
Structure and Functional Diversity of GCN5-Related N-Acetyltransferases (GNAT)


Abstract
:1. Introduction
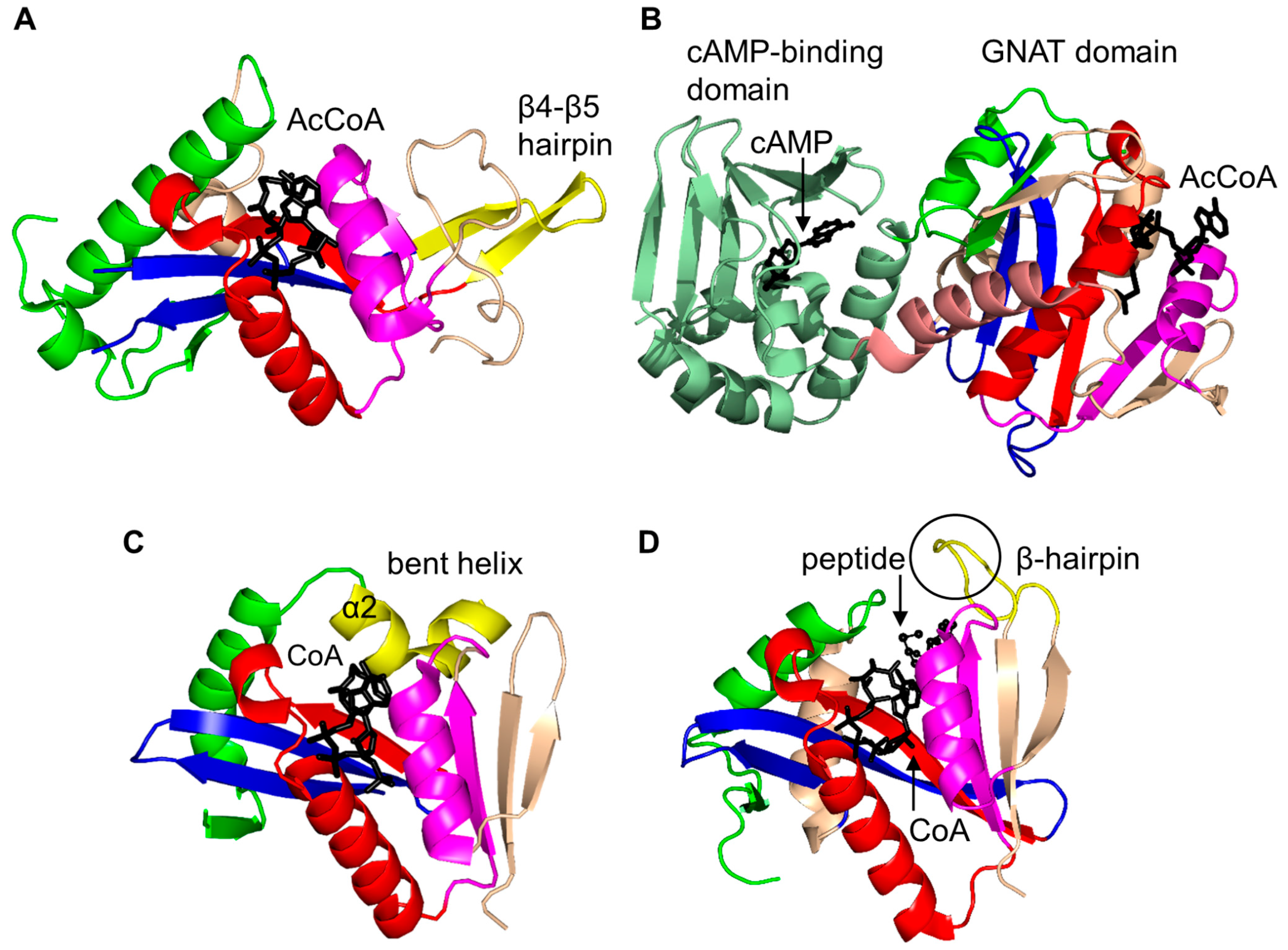
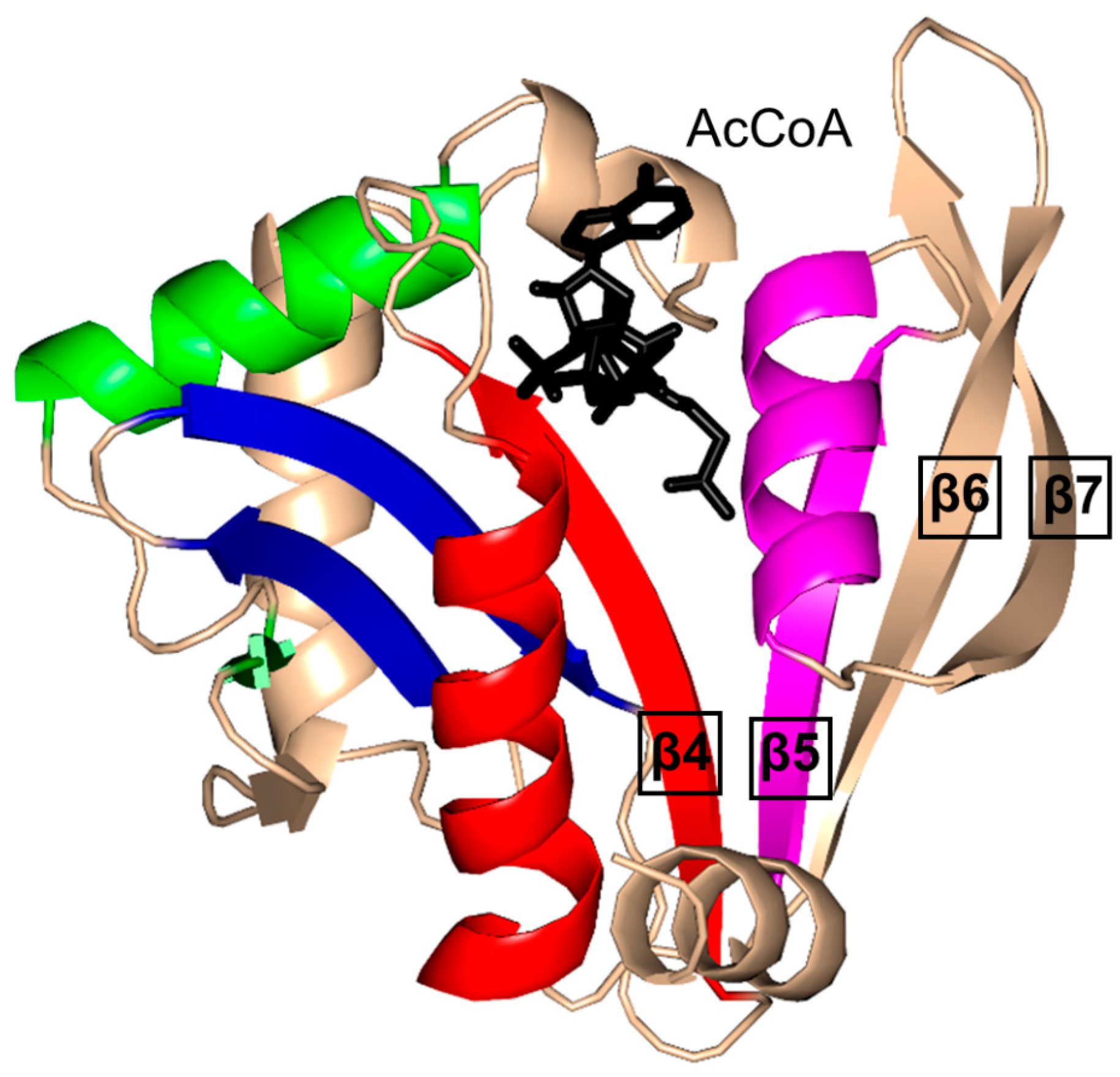
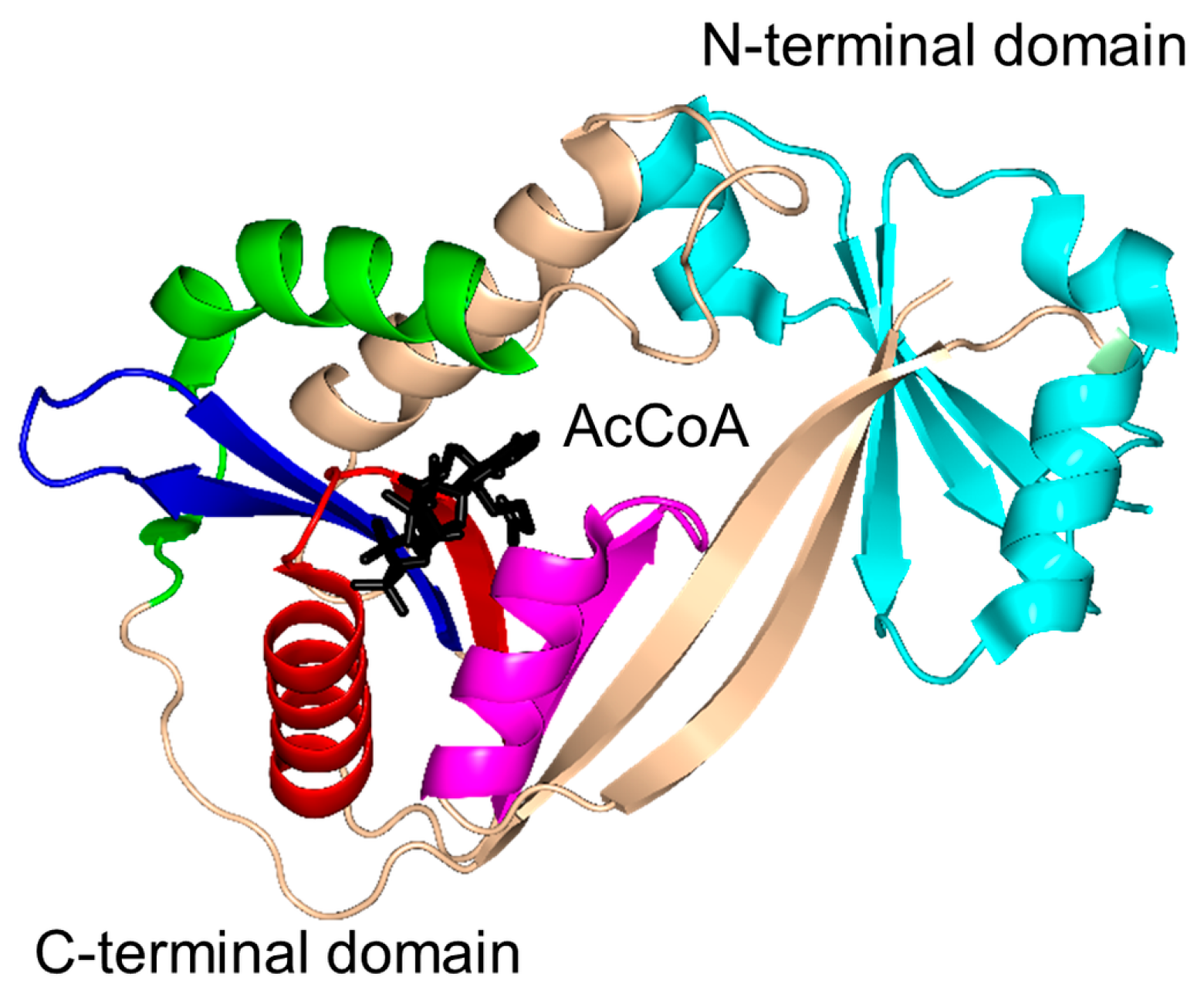
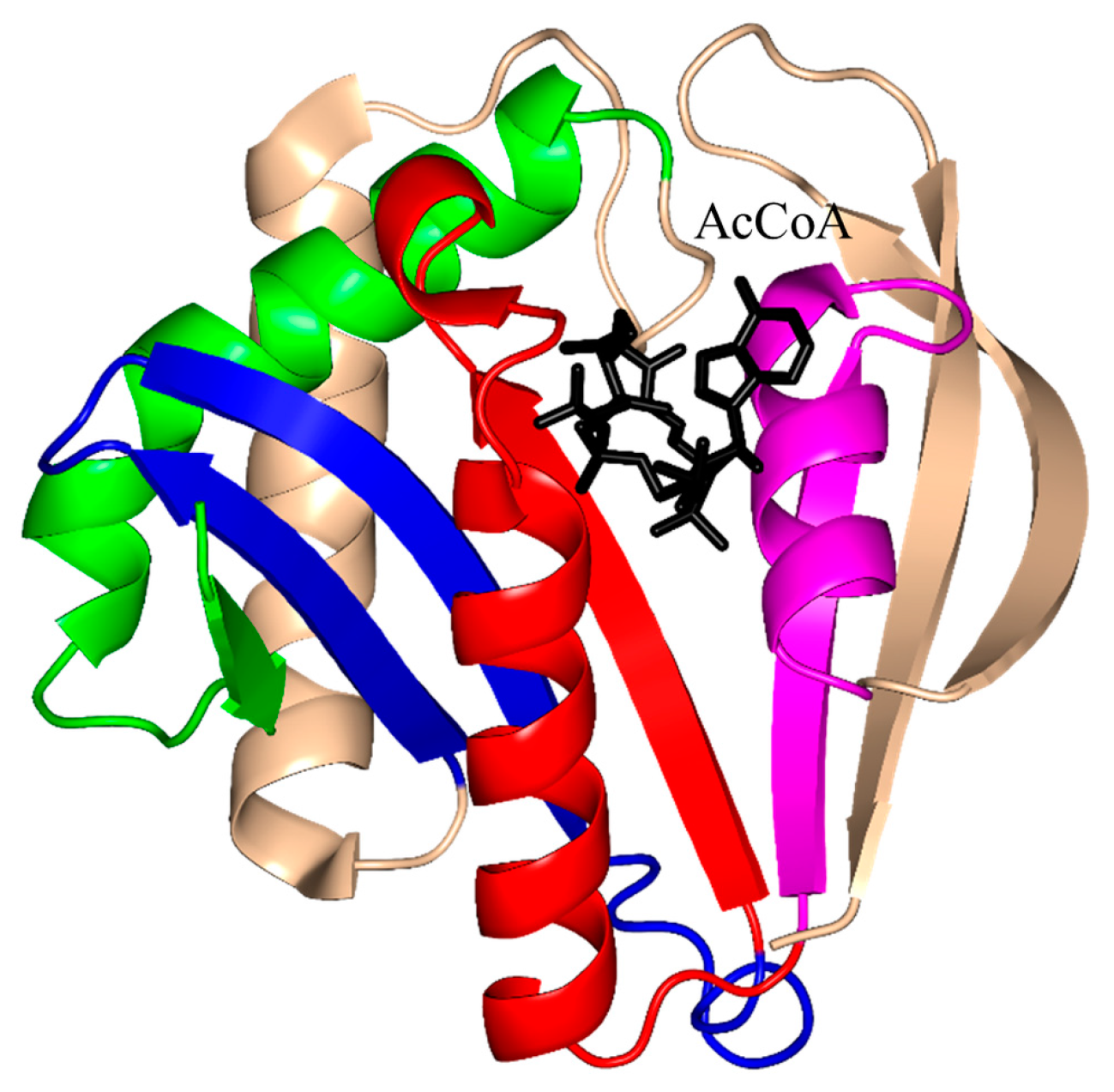
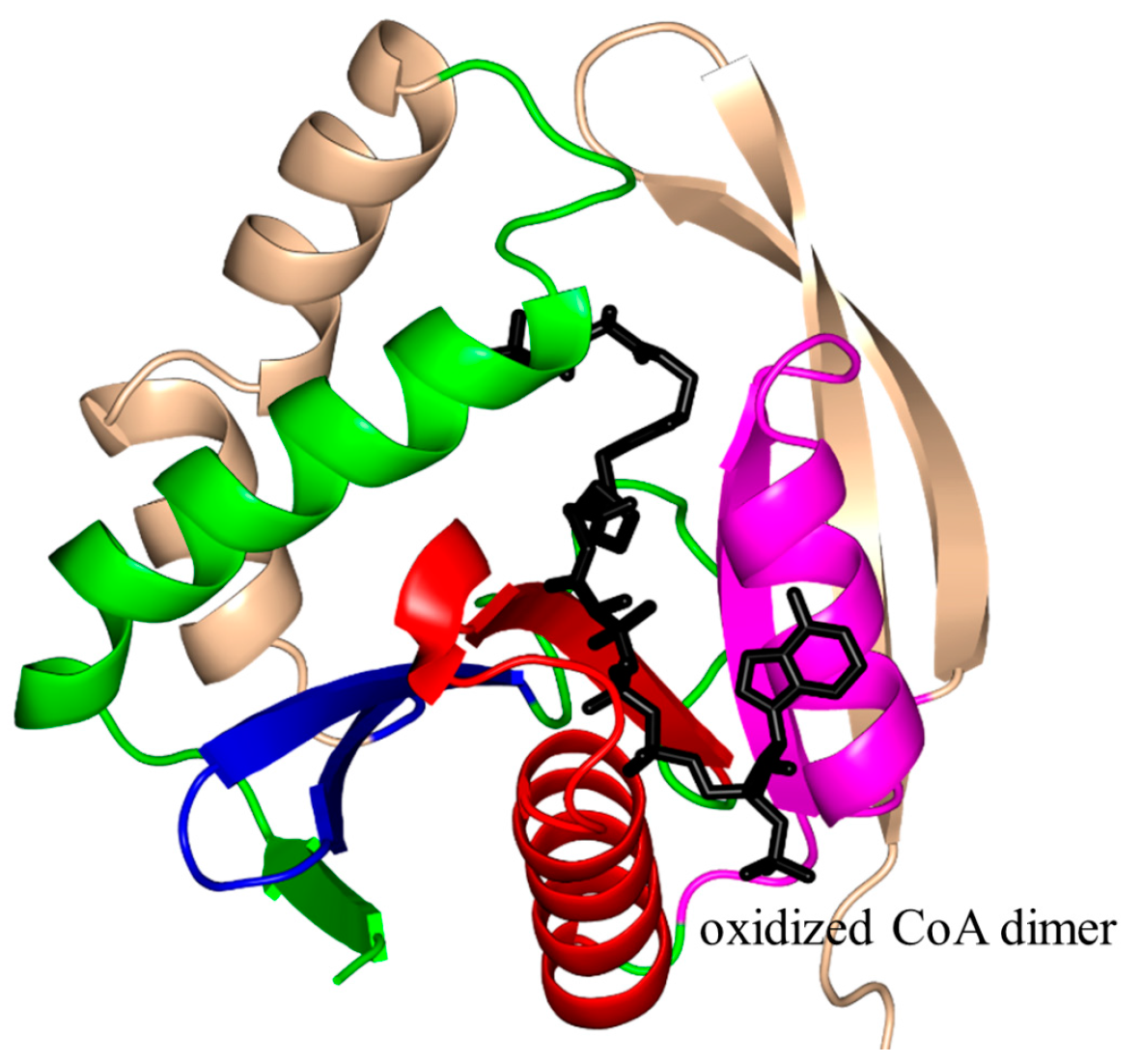
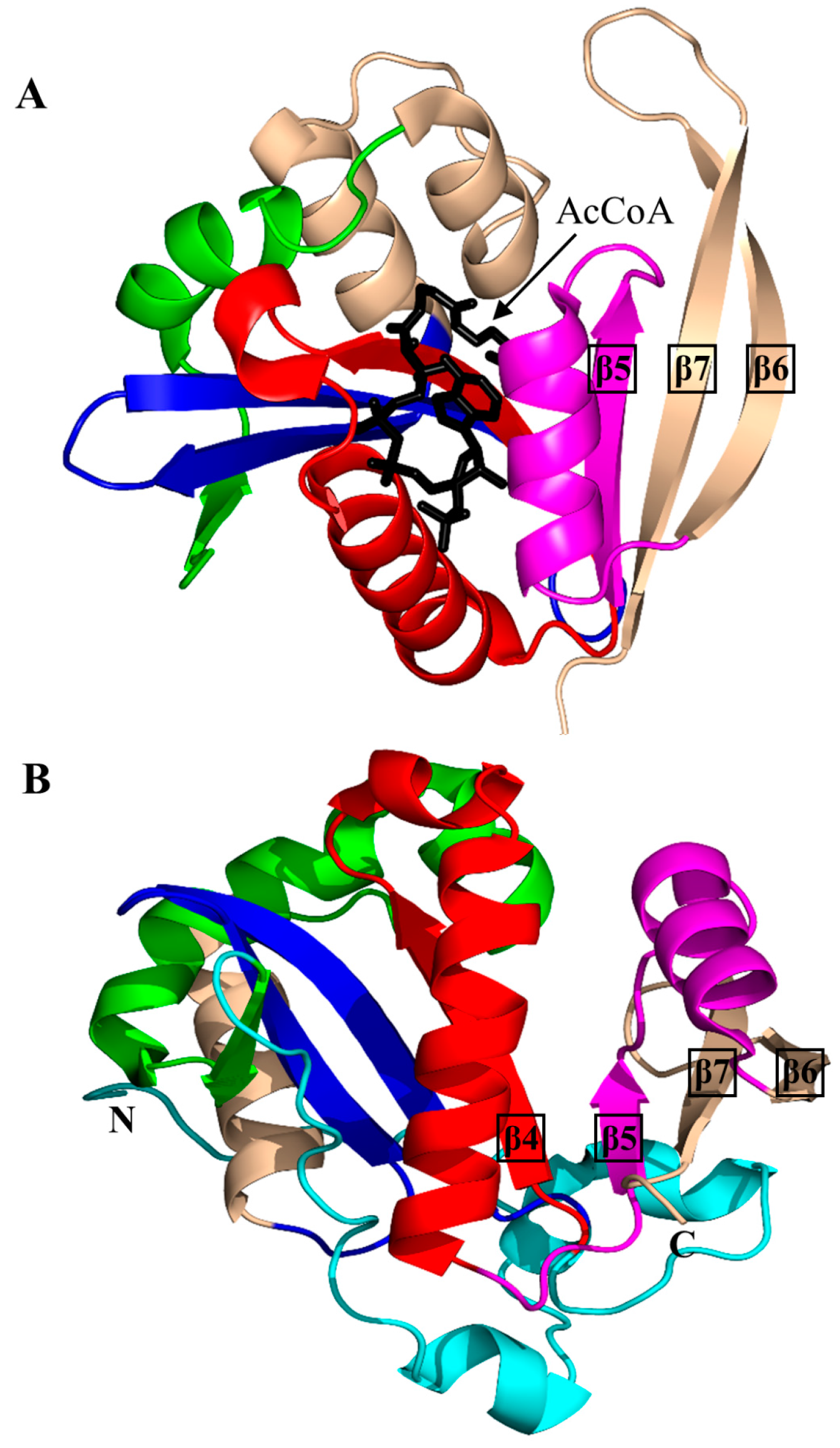
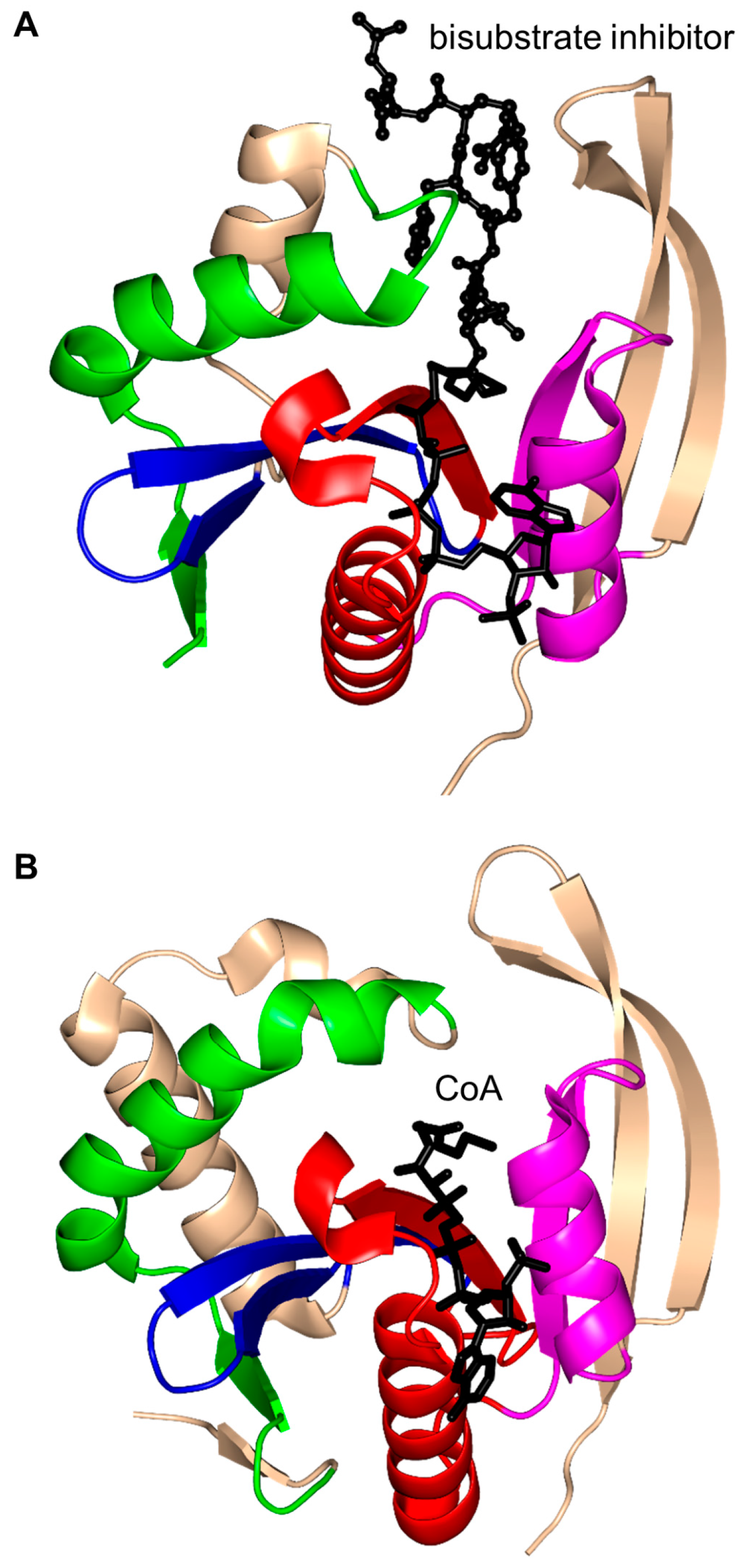
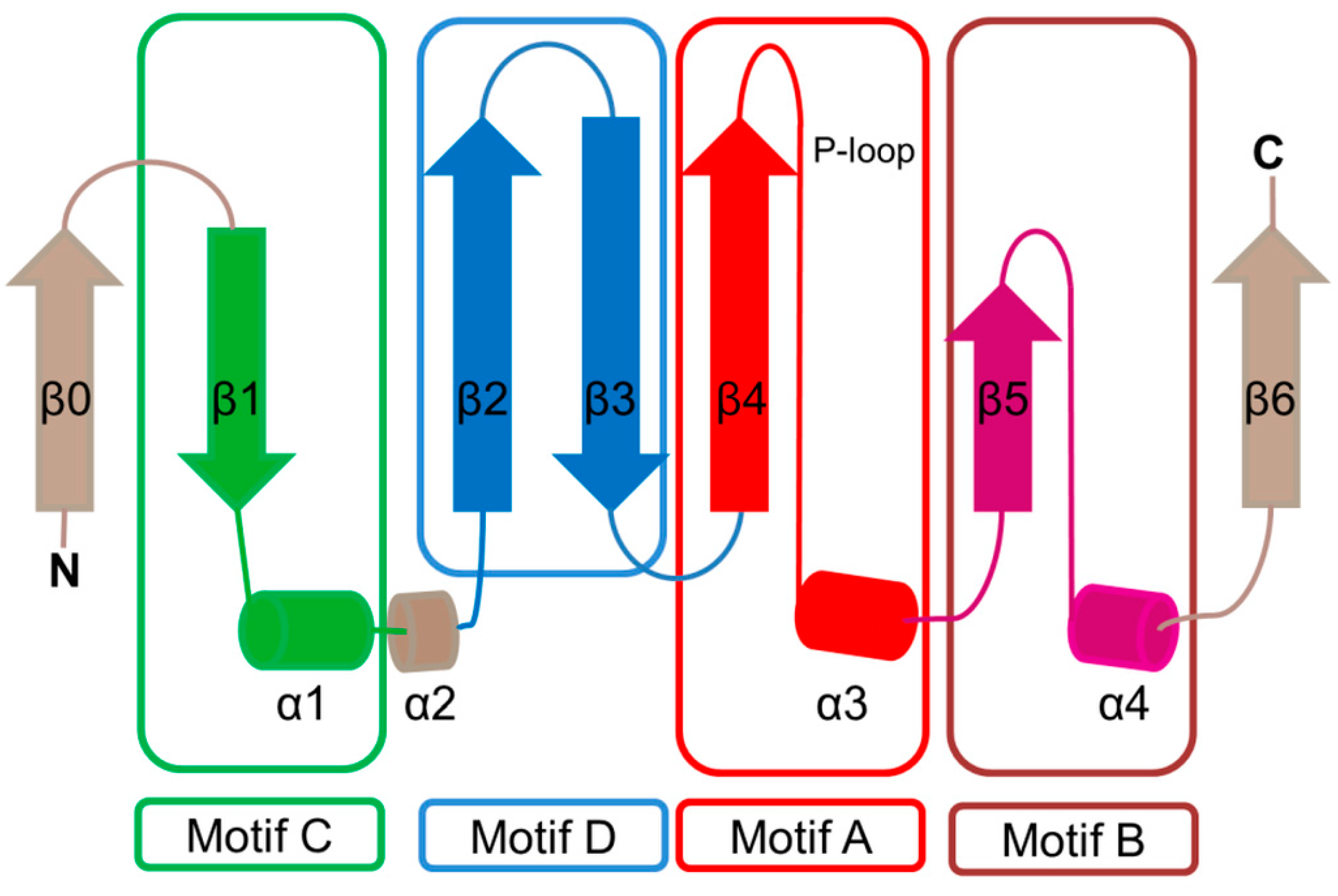
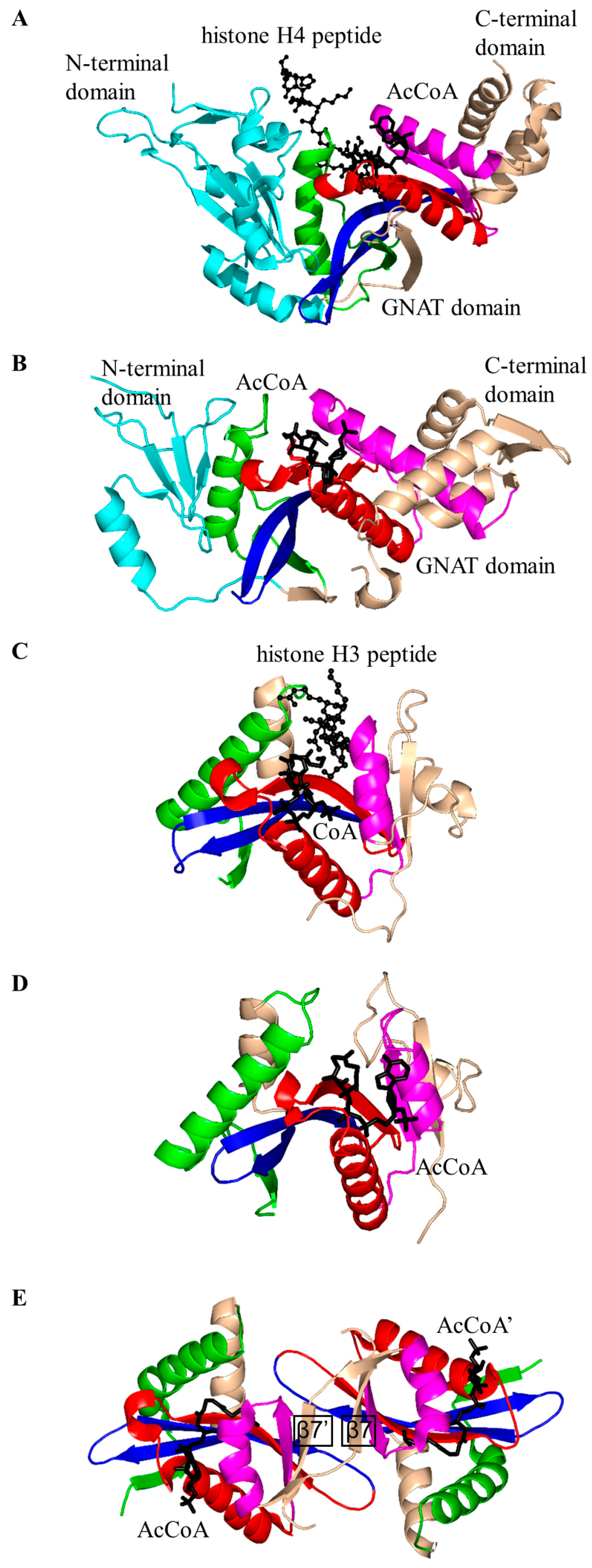
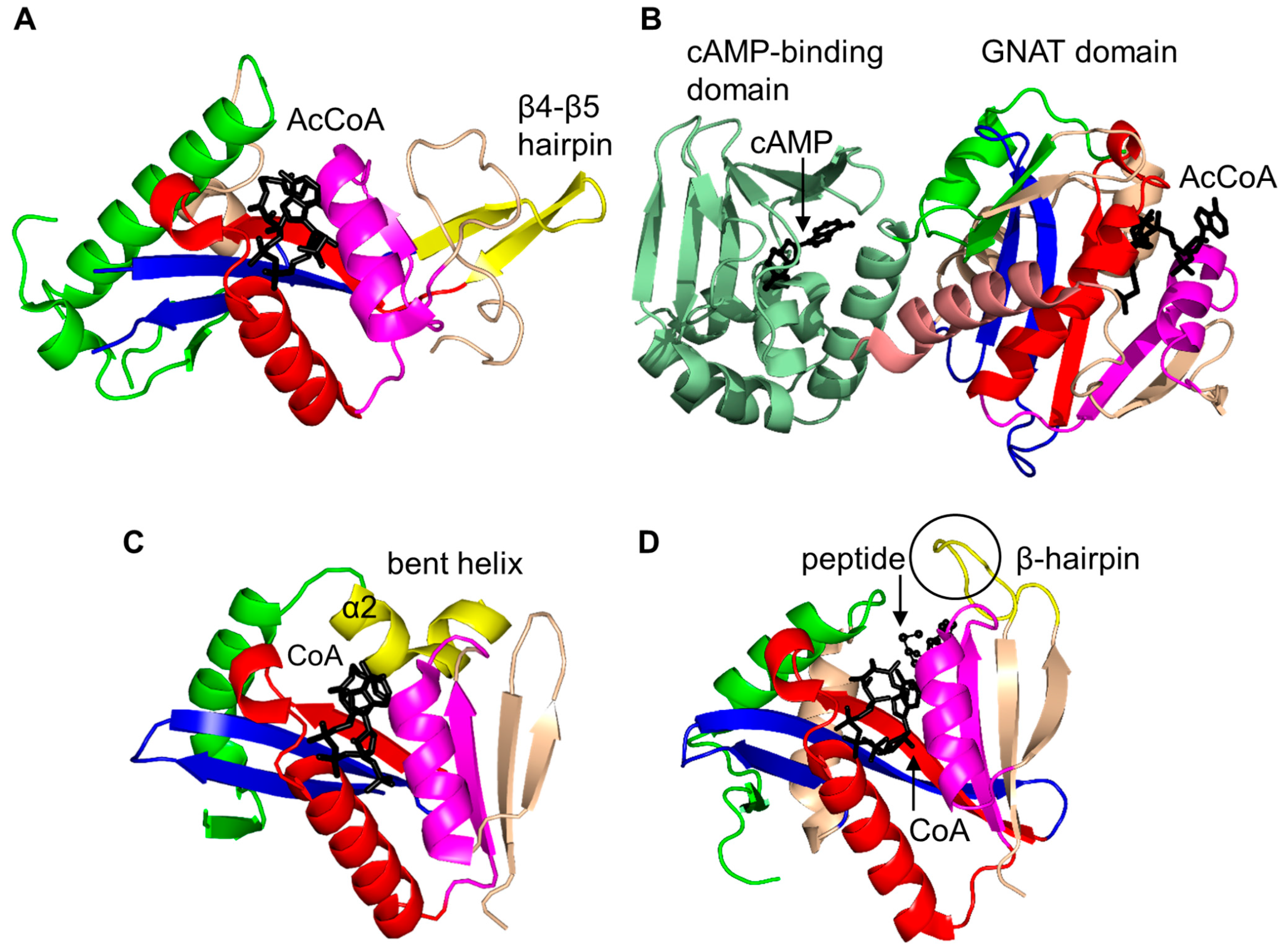
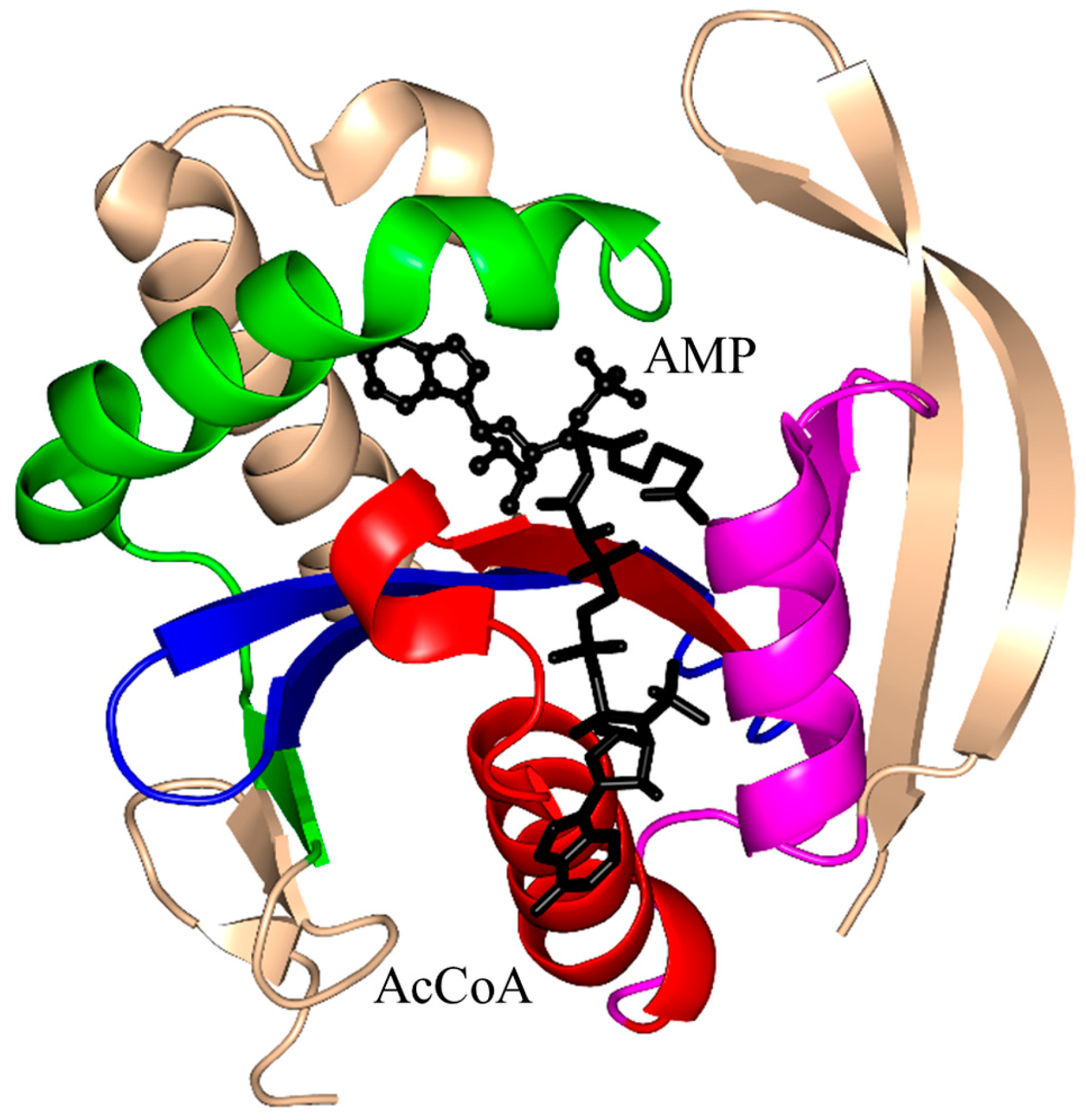
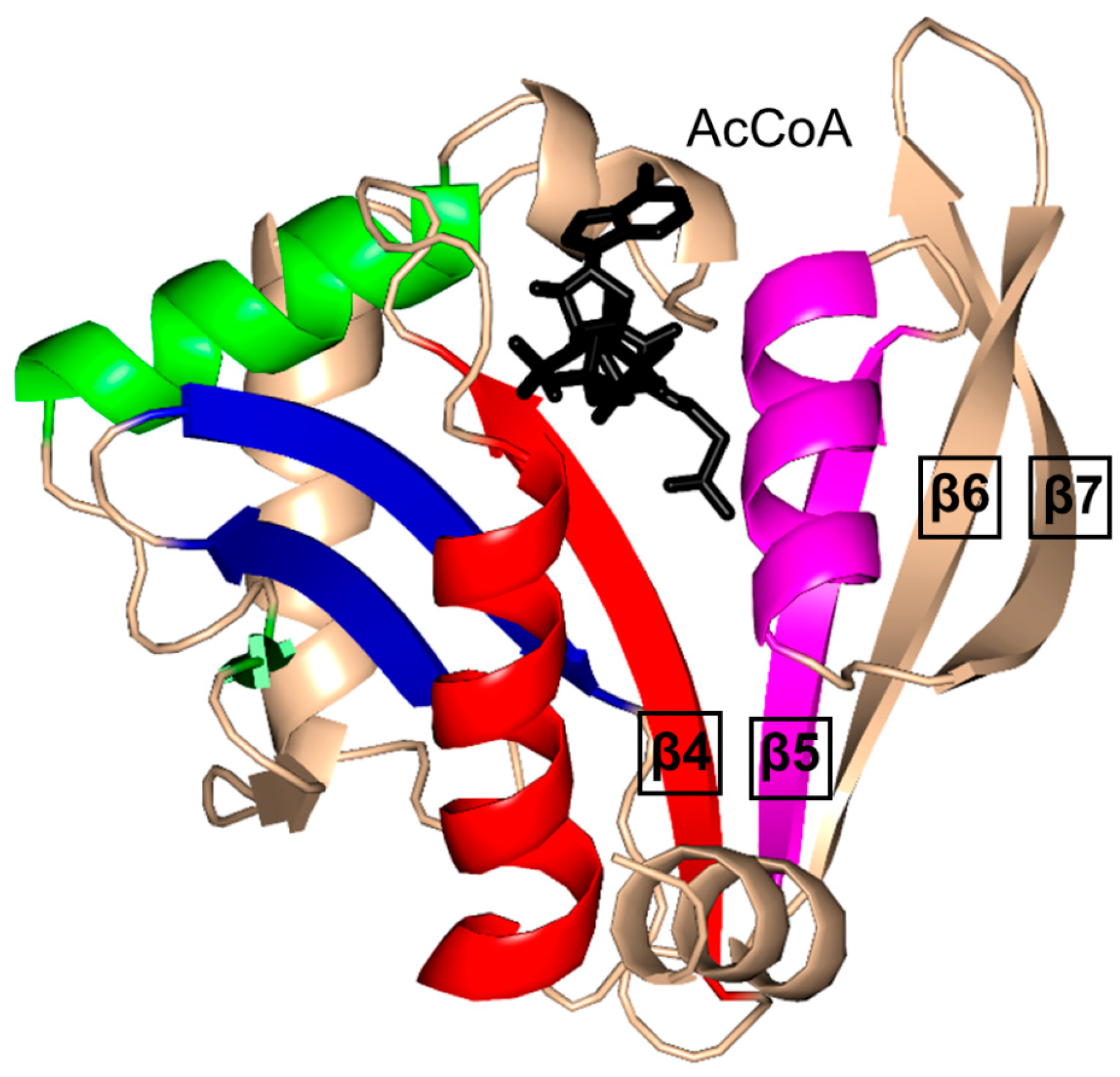
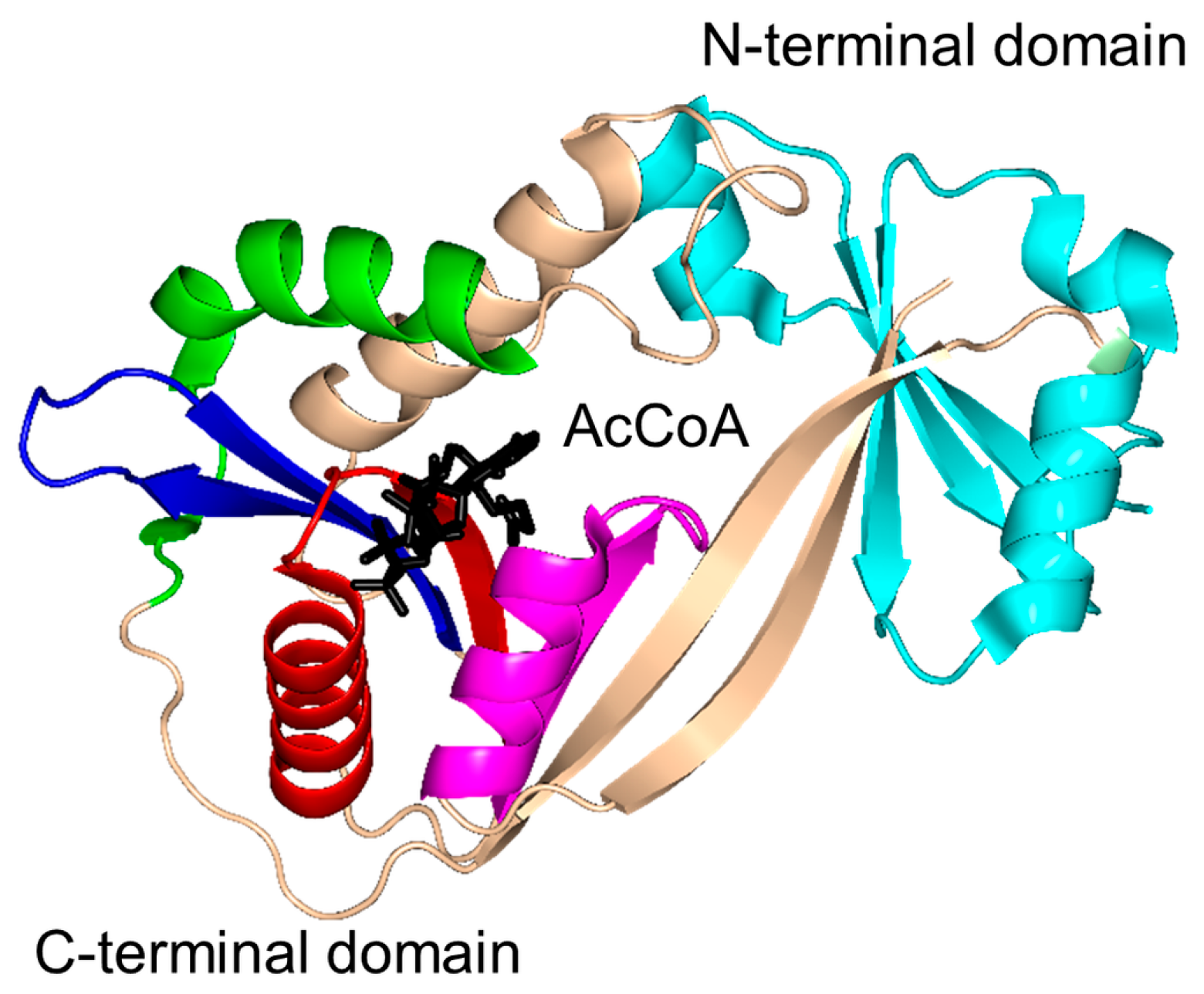
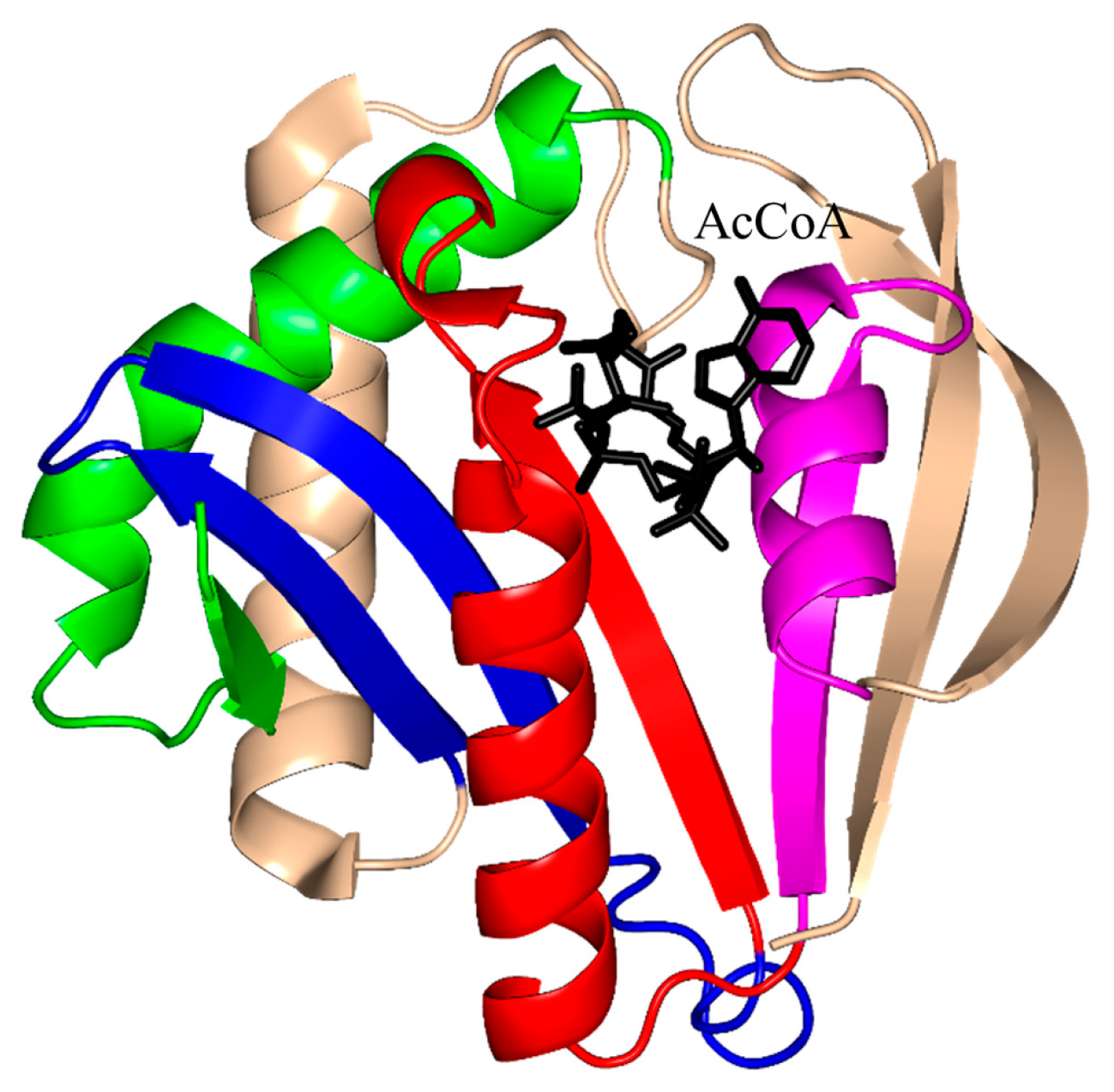
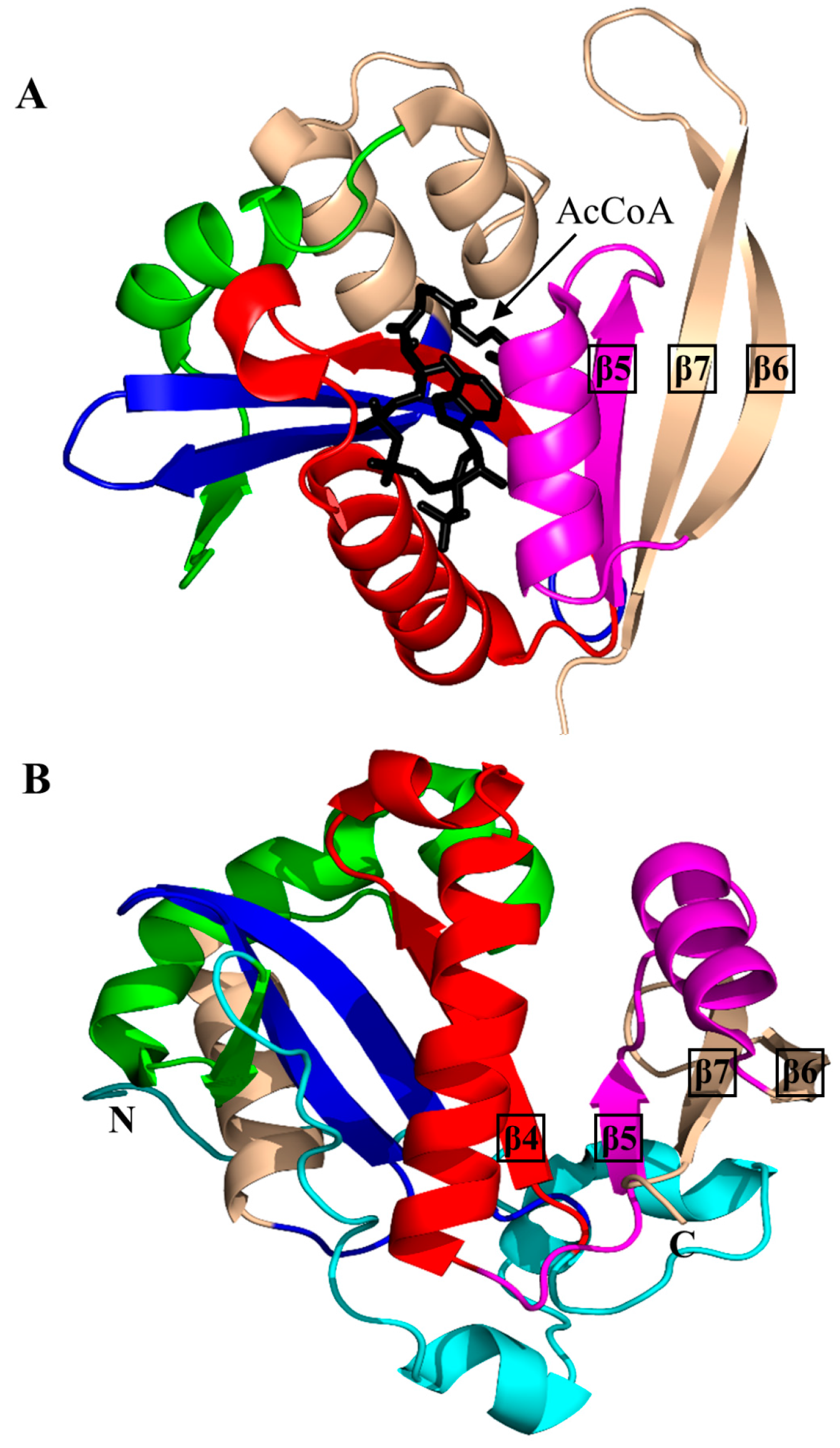
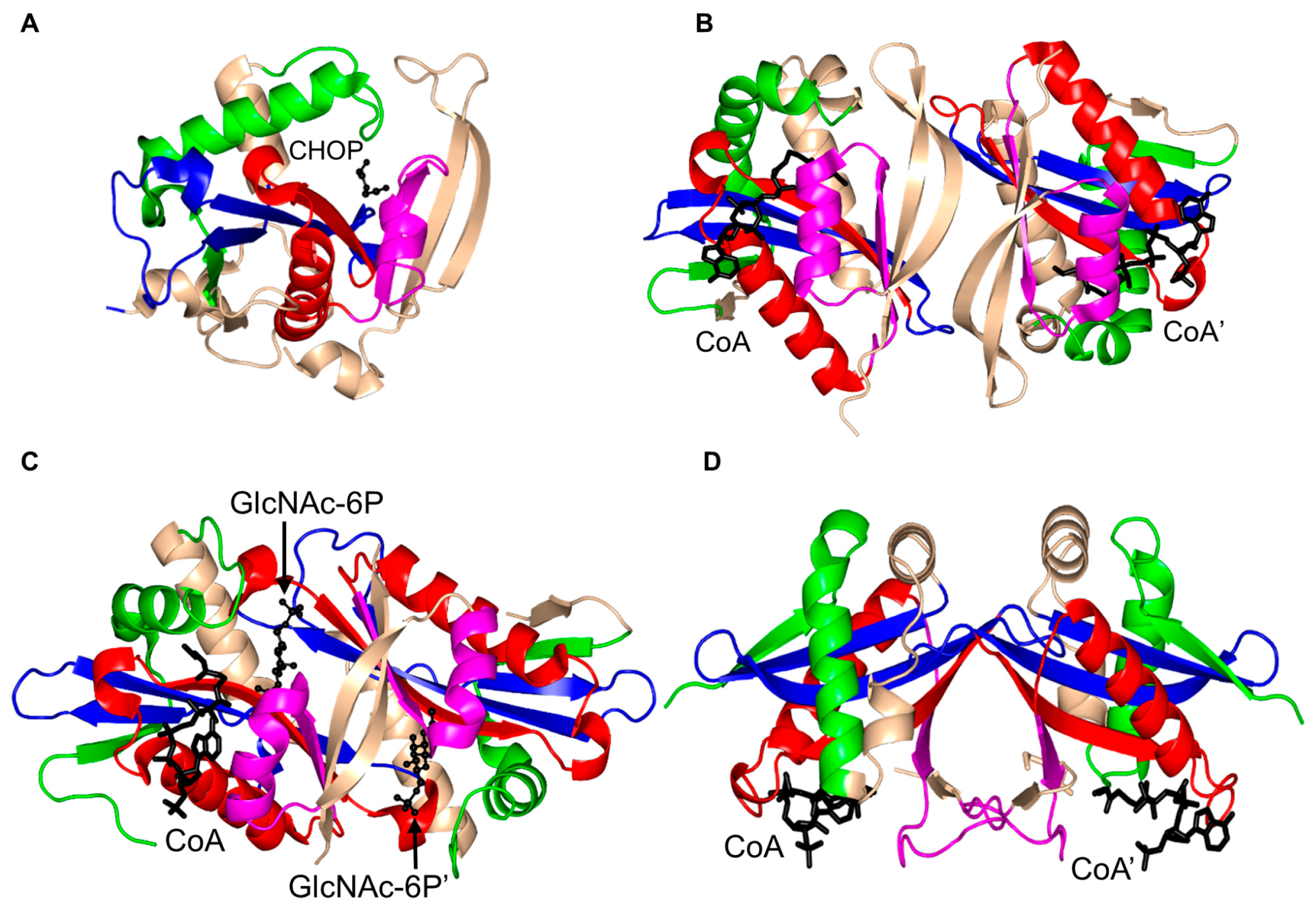
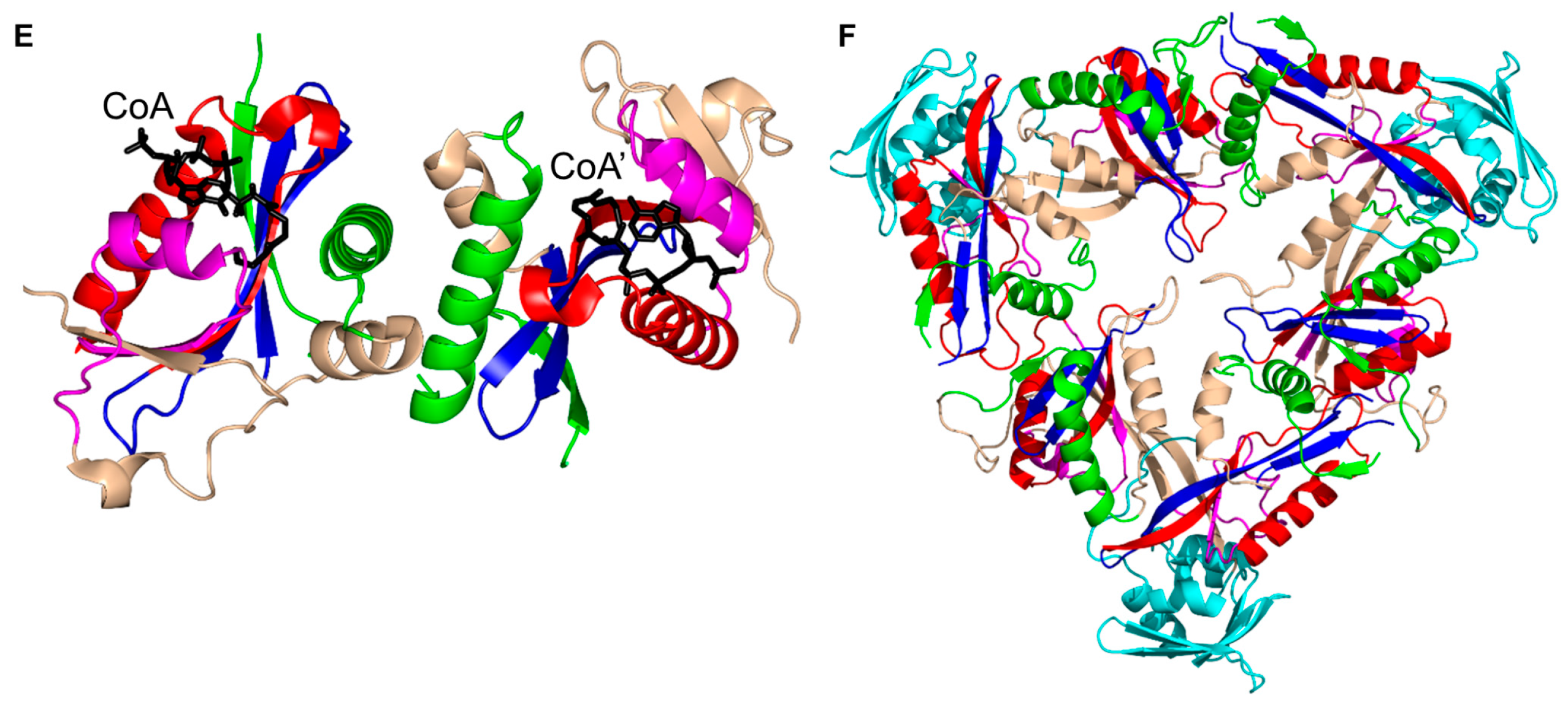
2. Overall Architecture of General Control Non-Repressible 5 (GCN5)-Related N-Acetyltransferase (GNAT) Superfamily Enzymes
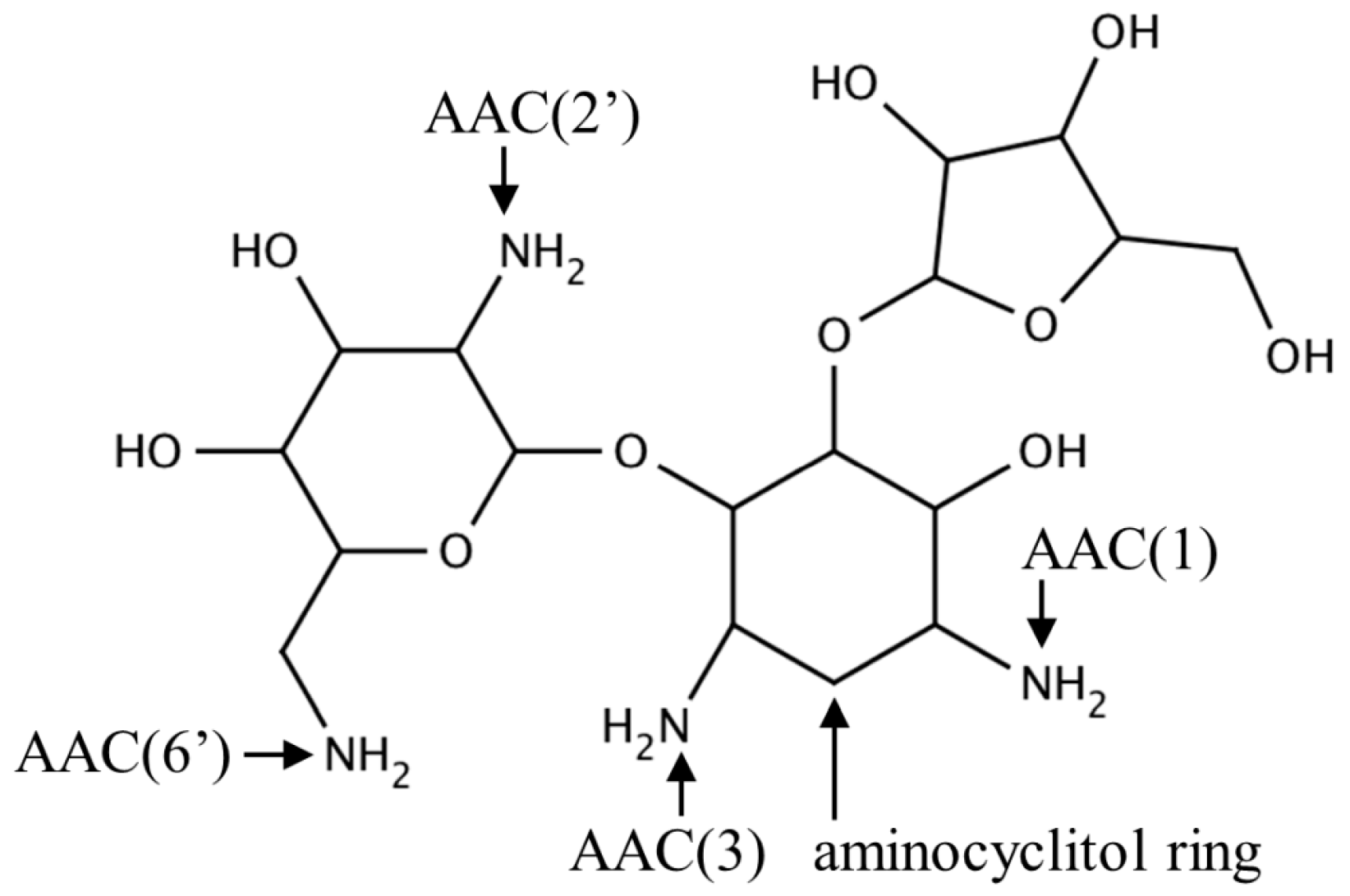
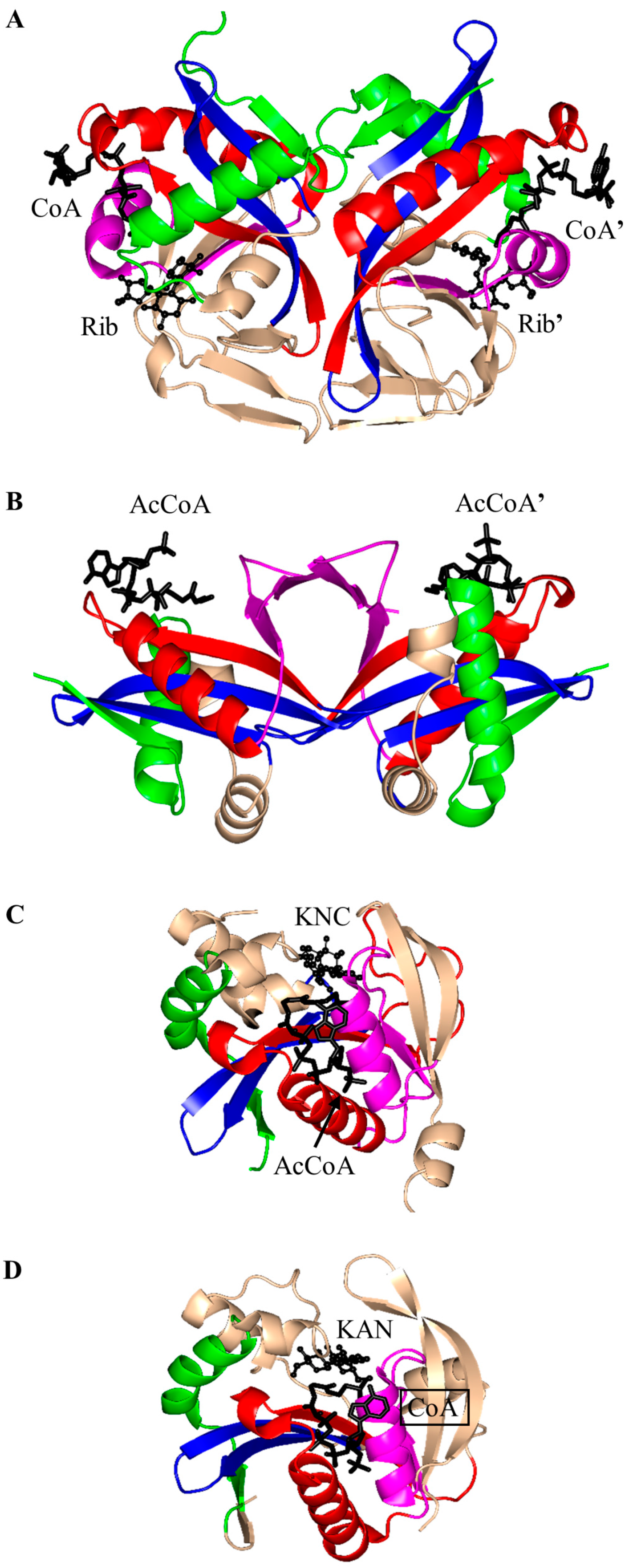
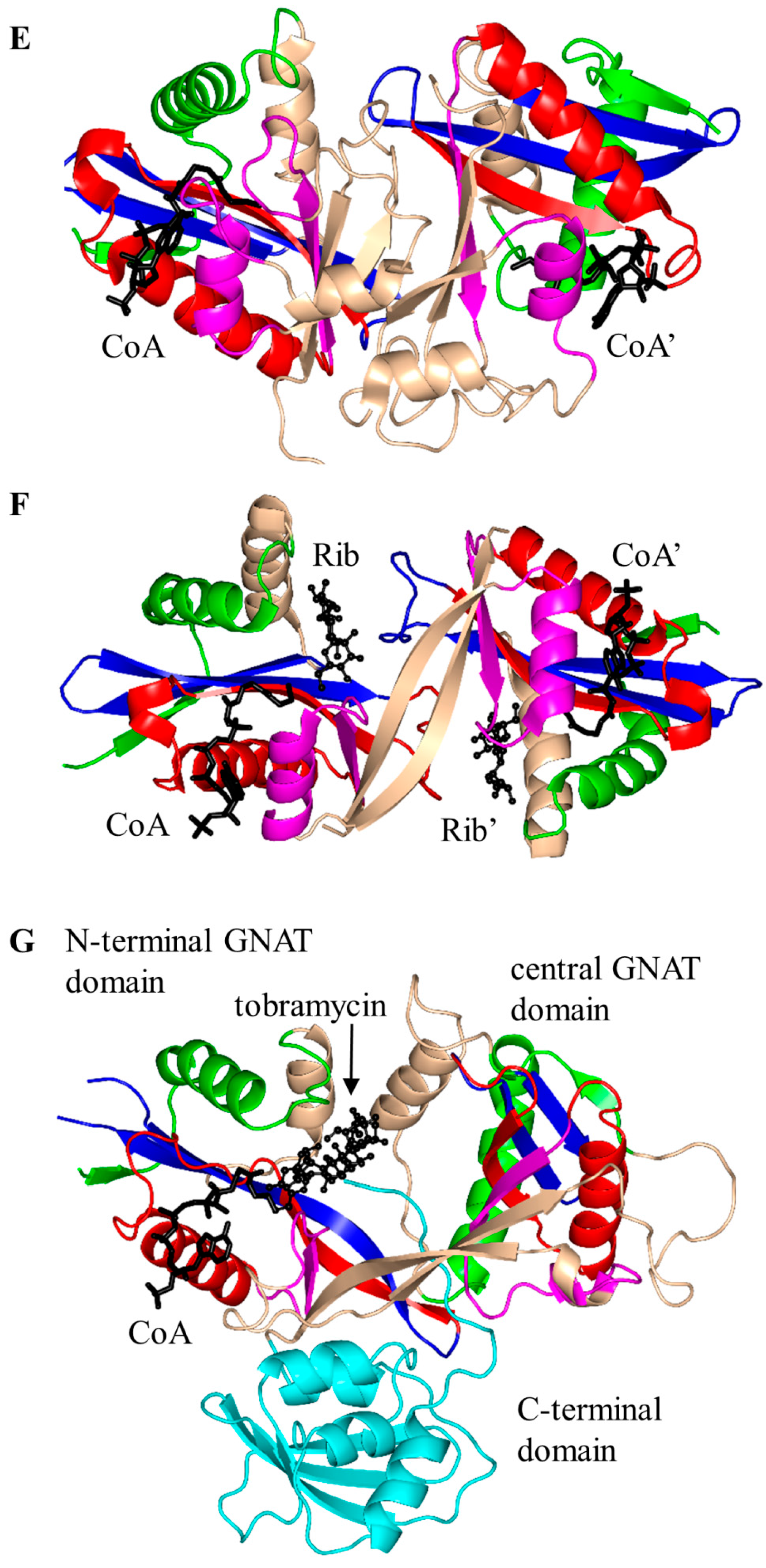
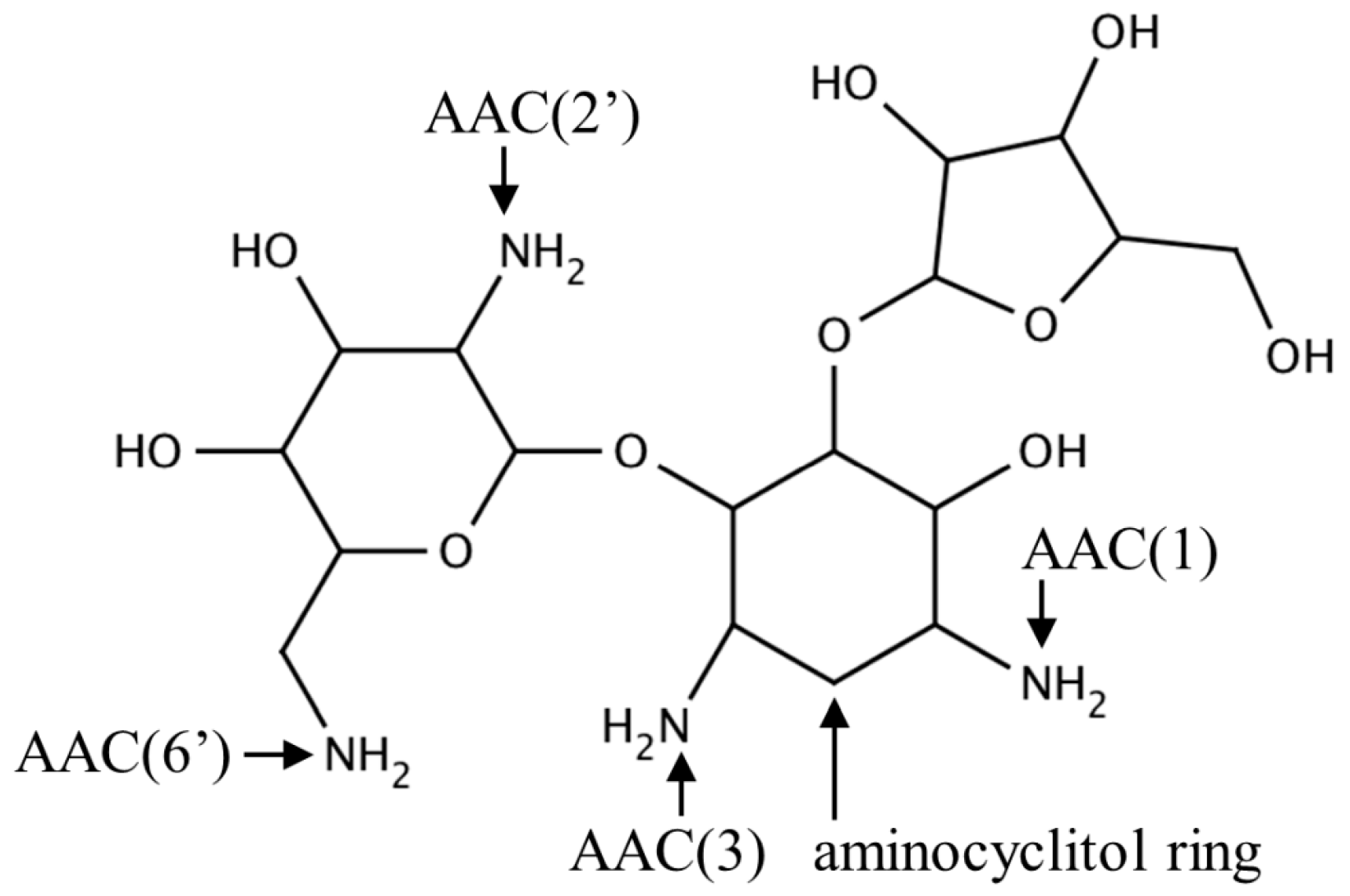
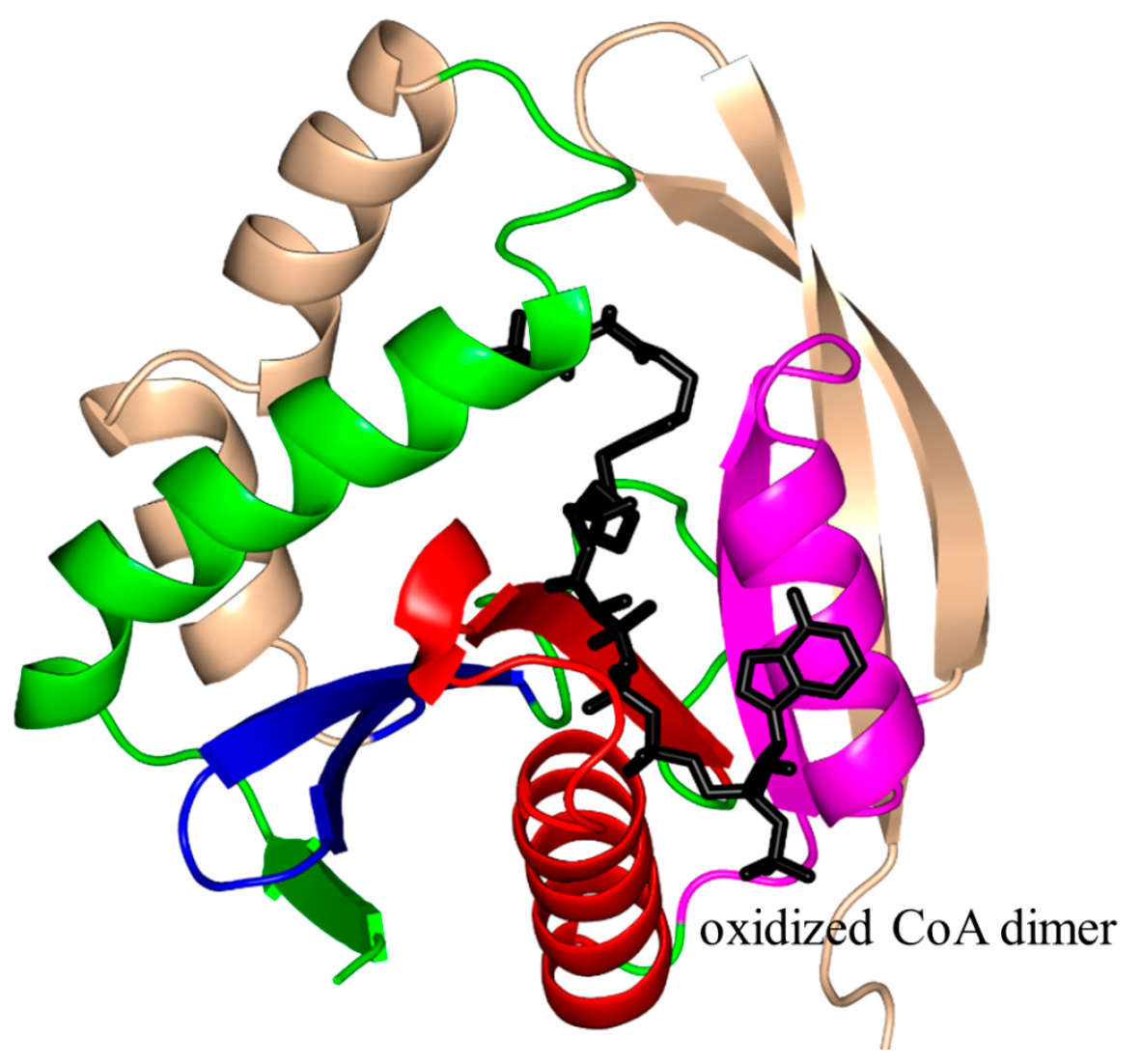
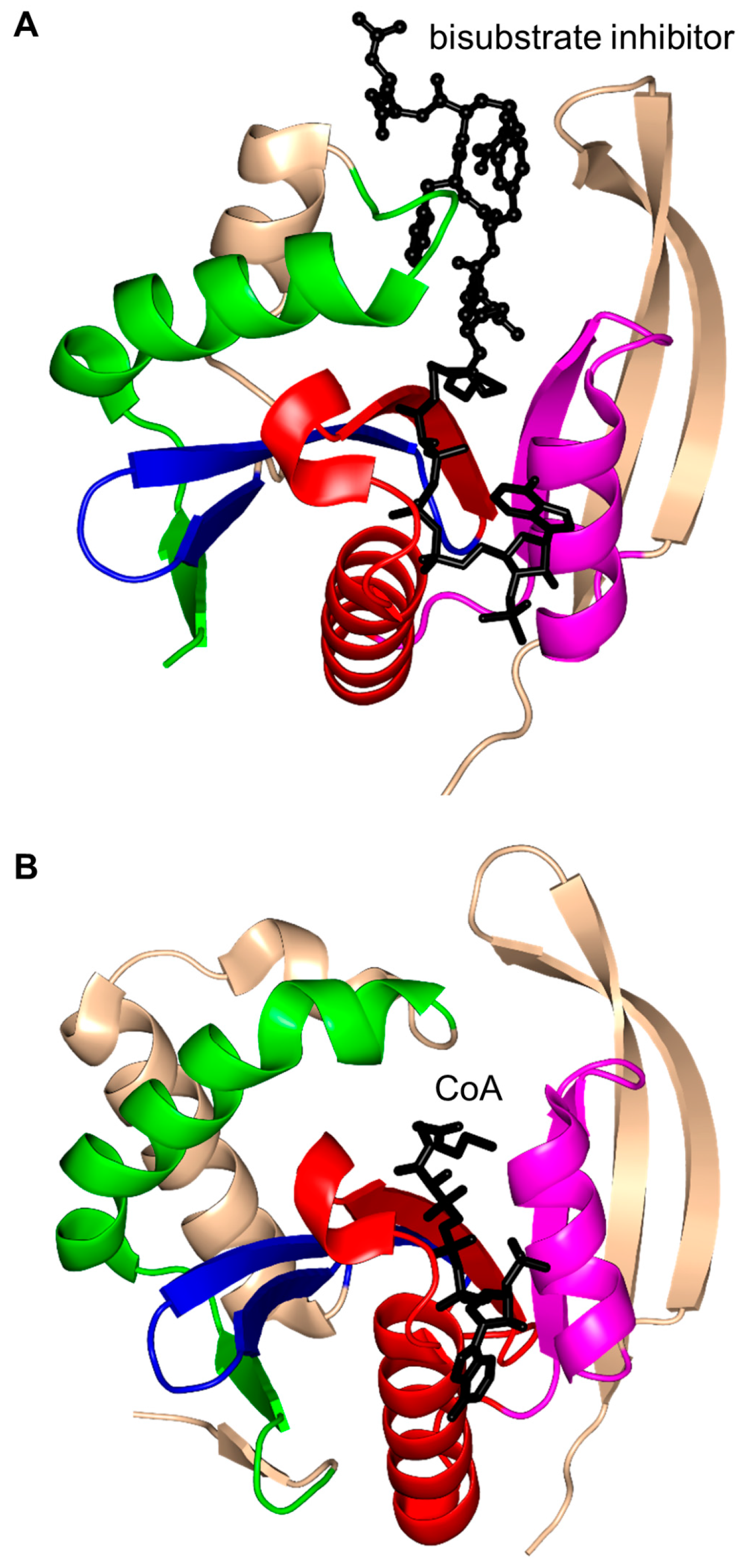
2.1. Aminoglycoside N-Acetyltransferases Family (AAC, EC 2.3.1.81)
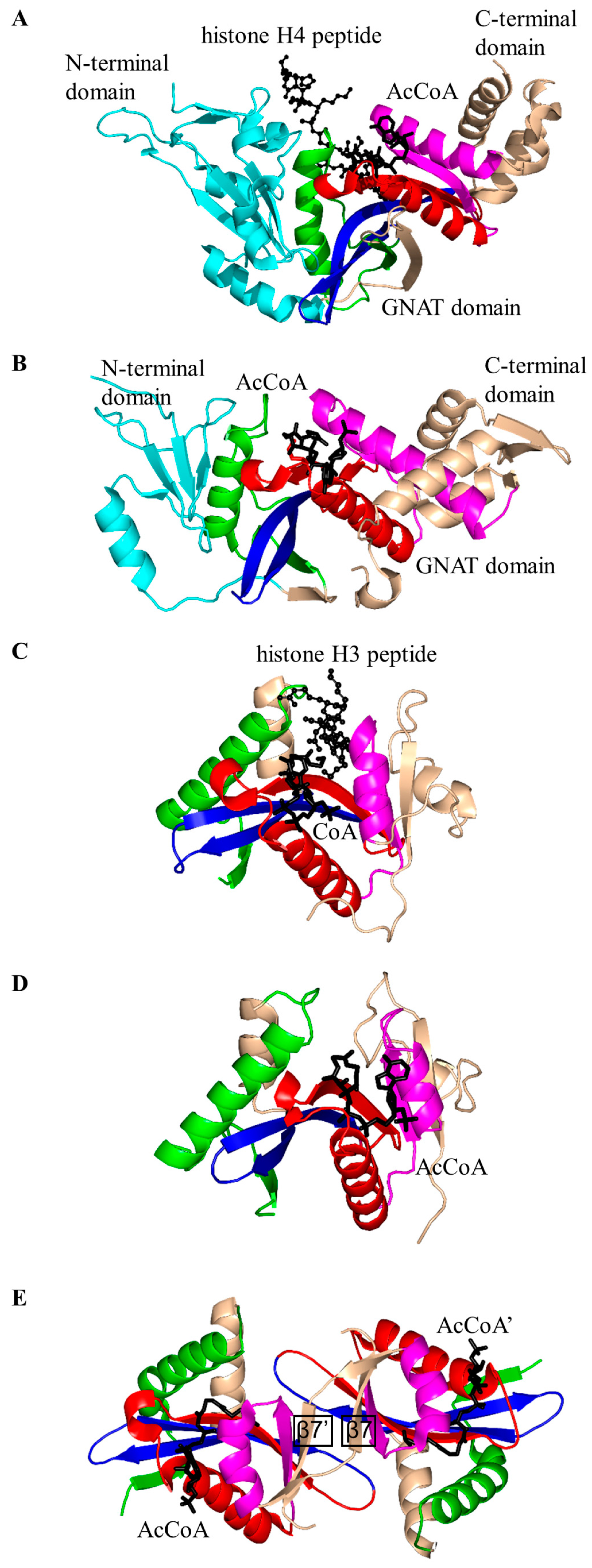
2.2. Histone N-Acetyltransferase Family (HATs, EC 2.3.1.48)
2.3. Non-Histone Protein N-Acetyltransferase Family
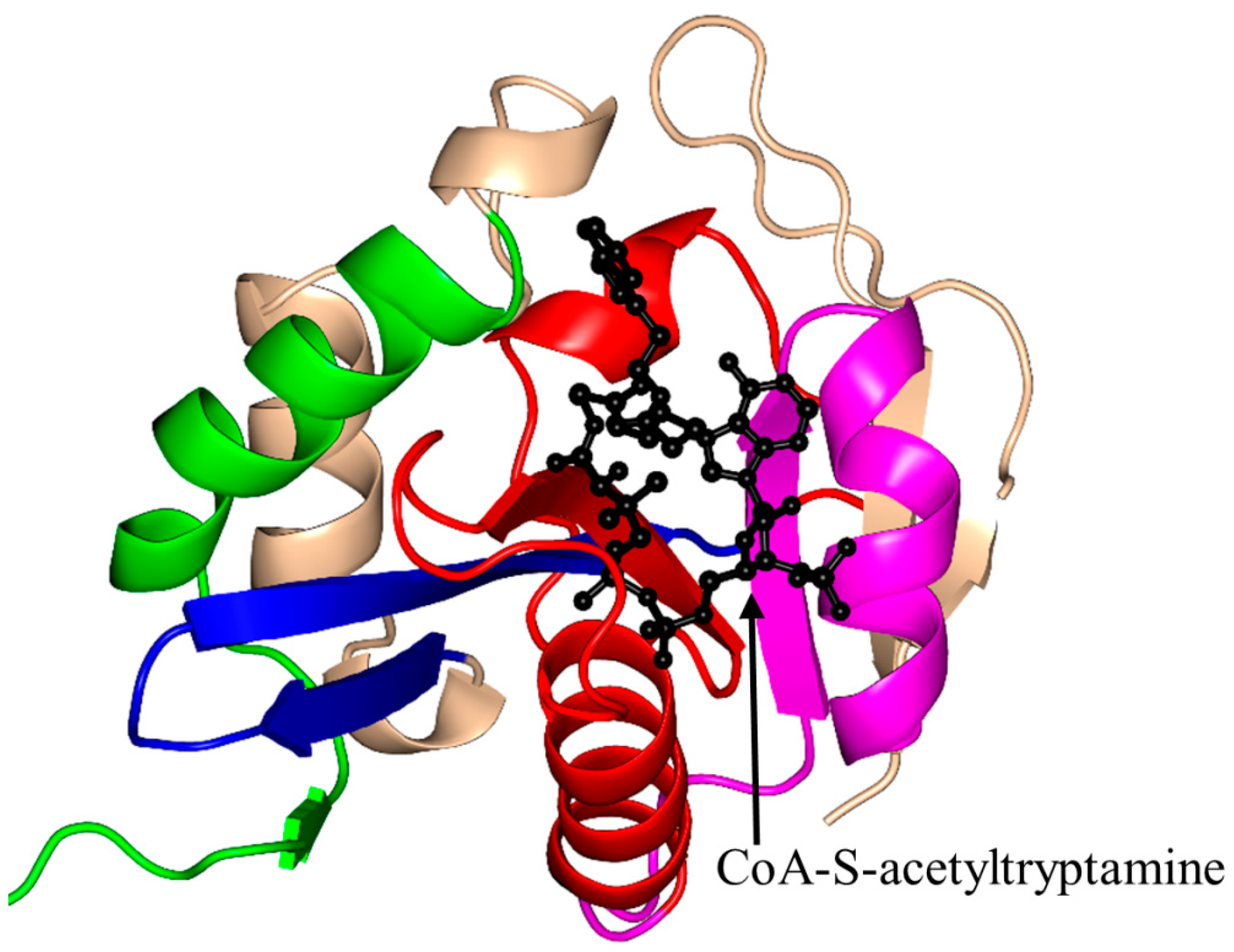
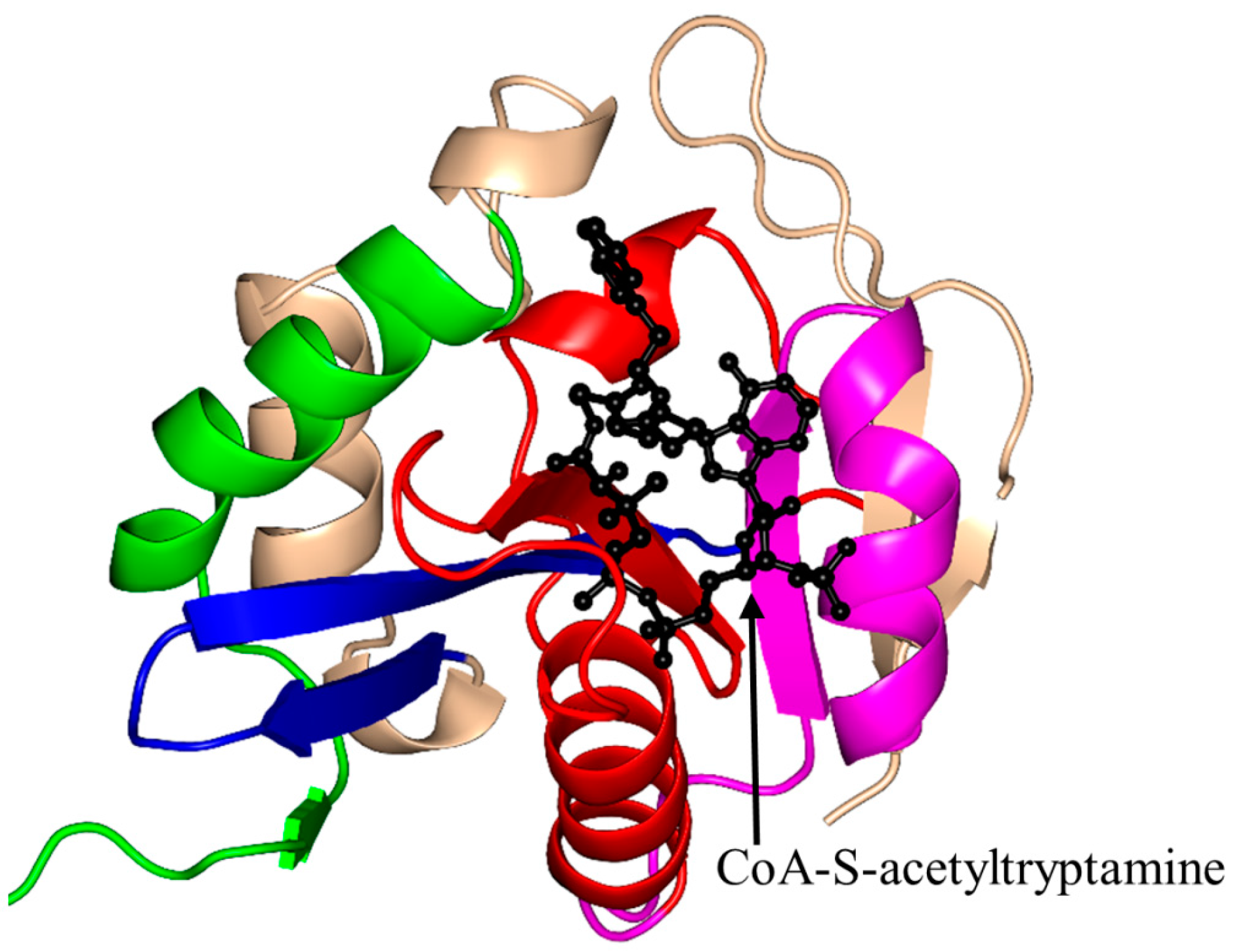
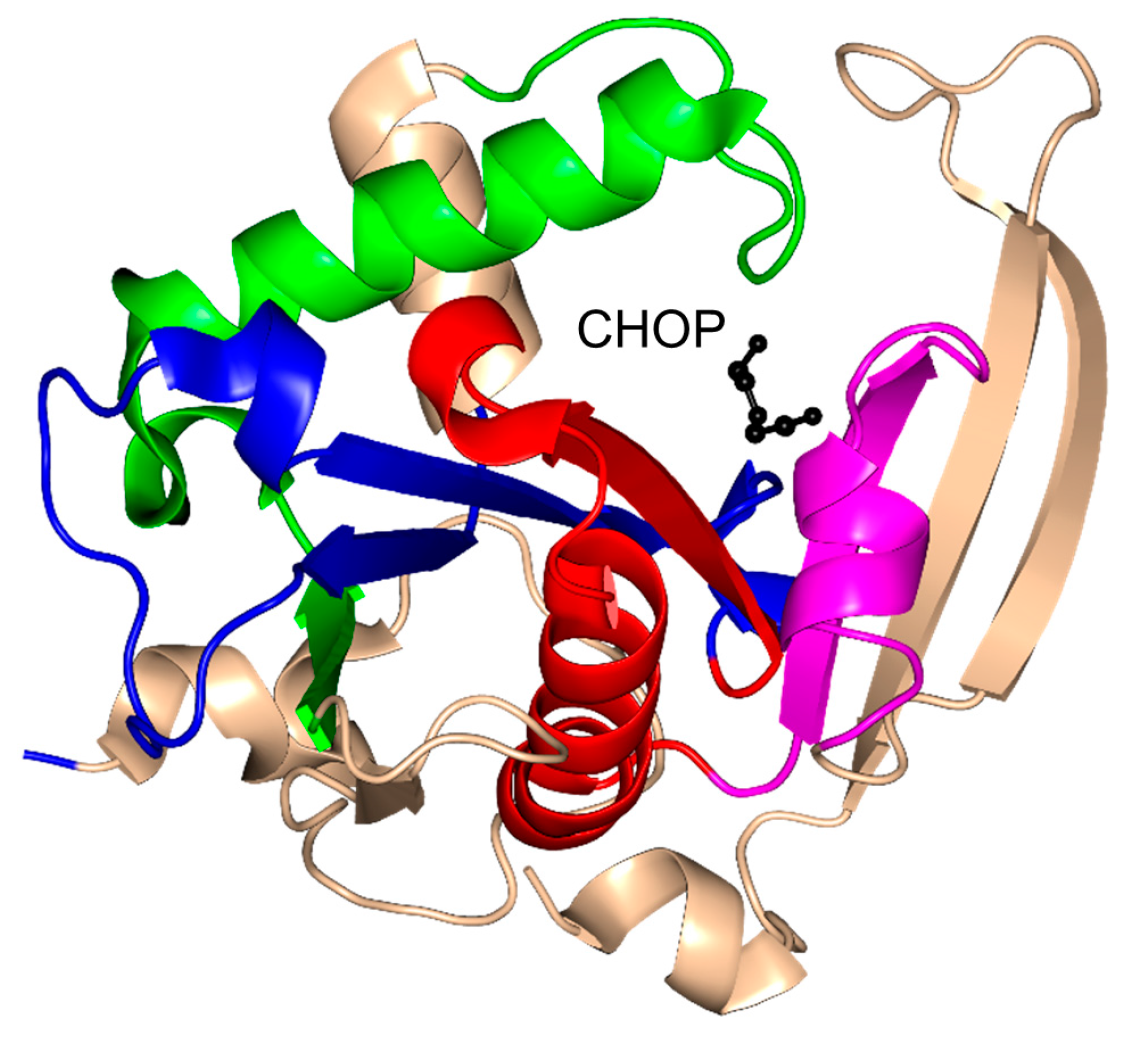
2.4. Arylalkylamine N-Acetyltransferase Family (AANAT, EC 2.3.1.87)
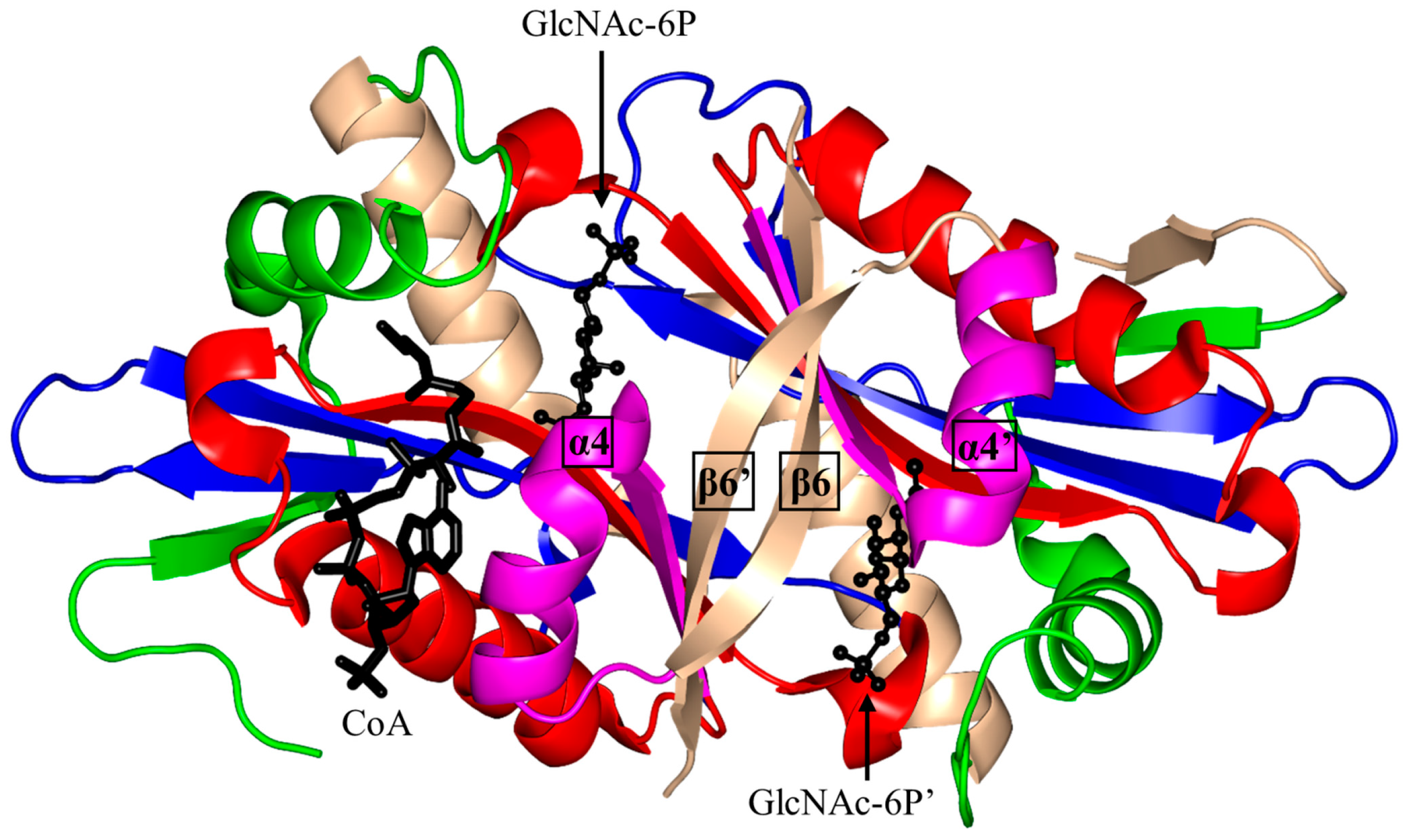
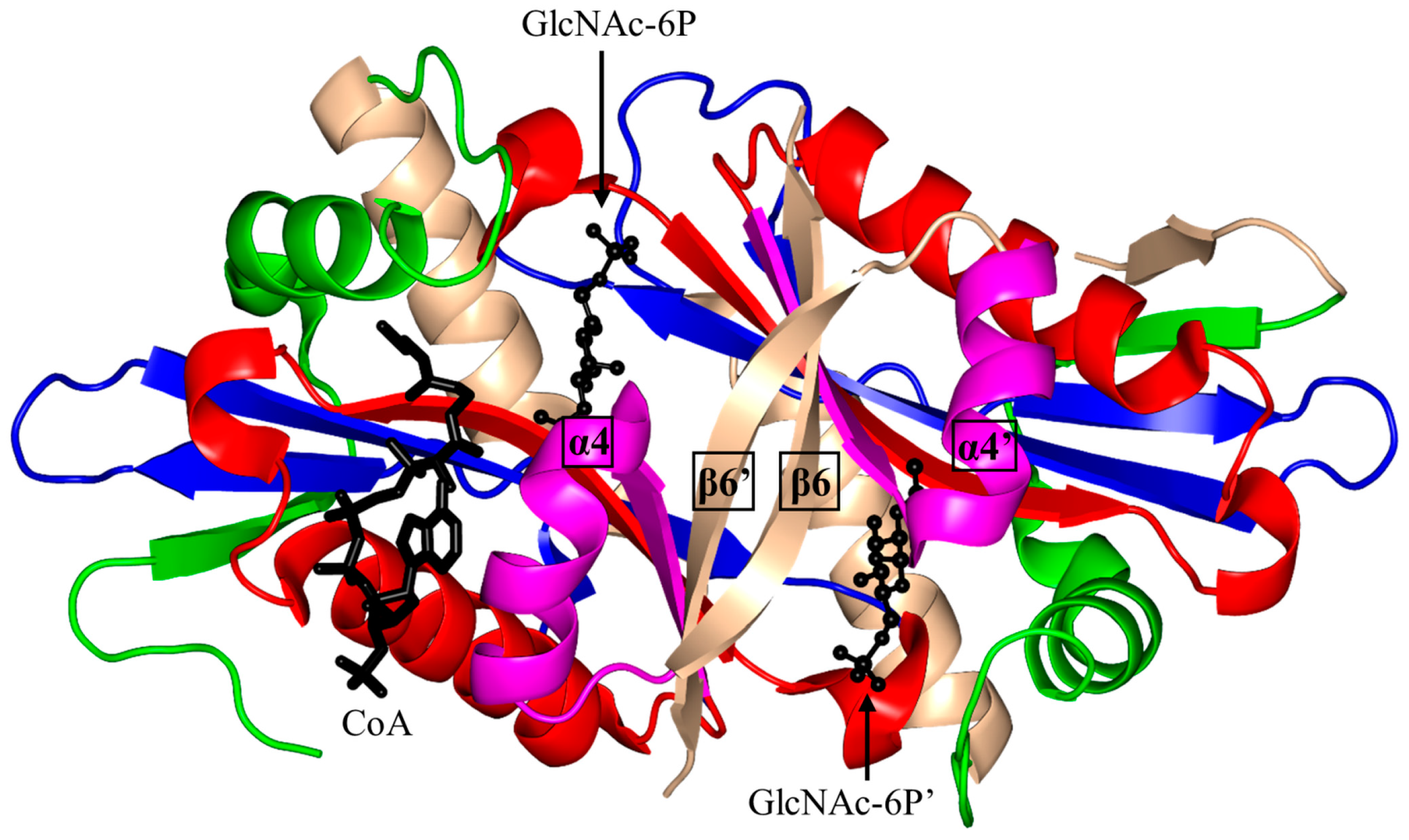
2.5. Glucosamine-6-Phosphate N-Acetyltransferase 1 Family (GNA1, EC 2.3.1.4)
2.6. Microcin C7 Self-Immunity Acetyltransferase Family (MccE)
2.7. Pseudaminic Acid Biosynthesis Protein H Family (PseH, EC 2.3.1.202)
2.8. Thymidine Diphosphate (TDP)-Fucosamine Acetyltransferase Family (WecD, EC 2.3.1.210)
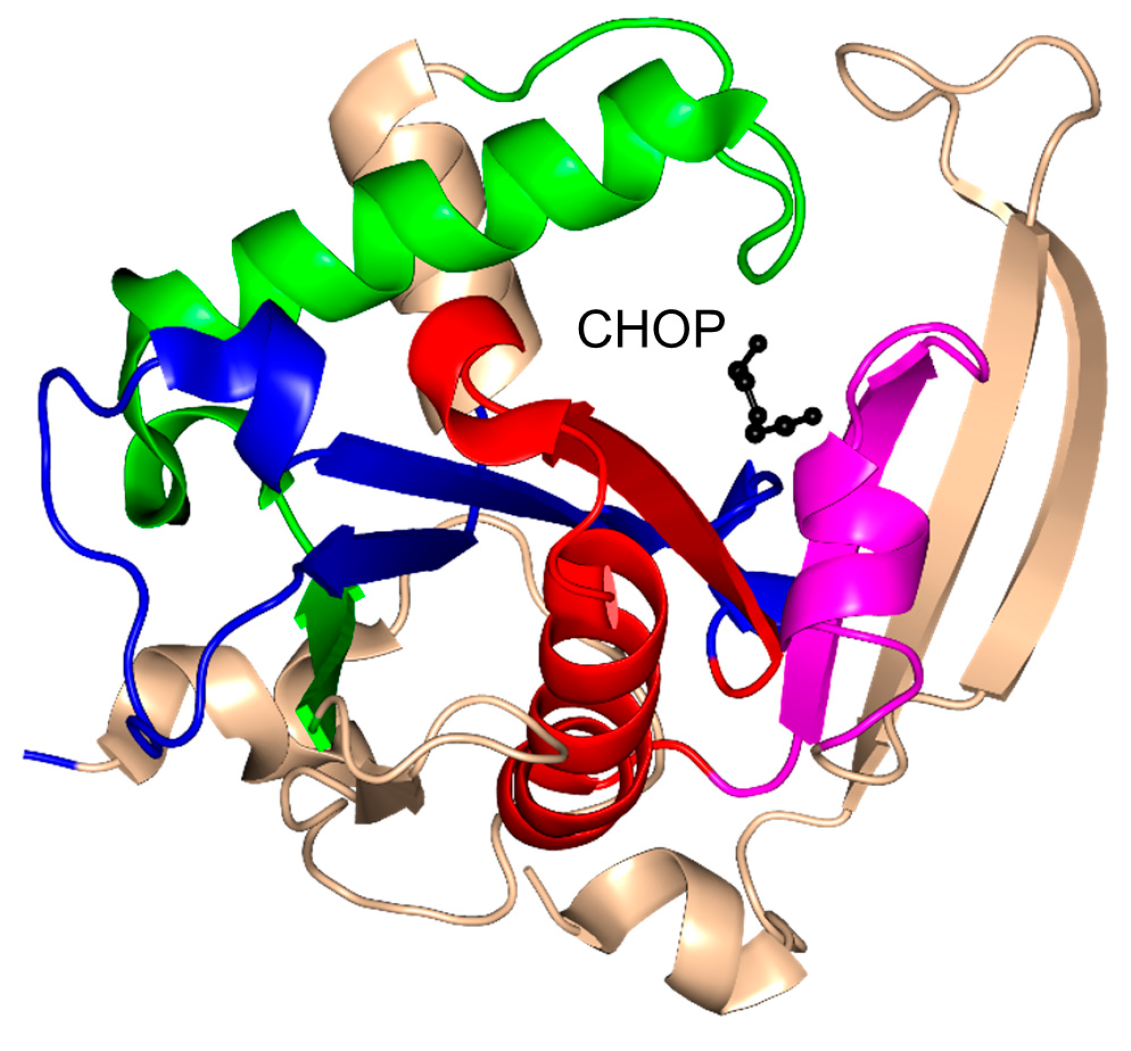
2.9. Tabtoxin Resistance Protein Family (TTR, EC 2.3.1.-)
2.10. Mpr1 Family (EC 3.4.1.-)
2.11. Spermidine/Spermine N1-Acetyltransferase Family (SSAT, EC 2.3.1.57)
2.12. C-Terminal Nε-Lysine Protein Acetyltransferase Family (EC 2.3.1.-)
2.13. Ribosomal Protein Nα-Acetyltransferase Family (EC 2.3.1.128)
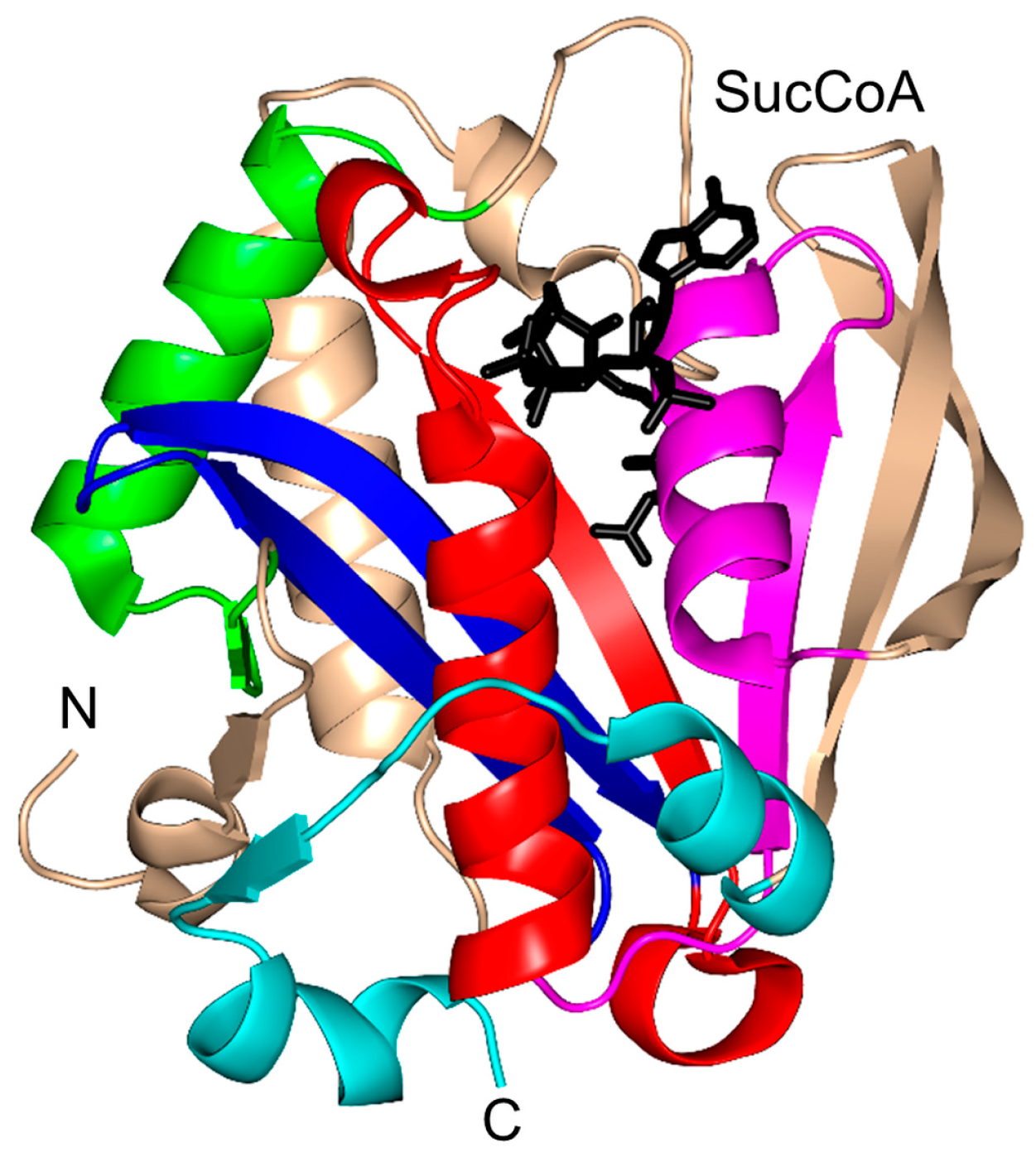
2.14. Succinyltransferase Family (EC 2.8.3.-)
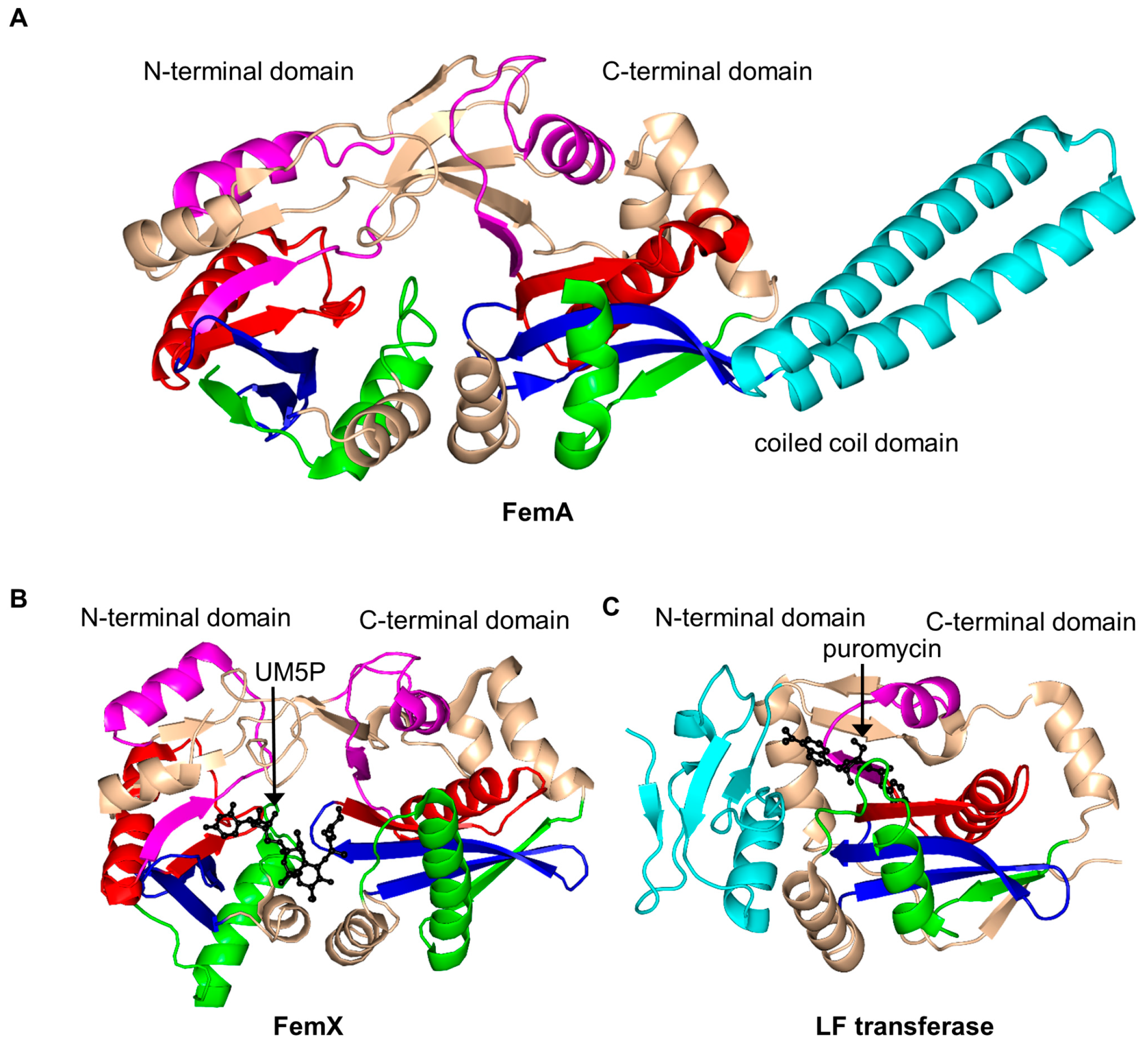
2.15. FemABX Aminoacyl Transferases Family (FemABX, EC 2.3.2.-)
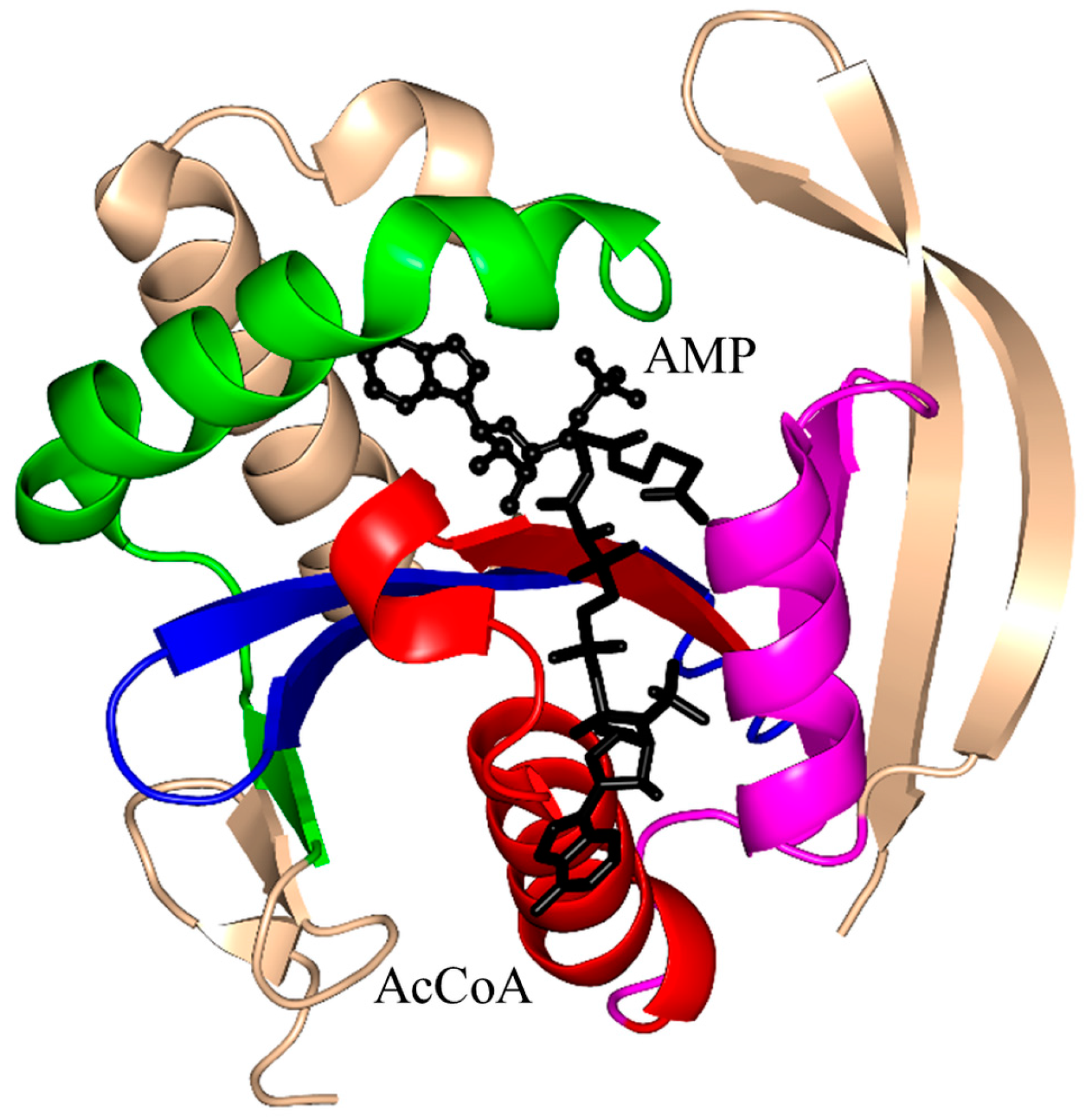
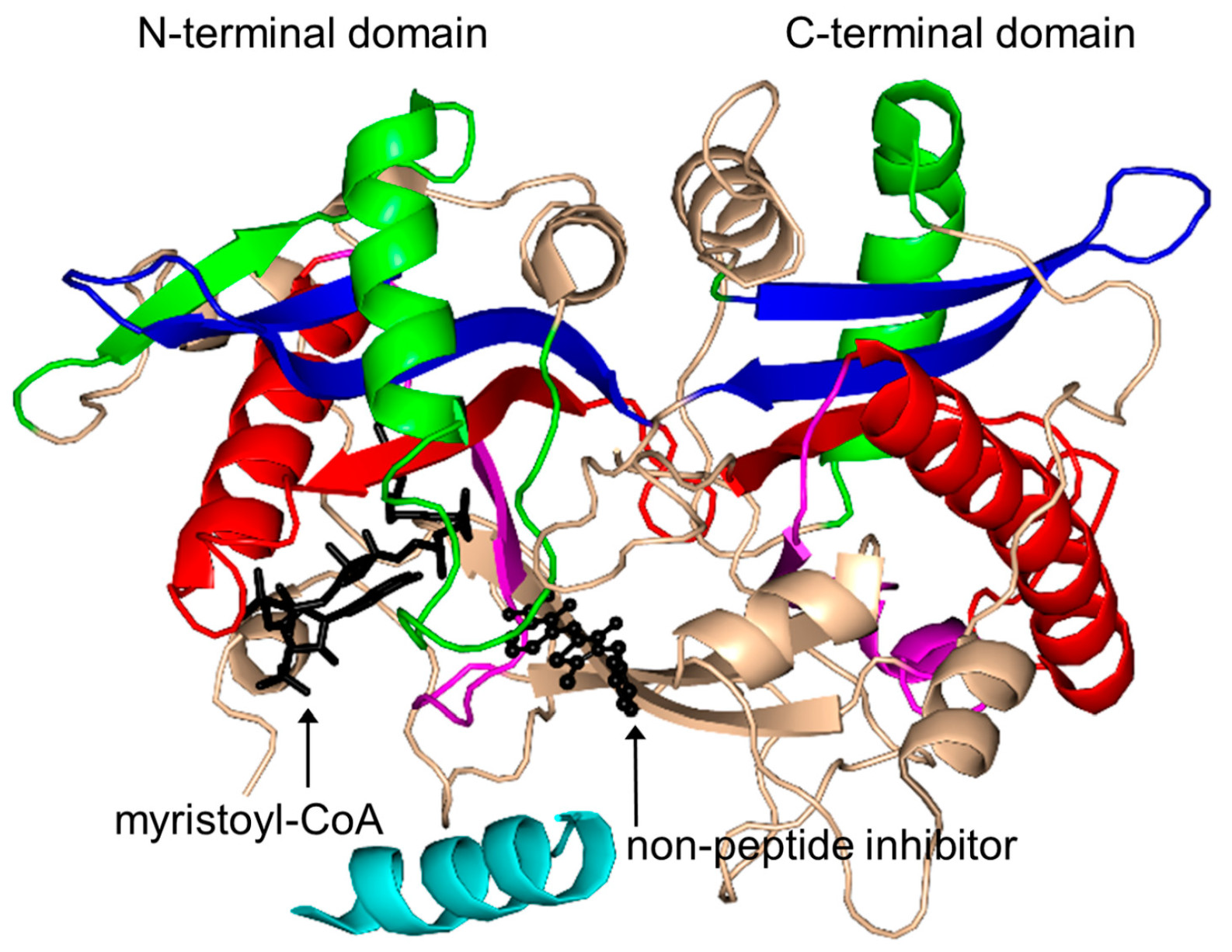
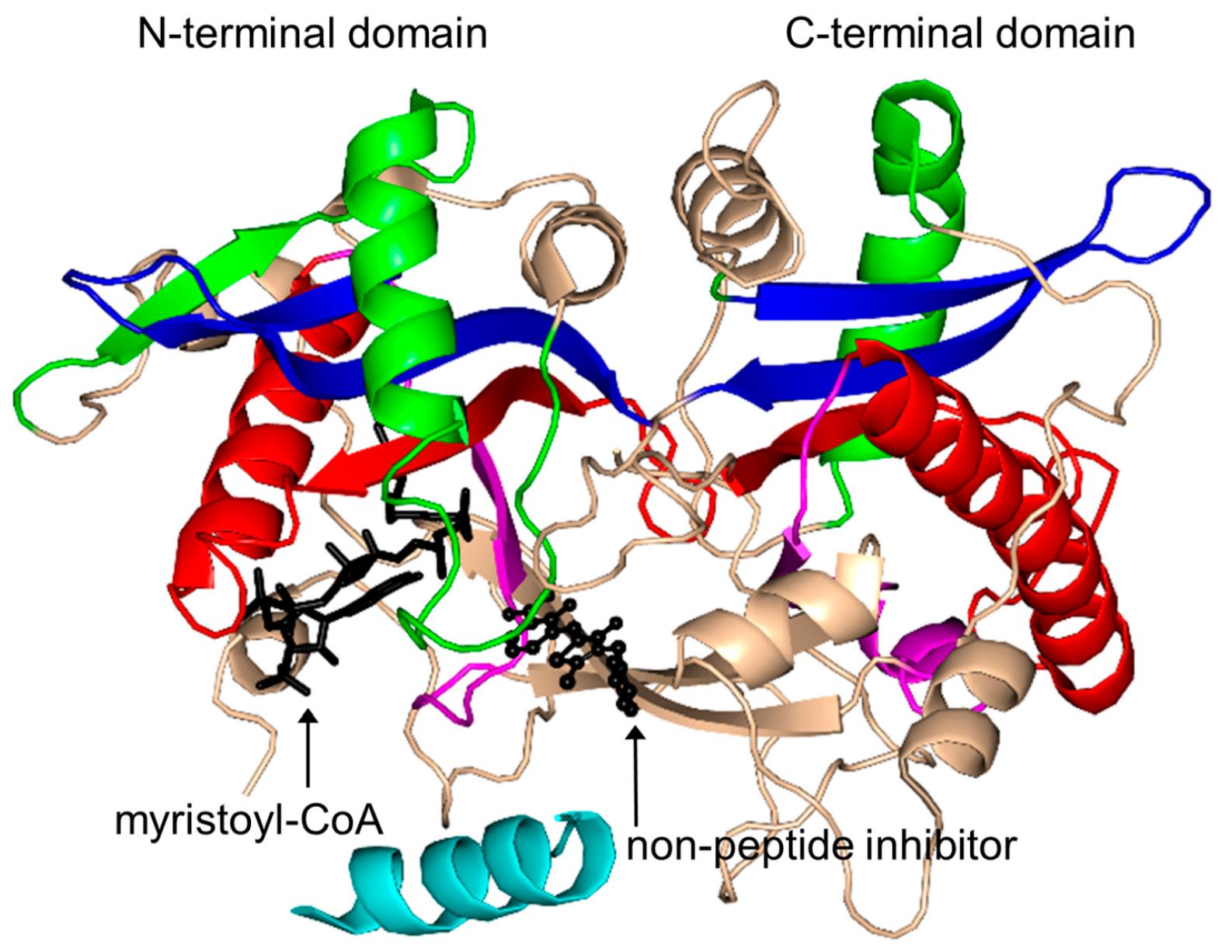
2.16. Protein N-Myristoyltransferase Family (NMT, EC 2.3.1.97)
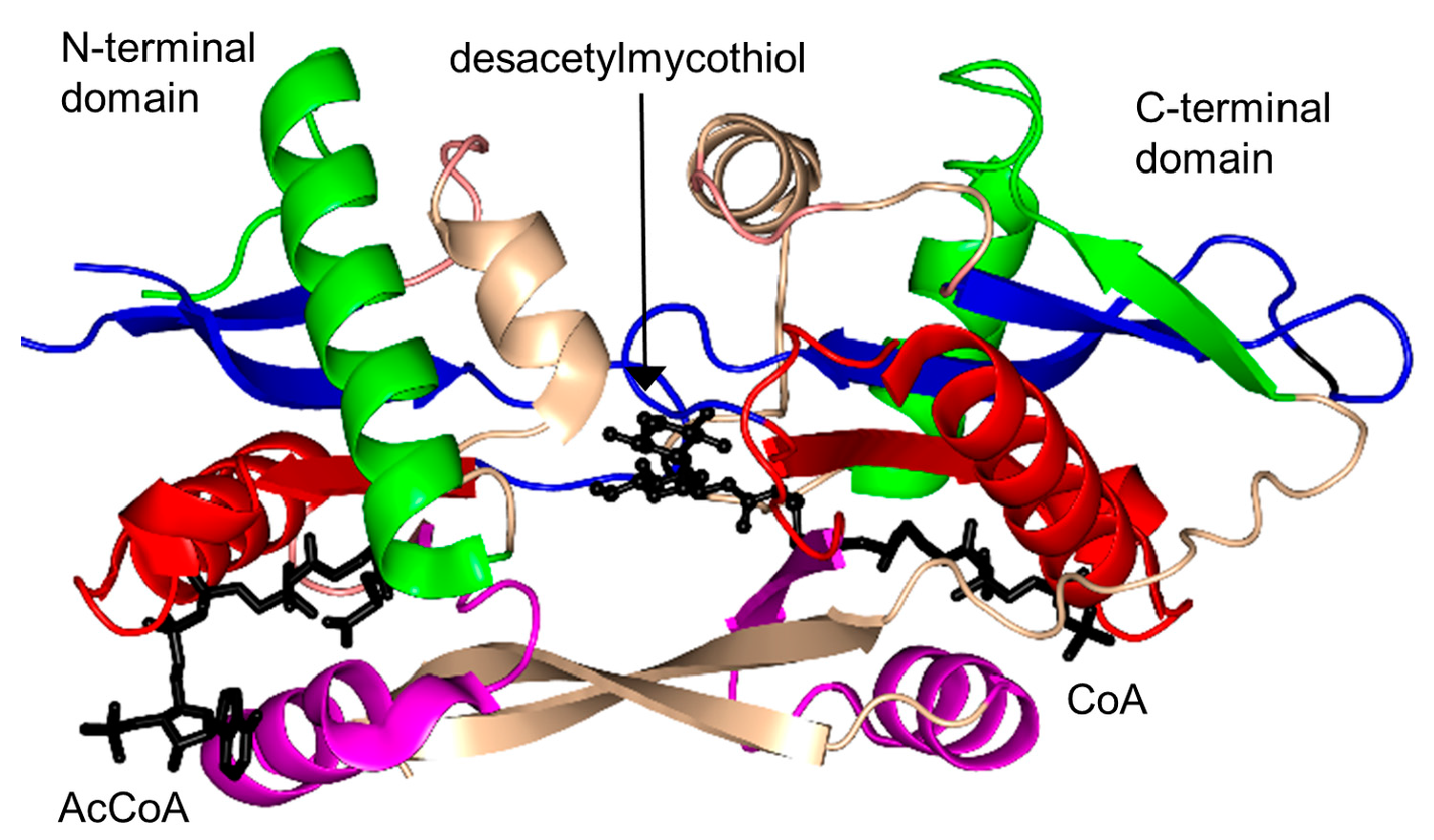
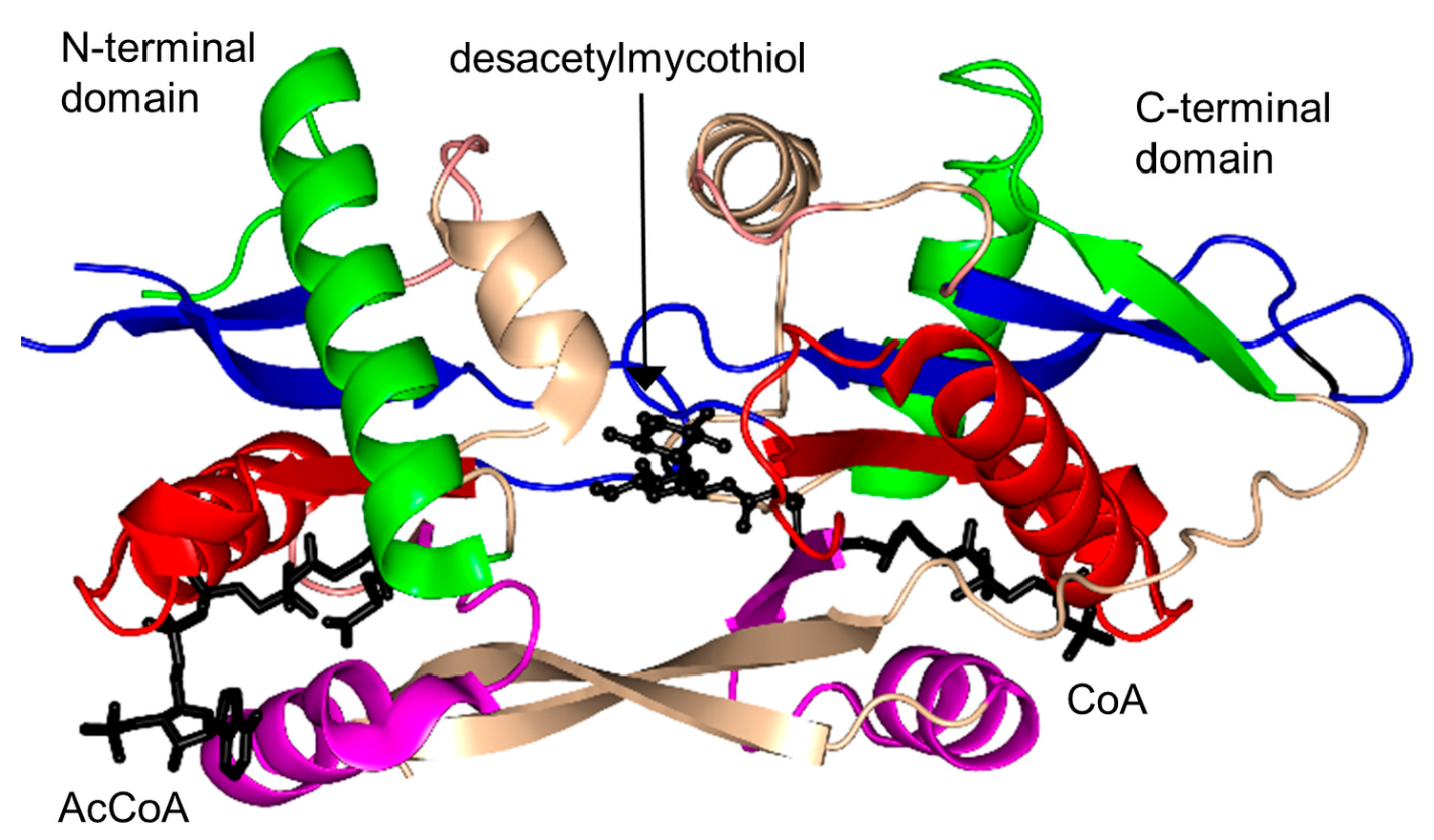
2.17. Mycothiol Synthase Family (MshD, EC 2.3.1.189)
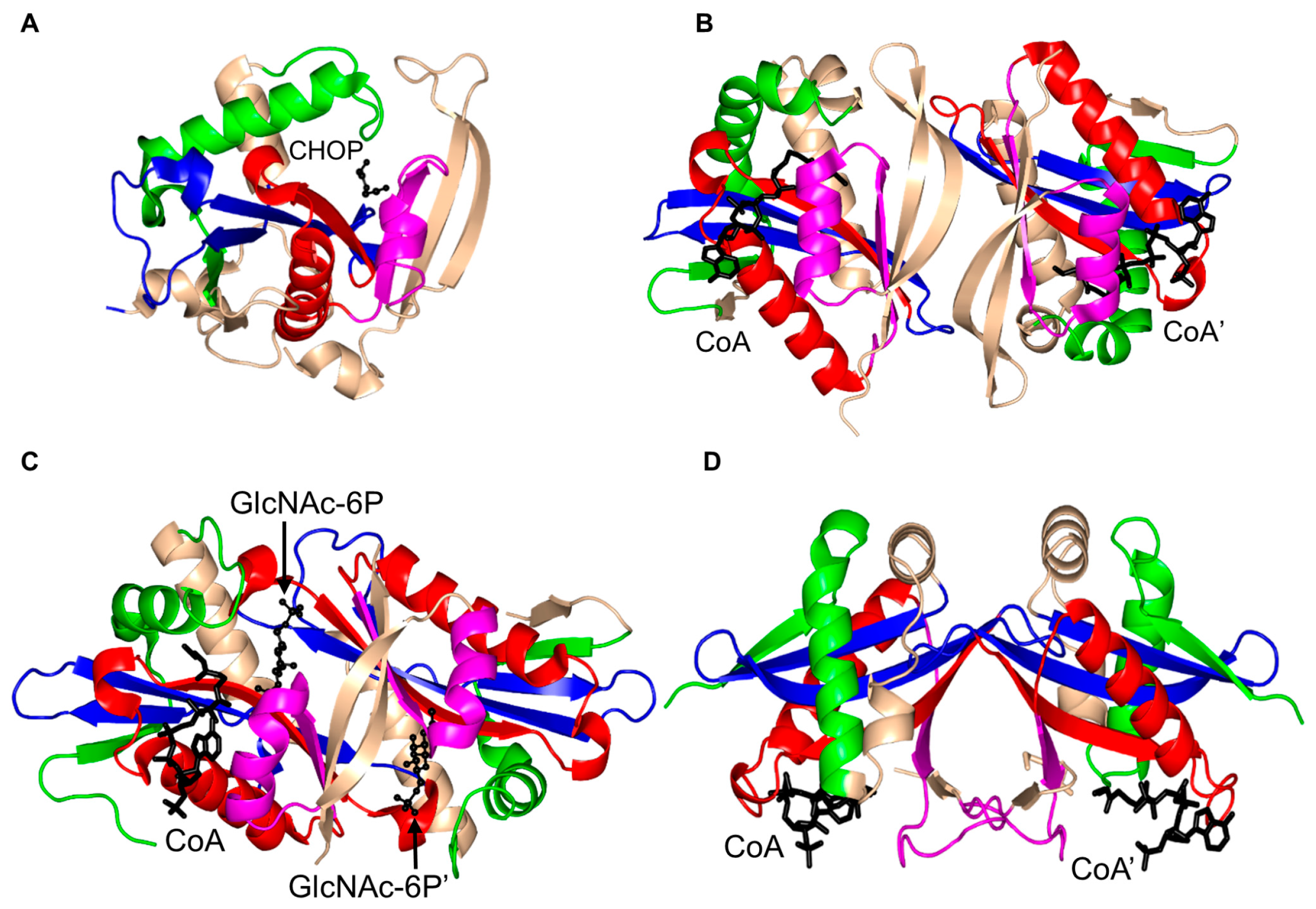
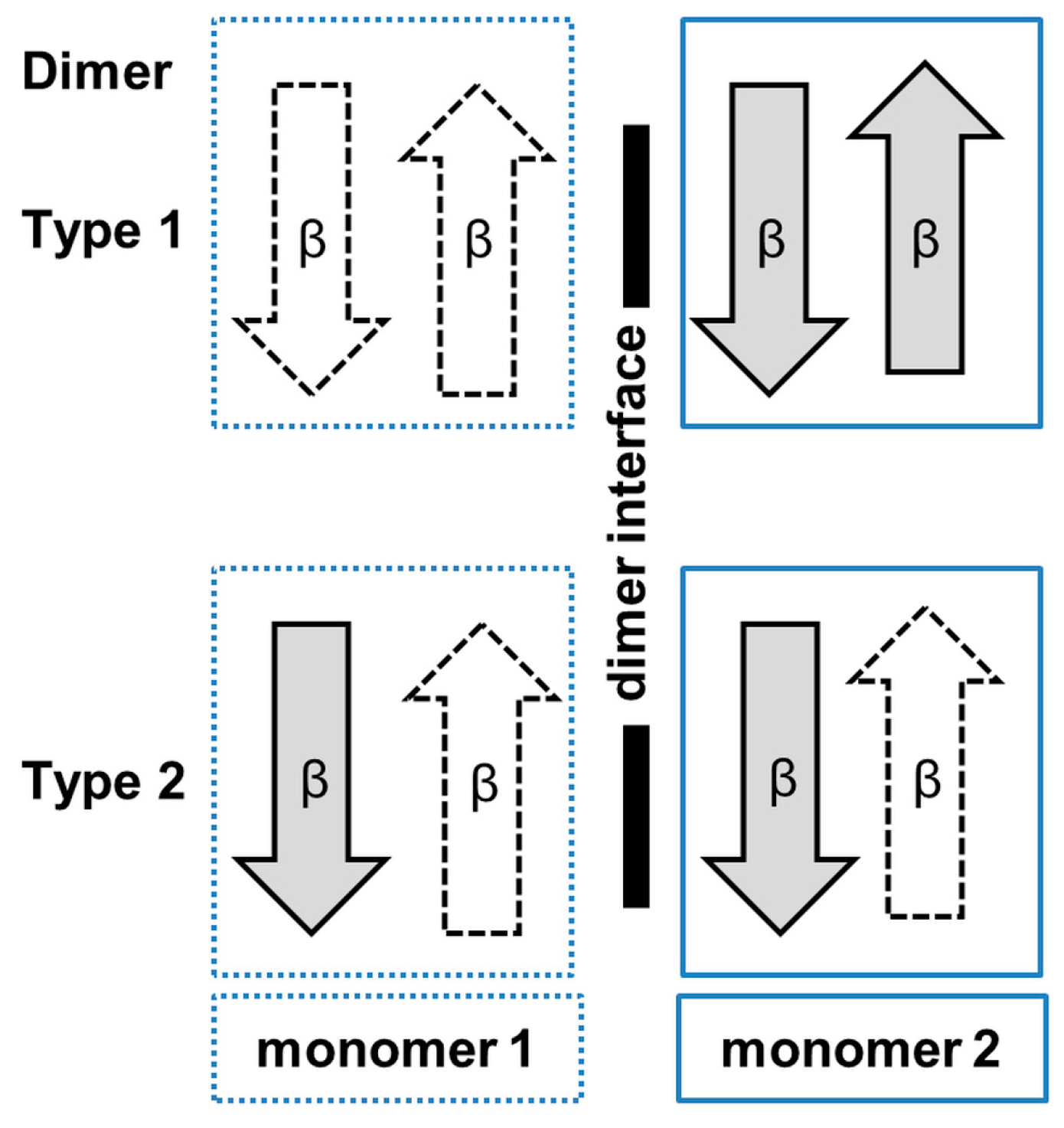
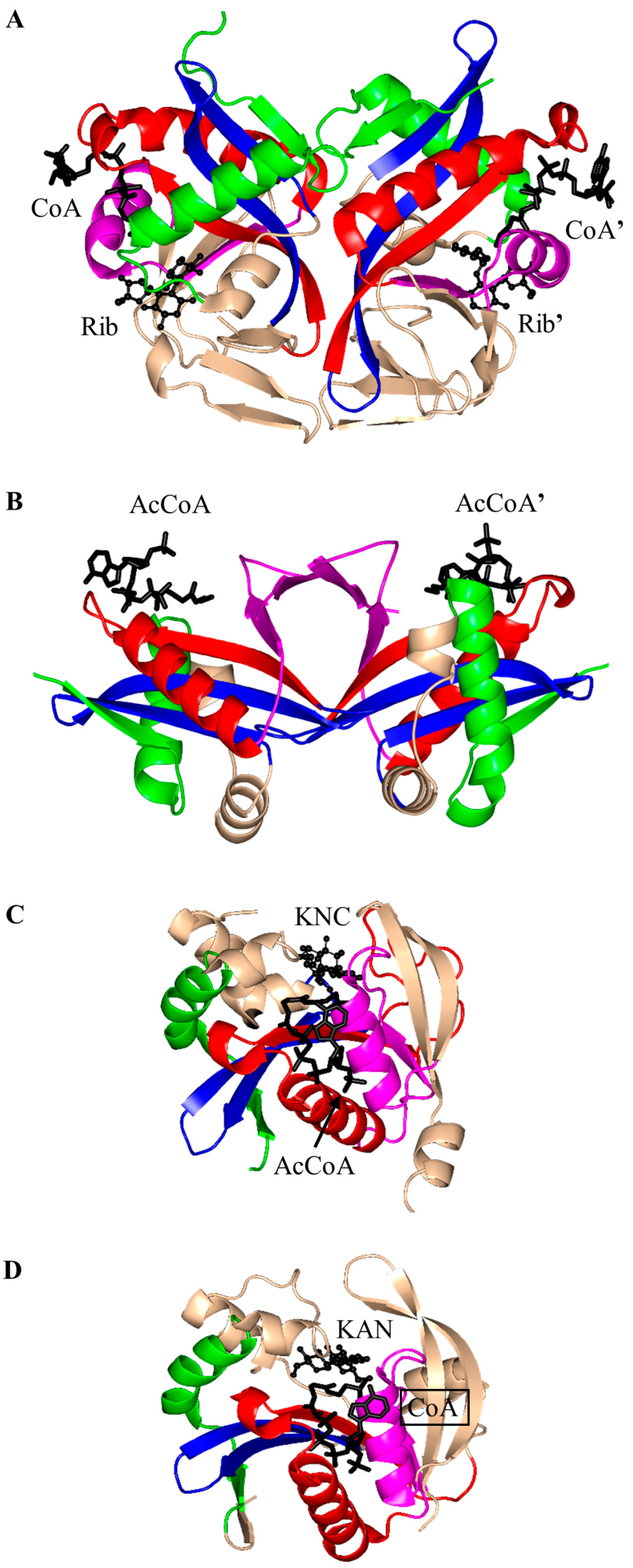
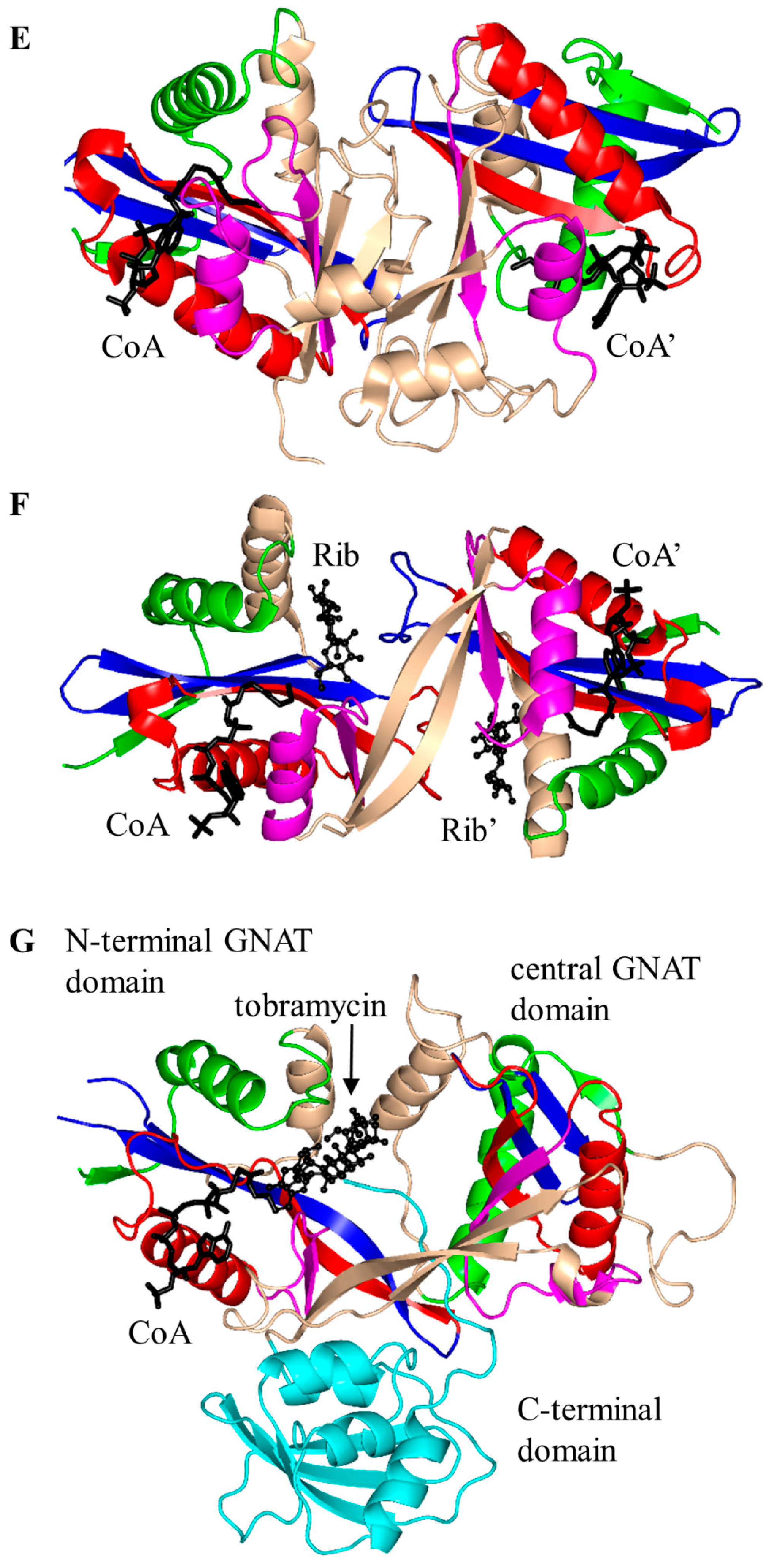
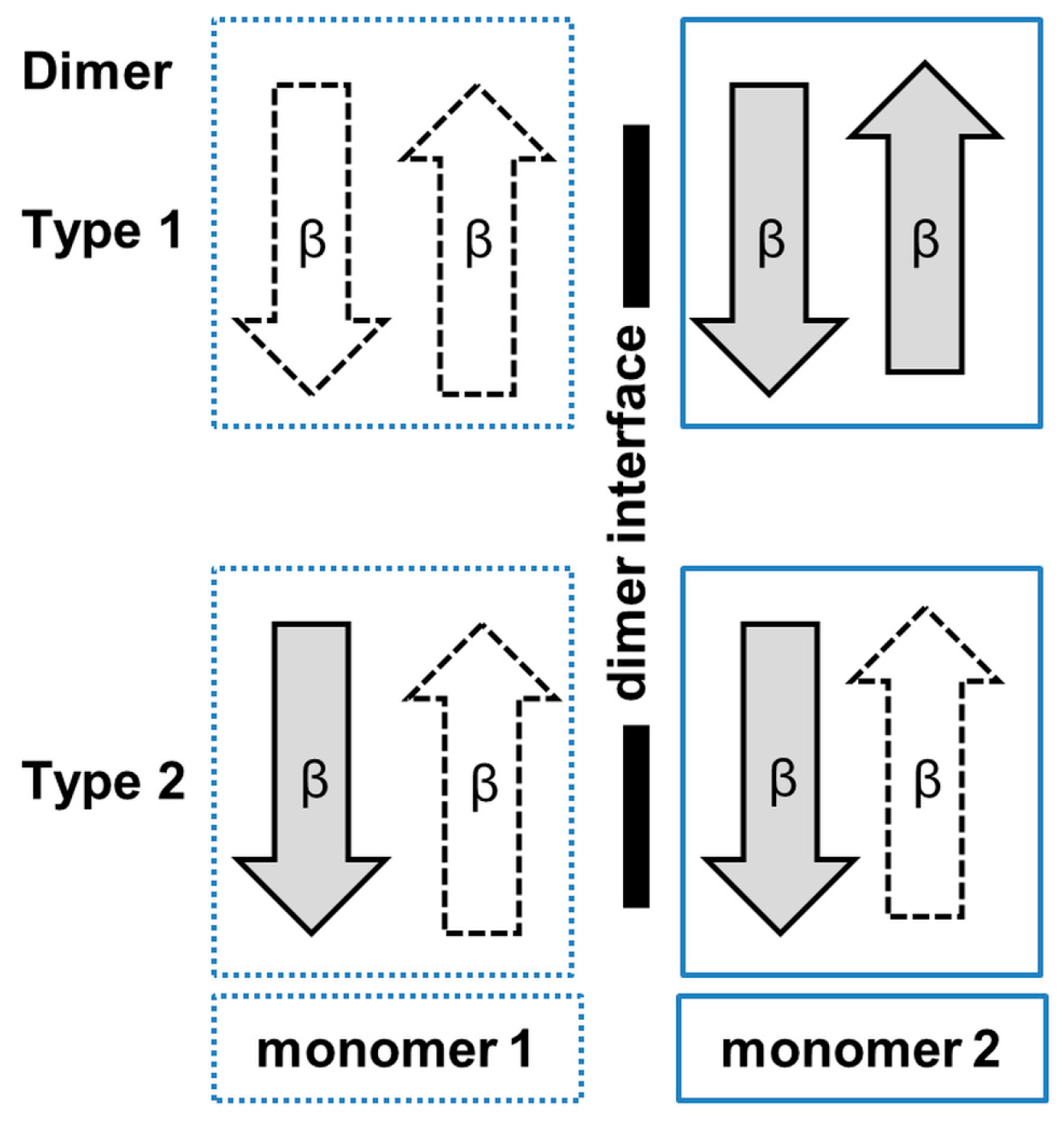
3. Oligomerization of GNAT Superfamily Enzymes
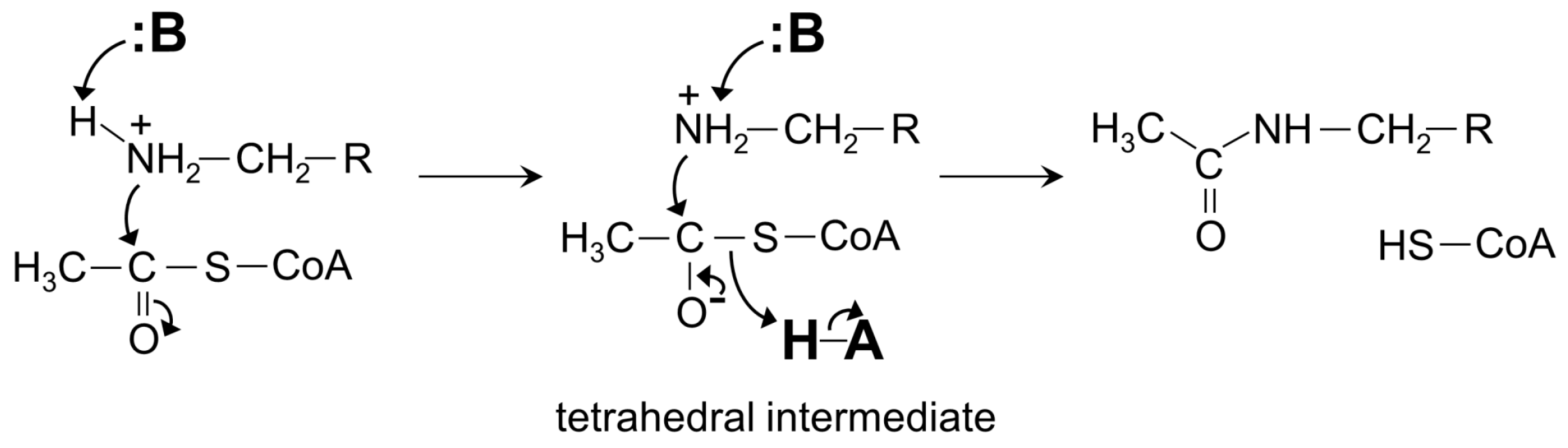
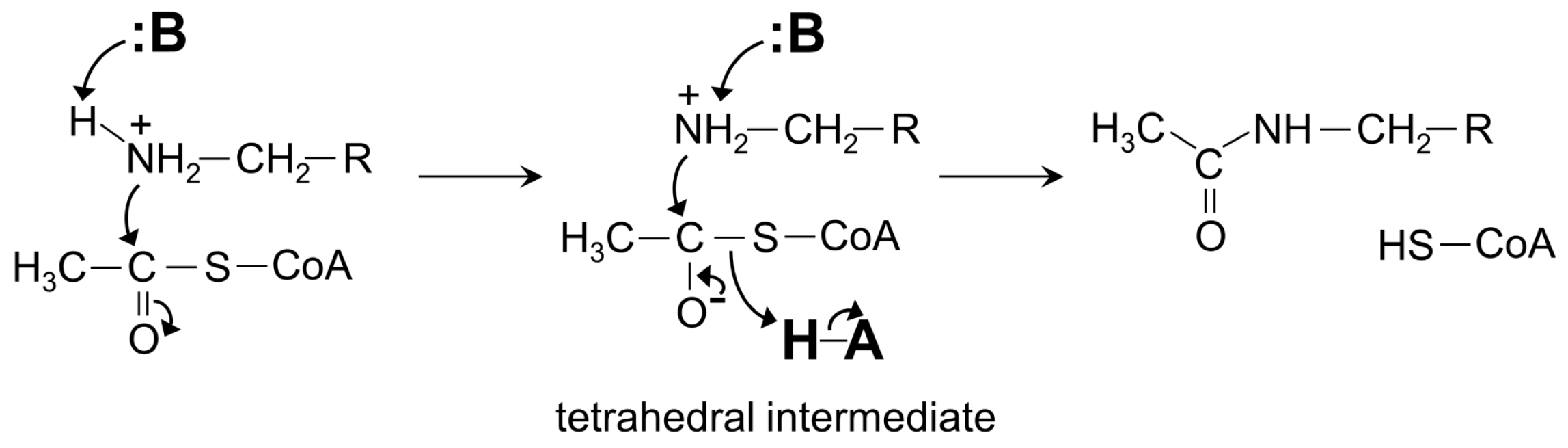
4. General Catalytic Mechanism of GNAT Superfamily Members
5. Association of Expression of GNAT Enzymes with Cancer
6. GNAT Enzymes as Potential Targets for Antimicrobial Agents
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Neuwald, A.F.; Landsnan, D. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Proc. Natl. Acad. Sci. USA 1997, 51, 154–155. [Google Scholar] [CrossRef]
- Dyda, F.; Klein, D.C.; Hickman, A.B. GCN5-related N-acetyltransferases: A structural overview. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 81–103. [Google Scholar] [CrossRef] [PubMed]
- Farazi, T.A.; Waksman, G.; Gordon, J.I. Structures of Saccharomyces cerevisiae N-myristoyltransferase with bound myristoylCoA and peptide provide insights about substrate recognition and catalysis. Biochemistry 2001, 40, 6335–6343. [Google Scholar] [CrossRef] [PubMed]
- Vetting, M.W.; Errey, J.C.; Blanchard, J.S. Rv0802c from Mycobacterium tuberculosis: The first structure of a succinyltransferase with the GNAT fold. Acta Crystallogr. F Struct. Biol. Commun. 2008, 64, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; Vassilev, A.; Makino, Y.; Sali, A.; Nakatani, Y.; Burley, S.K. Crystal structure of a GCN5-related N-acetyltransferase: Serratia marcescens aminoglycoside 3-N-acetyltransferase. Cell 1998, 94, 439–449. [Google Scholar] [CrossRef]
- Dutnall, R.N.; Tafrov, S.T.; Sternglanz, R.; Ramakrishnan, V. Structure of the histone acetyltransferase Hat1: A paradigm for the GCN5-related N-acetyltransferase superfamily. Cell 1998, 94, 427–438. [Google Scholar] [CrossRef]
- InterPro Database. Available online: http://www.ebi.ac.uk/interpro (accessed on 26 June 2016).
- Xie, L.; Zeng, J.; Luo, H.; Pan, W.; Xie, J. The roles of bacterial GCN5-related N-acetyltransferases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Favrot, L.; Blanchard, J.S.; Vergnolle, O. Bacterial GCN5-related N-acetyltransferases: From resistance to regulation. Biochemistry 2016, 55, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Vetting, M.W.; de Carvalho, L.P.S.; Yu, M.; Hegde, S.S.; Magnet, S.; Roderick, S.L.; Blanchard, J.S. Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 2005, 433, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Mio, T.; Yamada-Okabe, T.; Arisawa, M.; Yamada-Okabe, H. Saccharomyces cerevisiae GNA1, an essential gene encoding a novel acetyltransferase involved in UDP-N-acetylglucosamine synthesis. J. Chem. Biol. 1999, 274, 424–429. [Google Scholar] [CrossRef]
- Wallace, H.; Fraser, A.; Hughes, A. A perspective of polyamine metabolism. Biochem. J. 2003, 376, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wybenga-Groot, L.E.; Draker, K.; Wright, G.D.; Berghuis, A.M. Crystal structure of an aminoglycoside 6′-N-acetyltransferase: Defining the GCN5-related N-acetyltransferase superfamily fold. Structure 1999, 7, 497–507. [Google Scholar] [CrossRef]
- Bhatnagar, R.S.; Futterer, K.; Farazi, T.A.; Korolev, S.; Murray, C.L.; Jackson-Machelski, E.; Gokel, G.W.; Gordon, J.I.; Waksman, G. Structure of N-myristoyltransferase with bound myristoylCoA and peptide substrate analogs. Nat. Struct. Biol. 1998, 5, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.T.; Rossmann, M.G. Comparison of super-secondary structures in proteins. J. Mol. Biol. 1973, 76, 241–256. [Google Scholar] [CrossRef]
- Llano-Sotelo, B.; Azucena, E.F., Jr.; Kotra, L.P.; Mobashery, S.; Chow, C.S. Aminoglycosides modified by resistance enzymes display diminished binding to the bacterial ribosomal aminoacyl-tRNA site. Chem. Biol. 2002, 9, 455–463. [Google Scholar] [CrossRef]
- Magnet, S.; Blanchard, J.S. Molecular insights into aminoglycoside action and resistance. Chem. Rev. 2005, 105, 477–498. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Morlock, G.P.; Sikes, R.D.; Dalton, T.L.; Metchock, B.; Starks, A.M.; Hooks, D.P.; Cowan, L.S.; Plikaytis, B.B.; Posey, J.E. Molecular detection of mutations associated with first-and second-line drug resistance compared with conventional drug susceptibility testing of Mycobacterium tuberculosis. Antimicrob. Agents. Chemother. 2011, 55, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Houghton, J.L.; Green, K.D.; Pricer, R.E.; Mayhoub, A.S.; Garneau-Tsodikova, S. Unexpected N-acetylation of capreomycin by mycobacterial Eis enzymes. J. Antimicrob. Chemother. 2013, 68, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Zaunbrecher, M.A.; Sikes, R.D.; Metchock, B.; Shinnick, T.M.; Posey, J.E. Overexpression of the chromosomally encoded aminoglycoside acetyltransferase Eis confers kanamycin resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2009, 106, 20004–20009. [Google Scholar] [CrossRef] [PubMed]
- Burk, D.L.; Xiong, B.; Breitbach, C.; Berghuis, A.M. Structures of aminoglycoside acetyltransferase AAC(6′)-Ii in a novel crystal form: Structural and normal-mode analyses. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, M.L.; Vetting, M.W.; Gao, F.; Freiburger, L.; Auclair, K.; Blanchard, J.S. Kinetic and structural analysis of bisubstrate inhibition of the Salmonella enterica aminoglycoside 6′-N-acetyltransferase. Biochemistry 2008, 47, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Vetting, M.W.; Hegde, S.S.; Javid-Majd, F.; Blanchard, J.S.; Roderick, S.L. Aminoglycoside 2′-N-acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat. Struct. Biol. 2002, 9, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Toth, M.; Weiss, T.M.; Frase, H.; Vakulenko, S.B. Structure of the bifunctional aminoglycoside-resistance enzyme AAC(6′)-Ie-APH(2′′)-Ia revealed by crystallographic and small-angle X-ray scattering analysis. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 2754–2764. [Google Scholar] [CrossRef] [PubMed]
- Vetting, M.W.; Park, C.H.; Hegde, S.S.; Jacoby, G.A.; Hooper, D.C.; Blanchard, J.S. Mechanistic and structural analysis of aminoglycoside N-acetyltransferase AAC(6′)-Ib and its bifunctional, fluoroquinolone-active AAC(6′)-Ib-cr variant. Biochemistry 2008, 47, 9825–9835. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.S.; Nikolaidis, N.; Tolmasky, M.E. Rise and dissemination of aminoglycoside resistance: The aac(6′)-Ib paradigm. Front. Microbiol. 2013, 4, 6003–6009. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.W.; Northrop, D.B. Synthesis of a tight-binding, multisubstrate analog inhibitor of gentamicin acetyltransferase I. J. Antibiot. 1979, 32, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Vakulenko, S.B.; Mobashery, S. Versatility of aminoglycosides and prospects for their future. Clin. Microbiol. Rev. 2003, 16, 430–450. [Google Scholar] [CrossRef] [PubMed]
- Casin, I.; Hanau-Berçot, B.; Podglajen, I.; Vahaboglu, H.; Collatz, E. Salmonella enterica serovar Typhimurium blaPER-1-carrying plasmid pSTI1 encodes an extended-spectrum aminoglycoside 6′-N-acetyltransferase of type Ib. Antimicrob. Agents. Chemother. 2003, 47, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Maurice, F.; Broutin, I.; Podglajen, I.; Benas, P.; Collatz, E.; Dardel, F. Enzyme structural plasticity and the emergence of broad-spectrum antibiotic resistance. EMBO Rep. 2008, 9, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Pourreza, A.; Witherspoon, M.; Fox, J.; Newmark, J.; Bui, D.; Tolmasky, M.E. Mutagenesis analysis of a conserved region involved in acetyl coenzyme A binding in the aminoglycoside 6′-N-acetyltransferase type Ib encoded by plasmid pJHCMW1. Antimicrob. Agents. Chemother. 2005, 49, 2979–2982. [Google Scholar] [CrossRef] [PubMed]
- Chiem, K.; Jani, S.; Fuentes, B.; Lin, D.L.; Rasche, M.E.; Tolmasky, M.E. Identification of an inhibitor of the aminoglycoside 6′-N-acetyltransferase type Ib [AAC(6′)-Ib] by glide molecular docking. Med. Chem. Commun. 2016, 7, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D.; Ladak, P. Overexpression and characterization of the chromosomal aminoglycoside 6′-N-acetyltransferase from Enterococcus faecium. Antimicrob. Agents. Chemother. 1997, 41, 956–960. [Google Scholar] [PubMed]
- Burk, D.L.; Ghuman, N.; Wybenga-Groot, L.E.; Berghuis, A.M. X-ray structure of the AAC(6′)-Ii antibiotic resistance enzyme at 1.8 Å resolution; examination of oligomeric arrangements in GNAT superfamily members. Protein Sci. 2003, 12, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Draker, K.A.; Wright, G.D. Molecular mechanism of the enterococcal aminoglycoside 6′-N-acetyltransferase: Role of GNAT-conserved residues in the chemistry of antibiotic inactivation. Biochemistry 2004, 43, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Draker, K.-A.; Northrop, D.B.; Wright, G.D. Kinetic mechanism of the GCN5-related chromosomal aminoglycoside acetyltransferase AAC(6′)-Ii from Enterococcus faecium: Evidence of dimer subunit cooperativity. Biochemistry 2003, 42, 6565–6574. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Yan, X.; Baettig, O.M.; Berghuis, A.M.; Auclair, K. Regio-and chemoselective 6′-N-derivatization of aminoglycosides: Bisubstrate inhibitors as probes to study aminoglycoside 6′-N-acetyltransferases. Angew. Chem. 2005, 117, 7019–7022. [Google Scholar] [CrossRef]
- Gao, F.; Yan, X.; Shakya, T.; Baettig, O.M.; Ait-Mohand-Brunet, S.; Berghuis, A.M.; Wright, G.D.; Auclair, K. Synthesis and structure-activity relationships of truncated bisubstrate inhibitors of aminoglycoside 6′-N-acetyltransferases. J. Med. Chem. 2006, 49, 5273–5281. [Google Scholar] [CrossRef] [PubMed]
- Vetting, M.W.; Magnet, S.; Nieves, E.; Roderick, S.L.; Blanchard, J.S. A bacterial acetyltransferase capable of regioselective N-acetylation of antibiotics and histones. Chem. Biol. 2004, 11, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Green, K.D.; Tsodikov, O.V.; Garneau-Tsodikova, S. Aminoglycoside multiacetylating activity of the enhanced intracellular survival protein from Mycobacterium smegmatis and its inhibition. Biochemistry 2012, 51, 4959–4967. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; An, D.R.; Song, J.; Yoon, J.Y.; Kim, H.S.; Yoon, H.J.; Im, H.N.; Kim, J.; Lee, S.J.; Kim, K.H. Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc. Natl. Acad. Sci. USA 2012, 109, 7729–7734. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Biswas, T.; Porter, V.R.; Tsodikov, O.V.; Garneau Tsodikova, S. Unusual regioversatility of acetyltransferase Eis, a cause of drug resistance in XDR-TB. Proc. Natl. Acad. Sci. USA 2011, 108, 9804–9808. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Parthun, M. Hat1: The emerging cellular roles of a type B histone acetyltransferase. Oncogene 2007, 26, 5319–5328. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Moshkina, N.; Min, J.; Zeng, H.; Joshua, J.; Zhou, M.M.; Plotnikov, A.N. Structural basis for substrate specificity and catalysis of human histone acetyltransferase 1. Proc. Natl. Acad. Sci. USA 2012, 109, 8925–8930. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, L.; Liang, J.; Shi, L.; Yang, J.; Yi, X.; Zhang, D.; Han, X.; Yu, N.; Shang, Y. Histone acetyltransferase 1 promotes homologous recombination in DNA repair by facilitating histone turnover. J. Biol. Chem. 2013, 288, 18271–18282. [Google Scholar] [CrossRef] [PubMed]
- Decker, P.V.; David, Y.Y.; Iizuka, M.; Qiu, Q.; Smith, M.M. Catalytic-site mutations in the MYST family histone acetyltransferase Esa1. Genetics 2008, 178, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Allard, S.; Utley, R.T.; Savard, J.; Clarke, A.; Grant, P.; Brandl, C.J.; Pillus, L.; Workman, J.L.; Côté, J. NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. EMBO J. 1999, 18, 5108–5119. [Google Scholar] [CrossRef] [PubMed]
- Babiarz, J.E.; Halley, J.E.; Rine, J. Telomeric heterochromatin boundaries require NuA4-dependent acetylation of histone variant H2A. Z in Saccharomyces cerevisiae. Genes Dev. 2006, 20, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.S.; Lowell, J.E.; Jacobson, S.J.; Pillus, L. Esa1p is an essential histone acetyltransferase required for cell cycle progression. Mol. Cell. Biol. 1999, 19, 2515–2526. [Google Scholar] [CrossRef] [PubMed]
- Keogh, M.C.; Mennella, T.A.; Sawa, C.; Berthelet, S.; Krogan, N.J.; Wolek, A.; Podolny, V.; Carpenter, L.R.; Greenblatt, J.F.; Baetz, K. The Saccharomyces cerevisiae histone H2A variant Htz1 is acetylated by NuA4. Genes Dev. 2006, 20, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Millar, C.B.; Grunstein, M. Genome-wide patterns of histone modifications in yeast. Nat. Rev. Mol. Cell Biol. 2006, 7, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Eisen, A.; Gu, W.; Sattah, M.; Pannuti, A.; Zhou, J.; Cook, R.G.; Lucchesi, J.C.; Allis, C.D. ESA1 is a histone acetyltransferase that is essential for growth in yeast. Proc. Natl. Acad. Sci. USA 1998, 95, 3561–3565. [Google Scholar] [CrossRef] [PubMed]
- Durant, M.; Pugh, B.F. Genome-wide relationships between TAF1 and histone acetyltransferases in Saccharomyces cerevisiae. Mol. Cell. Biol. 2006, 26, 2791–2802. [Google Scholar] [CrossRef] [PubMed]
- Durant, M.; Pugh, B.F. NuA4-directed chromatin transactions throughout the Saccharomyces cerevisiae genome. Mol. Cell. Biol. 2007, 27, 5327–5335. [Google Scholar] [CrossRef] [PubMed]
- Lafon, A.; Chang, C.; Scott, E.; Jacobson, S.; Pillus, L. MYST opportunities for growth control: Yeast genes illuminate human cancer gene functions. Oncogene 2007, 26, 5373–5384. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Harper, S.; Speicher, D.W.; Marmorstein, R. The catalytic mechanism of the ESA1 histone acetyltransferase involves a self-acetylated intermediate. Nat. Struct. Biol. 2002, 9, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Barlev, N.A.; Haley, R.H.; Berger, S.L.; Marmorstein, R. Crystal structure of yeast Esa1 suggests a unified mechanism for catalysis and substrate binding by histone acetyltransferases. Mol. Cell. 2000, 6, 1195–1205. [Google Scholar] [CrossRef]
- Yuan, H.; Rossetto, D.; Mellert, H.; Dang, W.; Srinivasan, M.; Johnson, J.; Hodawadekar, S.; Ding, E.C.; Speicher, K.; Abshiru, N.; et al. MYST protein acetyltransferase activity requires active site lysine autoacetylation. EMBO J. 2012, 31, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, C.E.; Albaugh, B.N.; Tan, S.; Denu, J.M. Catalytic mechanism of a MYST family histone acetyltransferase. Biochemistry 2007, 46, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Pollard, K.J.; Peterson, C.L. Role for ADA/GCN5 products in antagonizing chromatin-mediated transcriptional repression. Mol. Cell. Biol. 1997, 17, 6212–6222. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, H.; Takami, Y.; Nakayama, T. GCN5: A supervisor in all-inclusive control of vertebrate cell cycle progression through transcription regulation of various cell cycle-related genes. Gene 2005, 347, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, A.G.; Fink, G.R. Positive regulation in the general amino acid control of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1983, 80, 5374–5378. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Edmondson, D.G.; Evrard, Y.A.; Wakamiya, M.; Behringer, R.R.; Roth, S.Y. Loss of Gcn5l2 leads to increased apoptosis and mesodermal defects during mouse development. Nat. Genet. 2000, 26, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Trievel, R.C.; Rojas, J.R.; Sterner, D.E.; Venkataramani, R.N.; Wang, L.; Zhou, J.; Allis, C.D.; Berger, S.L.; Marmorstein, R. Crystal structure and mechanism of histone acetylation of the yeast GCN5 transcriptional coactivator. Proc. Natl. Acad. Sci. USA 1999, 96, 8931–8936. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, A.; Bernstein, G.; Dong, A.; Antoshenko, T.; Wu, H.; Loppnau, P.; Bochkarev, A.; Plotnikov, A.N. Crystal structure of a binary complex between human GCN5 histone acetyltransferase domain and acetyl coenzyme A. Proteins 2007, 68, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Clements, A.; Poux, A.N.; Lo, W.S.; Pillus, L.; Berger, S.L.; Marmorstein, R. Structural basis for histone and phosphohistone binding by the GCN5 histone acetyltransferase. Mol. Cell 2003, 12, 461–473. [Google Scholar] [CrossRef]
- Lin, Y.; Fletcher, C.M.; Zhou, J.; Allis, C.D.; Wagner, G. Solution structure of the catalytic domain of GCN5 histone acetyltransferase bound to coenzyme A. Nature 1999, 400, 86–89. [Google Scholar] [PubMed]
- Poux, A.N.; Cebrat, M.; Kim, C.M.; Cole, P.A.; Marmorstein, R. Structure of the GCN5 histone acetyltransferase bound to a bisubstrate inhibitor. Proc. Natl. Acad. Sci. USA 2002, 99, 14065–14070. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.R.; Trievel, R.C.; Zhou, J.; Mo, Y.; Li, X.; Berger, S.L.; Allis, C.D.; Marmorstein, R. Structure of Tetrahymena GCN5 bound to coenzyme A and a histone H3 peptide. Nature 1999, 401, 93–98. [Google Scholar] [PubMed]
- Liu, L.; Scolnick, D.M.; Trievel, R.C.; Zhang, H.B.; Marmorstein, R.; Halazonetis, T.D.; Berger, S.L. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 1999, 19, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Lin, J.; Cai, Y.; Yu, J.; Hong, H.; Ji, K.; Downey, J.S.; Lu, X.; Chen, R.; Han, J.; et al. Dimeric structure of p300/CBP associated factor. BMC Struct. Biol. 2014, 14, 2. [Google Scholar] [CrossRef] [PubMed]
- Clements, A.; Rojas, J.R.; Trievel, R.C.; Wang, L.; Berger, S.L.; Marmorstein, R. Crystal structure of the histone acetyltransferase domain of the human PCAF transcriptional regulator bound to coenzyme A. EMBO J. 1999, 18, 3521–3532. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Edmondson, D.G.; Roth, S.Y. Mammalian GCN5 and P/CAF acetyltransferases have homologous amino-terminal domains important for recognition of nucleosomal substrates. Mol. Cell. Biol. 1998, 18, 5659–5669. [Google Scholar] [CrossRef] [PubMed]
- Sampath, V.; Liu, B.; Tafrov, S.; Srinivasan, M.; Rieger, R.; Chen, E.I.; Sternglanz, R. Biochemical characterization of Hpa2 and Hpa3, two small closely related acetyltransferases from Saccharomyces cerevisiae. J. Chem. Biol. 2013, 288, 21506–21513. [Google Scholar] [CrossRef] [PubMed]
- Angus-Hill, M.L.; Dutnall, R.N.; Tafrov, S.T.; Sternglanz, R.; Ramakrishnan, V. Crystal structure of the histone acetyltransferase Hpa2: A tetrameric member of the Gcn5-related N-acetyltransferase superfamily. J. Mol. Biol. 1999, 294, 1311–1325. [Google Scholar] [CrossRef] [PubMed]
- Hickman, A.B.; Namboodiri, M.; Klein, D.C.; Dyda, F. The structural basis of ordered substrate binding by serotonin N-acetyltransferase: Enzyme complex at 1.8 Å resolution with a bisubstrate analog. Cell 1999, 97, 361–369. [Google Scholar] [CrossRef]
- Brent, M.M.; Iwata, A.; Carten, J.; Zhao, K.; Marmorstein, R. Structure and biochemical characterization of protein acetyltransferase from Sulfolobus solfataricus. J. Chem. Biol. 2009, 284, 19412–19419. [Google Scholar] [CrossRef] [PubMed]
- Friedmann, D.R.; Aguilar, A.; Fan, J.; Nachury, M.V.; Marmorstein, R. Structure of the α-tubulin acetyltransferase, αTAT1, and implications for tubulin-specific acetylation. Proc. Natl. Acad. Sci. USA 2012, 109, 19655–19660. [Google Scholar] [CrossRef] [PubMed]
- Liszczak, G.; Arnesen, T.; Marmorstein, R. Structure of a ternary Naa50p (NAT5/SAN) N-terminal acetyltransferase complex reveals the molecular basis for substrate-specific acetylation. J. Chem. Biol. 2011, 286, 37002–37010. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lang, P.T.; Fortune, S.M.; Sassetti, C.M.; Alber, T. Cyclic AMP regulation of protein lysine acetylation in Mycobacterium tuberculosis. Nat. Struct. Mol. Biol. 2012, 19, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Zhang, G.; Hwa, Y.L.; Li, J.; Dowdy, S.C.; Jiang, S.W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 2010, 10, 935–954. [Google Scholar] [CrossRef] [PubMed]
- Friedmann, D.R.; Marmorstein, R. Structure and mechanism of non-histone protein acetyltransferase enzymes. FASEB J. 2013, 280, 5570–5581. [Google Scholar] [CrossRef] [PubMed]
- Kalebic, N.; Sorrentino, S.; Bolasco, G.; Martinez, C.; Heppenstall, P.A. αTAT1 is the major α-tubulin acetyltransferase in mice. Nat. Commun. 2013, 4, 1962. [Google Scholar] [CrossRef] [PubMed]
- Akella, J.S.; Wloga, D.; Kim, J.; Starostina, N.G.; Lyons-Abbott, S.; Morrissette, N.S.; Dougan, S.T.; Kipreos, E.T.; Gaertig, J. MEC-17 is an α-tubulin acetyltransferase. Nature 2010, 467, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Hickman, A.B.; Klein, D.C.; Dyda, F. Melatonin biosynthesis: The structure of serotonin N-acetyltransferase at 2.5 Å resolution suggests a catalytic mechanism. Mol. Cell 1999, 3, 23–32. [Google Scholar] [CrossRef]
- Davenport, A.M.; Collins, L.N.; Chiu, H.; Minor, P.J.; Sternberg, P.W.; Hoelz, A. Structural and functional characterization of the α-tubulin acetyltransferase MEC-17. J. Mol. Biol. 2014, 426, 2605–2616. [Google Scholar] [CrossRef] [PubMed]
- Nambi, S.; Basu, N.; Visweswariah, S.S. cAMP-regulated protein lysine acetylases in mycobacteria. J. Chem. Biol. 2010, 285, 24313–24323. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Hegde, S.S.; Blanchard, J.S. Reversible acetylation and inactivation of Mycobacterium tuberculosis acetyl-CoA synthetase is dependent on cAMP. Biochemistry 2011, 50, 5883–5892. [Google Scholar] [CrossRef] [PubMed]
- Starheim, K.K.; Gromyko, D.; Velde, R.; Varhaug, J.E.; Arnesen, T. Composition and Biological Significance of the Human Nα-Terminal Acetyltransferases; BioMed Central Ltd.: Bergen, Norway, 2009; p. S3. [Google Scholar]
- Klein, D.C. Arylalkylamine N-acetyltransferase:“The Timezyme”. J. Chem. Biol. 2007, 282, 4233–4237. [Google Scholar] [CrossRef] [PubMed]
- Khalil, E.M.; Cole, P.A. A potent inhibitor of the melatonin rhythm enzyme. J. Am. Chem. Soc. 1998, 120, 6195–6196. [Google Scholar] [CrossRef]
- Cheng, K.C.; Liao, J.-N.; Lyu, P.C. Crystal structure of the dopamine N-acetyltransferase–acetyl-CoA complex provides insights into the catalytic mechanism. Biochem. J. 2012, 446, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Robinson, H.; Ding, H.; Christensen, B.M.; Li, J. Evolution of insect arylalkylamine N-acetyltransferases: Structural evidence from the yellow fever mosquito, Aedes aegypti. Proc. Natl. Acad. Sci. USA 2012, 109, 11669–11674. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, D.R.; Jeffries, K.A.; Anderson, R.L.; Carpenter, A.M.; Opsina, S.R.; Merkler, D.J. Identification of an arylalkylamine N-acyltransferase from Drosophila melanogaster that catalyzes the formation of long-chain N-acylserotonins. FEBS Lett. 2014, 588, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Boehmelt, G.; Fialka, I.; Brothers, G.; McGinley, M.D.; Patterson, S.D.; Mo, R.; Hui, C.C.; Chung, S.; Huber, L.A.; Mak, T.W. Cloning and characterization of the murine glucosamine-6-phosphate acetyltransferase EMeg32. Differential expression and intracellular membrane association. J. Chem. Biol. 2000, 275, 12821–12832. [Google Scholar] [CrossRef]
- Herscovics, A.; Orlean, P. Glycoprotein biosynthesis in yeast. FASEB J. 1993, 7, 540–550. [Google Scholar] [PubMed]
- Wendland, J.; Schaub, Y.; Walther, A. N-acetylglucosamine utilization by Saccharomyces cerevisiae based on expression of Candida albicans NAG genes. Appl. Environ. Microbiol. 2009, 75, 5840–5845. [Google Scholar] [CrossRef] [PubMed]
- Peneff, C.; Mengin-Lecreulx, D.; Bourne, Y. The crystal structures of Apo and complexed Saccharomyces cerevisiae GNA1 shed light on the catalytic mechanism of an amino-sugar N-acetyltransferase. J. Chem. Biol. 2001, 276, 16328–16334. [Google Scholar] [CrossRef] [PubMed]
- Hurtado-Guerrero, R.; Raimi, O.G.; Min, J.; Zeng, H.; Vallius, L.; Shepherd, S.; Ibrahim, A.F.; Wu, H.; Plotnikov, A.N.; van Aalten, D.M. Structural and kinetic differences between human and Aspergillus fumigatus d-glucosamine-6-phosphate N-acetyltransferase. Biochem. J. 2008, 415, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Liang, Y.H.; Li, L.F.; Su, X.D. Acceptor substrate binding revealed by crystal structure of human glucosamine-6-phosphate N-acetyltransferase 1. FEBS Lett. 2008, 582, 2973–2978. [Google Scholar] [CrossRef] [PubMed]
- Riegler, H.; Herter, T.; Grishkovskaya, I.; Lude, A.; Ryngajllo, M.; Bolger, M.; Essigmann, B.; Usadel, B. Crystal structure and functional characterization of a glucosamine-6-phosphate N-acetyltransferase from Arabidopsis thaliana. Biochem. J. 2012, 443, 427. [Google Scholar] [CrossRef] [PubMed]
- Dorfmueller, H.C.; Fang, W.; Rao, F.V.; Blair, D.E.; Attrill, H.; van Aalten, D.M. Structural and biochemical characterization of a trapped coenzyme A adduct of Caenorhabditis elegans glucosamine-6-phosphate N-acetyltransferase 1. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Metlitskaya, A.; Severinov, K.; Nair, S.K. Structural basis for microcin C7 inactivation by the MccE acetyltransferase. J. Chem. Biol. 2011, 286, 21295–21303. [Google Scholar] [CrossRef] [PubMed]
- Novikova, M.; Kazakov, T.; Vondenhoff, G.H.; Semenova, E.; Rozenski, J.; Metlytskaya, A.; Zukher, I.; Tikhonov, A.; van Aerschot, A.; Severinov, K. MccE provides resistance to protein synthesis inhibitor microcin C by acetylating the processed form of the antibiotic. J. Chem. Biol. 2010, 285, 12662–12669. [Google Scholar] [CrossRef] [PubMed]
- Schoenhofen, I.C.; McNally, D.J.; Brisson, J.-R.; Logan, S.M. Elucidation of the CMP-pseudaminic acid pathway in Helicobacter pylori: Synthesis from UDP-N-acetylglucosamine by a single enzymatic reaction. Glycobiology 2006, 16, 8C–14C. [Google Scholar] [CrossRef] [PubMed]
- Ud-Din, A.I.; Liu, Y.C.; Roujeinikova, A. Crystal structure of Helicobacter pylori pseudaminic acid biosynthesis N-acetyltransferase PseH: Implications for substrate specificity and catalysis. PLoS ONE 2015, 10, e0115634. [Google Scholar] [CrossRef] [PubMed]
- Vetting, M.W.; de Carvalho, L.P.S.; Roderick, S.L.; Blanchard, J.S. A novel dimeric structure of the RimL Nα-acetyltransferase from Salmonella typhimurium. J. Chem. Biol. 2005, 280, 22108–22114. [Google Scholar] [CrossRef] [PubMed]
- Song, W.S.; Nam, M.S.; Namgung, B.; Yoon, S.I. Structural analysis of PseH, the Campylobacter jejuni N-acetyltransferase involved in bacterial O-linked glycosylation. Biochem. Biophys. Res. Commun. 2015, 458, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Morales, F.; Prieto, A.I.; Beuzón, C.R.; Holden, D.W.; Casadesús, J. Role for Salmonella enterica enterobacterial common antigen in bile resistance and virulence. J. Bacteriol. 2003, 185, 5328–5332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Radziejewska Lebrecht, J.; Krajewska Pietrasik, D.; Toivanen, P.; Skurnik, M. Molecular and chemical characterization of the lipopolysaccharide O-antigen and its role in the virulence of Yersinia enterocolitica serotype O: 8. Mol. Microbiol. 1997, 23, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.E.; Fedrigo, G.V.; Clementín, A.L.; Ielmini, M.V.; Feldman, M.F.; Véscovi, E.G. Enterobacterial common antigen integrity is a checkpoint for flagellar biogenesis in Serratia marcescens. J. Bacteriol. 2008, 190, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.-N.; Rangarajan, E.; Munger, C.; Nadeau, G.; Sulea, T.; Matte, A. Crystal structure of TDP-fucosamine acetyltransferase (WecD) from Escherichia coli, an enzyme required for enterobacterial common antigen synthesis. J. Bacteriol. 2006, 188, 5606–5617. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Ding, Y.; Bartlam, M.; Sun, F.; Le, Y.; Qin, X.; Tang, H.; Zhang, R.; Joachimiak, A.; Liu, J. Crystal structure of tabtoxin resistance protein complexed with acetyl coenzyme A reveals the mechanism for β-lactam acetylation. J. Mol. Biol. 2003, 325, 1019–1030. [Google Scholar] [CrossRef]
- Marek, E.T.; Dickson, R.C. Cloning and characterization of Saccharomyces cerevisiae genes that confer l-methionine sulfoximine and tabtoxin resistance. J. Bacteriol. 1987, 169, 2440–2448. [Google Scholar] [PubMed]
- Bender, C.L.; Alarcón-Chaidez, F.; Gross, D.C. Pseudomonas syringae phytotoxins: Mode of action, regulation, and biosynthesis by peptide and polyketide synthetases. Microbiol. Mol. Biol. Rev. 1999, 63, 266–292. [Google Scholar] [PubMed]
- Anzai, H.; Yoneyama, K.; Yamaguchi, I. Transgenic tobacco resistant to a bacterial disease by the detoxification of a pathogenic toxin. Mol. Gen. Genet. 1989, 219, 492–494. [Google Scholar] [CrossRef]
- Batchvarova, R.; Nikolaeva, V.; Slavov, S.; Bossolova, S.; Valkov, V.; Atanassova, S.; Guelemerov, S.; Atanassov, A.; Anzai, H. Transgenic tobacco cultivars resistant to Pseudomonas syringae pv. tabaci. Theor. Appl. Genet. 1998, 97, 986–989. [Google Scholar] [CrossRef]
- Nasuno, R.; Hirano, Y.; Itoh, T.; Hakoshima, T.; Hibi, T.; Takagi, H. Structural and functional analysis of the yeast N-acetyltransferase Mpr1 involved in oxidative stress tolerance via proline metabolism. Proc. Natl. Acad. Sci. USA 2013, 110, 11821–11826. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Takagi, H. N-acetyltransferase Mpr1 confers freeze tolerance on Saccharomyces cerevisiae by reducing reactive oxygen species. J. Bacteriol. 2005, 138, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Takagi, H. N-Acetyltransferase Mpr1 confers ethanol tolerance on Saccharomyces cerevisiae by reducing reactive oxygen species. Appl. Environ. Microbiol. 2007, 75, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Takagi, H. Role of the yeast acetyltransferase Mpr1 in oxidative stress: Regulation of oxygen reactive species caused by a toxic proline catabolism intermediate. Proc. Natl. Acad. Sci. USA 2004, 101, 12616–12621. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Spermidine/spermine-N1-acetyltransferase: A key metabolic regulator. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E995–E1010. [Google Scholar] [CrossRef] [PubMed]
- Perez-Leal, O.; Merali, S. Regulation of polyamine metabolism by translational control. Amino Acids 2012, 42, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Montemayor, E.J. Biochemical Studies of Spermidine/Spermine N1-Acetyltransferase, an Important Regulator of Cellular Polyamines; The University of Texas At Austin: Austin, TX, USA, 2008. [Google Scholar]
- Forouhar, F.; Lee, I.S.; Vujcic, J.; Vujcic, S.; Shen, J.; Vorobiev, S.M.; Xiao, R.; Acton, T.B.; Montelione, G.T.; Porter, C.W.; et al. Structural and functional evidence for Bacillus subtilis PaiA as a novel N1-spermidine/spermine acetyltransferase. J. Chem. Biol. 2005, 280, 40328–40336. [Google Scholar] [CrossRef] [PubMed]
- Filippova, E.V.; Kuhn, M.L.; Osipiuk, J.; Kiryukhina, O.; Joachimiak, A.; Ballicora, M.A.; Anderson, W.F. A novel polyamine allosteric site of SpeG from Vibrio cholerae is revealed by its dodecameric structure. J. Mol. Biol. 2015, 427, 1316–1334. [Google Scholar] [CrossRef] [PubMed]
- Montemayor, E.J.; Hoffman, D.W. The crystal structure of spermidine/spermine N1-acetyltransferase in complex with spermine provides insights into substrate binding and catalysis. Biochemistry 2008, 47, 9145–9153. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.Q.; Zhu, D.Y.; Yin, L.; Zhang, Y.; Vonrhein, C.; Wang, D.C. Crystal structure of human spermidine/spermine N1-acetyltransferase (hSSAT): The first structure of a new sequence family of transferase homologous superfamily. Proteins 2006, 63, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Filippova, E.V.; Shuvalova, L.; Minasov, G.; Kiryukhina, O.; Zhang, Y.; Clancy, S.; Radhakrishnan, I.; Joachimiak, A.; Anderson, W. Crystal structure of the novel PaiA N-acetyltransferase from Thermoplasma acidophilum involved in the negative control of sporulation and degradative enzyme production. Proteins 2011, 79, 2566–2577. [Google Scholar] [CrossRef] [PubMed]
- Filippova, E.V.; Weigand, S.; Osipiuk, J.; Kiryukhina, O.; Joachimiak, A.; Anderson, W.F. Substrate-induced allosteric change in the quaternary structure of the spermidine N-acetyltransferase SpeG. J. Mol. Biol. 2015, 427, 3538–3553. [Google Scholar] [CrossRef] [PubMed]
- Hegde, S.S.; Chandler, J.; Vetting, M.W.; Yu, M.; Blanchard, J.S. Mechanistic and structural analysis of human spermidine/spermine N1-acetyltransferase. Biochemistry 2007, 46, 7187–7195. [Google Scholar] [CrossRef] [PubMed]
- Bewley, M.C.; Graziano, V.; Jiang, J.; Matz, E.; Studier, F.W.; Pegg, A.E.; Coleman, C.S.; Flanagan, J.M. Structures of wild-type and mutant human spermidine/spermine N1-acetyltransferase, a potential therapeutic drug target. Proc. Natl. Acad. Sci. USA 2006, 103, 2063–2068. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.S.; Huang, H.; Pegg, A.E. Role of the carboxyl terminal MATEE sequence of spermidine/spermine N1-acetyltransferase in the activity and stabilization by the polyamine analog N1, N12-bis (ethyl) spermine. Biochemistry 1995, 34, 13423–13430. [Google Scholar] [CrossRef] [PubMed]
- Escalante-Semerena, J.C. Nε-lysine acetylation control conserved in all three different domains. Issues 2010. [Google Scholar] [CrossRef]
- Hu, L.I.; Chi, B.K.; Kuhn, M.L.; Filippova, E.V.; Walker-Peddakotla, A.J.; Bäsell, K.; Becher, D.; Anderson, W.F.; Antelmann, H.; Wolfe, A.J. Acetylation of the response regulator RcsB controls transcription from a small RNA promoter. J. Bacteriol. 2013, 195, 4174–4186. [Google Scholar] [CrossRef] [PubMed]
- Majorek, K.A.; Kuhn, M.L.; Chruszcz, M.; Anderson, W.F.; Minor, W. Structural, functional and inhibition studies of a GNAT superfamily protein PA4794: A new C-terminal lysine protein acetyltransferase from Pseudomonas aeruginosa. J. Chem. Biol. 2013, 288, 30223–30235. [Google Scholar] [CrossRef] [PubMed]
- Card, G.L.; Peterson, N.A.; Smith, C.A.; Rupp, B.; Schick, B.M.; Baker, E.N. The crystal structure of Rv1347c, a putative antibiotic resistance protein from Mycobacterium tuberculosis, reveals a GCN5-related fold and suggests an alternative function in siderophore biosynthesis. J. Chem. Biol. 2005, 280, 13978–13986. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.V.; Puri, R.V.; Chauhan, P.; Kar, R.; Rohilla, A.; Khera, A.; Tyagi, A.K. Disruption of mycobactin biosynthesis leads to attenuation of Mycobacterium tuberculosis for growth and virulence. J. Infect. Dis. 2013, 208, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Crouch, M.L.V.; Castor, M.; Karlinsey, J.E.; Kalhorn, T.; Fang, F.C. Biosynthesis and IroC-dependent export of the siderophore salmochelin are essential for virulence of Salmonella enterica serovar Typhimurium. Mol. Microbiol. 2008, 67, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, M.S.; O’Connor, C.; Miller, V.L. Yersiniabactin is a virulence factor for Klebsiella pneumoniae during pulmonary infection. Infect. Immun. 2007, 75, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Takase, H.; Nitanai, H.; Hoshino, K.; Otani, T. Impact of siderophore production on Pseudomonas aeruginosai infections in immunosuppressed mice. Infect. Immun. 2000, 68, 1834–1839. [Google Scholar] [CrossRef] [PubMed]
- Frankel, B.A.; Blanchard, J.S. Mechanistic analysis of Mycobacterium tuberculosis Rv1347c, a lysine Nε-acyltransferase involved in mycobactin biosynthesis. Arch. Biochem. Biophys. 2008, 477, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Polevoda, B.; Sherman, F. Nα-terminal acetylation of eukaryotic proteins. J. Chem. Biol. 2000, 275, 36479–36482. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, A.; Isono, S.; Sheback, A.; Isono, K. Cloning and nucleotide sequencing of the genes rimI and rimJ which encode enzymes acetylating ribosomal proteins S18 and S5 of Escherichia coli K12. Mol. Gen. Genet. 1987, 209, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Matsushita, Y.; Yoshikawa, A.; Isono, K. Cloning and molecular characterization of the gene rimL which encodes an enzyme acetylating ribosomal protein L 12 of Escherichia coli K 12. Mol. Gen. Genet. 1989, 217, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Vetting, M.W.; Bareich, D.C.; Yu, M.; Blanchard, J.S. Crystal structure of RimI from Salmonella typhimurium LT2, the GNAT responsible for Nα-acetylation of ribosomal protein S18. Protein Sci. 2008, 17, 1781–1790. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, T.; Kuznedelov, K.; Semenova, E.; Mukhamedyarov, D.; Datsenko, K.A.; Metlitskaya, A.; Vondenhoff, G.H.; Tikhonov, A.; Agarwal, V.; Nair, S. The RimL transacetylase provides resistance to translation inhibitor microcin C. J. Bacteriol. 2014, 196, 3377–3385. [Google Scholar] [CrossRef] [PubMed]
- Brunzelle, J.S.; Wu, R.; Korolev, S.V.; Collart, F.R.; Joachimiak, A.; Anderson, W.F. Crystal structure of Bacillus subtilis YdaF protein: A putative ribosomal N-acetyltransferase. Proteins 2004, 57, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Benson, T.E.; Prince, D.B.; Mutchler, V.T.; Curry, K.A.; Ho, A.M.; Sarver, R.W.; Hagadorn, J.C.; Choi, G.H.; Garlick, R.L. X-ray crystal structure of Staphylococcus aureus FemA. Structure 2002, 10, 1107–1115. [Google Scholar] [CrossRef]
- Rohrer, S.; Berger-Bächi, B. FemABX peptidyl transferases: A link between branched-chain cell wall peptide formation and β-lactam resistance in gram-positive cocci. Antimicrob. Agents Chemother. 2003, 47, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Biarrotte-Sorin, S.; Maillard, A.P.; Delettré, J.; Sougakoff, W.; Arthur, M.; Mayer, C. Crystal structures of Weissella viridescens FemX and its complex with UDP-MurNAc-pentapeptide: Insights into FemABX family substrates recognition. Structure 2004, 12, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Berger-Bächi, B.; Barberis-Maino, L.; Strässle, A.; Kayser, F.H. FemA, a host-mediated factor essential for methicillin resistance in Staphylococcus aureus: Molecular cloning and characterization. Mol. Gen. Genet. 1989, 219, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Kamiryo, T.; Matsuhashi, M. The biosynthesis of the cross-linking peptides in the cell wall peptidoglycan of Staphylococcus aureus. J. Chem. Biol. 1972, 247, 6306–6311. [Google Scholar]
- Maidhof, H.; Reinicke, B.; Blümel, P.; Berger-Bächi, B.; Labischinski, H. femA, which encodes a factor essential for expression of methicillin resistance, affects glycine content of peptidoglycan in methicillin-resistant and methicillin-susceptible Staphylococcus aureus strains. J. Bacteriol. 1991, 173, 3507–3513. [Google Scholar] [PubMed]
- Ling, B.; Berger-Bächi, B. Increased overall antibiotic susceptibility in Staphylococcus aureus femAB null mutants. Antimicrob. Agents. Chemother. 1998, 42, 936–938. [Google Scholar] [PubMed]
- Biou, V.; Yaremchuk, A.; Tukalo, M.; Cusack, S. The 2.9 A crystal structure of T. thermophilus seryl-tRNA synthetase complexed with tRNA (Ser). Science 1994, 263, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- Fonvielle, M.; Li de La Sierra-Gallay, I.; El-Sagheer, A.H.; Lecerf, M.; Patin, D.; Mellal, D.; Mayer, C.; Blanot, D.; Gale, N.; Brown, T. The structure of FemXWv in complex with a peptidyl-RNA conjugate: Mechanism of aminoacyl transfer from Ala-tRNAAla to peptidoglycan precursors. Angew. Chem. 2013, 125, 7419–7422. [Google Scholar] [CrossRef]
- Maillard, A.P.; Biarrotte-Sorin, S.; Villet, R.; Mesnage, S.; Bouhss, A.; Sougakoff, W.; Mayer, C.; Arthur, M. Structure-based site-directed mutagenesis of the UDP-MurNAc-pentapeptide-binding cavity of the FemX alanyl transferase from Weissella viridescens. J. Bacteriol. 2005, 187, 3833–3838. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Kashina, A. Posttranslational arginylation as a global biological regulator. Dev. Biol. 2011, 358, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Suto, K.; Shimizu, Y.; Watanabe, K.; Ueda, T.; Fukai, S.; Nureki, O.; Tomita, K. Crystal structures of leucyl/phenylalanyl-tRNA-protein transferase and its complex with an aminoacyl-tRNA analog. EMBO J. 2006, 25, 5942–5950. [Google Scholar] [CrossRef] [PubMed]
- Doerig, C.; Rayner, J.C.; Scherf, A.; Tobin, A.B. Post-translational protein modifications in malaria parasites. Nat. Rev. Microbiol. 2015, 13, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, D.; McWherter, C.; Rocque, W.; Lennon, P.; Getman, D.; Gordon, J. Kinetic and structural evidence for a sequential ordered Bi Bi mechanism of catalysis by Saccharomyces cerevisiae myristoyl-CoA: Protein N-myristoyltransferase. J. Chem. Biol. 1991, 266, 9732–9739. [Google Scholar]
- Towler, D.A.; Adams, S.; Eubanks, S.; Towery, D.; Jackson-Machelski, E.; Glaser, L.; Gordon, J. Myristoyl CoA: Protein N-myristoyltransferase activities from rat liver and yeast possess overlapping yet distinct peptide substrate specificities. J. Chem. Biol. 1988, 263, 1784–1790. [Google Scholar]
- Wright, M.H.; Heal, W.P.; Mann, D.J.; Tate, E.W. Protein myristoylation in health and disease. J. Chem. Biol. 2010, 3, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Maurer-Stroh, S.; Eisenhaber, F. Myristoylation of viral and bacterial proteins. Trends Microbiol. 2004, 12, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Nagatoshi, K.; Noriyasu, Y.; Okamura, T.; Takamitsu, E.; Suzuki, T.; Utsumi, T. Protein N-myristoylation plays a critical role in the endoplasmic reticulum morphological change induced by overexpression of protein lunapark, an integral membrane protein of the endoplasmic reticulum. PLoS ONE 2013, 8, e78235. [Google Scholar] [CrossRef] [PubMed]
- Weston, S.A.; Camble, R.; Colls, J.; Rosenbrock, G.; Taylor, I.; Egerton, M.; Tucker, A.D.; Tunnicliffe, A.; Mistry, A.; Mancia, F.; et al. Crystal structure of the anti-fungal target N-myristoyl transferase. Nat. Struct. Biol. 1998, 5, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Tao, Y.; Zhang, M.; Howard, M.H.; Gutteridge, S.; Ding, J. Crystal structures of Saccharomyces cerevisiae N-myristoyltransferase with bound myristoyl-CoA and inhibitors reveal the functional roles of the N-terminal region. J. Chem. Biol. 2007, 282, 22185–22194. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.R.; Bhatnagar, R.S.; Knoll, L.J.; Gordon, J.I. Genetic and biochemical studies of protein N-myristoylation. Annu. Rev. Biochem. 1994, 63, 869–914. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, D.; Johnson, R.; Gordon, J. Studies of the catalytic activities and substrate specificities of Saccharomyces cerevisiae myristoyl-coenzyme A: Protein N-myristoyltransferase deletion mutants and human/yeast Nmt chimeras in Escherichia coli and S. cerevisiae. J. Chem. Biol. 1992, 267, 23852–23861. [Google Scholar]
- Bhatnagar, R.S.; Fütterer, K.; Waksman, G.; Gordon, J.I. The structure of myristoyl-CoA: Protein N-myristoyltransferase. Biochim. Biophys. Acta 1999, 1441, 162–172. [Google Scholar] [CrossRef]
- Goncalves, V.; Brannigan, J.A.; Whalley, D.; Ansell, K.H.; Saxty, B.; Holder, A.A.; Wilkinson, A.J.; Tate, E.W.; Leatherbarrow, R.J. Discovery of Plasmodium vivax N-myristoyltransferase inhibitors: Screening, synthesis, and structural characterization of their binding mode. J. Med. Chem. 2012, 55, 3578–3582. [Google Scholar] [CrossRef] [PubMed]
- Rackham, M.D.; Brannigan, J.A.; Moss, D.K.; Yu, Z.; Wilkinson, A.J.; Holder, A.A.; Tate, E.W.; Leatherbarrow, R.J. Discovery of novel and ligand-efficient inhibitors of Plasmodium falciparum and Plasmodium vivax N-myristoyltransferase. J. Med. Chem. 2013, 56, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Rackham, M.D.; Brannigan, J.A.; Rangachari, K.; Meister, S.; Wilkinson, A.J.; Holder, A.A.; Leatherbarrow, R.J.; Tate, E.W. Design and synthesis of high affinity inhibitors of Plasmodium falciparum and Plasmodium vivax N-myristoyltransferases directed by ligand efficiency dependent lipophilicity (LELP). J. Med. Chem. 2014, 57, 2773–2788. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.H.; Clough, B.; Rackham, M.D.; Rangachari, K.; Brannigan, J.A.; Grainger, M.; Moss, D.K.; Bottrill, A.R.; Heal, W.P.; Broncel, M.; et al. Validation of N-myristoyltransferase as an antimalarial drug target using an integrated chemical biology approach. Nat. Chem. 2014, 6, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Brannigan, J.A.; Moss, D.K.; Brzozowski, A.M.; Wilkinson, A.J.; Holder, A.A.; Tate, E.W.; Leatherbarrow, R.J. Design and synthesis of inhibitors of Plasmodium falciparum N-myristoyltransferase, a promising target for antimalarial drug discovery. J. Med. Chem. 2012, 55, 8879–8890. [Google Scholar] [CrossRef] [PubMed]
- Brannigan, J.A.; Roberts, S.M.; Bell, A.S.; Hutton, J.A.; Hodgkinson, M.R.; Tate, E.W.; Leatherbarrow, R.J.; Smith, D.F.; Wilkinson, A.J. Diverse modes of binding in structures of Leishmania major N-myristoyltransferase with selective inhibitors. IUCrJ 2014, 1, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Hutton, J.A.; Goncalves, V.; Brannigan, J.A.; Paape, D.; Wright, M.H.; Waugh, T.M.; Roberts, S.M.; Bell, A.S.; Wilkinson, A.J.; Smith, D.F.; et al. Structure-based design of potent and selective Leishmania N-myristoyltransferase inhibitors. J. Med. Chem. 2014, 57, 8664–8670. [Google Scholar] [CrossRef] [PubMed]
- Olaleye, T.O.; Brannigan, J.A.; Roberts, S.M.; Leatherbarrow, R.J.; Wilkinson, A.J.; Tate, E.W. Peptidomimetic inhibitors of N-myristoyltransferase from human malaria and leishmaniasis parasites. Org. Biomol. Chem. 2014, 12, 8132–8137. [Google Scholar] [CrossRef] [PubMed]
- Brand, S.; Cleghorn, L.A.; McElroy, S.P.; Robinson, D.A.; Smith, V.C.; Hallyburton, I.; Harrison, J.R.; Norcross, N.R.; Spinks, D.; Bayliss, T. Discovery of a novel class of orally active trypanocidal N-myristoyltransferase inhibitors. J. Med. Chem. 2011, 55, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Frearson, J.A.; Brand, S.; McElroy, S.P.; Cleghorn, L.A.; Smid, O.; Stojanovski, L.; Price, H.P.; Guther, M.L.; Torrie, L.S.; Robinson, D.A.; et al. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature 2010, 464, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Robinson, D.A.; Raimi, O.G.; Blair, D.E.; Harrison, J.R.; Lockhart, D.E.; Torrie, L.S.; Ruda, G.F.; Wyatt, P.G.; Gilbert, I.H. N-myristoyltransferase is a cell wall target in Aspergillus fumigatus. ACS Chem. Biol. 2015, 10, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Thinon, E.; Serwa, R.A.; Broncel, M.; Brannigan, J.A.; Brassat, U.; Wright, M.H.; Heal, W.P.; Wilkinson, A.J.; Mann, D.J.; Tate, E.W. Global profiling of co- and post-translationally N-myristoylated proteomes in human cells. Nat. Commun. 2014, 5, 4919. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Vetting, M.W.; Frantom, P.A.; Blanchard, J.S. Structures and mechanisms of the mycothiol biosynthetic enzymes. Curr. Opin. Chem. Biol. 2009, 13, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Vetting, M.W.; Roderick, S.L.; Yu, M.; Blanchard, J.S. Crystal structure of mycothiol synthase (Rv0819) from Mycobacterium tuberculosis shows structural homology to the GNAT family of N-acetyltransferases. Protein Sci. 2003, 12, 1954–1959. [Google Scholar] [CrossRef] [PubMed]
- Vetting, M.W.; Yu, M.; Rendle, P.M.; Blanchard, J.S. The substrate-induced conformational change of Mycobacterium tuberculosis mycothiol synthase. J. Chem. Biol. 2006, 281, 2795–2802. [Google Scholar] [CrossRef] [PubMed]
- Tanner, K.G.; Trievel, R.C.; Kuo, M.H.; Howard, R.M.; Berger, S.L.; Allis, C.D.; Marmorstein, R.; Denu, J.M. Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J. Chem. Biol. 1999, 274, 18157–18160. [Google Scholar] [CrossRef]
- Kaypee, S.; Sudarshan, D.; Shanmugam, M.K.; Mukherjee, D.; Sethi, G.; Kundu, T.K. Aberrant lysine acetylation in tumorigenesis: Implications in the development of therapeutics. Pharmacol. Ther. 2016, 162, 98–119. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef] [PubMed]
- Dekker, F.J.; Haisma, H.J. Histone acetyl transferases as emerging drug targets. Drug Discov. Today 2009, 14, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenet. 2016, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Seiden-Long, I.; Brown, K.; Shih, W.; Wigle, D.A.; Radulovich, N.; Jurisica, I.; Tsao, M. Transcriptional targets of hepatocyte growth factor signaling and Ki-ras oncogene activation in colorectal cancer. Oncogene 2006, 25, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wei, T.; Si, X.; Wang, Q.; Li, Y.; Leng, Y.; Deng, A.; Chen, J.; Wang, G.; Zhu, S. Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, cyclin D1, and cyclin E1 expression. J. Biol. Chem. 2013, 288, 14510–14521. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.W.; Jin, H.J.; Zhao, W.; Gao, B.; Fang, J.; Wei, J.; Zhang, D.D.; Zhang, J.; Fang, D. The histone acetyltransferase GCN5 expression is elevated and regulated by c-Myc and E2F1 transcription factors in human colon cancer. Gene Exp. 2015, 16, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Gai, X.; Tu, K.; Li, C.; Lu, Z.; Roberts, L.; Zheng, X. Histone acetyltransferase PCAF accelerates apoptosis by repressing a GLI1/BCL2/BAX axis in hepatocellular carcinoma. Cell Death Dis. 2015, 6, e1712. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; DeMorrow, S.; Invernizzi, P.; Jing, Q.; Glaser, S.; Renzi, A.; Meng, F.; Venter, J.; Bernuzzi, F.; White, M. Melatonin exerts by an autocrine loop antiproliferative effects in cholangiocarcinoma; its synthesis is reduced favoring cholangiocarcinoma growth. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G623–G633. [Google Scholar] [CrossRef] [PubMed]
- Neidhart, M.; Karouzakis, E.; Jüngel, A.; Gay, R.E.; Gay, S. Inhibition of spermidine/spermine N1-acetyltransferase activity: A new therapeutic concept in rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Eickhoff, J.C.; Mehraein-Ghomi, F.; Church, D.R.; Wilding, G.; Basu, H.S. Expression of spermidine/spermine N1-acetyl transferase (SSAT) in human prostate tissues is related to prostate cancer progression and metastasis. Prostate 2015, 75, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Vosseller, K. O-GlcNAc in cancer biology. Amino Acids 2013, 45, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.; Jackson, S.; Shahriari, K.; Lynch, T.; Sethi, G.; Walker, S.; Vosseller, K.; Reginato, M. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene 2010, 29, 2831–2842. [Google Scholar] [CrossRef] [PubMed]
- Das, U.; Kumar, S.; Dimmock, J.R.; Sharma, R.K. Inhibition of protein N-myristoylation: A therapeutic protocol in developing anticancer agents. Curr. Cancer Drug Targets 2012, 12, 667–692. [Google Scholar] [CrossRef] [PubMed]
- Selvakumar, P.; Kumar, S.; Dimmock, J.R.; Sharma, R.K. NMT1 (N-myristoyltransferase 1). Atlas Genet. Cytogenet. Oncol. Haematol. 2011, 15, 570. [Google Scholar] [CrossRef] [PubMed]
- Lombès, T.; Bégis, G.; Maurice, F.; Turcaud, S.; Lecourt, T.; Dardel, F.; Micouin, L. NMR-guided fragment-based approach for the design of AAC(6′)-Ib ligands. ChemBioChem 2008, 9, 1368–1371. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, M.; Ebiike, H.; Kawasaki, K.-i.; Sogabe, S.; Morikami, K.; Shiratori, Y.; Tsujii, S.; Fujii, T.; Sakata, K.; Hayase, M. Synthesis and biological activities of benzofuran antifungal agents targeting fungal N-myristoyltransferase. Bioorg. Med. Chem. 2003, 11, 4463–4478. [Google Scholar] [CrossRef]
- Brand, S.; Norcross, N.R.; Thompson, S.; Harrison, J.R.; Smith, V.C.; Robinson, D.A.; Torrie, L.S.; McElroy, S.P.; Hallyburton, I.; Norval, S. Lead optimization of a pyrazole sulfonamide series of Trypanosoma brucei N-myristoyltransferase inhibitors: Identification and evaluation of CNS penetrant compounds as potential treatments for stage 2 human African trypanosomiasis. J. Med. Chem. 2014, 57, 9855–9869. [Google Scholar] [CrossRef] [PubMed]
- Herrera, L.J.; Brand, S.; Santos, A.; Nohara, L.L.; Harrison, J.; Norcross, N.R.; Thompson, S.; Smith, V.; Lema, C.; Varela-Ramirez, A. Validation of N-myristoyltransferase as potential chemotherapeutic target in mammal-dwelling stages of Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2016, 10, e0004540. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Toth, M.; Bhattacharya, M.; Frase, H.; Vakulenko, S.B. Structure of the phosphotransferase domain of the bifunctional aminoglycoside-resistance enzyme AAC(6′)-Ie-APH(2′′)-Ia. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Scheibner, K.A.; de Angelis, J.; Burley, S.K.; Cole, P.A. Investigation of the roles of catalytic residues in serotonin N-acetyltransferase. J. Biol. Chem. 2002, 277, 18118–18126. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; de Angelis, J.; Khalil, E.M.; Cole, P.A.; Burley, S.K. X-ray crystallographic studies of serotonin N-acetyltransferase catalysis and inhibition. J. Mol. Biol. 2002, 317, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Obsil, T.; Ghirlando, R.; Klein, D.C.; Ganguly, S.; Dyda, F. Crystal structure of the 14-3-3ζ: Serotonin N-acetyltransferase complex: A role for scaffolding in enzyme regulation. Cell 2001, 105, 257–267. [Google Scholar] [CrossRef]
- Hurtado-Guerrero, R.; Raimi, O.; Shepherd, S.; van Aalten, D.M. Glucose-6-phosphate as a probe for the glucosamine-6-phosphate N-acetyltransferase michaelis complex. FEBS Lett. 2007, 581, 5597–5600. [Google Scholar] [CrossRef] [PubMed]
- Mariño, K.; Güther, M.L.S.; Wernimont, A.K.; Qiu, W.; Hui, R.; Ferguson, M.A. Characterization, localization, essentiality, and high-resolution crystal structure of glucosamine 6-phosphate N-acetyltransferase from Trypanosoma brucei. Eukaryot. Cell 2011, 10, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Han, B.W.; Bingman, C.A.; Wesenberg, G.E.; Phillips, G.N., Jr. Crystal structure of Homo sapiens thialysine Nε-acetyltransferase (HsSSAT2) in complex with acetyl coenzyme A. Proteins 2006, 64, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Sogabe, S.; Masubuchi, M.; Sakata, K.; Fukami, T.A.; Morikami, K.; Shiratori, Y.; Ebiike, H.; Kawasaki, K.; Aoki, Y.; Shimma, N.; et al. Crystal structures of Candida albicans N-myristoyltransferase with two distinct inhibitors. Chem. Biol. 2002, 9, 1119–1128. [Google Scholar] [CrossRef]
- Brannigan, J.A.; Smith, B.A.; Yu, Z.; Brzozowski, A.M.; Hodgkinson, M.R.; Maroof, A.; Price, H.P.; Meier, F.; Leatherbarrow, R.J.; Tate, E.W. N-myristoyltransferase from Leishmania donovani: Structural and functional characterisation of a potential drug target for visceral leishmaniasis. J. Mol. Biol. 2010, 396, 985–999. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Name | EC No. | Source | Substrates | PBD IDs | References |
|---|---|---|---|---|---|---|
| Aminoglycoside N-acetyltransferases | MtAAC(2′)-Ic | 2.3.1.- | M. tuberculosis | aminoglycosides | 1M44/1M4D/1M4G/1M4I | [23] |
| SmAAC(3)-Ia | 2.3.1.81 | S. marcescens | aminoglycosides | 1BO4 | [5] | |
| EcAAC(6′)-Ib | 2.3.1.82 | E. coli | aminoglycosides | 1V0C/2BUE/2VQY | [25] | |
| SeAAC(6′)-Ib11 | 2.3.1.82 | S. enterica | aminoglycosides | 2PR8/2PRB/2QIR | [30] | |
| SwAAC(6′)-Ie | 2.3.1.- | S. warneri | aminoglycosides | 4QC6 | [208] | |
| EfAAC(6′)-Ii | 2.3.1.82 | E. faecium | aminoglycosides | 1B87/1N71/2A4N | [13,21,34] | |
| SeAAC(6′)-Iy | 2.3.1.82 | S. enterica | aminoglycosides | 1S60/1S3Z/1S5K/2VBQ | [22,39] | |
| MtEis | 2.3.1.- | M. tuberculosis | aminoglycosides | 3R1K/3RYO/3SXN/3UY5/4JD6 | [19,41,42] | |
| Histone N-acetyltransferases | hHAT1 | 2.3.1.48 | H. sapiens | histone H4 | 2P0W | [45] |
| ScHAT1 | 2.3.1.48 | S. cerevisiae | histone H4 | 1BOB | [6] | |
| ScEsa1 | 2.3.1.48 | S. cerevisiae | histone H4 | 1MJ9/1MJA/1MJB | [57,58] | |
| ScGCN5 | 2.3.1.48 | S. cerevisiae | histone H3, H4 | 1YGH | [65] | |
| TtGCN5 | 2.3.1.48 | T. thermophila | histone H3 | 1QSN/1QSR/1QST/5GCN/1M1D/1PU9/1PUA/1Q2C | [67,68,69,70] | |
| hGCN5 | 2.3.1.48 | H. sapiens | histone H3, H4, H2b | 1Z4R | [66] | |
| hPCAF | 2.3.1.48 | H. sapiens | histone H3, H4 | 1CM0/4NSQ | [72,73] | |
| ScHpa2 | 2.3.1.48 | S. cerevisiae | histone H3, H4 | 1QSM/1QSO | [76] | |
| Non-histone protein N-acetyltransferases | αTAT1 | 2.3.1.108 | H. sapiens | α-tubulin | 4GS4 | [79] |
| MtPAT | 3.4.1.- | M. tuberculosis | - | 4AVA /4AVB/4AVC | [81] | |
| SsPAT | 2.3.1- | S. solfataricus | - | 3F8K | [78] | |
| hNaa50p | 2.3.1.- | H. sapiens | peptides | 3TFY | [80] | |
| Arylalkylamine N-acetyltransferases | OaAANAT | 2.3.1.87 | O. aries | 2-arylethylamines | 1B6B/1CJW/1KUV/1KUX/1KUY/1L0C | [77,86,209,210] |
| hAANAT | 2.3.1.87 | H. sapiens | 2-arylethylamines | 1IB1 | [211] | |
| DmAANAT | 2.3.1.87 | D. melanogaster | 2-arylethylamines | 3TE4 | [93] | |
| AaAANAT | 2.3.1.87 | A. aegypti | histamine, arylalkylamines, hydrazine | 4FD5/4FD4,4FD6/4FD7 | [94] | |
| Glucosamine-6-phosphate N-acetyltransferases | ScGNA1 | 2.3.1.4 | S. cerevisiae | d-glucosamine 6-phosphate | 1I1D/1I12/1I21 | [99] |
| AfGNA1 | 2.3.1.4 | A. fumigatus | d-glucosamine 6-phosphate | 2VEZ/2VXK | [100,212] | |
| hGNA1 | 2.3.1.4 | H. sapiens | d-glucosamine 6-phosphate | 3CXP/3CXQ/3CXS | [101] | |
| TbGNA1 | 2.3.1.4 | T. brucei | d-glucosamine 6-phosphate | 3I3G | [213] | |
| AtGNA1 | 2.3.1.4 | A. thaliana | d-glucosamine 6-phosphate | 3T90 | [102] | |
| CeGNA1 | 2.3.1.4 | C. elegans | d-glucosamine 6-phosphate | 4AG7/4AG9 | [103] | |
| MccE | EcMccE | - | E. coli | aspartyl-tRNA synthetase | 3R95/3R96/3R9E/3R9F | [23] |
| Pseudaminic acid biosynthesis protein H | HpPseH | 2.3.1.202 | H. pylori | UDP—linked sugar | 4RI1 | [107] |
| CjPseH | 2.3.1.202 | C. jejuni | UDP—linked sugar | 4XPK/4XPL | [109] | |
| WecD | EcWecD | 2.3.1.210 | E. coli | dTDP-4-amino-4,6-dideoxy-α-d-galactose | 2FS5/2FT0 | [113] |
| Tabtoxin resistance protein | PsTTR | 2.3.1.- | P. syringae | tabtoxin | 1GHE | [114] |
| Mpr1 | ScMpr1 | 3.4.1.- | S. cerevisiae | l-azetidine-2-carboxylic acid | 3W6S/3W6X/3W91 | [119] |
| Spermidine/spermine N1-acetyltransferases | BsPaiA | 2.3.1.57 | B. subtilis | spermidine/spermine | 1TIQ | [126] |
| TaPaiA | 2.3.1.57 | T. acidophilum | spermidine/spermine | 3FIX/3FIX/3NE7/3NE7 | [130] | |
| hSSAT | 2.3.1.57 | H. sapiens | spermidine/spermine | 2BEI/2B3U/2B3V/2B58/2B4B/2B4D/2B5G/2F5I/2G3T/2JEV | [129,132,133,214] | |
| MmSSAT | 2.3.1.57 | M. musculus | spermidine/spermine | 3BJ7/3BJ8 | [128] | |
| VcSpeG | 2.3.1.57 | V. cholerae | spermidine/spermine | 4NCZ/4JJX/4MHD/4MI4/4R57/4R87 | [127] | |
| C-terminal Nε-lysine protein acetyltransferases | PA4794 | 2.3.1.- | P. aeruginosa | C-terminal lysine containing peptide | 3PGP/4KOS/4KOT/4KOU/4KOV/4KOW/4KOX/4KOY/4KUA/4L89 | [167] |
| MtRv1347c | 2.8.3.- | M. tuberculosis | - | 1YK3 | [138] | |
| Ribosomal protein Nα-acetyltransferases | SeRimI | 2.3.1.128 | S. enterica | ribosomal protein S18 | 2CNS/2CNT | [147] |
| StRimL | 2.3.1.- | S. typhimurium. | ribosomal protein L7/L12 | 1S7F/1S7K/1S7L/1S7N | [108] | |
| BsYadF | - | B. subtilis | ribosomal protein L12 | 1NSL | [149] | |
| Succinyltransferase | MtRv0802c | 2.8.3.- | M. tuberculosis | - | 2VZY/2VZZ | [4] |
| FemABX aminoacyl transferases | SaFemA | 2.3.2.17 | S. aureus | peptidoglycan precursor, peptides | 1LRZ | [150] |
| WvFemX | 2.3.2.10 | W. viridescens | peptidoglycan precursor, peptides | 1P4N/1NE9/1XE4/1XF8/1XIX/4II9 | [152,158,159] | |
| EcLFT | 2.3.2.6 | E. coli | N-terminal Arg/Lys containing proteins | 2DPS/2DPT | [161] | |
| Protein N-myristoyltransferases | CaNMT | 2.3.1.97 | C. albicans | N-terminal glycyl-peptides | 1NMT/1IYK/1IYL | [168,215] |
| LdNMT | 2.3.1.97 | L. donovani | N-terminal glycyl-peptides | 2WUU | [216] | |
| LmNMT | 2.3.1.97 | L. major | N-terminal glycyl-peptides | 2WSA/3H5Z/4A2Z/4A30/4A31/4A32/4A33/4CGL/4CGM/4CGN/4CGO/4CGP/4C68/4C7H/4C7I/4CYN/4CYO/4CYP/4CYQ | [178,179,180,181] | |
| ScNMT | 2.3.1.97 | S. cerevisiae | N-terminal glycyl-peptides | 2NMT/1IIC/1IID/2P6E/2P6F/2P6G | [3,169,172] | |
| PvNMT | 2.3.1.97 | P. vivax | N-terminal glycyl-peptides | 4B10/4B11/4B12/4B13/4B14/4A95/4BBH/2YNC/2YND/2YNE/4CAE/4CAF | [173,174,175,176,177] | |
| TbNMT | 2.3.1.97 | T. brucei | N-terminal glycyl-peptides | 2WSA/3H5Z/4A2Z | [182] | |
| AfNMT | 2.3.1.97 | A. fumigatus | N-terminal glycyl-peptides | 4CAX/4CAV/4CAW | [183] | |
| hNMT | 2.3.1.97 | H. sapiens | N-terminal glycyl-peptides | 4C2X/4C2Y/4C2Z | [184] | |
| Mycothiol synthase | MtMshD | 2.3.1.189 | M. tuberculosis | des-acetylmycothiol | 1OZP/1P0H/2C27 | [186,187] |
| Family | Functions | References |
|---|---|---|
| Aminoglycoside N-acetyltransferases | resistance to aminoglycoside antibiotics | [10,19,41,42] |
| Histone N-acetyltransferases | histone deposition, transcription activation, chromatin assembly, DNA repair, promoting cancer cell growth and apoptosis, amino acid biosynthesis in yeast | [43,44,46,54,55,56,63] |
| Non-histone protein N-acetyltransferases | chromatin regulation, maintaining genome integrity, mRNA and protein stability | [78,82,83,90] |
| Arylalkylamine N-acetyltransferases | xenobiotic and folate metabolism, biosynthesis of melatonin, sclerotization and neurotransmitter inactivation | [91,92,95] |
| Glucosamine-6-phosphate N-acetyltransferases | biosynthesis of peptidoglycan, chitin, hexosamine and glycophosphatidylinositol; amino sugar and nucleotide sugar metabolism | [10,11,96,97,98] |
| MccE | inactivation of antibiotic microcin C7 | [104,105] |
| Pseudaminic acid biosynthesis protein H | flagellin glycosylation | [106] |
| WecD | biosynthesis of enterobacterial common antigen and flagella | [110,111,112] |
| Tabtoxin resistance protein | inactivation of tabtoxin and prevention of wildfire disease | [117,118] |
| Mpr1 | l-proline and l-arginine metabolism, oxidative stresses resistance and freeze tolerance | [119,120,121,122] |
| Spermidine/spermine N1-acetyltransferases | regulation of cellular polyamine levels and polyamine metabolism | [123,124,128] |
| C-terminal Nε-lysine protein acetyltransferases | regulation of gene expression | [135,136] |
| Ribosomal protein Nα-acetyltransferases | regulation of proteins function and stability, inactivation of antibiotic microcin C7 | [145,146,147] |
| Succinyltransferase | unknown | [4] |
| FemABX aminoacyl transferases | peptidoglycan biosynthesis and methicillin resistance | [151,153,154] |
| Protein N-myristoyltransferases | regulation of protein–protein and protein-cellular membrane interactions, enhancement of protein stability | [165,176] |
| Mycothiol synthase | biosynthesis of organosulfur compounds, neutralization of electrophiles, regulation of oxidative stress, antibiotic resistance and oxidation of formaldehyde | [10,185] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salah Ud-Din, A.I.M.; Tikhomirova, A.; Roujeinikova, A. Structure and Functional Diversity of GCN5-Related N-Acetyltransferases (GNAT). Int. J. Mol. Sci. 2016, 17, 1018. https://doi.org/10.3390/ijms17071018
Salah Ud-Din AIM, Tikhomirova A, Roujeinikova A. Structure and Functional Diversity of GCN5-Related N-Acetyltransferases (GNAT). International Journal of Molecular Sciences. 2016; 17(7):1018. https://doi.org/10.3390/ijms17071018
Chicago/Turabian StyleSalah Ud-Din, Abu Iftiaf Md, Alexandra Tikhomirova, and Anna Roujeinikova. 2016. "Structure and Functional Diversity of GCN5-Related N-Acetyltransferases (GNAT)" International Journal of Molecular Sciences 17, no. 7: 1018. https://doi.org/10.3390/ijms17071018
APA StyleSalah Ud-Din, A. I. M., Tikhomirova, A., & Roujeinikova, A. (2016). Structure and Functional Diversity of GCN5-Related N-Acetyltransferases (GNAT). International Journal of Molecular Sciences, 17(7), 1018. https://doi.org/10.3390/ijms17071018