
Aberrant Lipid Metabolism Promotes Prostate Cancer: Role in Cell Survival under Hypoxia and Extracellular Vesicles Biogenesis


Abstract
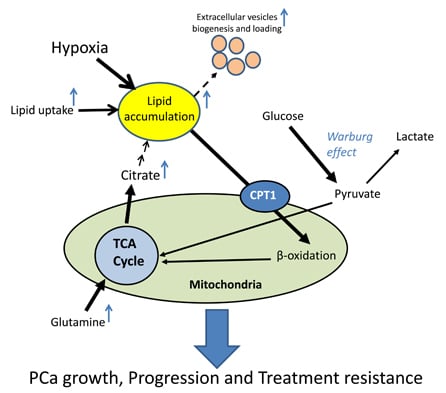
:
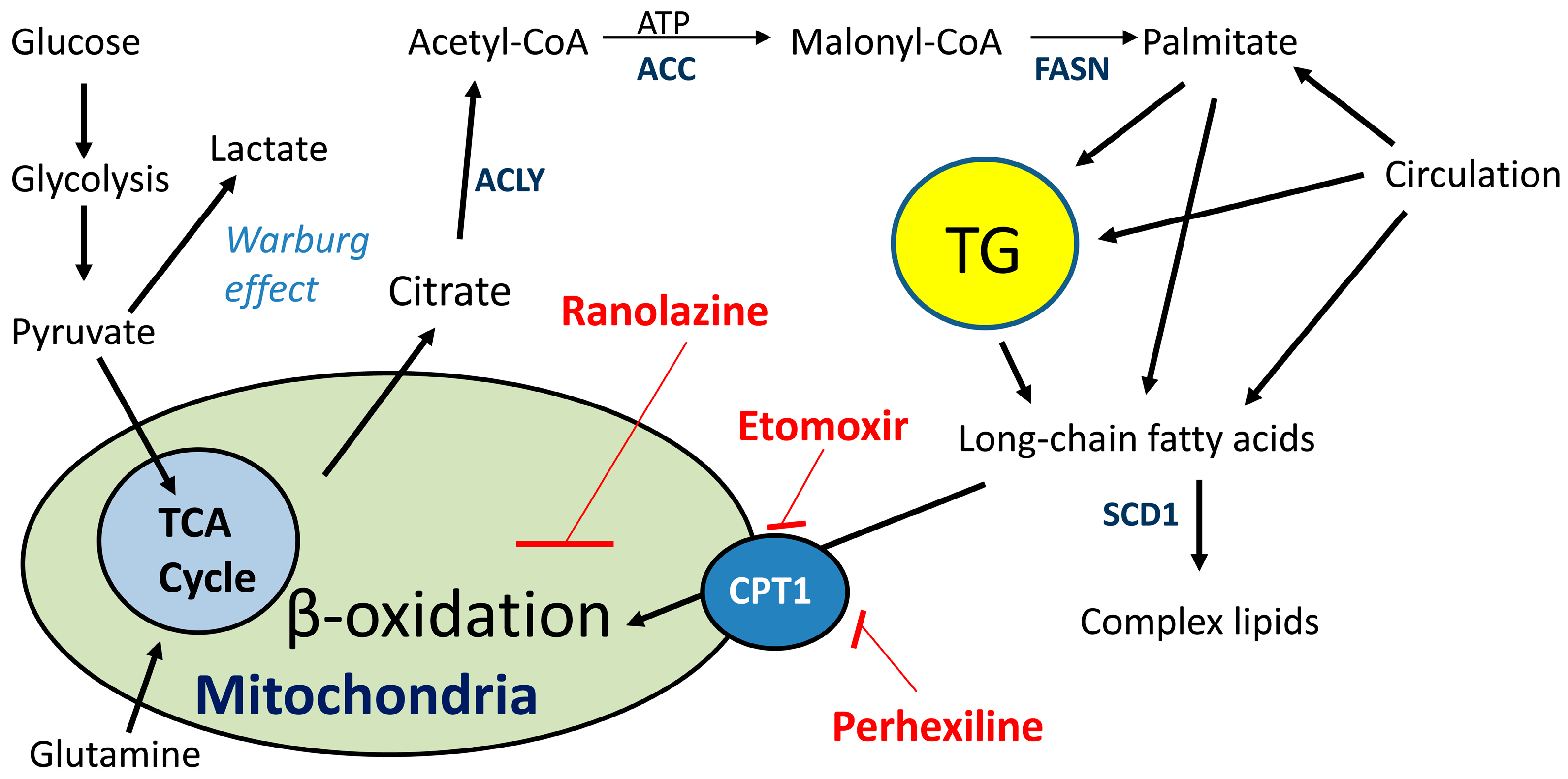
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Aberrant Lipid Metabolism in PCa (Prostate Cancer)
2.1. De Novo Lipid Synthesis Is a Characteristic of Cancer Cells
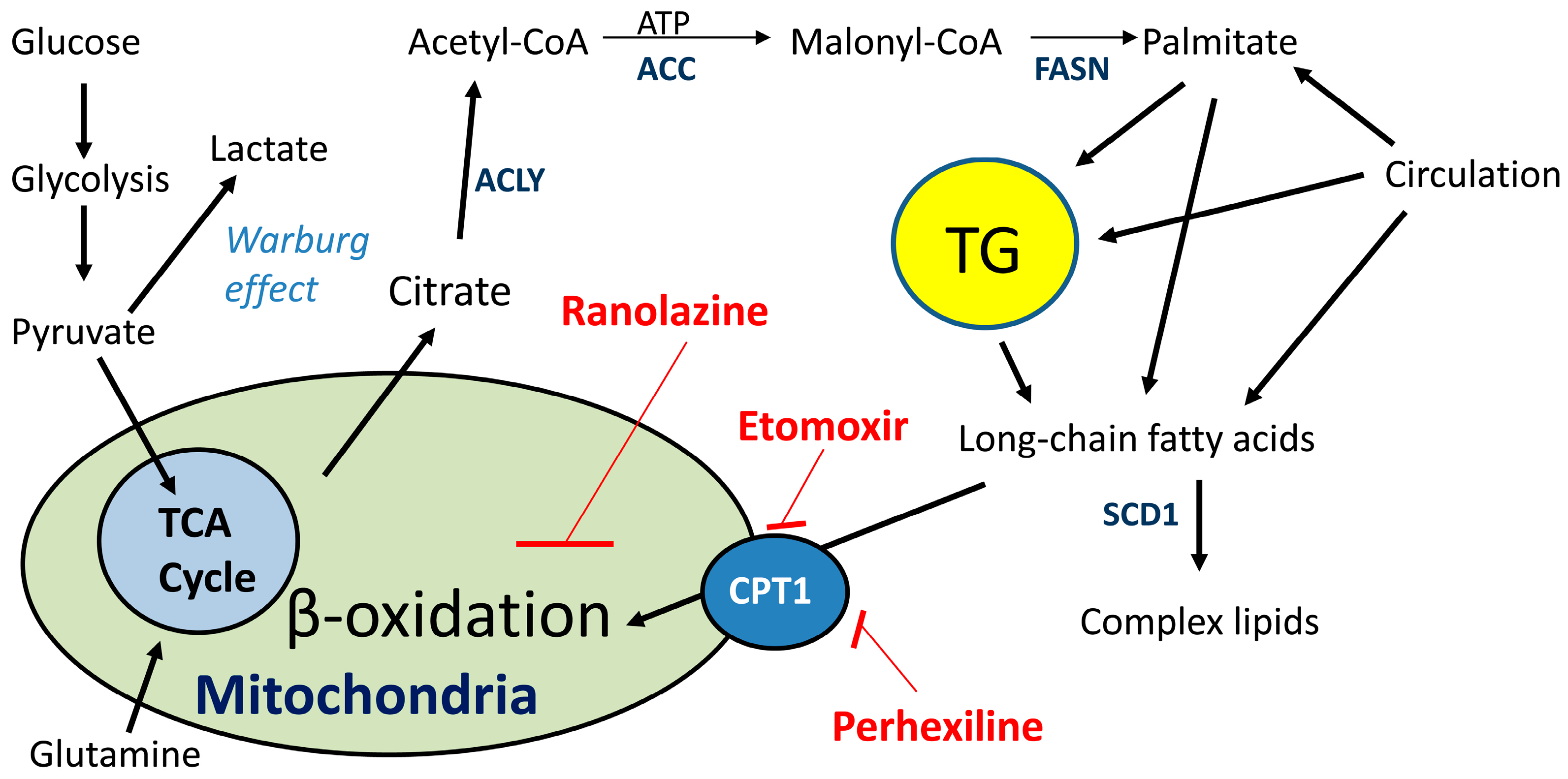
2.2. Lipid Oxidation Supports Cancer Growth and Survival
3. Hypoxia and Lipid Metabolism in PCa
3.1. Role of Hypoxia in PCa
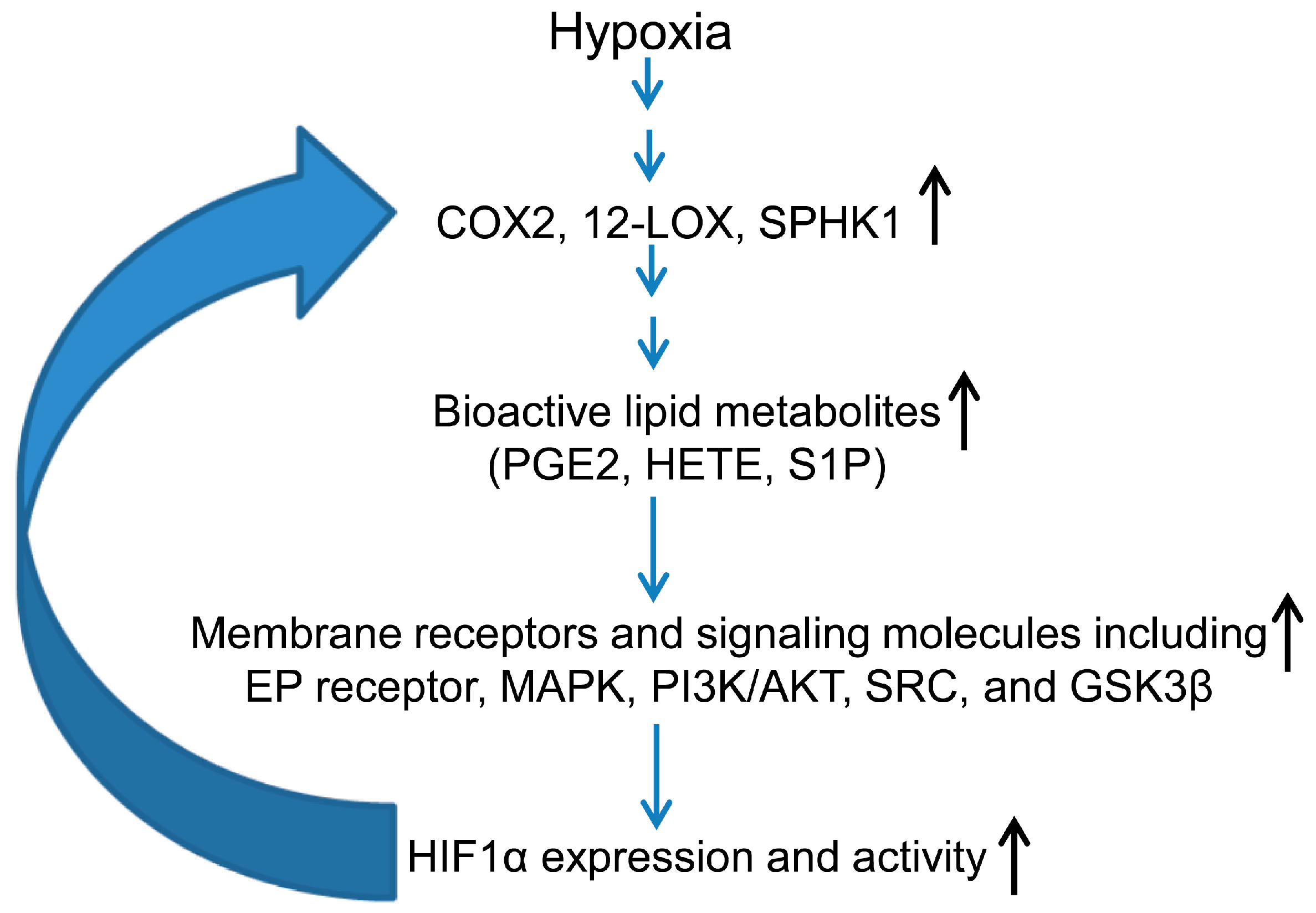
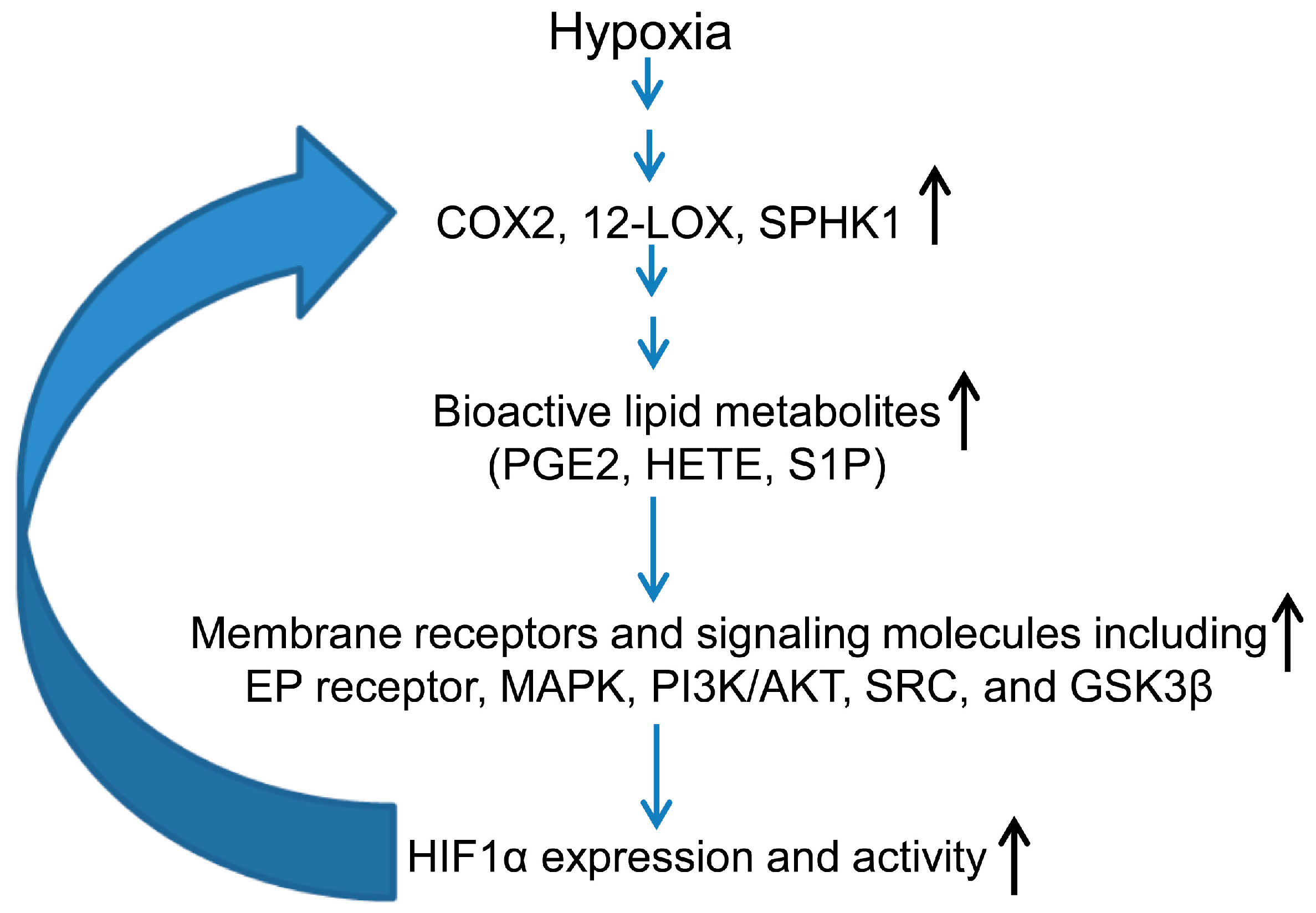
3.2. Hypoxia-Inducible Factor (HIF) and Lipid Mediators Exert a Positive Feedback Loop in Hypoxic PCa Cells
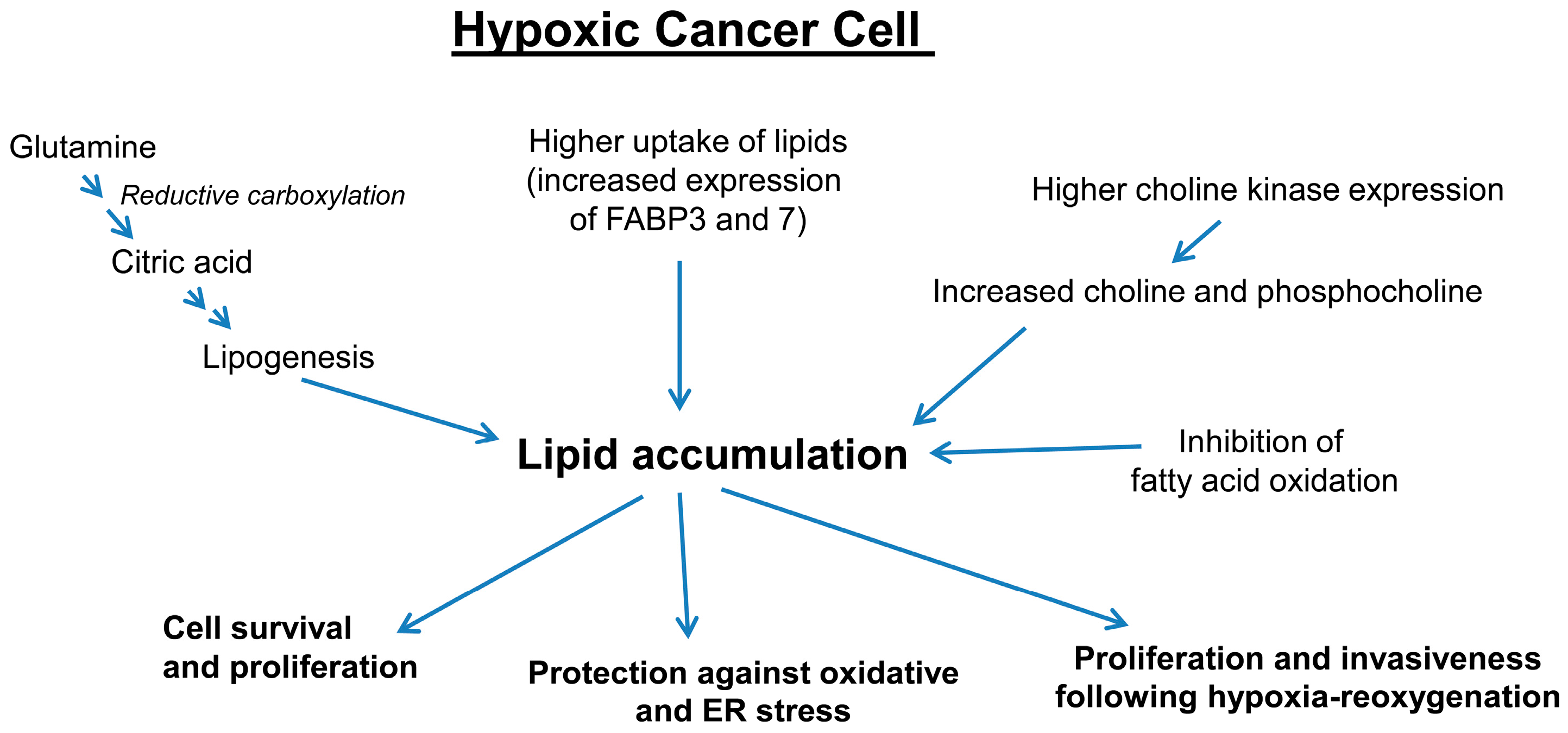
3.3. Hypoxia Promotes Lipid Accumulation in Cancer Cells
3.4. Role of Hypoxia-Induced Lipid Accumulation in Extracellular Vesicle Biogenesis
4. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yue, S.; Li, J.; Lee, S.Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Fritz, V.; Benfodda, Z.; Rodier, G.; Henriquet, C.; Iborra, F.; Avances, C.; Allory, Y.; de la Taille, A.; Culine, S.; Blancou, H.; et al. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol. Cancer Ther. 2010, 9, 1740–1754. [Google Scholar] [CrossRef] [PubMed]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; van Veldhoven, P.P.; Waltregny, D.; Daniels, V.W.; Machiels, J.; et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Nambiar, D.K.; Ramteke, A.; Kumar, R.; Dhar, D.; Agarwal, C.; Bergman, B.; Graner, M.; Maroni, P.; Singh, R.P.; et al. Hypoxia induces triglycerides accumulation in prostate cancer cells and extracellular vesicles supporting growth and invasiveness following reoxygenation. Oncotarget 2015, 6, 22836–22856. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar] [PubMed]
- Kuhajda, F.P. Fatty acid synthase and cancer: New application of an old pathway. Cancer Res. 2006, 66, 5977–5980. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, J.V.; van Veldhoven, P.P.; Esquenet, M.; Heyns, W.; Verhoeven, G. Androgens markedly stimulate the accumulation of neutral lipids in the human prostatic adenocarcinoma cell line LNCaP. Endocrinology 1996, 137, 4468–4474. [Google Scholar] [PubMed]
- Cahoon, E.K.; Kitahara, C.M.; Ntowe, E.; Bowen, E.M.; Doody, M.M.; Alexander, B.H.; Lee, T.; Little, M.P.; Linet, M.S.; Freedman, D.M. Female estrogen-related factors and incidence of basal cell carcinoma in a nationwide US cohort. J. Clin. Oncol. 2015, 33, 4058–4065. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T.; Anderson, G.L.; Gass, M.; Lane, D.S.; Aragaki, A.K.; Kuller, L.H.; Manson, J.E.; Stefanick, M.L.; Ockene, J.; Sarto, G.E.; et al. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 2010, 304, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Chlebowski, R.T.; Stefanick, M.L.; Aragaki, A.K.; Rossouw, J.E.; Prentice, R.L.; Anderson, G.; Howard, B.V.; Thomson, C.A.; LaCroix, A.Z.; et al. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the women’s health initiative randomized trials. JAMA 2013, 310, 1353–1368. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Glode, L.M.; Hitz, C.A.; Pac, C.T.; Boyle, K.E.; Maroni, P.; Deep, G.; Agarwal, R.; Lucia, S.M.; Cramer, S.D.; et al. Inhibition of lipid oxidation increases glucose metabolism and enhances 2-deoxy-2-[18F]Fluoro-d-glucose uptake in prostate cancer mouse xenografts. Mol. Imaging Biol. 2015, 17, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Photopoulos, C.; Loda, M. The fat side of prostate cancer. Biochim. Biophys. Acta 2013, 1831, 1518–1532. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.; Vickers, A.J.; Roobol, M.; Eastham, J.; Scardino, P.; Lilja, H.; Hugosson, J. Prostate cancer screening: Facts, statistics, and interpretation in response to the US Preventive Services Task Force Review. J. Clin. Oncol. 2012, 30, 2581–2584. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.E.; Scher, H.I. Starving the addiction: New opportunities for durable suppression of AR signaling in prostate cancer. Clin. Cancer Res. 2009, 15, 4792–4798. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, J.V.; Brusselmans, K.; Verhoeven, G. Increased lipogenesis in cancer cells: New players, novel targets. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Suburu, J.; Chen, Y.Q. Lipids and prostate cancer. Prostaglandins Other Lipid Mediat. 2012, 98, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, S.L.; Sobel, R.; Whitmore, T.G.; Akbari, M.; Bradley, D.R.; Gleave, M.E.; Nelson, C.C. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res. 2004, 64, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Vavere, A.L.; Kridel, S.J.; Wheeler, F.B.; Lewis, J.S. 1-11C-acetate as a PET radiopharmaceutical for imaging fatty acid synthase expression in prostate cancer. J. Nucl. Med. 2008, 49, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.K. Dissecting lipid raft facilitated cell signaling pathways in cancer. Biochim. Biophys. Acta 2008, 1785, 182–206. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, N.; Chisholm, J.W.; Igal, R.A. Inhibition of stearoylCoA desaturase-1 inactivates acetyl-CoA carboxylase and impairs proliferation in cancer cells: Role of AMPK. PLoS ONE 2009, 4, e6812. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Hitz, C.A.; Gijon, M.A.; Bergman, B.C.; Eckel, R.H.; Jacobsen, B.M. Progestin modulates the lipid profile and sensitivity of breast cancer cells to docetaxel. Mol. Cell. Endocrinol. 2012, 363, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.E.; Antoniou, A.; Villalobos-Menuey, E.; Russo, A.; Trauger, R.; Vendemelio, M.; George, A.; Bartholomew, R.; Carlo, D.; Shaikh, A.; et al. Characterization of a novel metabolic strategy used by drug-resistant tumor cells. FASEB J. 2002, 16, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta 2011, 1807, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Gatza, M.L.; Silva, G.O.; Parker, J.S.; Fan, C.; Perou, C.M. An integrated genomics approach identifies drivers of proliferation in luminal-subtype human breast cancer. Nat. Genet. 2014, 46, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Yu, J.; Laxman, B.; Rhodes, D.R.; Mehra, R.; Tomlins, S.A.; Shah, R.B.; Chandran, U.; Monzon, F.A.; Becich, M.J.; et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005, 8, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, M.R.; Mirabilii, S.; Allegretti, M.; Licchetta, R.; Calarco, A.; Torrisi, M.R.; Foa, R.; Nicolai, R.; Peluso, G.; Tafuri, A. Targeting the leukemia cell metabolism by the CPT1α inhibition: Functional preclinical effects in leukemias. Blood 2015, 126, 1925–1929. [Google Scholar] [CrossRef] [PubMed]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Hernlund, E.; Ihrlund, L.S.; Khan, O.; Ates, Y.O.; Linder, S.; Panaretakis, T.; Shoshan, M.C. Potentiation of chemotherapeutic drugs by energy metabolism inhibitors 2-deoxyglucose and etomoxir. Int. J. Cancer 2008, 123, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijon, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glode, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Amin, M.B.; Beltran, H.; Lotan, T.L.; Mosquera, J.M.; Reuter, V.E.; Robinson, B.D.; Troncoso, P.; Rubin, M.A. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am. J. Surg. Pathol. 2014, 38, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Shappell, S.B.; Boeglin, W.E.; Olson, S.J.; Kasper, S.; Brash, A.R. 15-lipoxygenase-2 (15-LOX-2) is expressed in benign prostatic epithelium and reduced in prostate adenocarcinoma. Am. J. Pathol. 1999, 155, 235–245. [Google Scholar] [CrossRef]
- Tang, S.; Bhatia, B.; Maldonado, C.J.; Yang, P.; Newman, R.A.; Liu, J.; Chandra, D.; Traag, J.; Klein, R.D.; Fischer, S.M.; et al. Evidence that arachidonate 15-lipoxygenase 2 is a negative cell cycle regulator in normal prostate epithelial cells. J. Biol. Chem. 2002, 277, 16189–16201. [Google Scholar] [CrossRef] [PubMed]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Abozguia, K.; Clarke, K.; Lee, L.; Frenneaux, M. Modification of myocardial substrate use as a therapy for heart failure. Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 490–498. [Google Scholar] [CrossRef] [PubMed]
- DeGrado, T.R.; Bhattacharyya, F.; Pandey, M.K.; Belanger, A.P.; Wang, S. Synthesis and preliminary evaluation of 18-18F-fluoro-4-thia-oleate as a PET probe of fatty acid oxidation. J. Nucl. Med. 2010, 51, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Schweda, S.; Holubarsch, C. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin. Sci. 2000, 99, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, V.; Stanley, W.C.; Recchia, F.A. Modulating fatty acid oxidation in heart failure. Cardiovasc. Res. 2011, 90, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; Horowitz, J.D.; Frenneaux, M.P. Perhexiline. Cardiovasc. Drug Rev. 2007, 25, 76–97. [Google Scholar] [CrossRef] [PubMed]
- Maly, D.J.; Papa, F.R. Druggable sensors of the unfolded protein response. Nat. Chem. Biol. 2014, 10, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.E.; Chauhan, V.; Koong, A.C. The unfolded protein response: A novel component of the hypoxic stress response in tumors. Mol. Cancer Res. 2005, 3, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Holz, M.S.; Janning, A.; Renne, C.; Gattenlohner, S.; Spieker, T.; Brauninger, A. Induction of endoplasmic reticulum stress by sorafenib and activation of NF-κB by lestaurtinib as a novel resistance mechanism in Hodgkin lymphoma cell lines. Mol. Cancer Ther. 2013, 12, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Koong, A.C.; Chauhan, V.; Romero-Ramirez, L. Targeting XBP-1 as a novel anti-cancer strategy. Cancer Biol. Ther. 2006, 5, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wei, Y.; Pagliassotti, M.J. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology 2006, 147, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Pagliassotti, M.J. Saturated fatty acid-mediated endoplasmic reticulum stress and apoptosis are augmented by trans-10, cis-12-conjugated linoleic acid in liver cells. Mol. Cell. Biochem. 2007, 303, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Gentile, C.L.; Pagliassotti, M.J. Reduced endoplasmic reticulum luminal calcium links saturated fatty acid-mediated endoplasmic reticulum stress and cell death in liver cells. Mol. Cell. Biochem. 2009, 331, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, M.; Warde, P.; Menard, C.; Chung, P.; Toi, A.; Ishkanian, A.; McLean, M.; Pintilie, M.; Sykes, J.; Gospodarowicz, M.; et al. Tumor hypoxia predicts biochemical failure following radiotherapy for clinically localized prostate cancer. Clin. Cancer Res. 2012, 18, 2108–2114. [Google Scholar] [CrossRef] [PubMed]
- Ragnum, H.B.; Vlatkovic, L.; Lie, A.K.; Axcrona, K.; Julin, C.H.; Frikstad, K.M.; Hole, K.H.; Seierstad, T.; Lyng, H. The tumour hypoxia marker pimonidazole reflects a transcriptional programme associated with aggressive prostate cancer. Br. J. Cancer 2015, 112, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, W.K.; Xiao, L.; Kovac, S.; Chang, M.; Michiels, C.; Bolton, D.; Shulkes, A.; Baldwin, G.S.; Patel, O. The role of hypoxia-inducible factor 1α in determining the properties of castrate-resistant prostate cancers. PLoS ONE 2013, 8, e54251. [Google Scholar] [CrossRef] [PubMed]
- Vergis, R.; Corbishley, C.M.; Norman, A.R.; Bartlett, J.; Jhavar, S.; Borre, M.; Heeboll, S.; Horwich, A.; Huddart, R.; Khoo, V.; et al. Intrinsic markers of tumour hypoxia and angiogenesis in localised prostate cancer and outcome of radical treatment: A retrospective analysis of two randomised radiotherapy trials and one surgical cohort study. Lancet Oncol. 2008, 9, 342–351. [Google Scholar] [CrossRef]
- Zhong, H.; Semenza, G.L.; Simons, J.W.; de Marzo, A.M. Up-regulation of hypoxia-inducible factor 1α is an early event in prostate carcinogenesis. Cancer Detect. Prev. 2004, 28, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; de Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar] [PubMed]
- Yasuda, M.; Shimizu, M.; Fujita, M.; Miyazawa, M.; Tang, X.; Kajiwara, H.; Osamura, R.Y.; Shoji, S.; Tokunaga, M.; Terachi, T. Usefulness of hypoxia inducible factor-1α in evaluating the prostatic adenocarcinoma viability following neoadjuvant hormone therapy. Cancer Detect. Prev. 2007, 31, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, W.K.; Sengupta, S.; Williams, S.; Chang, M.; Shulkes, A.; Bolton, D.M.; Baldwin, G.; Patel, O. The effects of nonspecific HIF1α inhibitors on development of castrate resistance and metastases in prostate cancer. Cancer Med. 2014, 3, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Platz, E.A.; Yegnasubramanian, S.; Liu, J.O.; Chong, C.R.; Shim, J.S.; Kenfield, S.A.; Stampfer, M.J.; Willett, W.C.; Giovannucci, E.; Nelson, W.G. A novel two-stage, transdisciplinary study identifies digoxin as a possible drug for prostate cancer treatment. Cancer Discov. 2011, 1, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, Y.; Miyazaki, K. The critical impact of HIF-1α on gastric cancer biology. Cancers 2013, 5, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Pouyssegur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.D.; Ross, J.A.; McLaren, D.B.; Parker, C.C.; Habib, F.K.; Riddick, A.C. The relevance of a hypoxic tumour microenvironment in prostate cancer. BJU Int. 2010, 105, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Deep, G.; Panigrahi, G.K. Hypoxia-induced signaling promotes prostate cancer progression: Exosomes role as messenger of hypoxic response in tumor microenvironment. Crit. Rev. Oncog. 2015, 20, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Kelly, B.; Semenza, G.L. Vascular endothelial growth factor gene expression in colon cancer cells exposed to prostaglandin E2 is mediated by hypoxia-inducible factor 1. Cancer Res. 2003, 63, 2330–2334. [Google Scholar] [PubMed]
- Krishnamoorthy, S.; Jin, R.; Cai, Y.; Maddipati, K.R.; Nie, D.; Pages, G.; Tucker, S.C.; Honn, K.V. 12-Lipoxygenase and the regulation of hypoxia-inducible factor in prostate cancer cells. Exp. Cell Res. 2010, 316, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, S.; Honn, K.V. Eicosanoids and other lipid mediators and the tumor hypoxic microenvironment. Cancer Metastasis Rev. 2011, 30, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Kirschenbaum, A.; Yao, S.; Stearns, M.E.; Holland, J.F.; Claffey, K.; Levine, A.C. Upregulation of vascular endothelial growth factor by cobalt chloride-simulated hypoxia is mediated by persistent induction of cyclooxygenase-2 in a metastatic human prostate cancer cell line. Clin. Exp. Metastasis 1999, 17, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Kirschenbaum, A.; Lu, M.; Yao, S.; Dosoretz, A.; Holland, J.F.; Levine, A.C. Prostaglandin E2 induces hypoxia-inducible factor-1α stabilization and nuclear localization in a human prostate cancer cell line. J. Biol. Chem. 2002, 277, 50081–50086. [Google Scholar] [CrossRef] [PubMed]
- Ader, I.; Brizuela, L.; Bouquerel, P.; Malavaud, B.; Cuvillier, O. Sphingosine kinase 1: A new modulator of hypoxia inducible factor 1α during hypoxia in human cancer cells. Cancer Res. 2008, 68, 8635–8642. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of AKT and sterol regulatory element binding protein-1. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Sembongi, H.; Befani, C.; Liakos, P.; Siniossoglou, S.; Simos, G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J. Cell Sci. 2012, 125, 3485–3493. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Greenwood, T.R.; Artemov, D.; Raman, V.; Winnard, P.T., Jr.; Heeren, R.M.; Bhujwalla, Z.M.; Glunde, K. Localized hypoxia results in spatially heterogeneous metabolic signatures in breast tumor models. Neoplasia 2012, 14, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Glunde, K.; Shah, T.; Winnard, P.T., Jr.; Raman, V.; Takagi, T.; Vesuna, F.; Artemov, D.; Bhujwalla, Z.M. Hypoxia regulates choline kinase expression through hypoxia-inducible factor-1 α signaling in a human prostate cancer model. Cancer Res. 2008, 68, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Kourti, M.; Ikonomou, G.; Giakoumakis, N.N.; Rapsomaniki, M.A.; Landegren, U.; Siniossoglou, S.; Lygerou, Z.; Simos, G.; Mylonis, I. CK1δ restrains lipin-1 induction, lipid droplet formation and cell proliferation under hypoxia by reducing HIF-1α/ARNT complex formation. Cell Signal. 2015, 27, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Kalousi, A.; Mylonis, I.; Politou, A.S.; Chachami, G.; Paraskeva, E.; Simos, G. Casein kinase 1 regulates human hypoxia-inducible factor HIF-1. J. Cell Sci. 2010, 123, 2976–2986. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Ackerman, D.; Sanchez, D.J.; Li, B.; Ochocki, J.D.; Grazioli, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; Simon, M.C. HIF2α-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov. 2015, 5, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Bao, B.; Sarkar, F.H. Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Rev. 2013, 32, 623–642. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic, M.M.; Molina, H.; Kohsaka, S.; Di, G.A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Ramteke, A.; Ting, H.; Agarwal, C.; Mateen, S.; Somasagara, R.; Hussain, A.; Graner, M.; Frederick, B.; Agarwal, R.; Deep, G. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol. Carcinog. 2015, 54, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, H.; Umezu, T.; Ohyashiki, K.; Hirano, T.; Ohyashiki, J.H. Exosomes derived from hypoxic leukemia cells enhance tube formation in endothelial cells. J. Biol. Chem. 2013, 288, 34343–34351. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Tadokoro, H.; Azuma, K.; Yoshizawa, S.; Ohyashiki, K.; Ohyashiki, J.H. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood 2014, 124, 3748–3757. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Gilkes, D.M.; Takano, N.; Xiang, L.; Luo, W.; Bishop, C.J.; Chaturvedi, P.; Green, J.J.; Semenza, G.L. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. USA 2014, 111, E3234–E3242. [Google Scholar] [CrossRef] [PubMed]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012, 12, 421. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, X.; Yang, Y.; Zhu, H.; Chen, X.; Zhang, H.; Wang, F.; Qin, Q.; Cheng, H.; Sun, X. Exosomes: A promising factor involved in cancer hypoxic microenvironments. Curr. Med. Chem. 2015, 22, 4189–4195. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Izumi, Y.; Yamaguchi, T.; Yamazaki, T.; Tanaka, M.; Shiota, M.; Osada-Oka, M.; Nakamura, Y.; Wei, M.; Wanibuchi, H.; et al. Lipid synthesis is promoted by hypoxic adipocyte-derived exosomes in 3T3-L1 cells. Biochem. Biophys. Res. Commun. 2014, 445, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, C.; Wang, S.; Wang, Z.; Jiang, J.; Wang, W.; Li, X.; Chen, J.; Liu, K.; Li, C.; et al. Exosomes derived from hypoxic oral squamous cell carcinoma cells deliver miR-21 to normoxic cells to elicit a prometastatic phenotype. Cancer Res. 2016, 76, 1770–1780. [Google Scholar] [CrossRef] [PubMed]
- Berchem, G.; Noman, M.Z.; Bosseler, M.; Paggetti, J.; Baconnais, S.; Le, C.E.; Nanbakhsh, A.; Moussay, E.; Mami-Chouaib, F.; Janji, B.; et al. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-β and miR23a transfer. Oncoimmunology 2016, 5, e1062968. [Google Scholar] [CrossRef] [PubMed]
- Record, M.; Carayon, K.; Poirot, M.; Silvente-Poirot, S. Exosomes as new vesicular lipid transporters involved in cell–cell communication and various pathophysiologies. Biochim. Biophys. Acta 2014, 1841, 108–120. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deep, G.; Schlaepfer, I.R. Aberrant Lipid Metabolism Promotes Prostate Cancer: Role in Cell Survival under Hypoxia and Extracellular Vesicles Biogenesis. Int. J. Mol. Sci. 2016, 17, 1061. https://doi.org/10.3390/ijms17071061
Deep G, Schlaepfer IR. Aberrant Lipid Metabolism Promotes Prostate Cancer: Role in Cell Survival under Hypoxia and Extracellular Vesicles Biogenesis. International Journal of Molecular Sciences. 2016; 17(7):1061. https://doi.org/10.3390/ijms17071061
Chicago/Turabian StyleDeep, Gagan, and Isabel R. Schlaepfer. 2016. "Aberrant Lipid Metabolism Promotes Prostate Cancer: Role in Cell Survival under Hypoxia and Extracellular Vesicles Biogenesis" International Journal of Molecular Sciences 17, no. 7: 1061. https://doi.org/10.3390/ijms17071061
APA StyleDeep, G., & Schlaepfer, I. R. (2016). Aberrant Lipid Metabolism Promotes Prostate Cancer: Role in Cell Survival under Hypoxia and Extracellular Vesicles Biogenesis. International Journal of Molecular Sciences, 17(7), 1061. https://doi.org/10.3390/ijms17071061