Marklund, G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur. J. Biochem. 1974, 47, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Aebi, H. Catalase in vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar] [PubMed]
- Lawrence, R.A.; Burk, R.F. Glutathione peroxidase activity in selenium deficient rat liver. Biochem. Biophys. Res. Commun. 1976, 71, 952–958. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Control | LPS | NAC + LPS | Ast + LPS | LA-2 + LPS | LA-5 + LPS | LA-10 + LPS |
|---|---|---|---|---|---|---|---|
| Body weight (g) Initial Final | 254.50 ± 8.35 a 298.12 ± 6.76 a | 259.25 ± 4.57 a 302.00 ± 2.58 a | 258.50 ± 8.58 a 282.50 ± 7.54 b | 260.75 ± 10.01 a 295.75 ± 4.99 a | 259.75 ± 2.21 a 302.25 ± 9.21 a | 259.50 ± 6.24 a 303.50 ± 7.14 a | 257.75 ± 7.32 a 310.50 ± 10.14 a |
| Liver weight (g) | 12.75 ± 0.64 a | 12.62 ± 2.30 a | 11.75 ± 1.07 a | 14.02 ± 1.34 a | 12.55 ± 0.60 a | 13.05 ± 1.04 a | 14.07 ± 0.41 a |
| Liver weight/Body weight (%) | 4.41 ± 0.21 a | 4.21 ± 0.36 a | 4.15 ± 0.31 a | 4.84 ± 0.46 a | 4.15 ± 0.27 a | 4.29 ± 0.26 a | 4.53 ± 0.12 a |
| Kidney weight/Body weight (%) | 0.89 ± 0.11 a | 0.95 ± 0.12 a | 0.81 ± 0.13 a | 0.87 ± 0.28 a | 0.91 ± 0.07 a | 0.91 ± 0.03 a | 0.90 ± 0.05 a |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, C.-H.; Chang, C.-C.; Lin, S.-T.; Chyau, C.-C.; Peng, R.Y. Improved Hepatoprotective Effect of Liposome-Encapsulated Astaxanthin in Lipopolysaccharide-Induced Acute Hepatotoxicity. Int. J. Mol. Sci. 2016, 17, 1128. https://doi.org/10.3390/ijms17071128
Chiu C-H, Chang C-C, Lin S-T, Chyau C-C, Peng RY. Improved Hepatoprotective Effect of Liposome-Encapsulated Astaxanthin in Lipopolysaccharide-Induced Acute Hepatotoxicity. International Journal of Molecular Sciences. 2016; 17(7):1128. https://doi.org/10.3390/ijms17071128
Chicago/Turabian StyleChiu, Chun-Hung, Chun-Chao Chang, Shiang-Ting Lin, Charng-Cherng Chyau, and Robert Y. Peng. 2016. "Improved Hepatoprotective Effect of Liposome-Encapsulated Astaxanthin in Lipopolysaccharide-Induced Acute Hepatotoxicity" International Journal of Molecular Sciences 17, no. 7: 1128. https://doi.org/10.3390/ijms17071128
APA StyleChiu, C.-H., Chang, C.-C., Lin, S.-T., Chyau, C.-C., & Peng, R. Y. (2016). Improved Hepatoprotective Effect of Liposome-Encapsulated Astaxanthin in Lipopolysaccharide-Induced Acute Hepatotoxicity. International Journal of Molecular Sciences, 17(7), 1128. https://doi.org/10.3390/ijms17071128