
Immune Mechanisms in Myelodysplastic Syndrome

 ,
,
Abstract
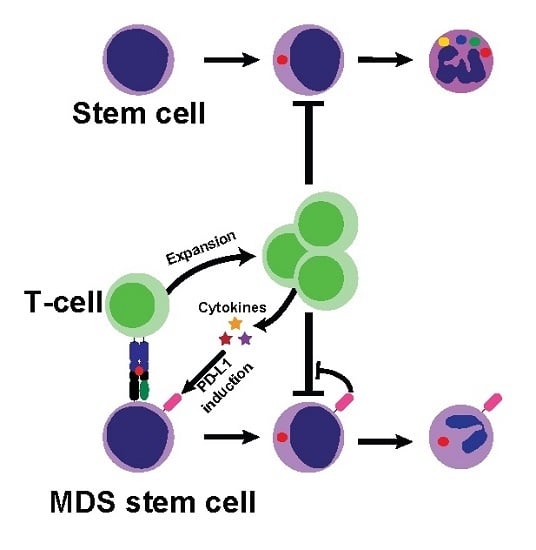
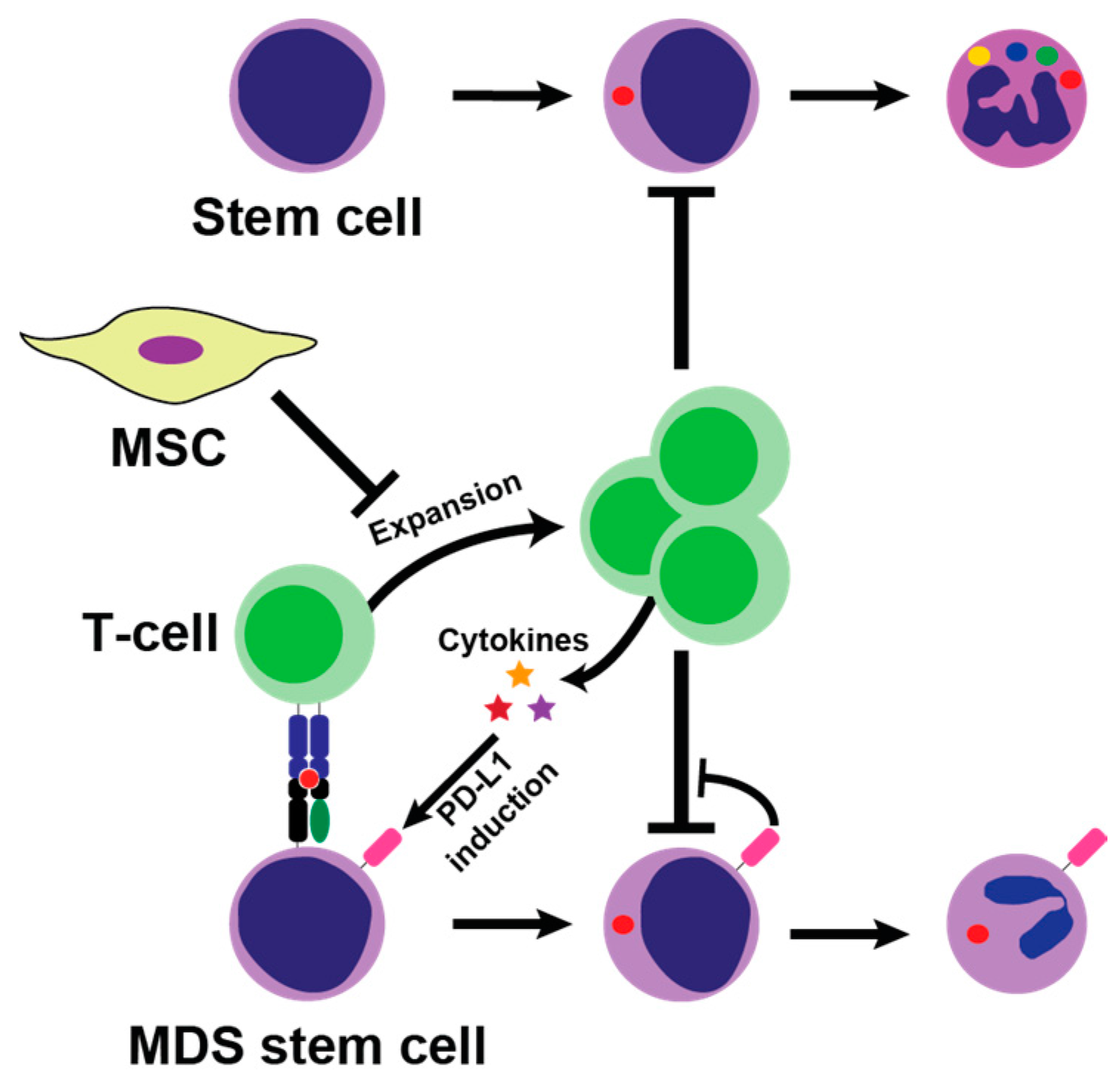
:
{kind=link}
{kind=link}
1. Introduction
2. Association with Autoimmune Disease
3. Immune Dysregulation in MDS
3.1. T-Cell Mediated Bone Marrow Suppression
3.2. Cytokines
3.3. Innate Immunity Activity
3.4. Mesenchymal Stromal Cells (MSC)
4. Immune Manipulation as Treatment of MDS
4.1. Which Low-Risk MDS Patients Are Subceptible to Immunosuppressive Therapy
4.2. Cyclosporine, Anti-Thymocyte Globulin, and Mycophenolate Mofetil
4.3. Alemtuzumab
4.4. Novel Immune Pathway Inhibitors
4.5. Epigenetic Modulation and Induction of Adaptive Immunity in Higher Risk MDS
5. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| ATG | Anti-thymocyte globulin |
| ERVs | Endogenous retroviruses |
| FasL | Fas ligand |
| FasR | Fas receptor |
| HMA | Hypomethylating agent ICUS Idiopathic cytopenia of undetermined significance |
| IDO | Indoleamine 2,3-dioxygenase |
| IFN-γ | Interferon-γ IPSS International Prognostic Scoring System |
| IPSS-R | Revised International Prognostic Scoring System |
| ITP | Idiopathic thrombocytopenic purpura |
| MAPK | Mitogen-activated protein kinase |
| MDS | Myelodysplastic syndrome MHC Major histocompatibility complex MMF Mycophenolate Mofetil |
| MSC | Mesenchymal stromal cells |
| NF-κB | Nuclear factor k-light-chain-enhancer of activated B cells |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| RA | Refactory anemia |
| RCMD | Refractory anemia with multilineage dysplasia |
| TCR | T-cell receptor |
| TNF-α | Tumor necrosis factor α |
| TRAIL | Tumor necrosis factor–related apoptosis-inducing ligand |
References
- Corey, S.J.; Minden, M.D.; Barber, D.L.; Kantarjian, H.; Wang, J.C.Y.; Schimmer, A.D. Myelodysplastic syndromes: The complexity of stem-cell diseases. Nat. Rev. Cancer 2007, 7, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia. Blood 2016, 127. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 Mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Aslan, D.; Garde, C.; Nygaard, M.K.; Helbo, A.S.; Dimopoulos, K.; Hansen, J.W.; Severinsen, M.T.; Treppendahl, M.B.; Sjø, L.D.; Grønbæk, K.; et al. Tumor suppressor microRNAs are downregulated in myelodysplastic syndrome with spliceosome mutations. Oncotarget 2016, 7, 9951–9963. [Google Scholar] [PubMed]
- Treppendahl, M.B.; Kristensen, L.S.; Groønb, K. Predicting response to epigenetic therapy. J. Clin. Investig. 2014, 124, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Kwok, B.; Hall, J.M.; Witte, J.S.; Xu, Y.; Reddy, P.; Lin, K.; Flamholz, R.; Dabbas, B.; Yung, A.; Al-Hafidh, J.; et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood 2015, 126, 2355–2362. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Fenaux, P. Myelodysplastic Syndromes (MDS) and autoimmune disorders (AD): Cause or consequence? Best Pract. Res. Clin. Haematol. 2013, 26, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.E.; Mufti, G.J.; Rasool, F.; Mijovic, A.; Devereux, S.; Pagliuca, A. The role of apoptosis, proliferation, and the Bcl-2-related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood 2000, 96, 3932–3938. [Google Scholar] [PubMed]
- Gañán-Gómez, I.; Wei, Y.; Starczynowski, D.T.; Colla, S.; Yang, H.; Cabrero-Calvo, M.; Bohannan, Z.S.; Verma, A.; Steidl, U.; Garcia-Manero, G. Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia 2015, 29, 1458–1469. [Google Scholar] [CrossRef] [PubMed]
- Olnes, M.J.; Sloand, E.M. Targeting immune dysregulation in myelodysplastic syndromes. JAMA 2011, 305, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.; Pfeiffer, R.M.; Landgren, O.; Gadalla, S.; Berndt, S.I.; Engels, E.A. Risks of myeloid malignancies in patients with autoimmune conditions. Br. J. Cancer 2009, 100, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Mekinian, A.; Grignano, E.; Braun, T.; Decaux, O.; Liozon, E.; Costedoat-chalumeau, N.; Kahn, J.-E.; Hamidou, M.; Park, S.; Puéchal, X.; et al. Systemic inflammatory and autoimmune manifestations associated with myelodysplastic syndromes and chronic myelomonocytic leukaemia: A French multicentre retrospective study. Rheumatology (Oxford) 2016, 55, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Kristinsson, S.Y.; Björkholm, M.; Hultcrantz, M.; Derolf, Å.R.; Landgren, O.; Goldin, L.R. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J. Clin. Oncol. 2011, 29, 2897–903. [Google Scholar] [CrossRef] [PubMed]
- Komrokji, R.S.; Kulasekararaj, A.; Al Ali, N.H.; Kordasti, S.; Bart-Smith, E.; Craig, B.M.; Padron, E.; Zhang, L.; Lancet, J.E.; Pinilla-Ibarz, J.; et al. Autoimmune diseases and myelodysplastic syndromes. Am. J. Hematol. 2016, 91, E280–E283. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.B.; Neogi, T.; Prout, M.; Jick, S. Relative risk of myelodysplastic syndromes in patients with autoimmune disorders in the general practice research database. Cancer Epidemiol. 2014, 38, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Dalamaga, M.; Petridou, E.; Cook, F.E.; Trichopoulos, D. Risk factors for myelodysplastic syndromes: A case-control study in Greece. Cancer Causes Control 2002, 13, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Pedersen-Bjergaard, J.; Andersen, M.K.; Christiansen, D.H.; Nerlov, C. Genetic pathways in therapy-related myelodysplasia and acute myeloid leukemia. Blood 2002, 99, 1909–1912. [Google Scholar] [CrossRef] [PubMed]
- Kwong, Y.L.; Au, W.Y.; Liang, R.H. Acute myeloid leukemia after azathioprine treatment for autoimmune diseases: Association with -7/7q-. Cancer Genet. Cytogenet. 1998, 104, 94–97. [Google Scholar] [CrossRef]
- Okamoto, H.; Teramura, M.; Kamatani, N. Myelodysplastic syndrome associated with low-dose methotrexate in rheumatoid arthritis. Ann. Pharmacother. 2004, 38, 172–173. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.W.; Westman, M.K.; Sjö, L.D.; Saft, L.; Kristensen, L.S.; Ørskov, A.D.; Treppendahl, M.; Andersen, M.K.; Grønbæk, K. Frequent Mutations in Epigenetic Regulators in Cytopenia of Undetermined Significance: Association with Risk of Progression to Myelodysplastic Syndrome; European Hematology Association 21st Congress: Copenhagen, Denmark, 9 June 2016; p. 257. [Google Scholar]
- Yang, L.; Qian, Y.; Eksioglu, E.; Epling-Burnette, P.K.; Wei, S. The inflammatory microenvironment in MDS. Cell. Mol. Life Sci. 2015, 72, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Mailloux, A.; Rollison, D.E.; Painter, J.S.; Maciejewski, J.; Paquette, R.L.; Loughran, T.P.; McGraw, K.; Makishima, H.; Radhakrishnan, R.; et al. Naive T-cells in myelodysplastic syndrome display intrinsic human telomerase reverse transcriptase (hTERT) deficiency. Leukemia 2013, 27, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Epperson, D.E.; Nakamura, R.; Saunthararajah, Y.; Melenhorst, J.; Barrett, A.J. Oligoclonal T cell expansion in myelodysplastic syndrome: Evidence for an autoimmune process. Leuk. Res. 2001, 25, 1075–1083. [Google Scholar] [CrossRef]
- Vercauteren, S.M.; Starczynowski, D.T.; Sung, S.; McNeil, K.; Salski, C.; Jensen, C.-L.; Bruyere, H.; Lam, W.L.; Karsan, A. T cells of patients with myelodysplastic syndrome are frequently derived from the malignant clone. Br. J. Haematol. 2012, 156, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Qianqiao, Z.; Qi, H.; Feng, X.; Chunkang, C.; Xiao, L. In vitro deprivation of CD8+ CD57+ T cells promotes the malignant growth of bone marrow colony cells in patients with lower-risk myelodysplastic syndrome. Exp. Hematol. 2010, 38, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Baumann, I.; Scheid, C.; Koref, M.S.; Swindell, R.; Stern, P.; Testa, N.G. Autologous lymphocytes inhibit hemopoiesis in long-term culture in patients with myelodysplastic syndrome. Exp. Hematol. 2002, 30, 1405–1411. [Google Scholar] [CrossRef]
- Molldrem, J.J.; Jiang, Y.Z.; Stetler-Stevenson, M.; Mavroudis, D.; Hensel, N.; Barrett, A.J. Haematological response of patients with myelodysplastic syndrome to antithymocyte globulin is associated with a loss of lymphocyte-mediated inhibition of CFU-GM and alterations in T-cell receptor Vbeta profiles. Br. J. Haematol. 1998, 102, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Sloand, E.M.; Melenhorst, J.J.; Tucker, Z.C.G.; Pfannes, L.; Brenchley, J.M.; Yong, A.; Visconte, V.; Wu, C.; Gostick, E.; Scheinberg, P.; et al. T-cell immune responses to Wilms tumor 1 protein in myelodysplasia responsive to immunosuppressive therapy. Blood 2011, 117, 2691–2699. [Google Scholar] [CrossRef] [PubMed]
- Gang, A.O.; Frøsig, T.M.; Brimnes, M.K.; Lyngaa, R.; Treppendahl, M.B.; Grønbæk, K.; Dufva, I.H.; Straten, P.T.; Hadrup, S.R. 5-Azacytidine treatment sensitizes tumor cells to T-cell mediated cytotoxicity and modulates NK cells in patients with myeloid malignancies. Blood Cancer J. 2014, 4, e197. [Google Scholar] [CrossRef] [PubMed]
- Sloand, E.M.; Wu, C.O.; Greenberg, P.; Young, N.; Barrett, J. Factors affecting response and survival in patients with myelodysplasia treated with immunosuppressive therapy. J. Clin. Oncol. 2008, 26, 2505–2511. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bueso-Ramos, C.; Dinardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.-R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2013, 28, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, W.; Ogawara, H.; Handa, H.; Tsukamoto, N.; Nojima, Y.; Murakami, H. Clinical significance of regulatory T cells in patients with myelodysplastic syndrome. Eur. J. Haematol. 2009, 82, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.-J.; Bourgeois, E.; Lobe, I.; Braun, T.; Visentin, G.; Bourhis, J.-H.; Fenaux, P.; Chouaib, S. Caignard, a Cytolytic function and survival of natural killer cells are severely altered in myelodysplastic syndromes. Leukemia 2006, 20, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Medyouf, H.; Mossner, M.; Jann, J.-C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and Ipilimumab versus Ipilimumab in untreated melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Yamashita, T.; Tamura, H.; Zhao, W.; Tsuji, T.; Shimizu, M.; Shinya, E.; Takahashi, H.; Tamada, K.; Chen, L.; et al. Interferon-γ and tumor necrosis factor-α induce an immunoinhibitory molecule, B7-H1, via nuclear factor κB activation in blasts in myelodysplastic syndromes. Blood 2010, 116, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Ørskov, A.D.; Treppendahl, M.B.; Skovbo, A.; Holm, M.S.; Friis, L.S.; Hokland, M.; Grønbæk, K. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients : A rationale for combined targeting of PD-1 and DNA methylation. Oncotarget 2015, 6, 9612–9626. [Google Scholar] [CrossRef] [PubMed]
- Young, N.S.; Scheinberg, P.; Calado, R.T. Aplastic anemia. Curr. Opin. Hematol. 2008, 15, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Yoshizato, T.; Dumitriu, B.; Hosokawa, K.; Makishima, H.; Yoshida, K.; Townsley, D.; Sato-Otsubo, A.; Sato, Y.; Liu, D.; Suzuki, H.; et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N. Engl. J. Med. 2015, 373, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Saito, I.; Kuwata, T.; Yoshida, S.; Yamaguchi, S.; Takahashi, M.; Tanizawa, T.; Kamiyama, R.; Hirokawa, K. Overexpression of tumor necrosis factor (TNF)-α and interferon (IFN)-gamma by bone marrow cells from patients with myelodysplastic syndromes. Leukemia 1997, 11, 2049–2054. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, G.E.; De Schouwer, P.; Ceuppens, J.L.; Van Damme, J.; Goossens, W.; Boogaerts, M.A. Measurement of serum cytokine levels in patients with myelodysplastic syndromes. Leukemia 1992, 6, 1268–1272. [Google Scholar] [PubMed]
- Kitagawa, M.; Takahashi, M.; Yamaguchi, S.; Inoue, M.; Ogawa, S.; Hirokawa, K.; Kamiyama, R. Expression of inducible nitric oxide synthase (NOS) in bone marrow cells of myelodysplastic syndromes. Leukemia 1999, 13, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Molnár, L.; Berki, T.; Hussain, A.; Németh, P.; Losonczy, H. Detection of TNF α Expression in the bone marrow and determination of TNF α production of peripheral blood mononuclear cells in myelodysplastic syndrome. Pathol. Oncol. Res. 2000, 6, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Stifter, G.; Heiss, S.; Gastl, G.; Tzankov, A.; Stauder, R. Over-expression of tumor necrosis factor-α in bone marrow biopsies from patients with myelodysplastic syndromes: Relationship to anemia and prognosis. Eur. J. Haematol. 2005, 75, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Miyazato, A.; Chen, G.; Kajigaya, S.; Young, N.S.; Maciejewski, J.P. Interferon-γ-induced gene expression in CD34 cells: Identification of pathologic cytokine-specific signature profiles. Blood 2006, 107, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Maciejewski, J.; Selleri, C.; Anderson, S.; Young, N.S. Fas antigen expression on CD34+ human marrow cells is induced by interferon γ and tumor necrosis factor alpha and potentiates cytokine-mediated hematopoietic suppression in vitro. Blood 1995, 85, 3183–3190. [Google Scholar] [PubMed]
- Zang, D.Y.; Goodwin, R.G.; Loken, M.R.; Bryant, E.; Deeg, H.J. Expression of tumor necrosis factor-related apoptosis-inducing ligand, Apo2L, and its receptors in myelodysplastic syndrome: Effects on in vitro hemopoiesis. Blood 2001, 98, 3058–3065. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Starczynowski, D.T.; Kuchenbauer, F.; Argiropoulos, B.; Sung, S.; Morin, R.; Muranyi, A.; Hirst, M.; Hogge, D.; Marra, M.; Wells, R.A.; et al. Identification of miR-145 and miR-146a as mediators of the 5q-syndrome phenotype. Nat. Med. 2010, 16, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.T.; Häger, M.; Glenthøj, A.; Asmar, F.; Clemmensen, S.N.; Mora-Jensen, H.; Borregaard, N.; Cowland, J.B. miRNA-130a regulates C/EBP-ε expression during granulopoiesis. Blood 2014, 123, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Dazzi, F.; Ramasamy, R.; Glennie, S.; Jones, S.P.; Roberts, I. The role of mesenchymal stem cells in haemopoiesis. Blood Rev. 2006, 20, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiao, Z. Mesenchymal stem cells in pathogenesis of myelodysplastic syndromes. Stem Cell Investig. 2014, 1, 16–19. [Google Scholar]
- Santamaría, C.; Muntión, S.; Rosón, B.; Blanco, B.; López-Villar, O.; Carrancio, S.; Sánchez-Guijo, F.M.; Díez-Campelo, M.; Alvarez-Fernández, S.; Sarasquete, M.E.; et al. Impaired expression of DICER, DROSHA, SBDS and some microRNAs in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica 2012, 97, 1218–1224. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.H.G.P.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, Z.; Li, Q.; Li, W.; You, Y.; Zou, P. The different immunoregulatory functions of mesenchymal stem cells in patients with low-risk or high-risk myelodysplastic syndromes. PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef] [PubMed]
- Tse, W.T.; Pendleton, J.D.; Beyer, W.M.; Egalka, M.C.; Guinan, E.C. Suppression of allogeneic T-cell proliferation by human marrow stromal cells: Implications in transplantation. Transplantation 2003, 75, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Glennie, S.; Soeiro, I.; Dyson, P.; Lam, E.; Dazzi, F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood 2005, 105, 2821–2828. [Google Scholar] [CrossRef] [PubMed]
- Kyurkchiev, D. Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J. Stem Cells 2014, 6, 552. [Google Scholar] [CrossRef] [PubMed]
- Kordasti, S.Y.; Ingram, W.; Hayden, J.; Darling, D.; Barber, L.; Afzali, B.; Lombardi, G.; Wlodarski, M.W.; Maciejewski, J.P.; Farzaneh, F.; Mufti, G.J. CD4+ CD25high Foxp3+ regulatory T cells in myelodysplastic syndrome (MDS). Blood 2007, 110, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Saunthararajah, Y.; Nakamura, R.; Nam, J.-M.; Robyn, J.; Loberiza, F.; Maciejewski, J.P.; Simonis, T.; Molldrem, J.; Young, N.S.; Barrett, A.J. HLA-DR15 (DR2) is overrepresented in myelodysplastic syndrome and aplastic anemia and predicts a response to immunosuppression in myelodysplastic syndrome. Blood 2002, 100, 1570–1574. [Google Scholar] [PubMed]
- Lim, Z.Y.; Killick, S.; Germing, U.; Cavenagh, J.; Culligan, D.; Bacigalupo, A.; Marsh, J.; Mufti, G.J. Low IPSS score and bone marrow hypocellularity in MDS patients predict hematological responses to antithymocyte globulin. Leukemia 2007, 21, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Komrokji, R.S.; Mailloux, A.W.; Chen, D.-T.; Sekeres, M.A.; Paquette, R.; Fulp, W.J.; Sugimori, C.; Paleveda-Pena, J.; Maciejewski, J.P.; List, A.F.; et al. A phase II multicenter rabbit anti-thymocyte globulin trial in patients with myelodysplastic syndromes identifying a novel model for response prediction. Haematologica 2014, 99, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Koyasu, S. Mechanisms of action of cyclosporine. Immunopharmacology 2000, 47, 119–125. [Google Scholar] [CrossRef]
- Shimamoto, T.; Tohyama, K.; Okamoto, T.; Uchiyama, T.; Mori, H.; Tomonaga, M.; Asano, Y.; Niho, Y.; Teramura, M.; Mizoguchi, H.; et al. Cyclosporin A therapy for patients with myelodysplastic syndrome: Multicenter pilot studies in Japan. Leuk. Res. 2003, 27, 783–788. [Google Scholar] [CrossRef]
- Jonásova, A.; Neuwirtová, R.; Cermák, J.; Vozobulová, V.; Mociková, K.; Sisková, M.; Hochová, I. Cyclosporin A therapy in hypoplastic MDS patients and certain refractory anaemias without hypoplastic bone marrow. Br. J. Haematol. 1998, 100, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Sloand, E.M.; Rezvani, K. The Role of the immune system in myelodysplasia: Implications for therapy. Semin. Hematol. 2008, 45, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Jilg, W.; Hannig, K. Lymphocyte surface proteins recognized by an anti-thymocyte-globulin. Hoppe-Seyler’s Z. Physiol. Chem. 1981, 362, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.; Clarkson, M.R.; Albin, M.; Sayegh, M.H.; Najafian, N. A novel mechanism of action for anti-thymocyte globulin: Induction of CD4+ CD25+ Foxp3+ regulatory T cells. J. Am. Soc. Nephrol. 2006, 17, 2844–2853. [Google Scholar] [CrossRef] [PubMed]
- Haidinger, M.; Geyeregger, R.; Poglitsch, M.; Weichhart, T.; Zeyda, M.; Vodenik, B.; Stulnig, T.M.; Böhmig, G.A.; Hörl, W.H.; Säemann, M.D. Antithymocyte globulin impairs T-cell/antigen-presenting cell interaction: Disruption of immunological synapse and conjugate formation. Transplantation 2007, 84, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, E.; Devergie, A.; Poros, A.; Degoulet, P. Results of immunosuppression in 170 cases of severe aplastic anaemia. Report of the european group of bone marrow transplant (EGBMT). Br. J. Haematol. 1982, 51, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Scheinberg, P.; Nunez, O.; Weinstein, B.; Scheinberg, P.; Biancotto, A.; Wu, C.O.; Young, N.S. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N. Engl. J. Med. 2011, 365, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Molldrem, J.J.; Caples, M.; Mavroudis, D.; Plante, M.; Young, N.S.; Barrett, A.J. Antithymocyte globulin for patients with myelodysplastic syndrome. Br. J. Haematol. 1997, 99, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.R.; Olnes, M.J.; Barrett, A.J. Immunomodulatory treatment of myelodysplastic syndromes: Antithymocyte globulin, cyclosporine, and alemtuzumab. Semin. Hematol. 2012, 49, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Broliden, P.A.; Dahl, I.-M.; Hast, R.; Johansson, B.; Juvonen, E.; Kjeldsen, L.; Porwit-MacDonald, A.; Sjoo, M.; Tangen, J.-M.; Uggla, B.; et al. Antithymocyte globulin and cyclosporine A as combination therapy for low-risk non-sideroblastic myelodysplastic syndromes. Haematologica 2006, 91, 667–670. [Google Scholar] [PubMed]
- Ransom, J.T. Mechanism of action of mycophenolate mofetil. Ther. Drug Monit. 1995, 17, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Remacha, A.F.; Arrizabalaga, B.; Bueno, J.; Muñoz, J.; Bargay, J.; Pedro, C. Treatment with mycophenolate mofetil followed by recombinant human erythropoietin in patients with low-risk myelodysplastic syndromes resistant to erythropoietin treatment. Haematologica 2010, 95, 339–340. [Google Scholar] [CrossRef] [PubMed]
- Sloand, E.M.; Olnes, M.J.; Shenoy, A.; Weinstein, B.; Boss, C.; Loeliger, K.; Wu, C.O.; More, K.; Barrett, A.J.; Scheinberg, P.; et al. Alemtuzumab treatment of intermediate-1 myelodysplasia patients is associated with sustained improvement in blood counts and cytogenetic remissions. J. Clin. Oncol. 2010, 28, 5166–5173. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.; Garg, S.; Cleary, M.M.; Edwards, D.K.; Loriaux, M.; Winski, S.L.; Cable, L.; Tyner, J.W.; Agarwal, A. p38MAPK inhibition blocks inflammatory signaling in acute myeloid leukemia. Blood 2015, 126, 2603. [Google Scholar]
- Rivera, G.A.; Saramipoor Behbahan, I.; Greenberg, P.L. Immune checkpoint pathways: Perspectives on myeloid malignancies. Leuk. Lymphoma 2016, 8194, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Bakker, C.Y.; Bluestone, J.A. CTLA-4 ligation blocks CD28-dependent T cell activation. J. Exp. Med. 1996, 183, 2541–2550. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Sun, Y.; Yin, Z.; Feng, S.; Sun, L.; Li, Z. Research progress of indoleamine 2,3-dioxygenase inhibitors. Future Med. Chem. 2015, 7, 185–201. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Héninger, E.; Krueger, T.E.G.; Lang, J.M. Augmenting antitumor immune responses with epigenetic modifying agents. Front. Immunol. 2015, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Karpf, A.R.; Lasek, A.W.; Ririe, T.O.; Hanks, A.N.; Grossman, D.; Jones, D.A. Limited gene activation in tumor and normal epithelial cells treated with the DNA methyltransferase inhibitor 5-aza-2’-deoxycytidine. Mol. Pharmacol. 2004, 65, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chiappinelli, K.B.; Guzzetta, A.A.; Easwaran, H.; Yen, R.-W.C.; Vatapalli, R.; Topper, M.J.; Luo, J.; Connolly, R.M.; Azad, N.S.; et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 2014, 5, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Vendetti, F.; Vancriekinge, W.; Demeyer, T.; Du, Z.; Parsana, P.; et al. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Dear, A.E. Epigenetic Modulators and the New Immunotherapies. N. Engl. J. Med. 2016, 374, 684–686. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Zahnow, C.A.; Ahuja, N.; Baylin, S.B. Combining epigenetic and immunotherapy to combat cancer. Cancer Res. 2016, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Goodyear, O.; Agathanggelou, A.; Novitzky-Basso, I.; Siddique, S.; McSkeane, T.; Ryan, G.; Vyas, P.; Cavenagh, J.; Stankovic, T.; Moss, P.; et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood 2010, 116, 1908–1918. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.; Tuechler, H. Revised international prognostic scoring system for myelodysplastic syndromes. 2012; 120, 2454–2465. [Google Scholar]
- Greenberg, P.; Cox, C.; LeBeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997, 89, 2079–2088. [Google Scholar] [PubMed]
- Sloand, E.M.; Mainwaring, L.; Fuhrer, M.; Ramkissoon, S.; Risitano, A.M.; Keyvanafar, K.; Lu, J.; Basu, A.; Barrett, A.J.; Young, N.S. Preferential suppression of trisomy 8 compared with normal hematopoietic cell growth by autologous lymphocytes in patients with trisomy 8 myelodysplastic syndrome. Blood 2005, 106, 841–851. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glenthøj, A.; Ørskov, A.D.; Hansen, J.W.; Hadrup, S.R.; O’Connell, C.; Grønbæk, K. Immune Mechanisms in Myelodysplastic Syndrome. Int. J. Mol. Sci. 2016, 17, 944. https://doi.org/10.3390/ijms17060944
Glenthøj A, Ørskov AD, Hansen JW, Hadrup SR, O’Connell C, Grønbæk K. Immune Mechanisms in Myelodysplastic Syndrome. International Journal of Molecular Sciences. 2016; 17(6):944. https://doi.org/10.3390/ijms17060944
Chicago/Turabian StyleGlenthøj, Andreas, Andreas Due Ørskov, Jakob Werner Hansen, Sine Reker Hadrup, Casey O’Connell, and Kirsten Grønbæk. 2016. "Immune Mechanisms in Myelodysplastic Syndrome" International Journal of Molecular Sciences 17, no. 6: 944. https://doi.org/10.3390/ijms17060944
APA StyleGlenthøj, A., Ørskov, A. D., Hansen, J. W., Hadrup, S. R., O’Connell, C., & Grønbæk, K. (2016). Immune Mechanisms in Myelodysplastic Syndrome. International Journal of Molecular Sciences, 17(6), 944. https://doi.org/10.3390/ijms17060944