
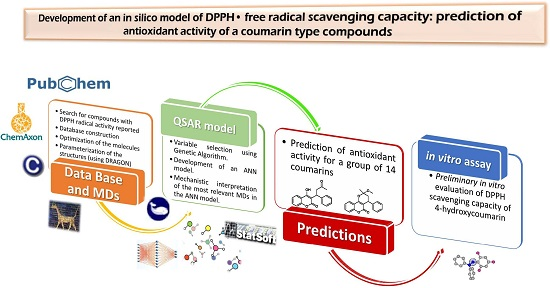
Development of an in Silico Model of DPPH• Free Radical Scavenging Capacity: Prediction of Antioxidant Activity of Coumarin Type Compounds

 ,
, 

Abstract
:
1. Introduction
2. Results
2.1. Modeling
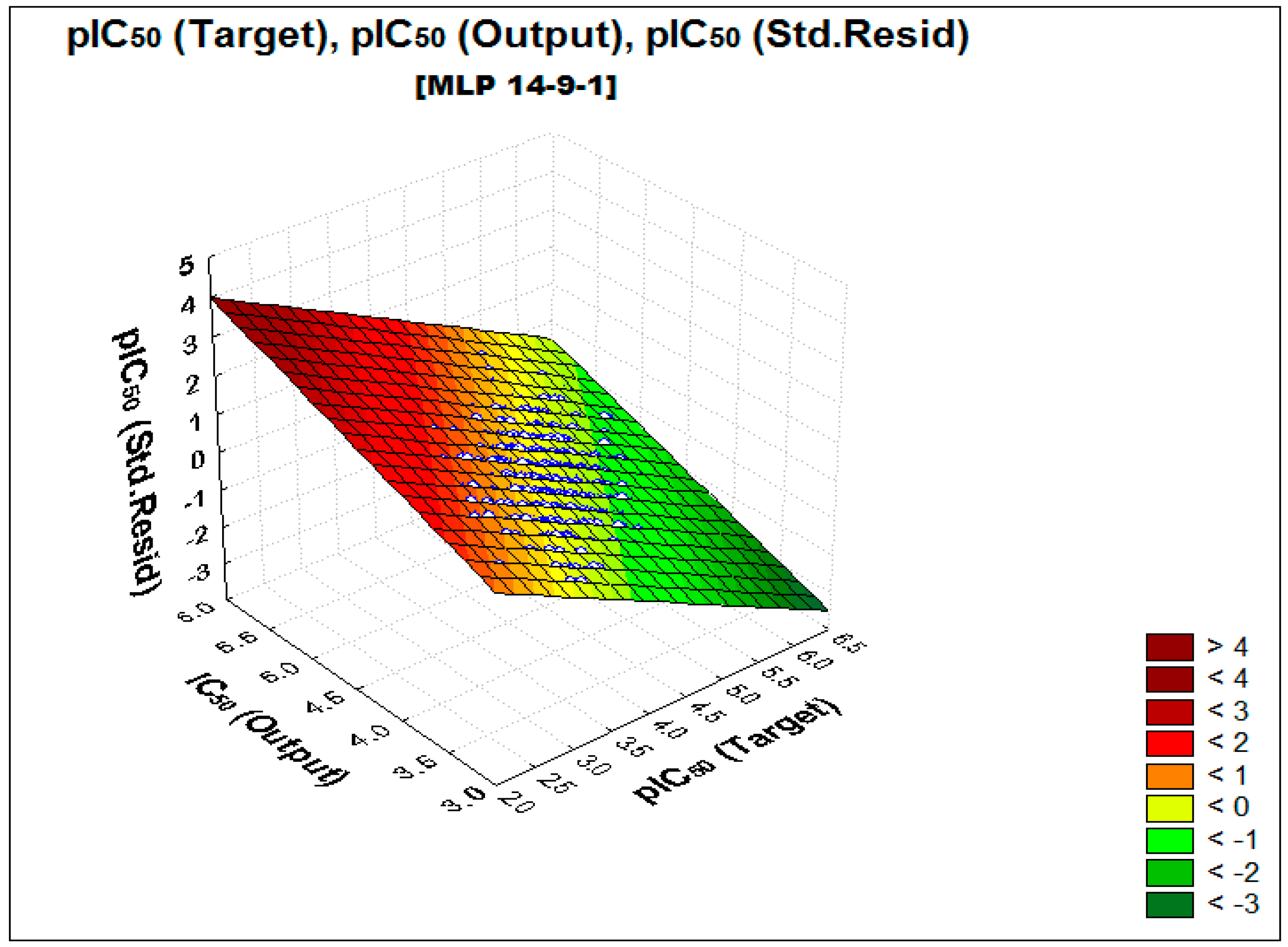
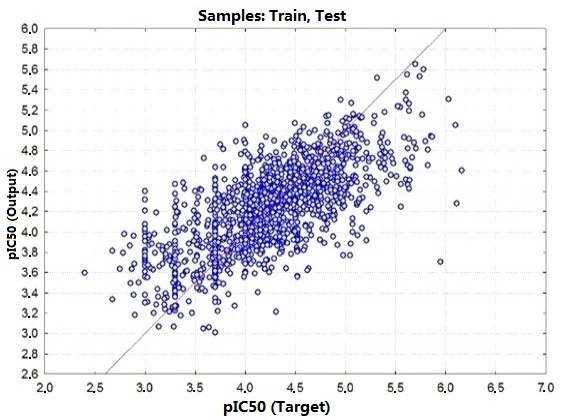
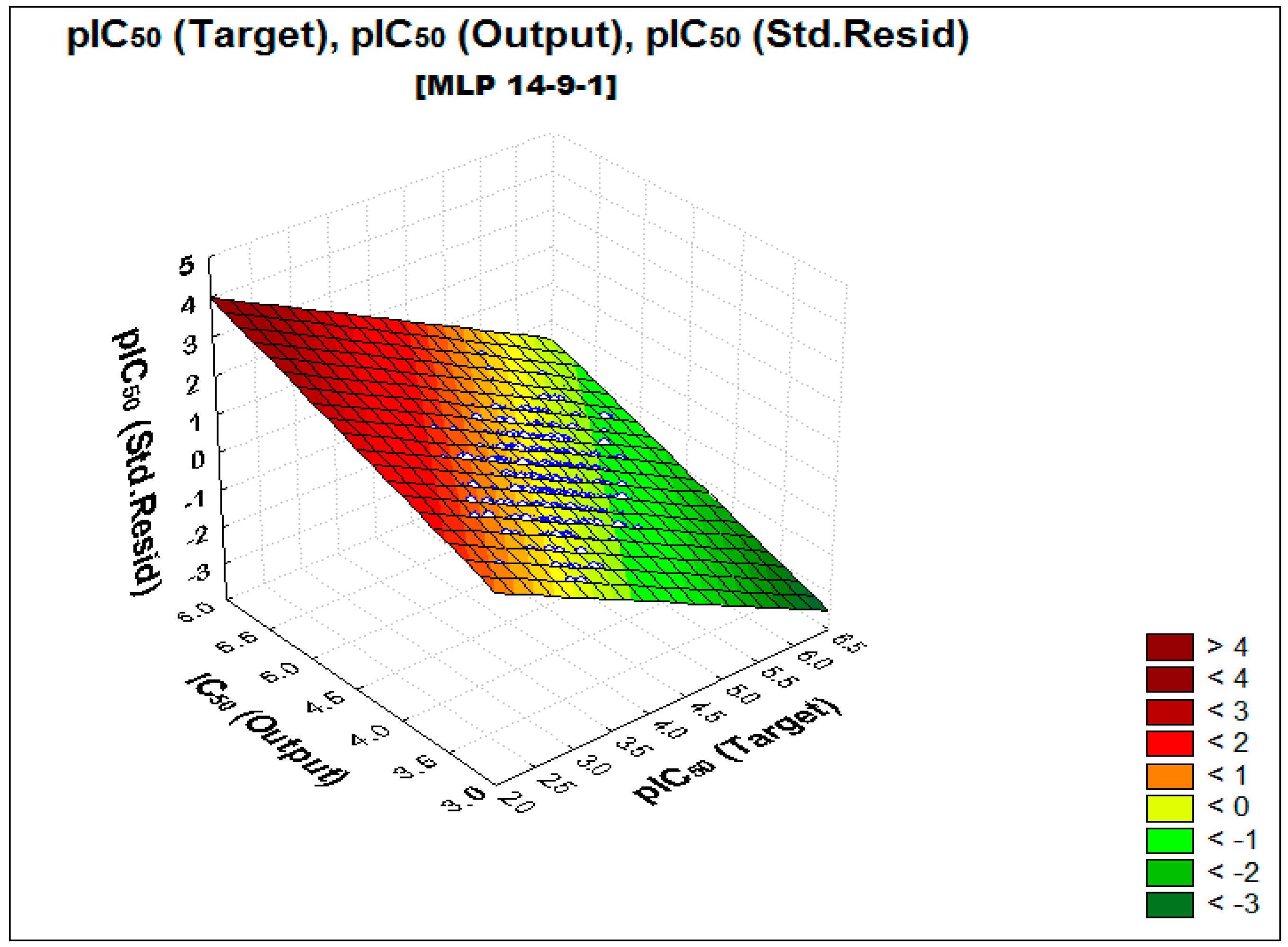
2.2. Performance and Predictive Capacity of the Model
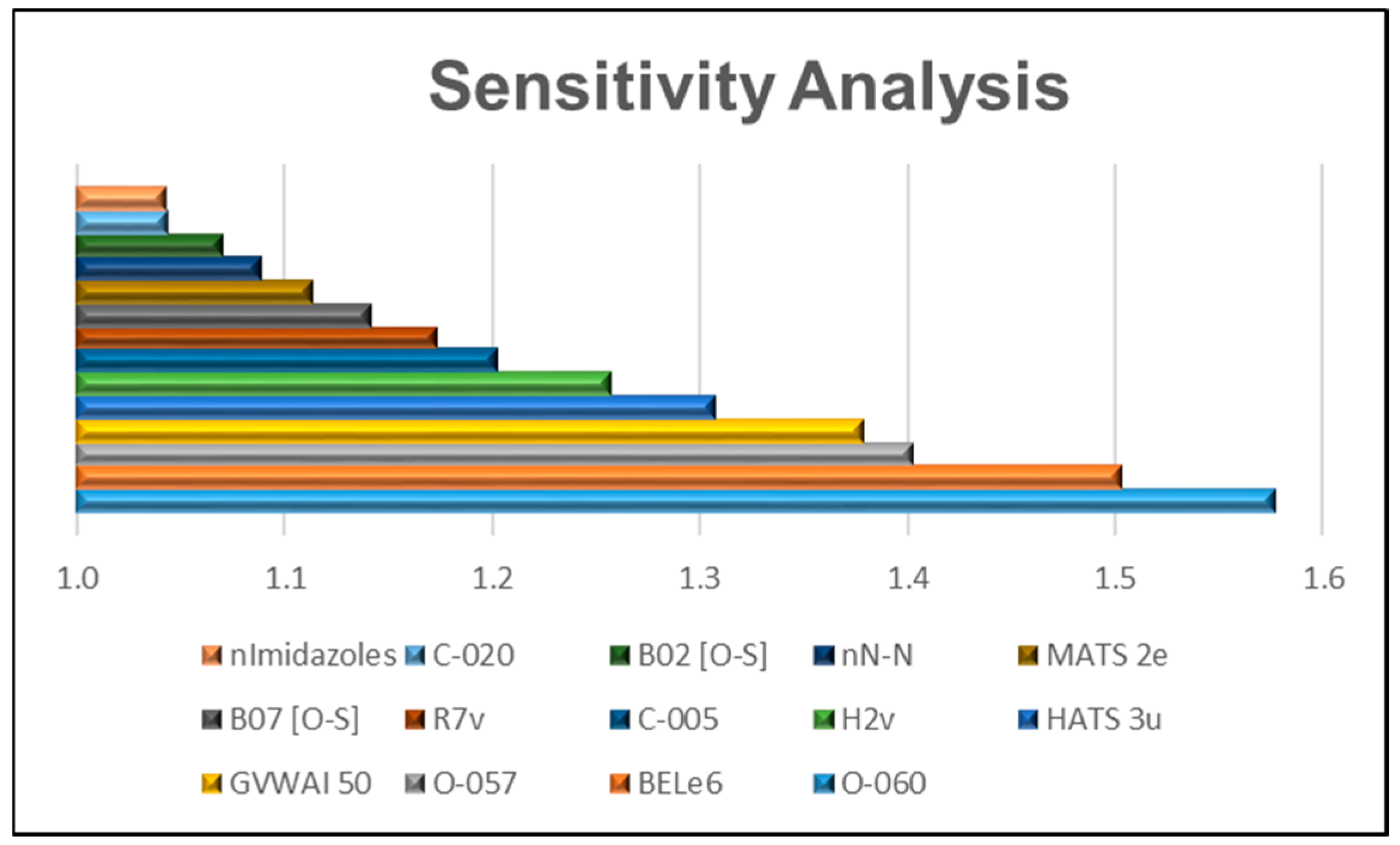
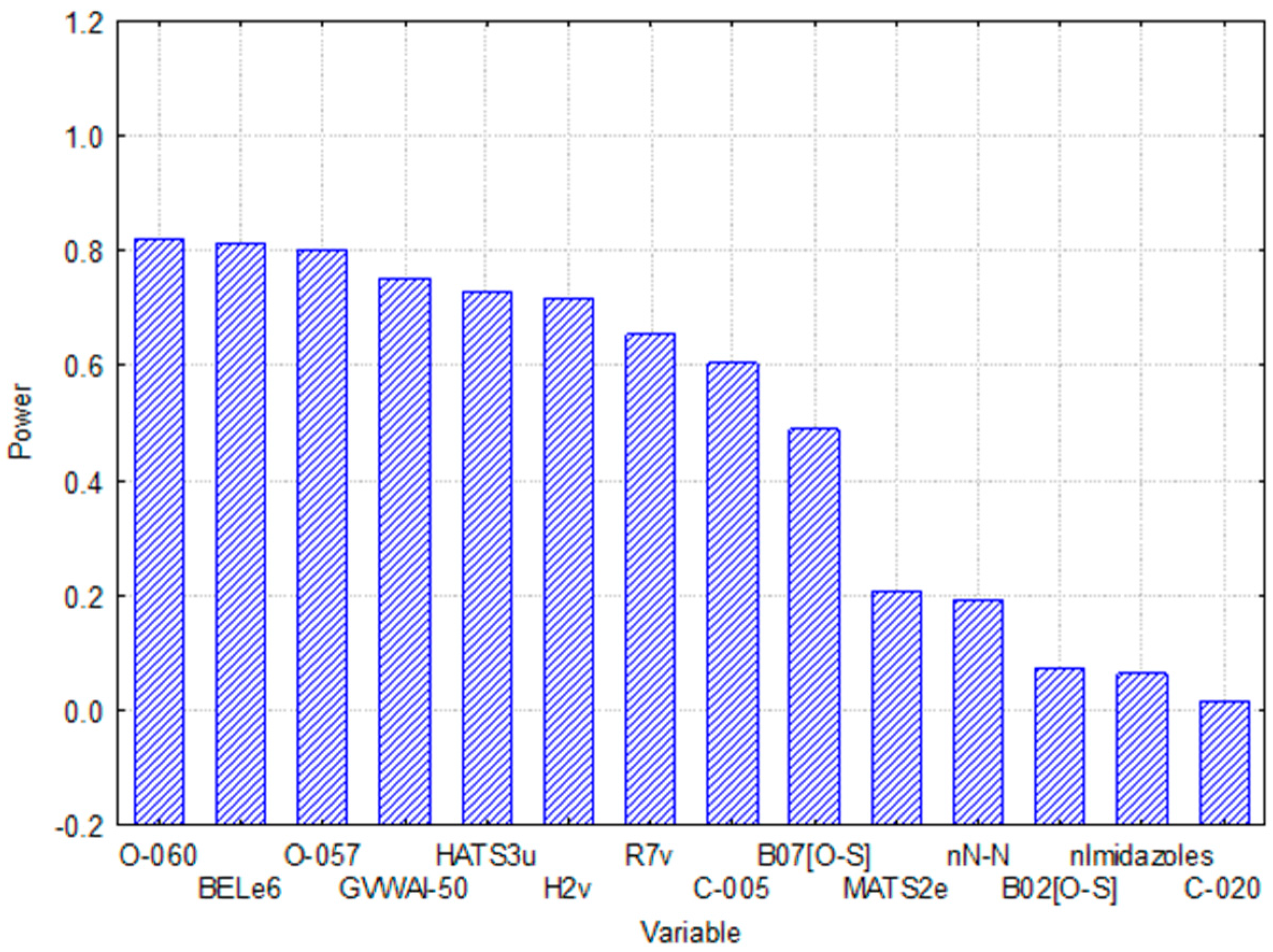
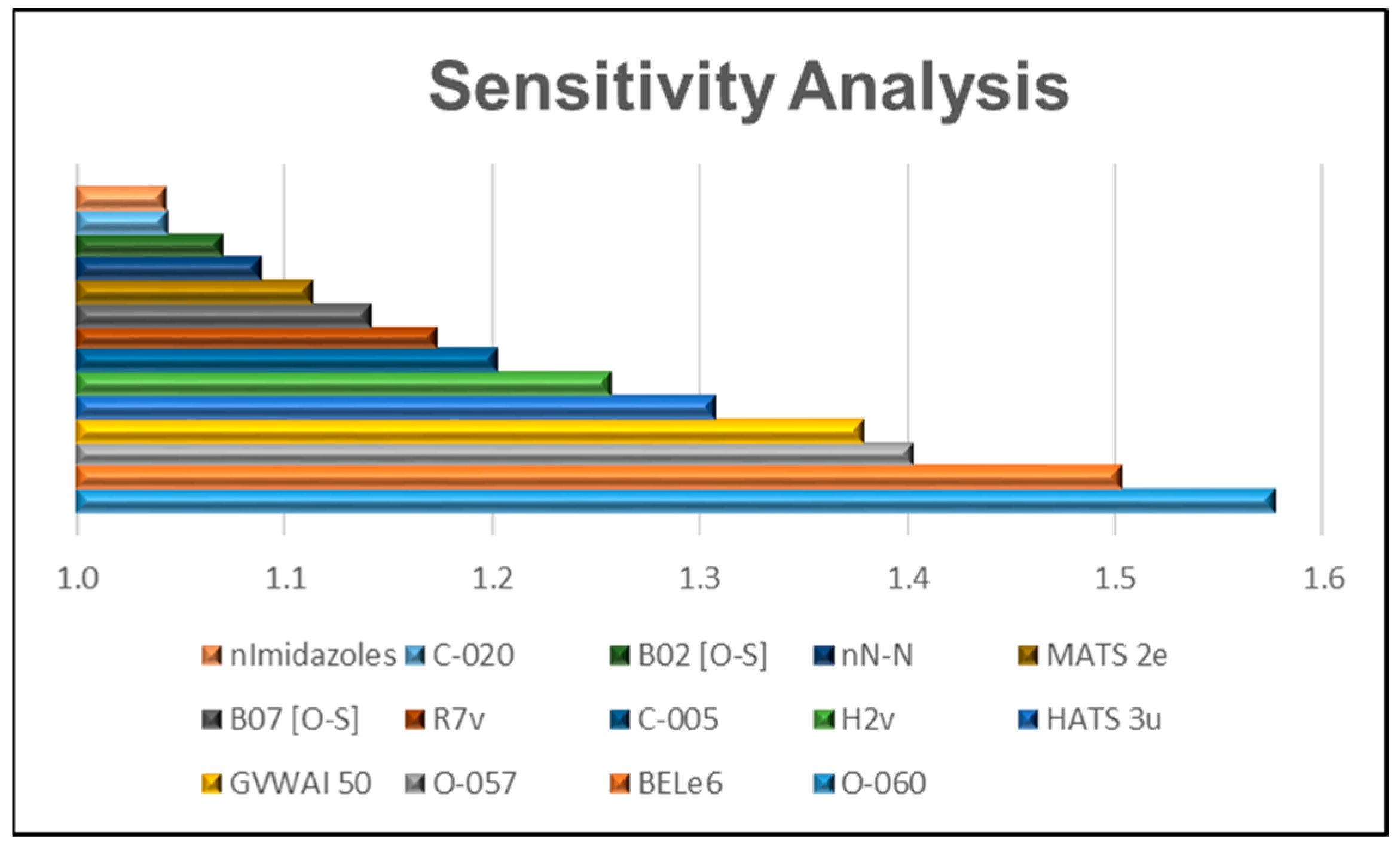
2.3. Relative Importance of the Variables in the Model
2.4. Prediction of Coumarin Derivatives Scavenging
2.5. In Vitro Assay
3. Discussion
3.1. Database and Neural Network
3.2. Analysis of the Molecular Descriptors
- -
- All the one-dimensional (1D) molecular descriptors included in the 14 variables, O-060, O-057, C-005, C-020, nN-N, n-imidazoles, are “Atom-centered fragments” or “Functional group counts” descriptors based in functional groups with the presence of some electronegative atom (O, N, S, Se, halogens) [20].
- -
- On the other hand, 2D molecular descriptors are related with the presence or absence of the O-S bond (in the case of B02 (O-S) and B07 (O-S)) or, such as MATS2e y BELe6, described properties correlated with Sanderson atomic electronegativity [20].
- -
- The 3D selected MD (HATS3u, H2v y R7v) are GATEWAY descriptors weighted by atomic van der Waals volumes [20].
- -
- The GVWAI-50 is a drug-like molecular properties descriptor, and its values are provided only for compounds having C, H, O, N, S, Se, P, B, Si, and halogens [20].
3.3. Prediction of Antioxidant Activities of a Group of Coumarins
3.4. In Vitro Assay
4. Materials and Methods
4.1. Preparation of Cases and Variables
4.2. Development of ANN Model
4.3. In Vitro DPPH• Assay
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, L.M.; Segundo, M.A.; Reis, S.; Lima, J.L.F.C. Methodological aspects about in vitro evaluation of antioxidant properties. Anal. Chim. Acta 2008, 613, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Ou, B.; Prior, R.L. The chemistry behind antioxidant capacity assays. J. Agric. Food Chem. 2005, 53, 1841–1856. [Google Scholar] [CrossRef] [PubMed]
- Scalzo, J.; Mezzetti, B.; Battino, M. Total antioxidant capacity evaluation: Critical steps for assaying berry antioxidant features. BioFactors 2005, 23, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Niki, E. Assessment of Antioxidant Capacity in vitro and in vivo. Free Rad. Biol. Med. 2010, 49, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Gunars, T.; Grzegorz, B. Determination of antiradical and antioxidant activity: Basic principles and new insights. Acta Biochim. Pol. 2010, 57, 139–142. [Google Scholar]
- Kedare, S.; Singh, R. Genesis and development of DPPH method of antioxidant assay. J. Food Sci. Technol. 2011, 48, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Naceur, H.; Fischmeister, C.; Puerta, M.; Valerga, P. A rapid access to new coumarinyl chalcone and substituted chromeno[4,3-c]pyrazol-4(1H)-ones and their antibacterial and DPPH radical scavenging activities. Med. Chem. Res. 2011, 20, 522–530. [Google Scholar]
- Puerta, M.; Naceur, H.; Valerga, P. Synthesis, structure, antimicrobial and antioxidant investigations of dicoumarol and related compounds. Eur. J. Med. Chem. 2008, 43, 2541–2548. [Google Scholar]
- Fylaktakidou, K.; Hadjipavlou-Litina, D.; Litinas, K.; Nicolaides, D. Natural and synthetic coumarin derivatives with antiinflammatory/antioxidant activities. Curr. Pharm. 2004, 10, 3813–3833. [Google Scholar] [CrossRef]
- Razo-Hernández, R.; Pineda-Urbina, K.; Velazco-Medel, M.; Villanueva-García, M.; Sumaya-Martínez, T.; Martínez-Martínez, F.; Gómez-Sandoval, Z. QSAR study of the DPPH• radical scavenging activity of coumarin derivatives and xanthine oxidase inhibition by molecular docking. Cent. Eur. J. Chem. 2014, 12, 1067–1080. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V.; Gramatica, P. Chemometrics in QSAR. In Comprehensive Chemometrics Chemical and Biochemical Data Analysis; Brown, S., Tauler, R., Walczak, B., Eds.; Elsevier: Ámsterdam, The Netherlands, 2009; Volume 4, pp. 129–172. [Google Scholar]
- Eriksson, L.; Jaworska, J.; Worth, A.; Cronin, M.; McDowell, R.; Gramatica, P. Methods for reliability and uncertainty assessment and for applicability evaluations of classification- and regression-based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Suzuki, Y.; Ichikawa, H. Neural networks applied to structure-activity relationships. J. Med. Chem. 1990, 33, 905–908. [Google Scholar] [CrossRef] [PubMed]
- CORINA Classic (software); Molecular Networks GmbH: Erlangen, Germany, 2015; Available online: http://www.molecular-networks.com (accessed on 9 March 2015).
- TALETE srl. MobyDigs (software), v1.0; Milano, Italy, 2004; Available online: http://www.talete.mi.it/mobydigs.htm (accessed on 2 June 2015).
- StatSoft, Inc. STATISTICA (software), v8.0; Tulsa, OK, USA, 2007; Available online: http://www.statsoft.com (accessed on 7 June 2015).
- Jeliazkova, N. Ambit Discovery (software), v0.04; Sofia, Bulgaria, 2005; Available online: http://www.ambit.acad.bg (accessed on 5 June 2015).
- Tropsha, A. Best Practices for QSAR model development, validation, and exploitation. Mol. Inf. 2010, 29, 476–488. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors, 1st ed.; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Wright, J.; Johnson, E.; DiLabio, G. Predicting the activity of phenolic antioxidants: Theoretical method, analysis of substituent effects, and application to major families of antioxidants. J. Am. Chem. Soc. 2001, 123, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xiao, H.; Zheng, J.; Liang, G. Structure-thermodynamics-antioxidant activity relationships of selected natural phenolic acids and derivatives: An experimental and theoretical evaluation. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Matthew, S.; Abreu, P.M.; Aires-de-Sousa, J. QSAR analysis of phenolic antioxidants using MOLMAP descriptors of local properties. Bioorg. Med. Chem. 2006, 14, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Jing, P.; Zhao, S.-J.; Jian, W.-J.; Qian, B.-J.; Dong, Y.; Pang, J. Quantitative studies on structure-DPPH scavenging activity relationships of food phenolic acids. Molecules 2012, 17, 12910–12924. [Google Scholar] [CrossRef] [PubMed]
- Khlebnikov, A.I.; Schepetkin, I.A.; Domina, N.G.; Kirpotina, L.N.; Quinn, M.T. Improved quantitative structure-activity relationship models to predict antioxidant activity of flavonoids in chemical, enzymatic, and cellular systems. Bioorg. Med. Chem. 2007, 15, 1749–1770. [Google Scholar] [CrossRef] [PubMed]
- Mitra, I.; Saha, A.; Roy, K. Quantitative structure-activity relationship modeling of antioxidant activities of hidroxybenzalacetones using quantum chemical, physicochemical and spatial descriptors. Chem. Biol. Drug. Des. 2009, 73, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Mitra, I.; Saha, A.; Roy, K. Chemometric modeling of free radical scavenging activiti of flavone derivatives. Eur. J. Med. Chem. 2010, 45, 5071–5079. [Google Scholar] [CrossRef] [PubMed]
- Worachartcheewan, A.; Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Prachaiasittikul, S.; Prachaiasittikul, V. Predicting the free radical scavenging activity of curcumin derivatives. Chemometr. Intell. Lab. Syst. 2011, 109, 207–216. [Google Scholar] [CrossRef]
- Yamagami, C.; Motohashi, N.; Emoto, T.; Hamasaki, A.; Tanahashi, T.; Nagakura, N.; Takeuchi, Y. Quantitative structure-activity relationship analyses of antioxidant and free radical scavenging activities for hydroxybenzalacetones. Bioorg. Med. Chem. Lett. 2004, 14, 5629–5633. [Google Scholar] [CrossRef] [PubMed]
- Kadhum, A.; Al-Amiery, A.; Musa, A.; Mohamad, A. The antioxidant activity of new coumarin derivatives. Int. J. Mol. Sci. 2011, 12, 5747–5761. [Google Scholar] [CrossRef] [PubMed]
- McInnes, C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007, 11, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Cseke, L.J.; Kirakosyan, A.; Kaufman, P.B.; Warber, S.; Duke, J.A.; Brielmann, H.L. Natural Products from Plants, 2nd ed.; CRC Press: New York, NY, USA, 2006. [Google Scholar]
- ChemAxon Ltd. ChemAxon, 6.1.0 (software); Budapest, Hungary, 2013; Available online: http://www.chemaxon.com (accessed on 10 March 2015).
- TALETE srl. DRAGON for Windows (Software for Molecular Descriptor Calculations), V 5.5; Milano, Italy, 2007; Available online: http://www.talete.mi.it/ (accessed on 15 March 2015).
- Blois, M. Antioxidant determinations by the use of a stable free radical. Nature 1958, 181, 1199–1200. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N° | Structures | IUPAC Name | Predicted pIC50 |
|---|---|---|---|
| 1 |  | 4-(4-(trifluoromethyl)phenyl)-3,4-dihydro-2-methoxy-2-methylpyrano[3,2-c]chromen-5(2H)-one | 3.881 |
| 2 | 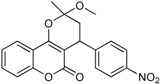 | 3,4-dihydro-2-methoxy-2-methyl-4-(4-nitrophenyl)pyrano[3,2-c]chromen-5(2H)-one | 3.951 |
| 3 | 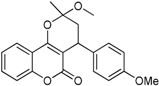 | 3,4-dihydro-2-methoxy-4-(4-methoxyphenyl)-2-methylpyrano[3,2-c]chromen-5(2H)-one | 3.829 |
| 4 | 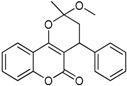 | 3,4-dihydro-2-methoxy-2-methyl-4-phenylpyrano[3,2-c]chromen-5(2H)-one | 3.944 |
| 5 | 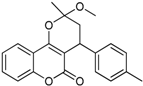 | 3,4-dihydro-2-methoxy-2-methyl-4-p-tolylpyrano[3,2-c]chromen-5(2H)-one | 3.946 |
| 6 | 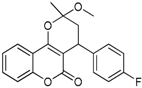 | 4-(4-fluorophenyl)-3,4-dihydro-2-methoxy-2-methylpyrano[3,2-c]chromen-5(2H)-one | 3.959 |
| 7 | 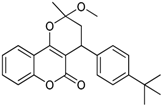 | 4-(4-tert-butylphenyl)-3,4-dihydro-2-methoxy-2-methylpyrano[3,2-c]chromen-5(2H)-one | 3.903 |
| 8 | 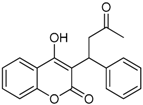 | 4-hydroxy-3-(3-oxo-1-phenylbutyl)-2H-chromen-2-one | 2.326 |
| 9 | 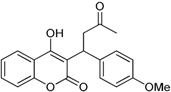 | 4-hydroxy-3-(1-(4-methoxyphenyl)-3-oxobutyl)-2H-chromen-2-one | 2.291 |
| 10 |  | 3-(1-(4-(trifluoromethyl)phenyl)-3-oxobutyl)-4-hydroxy-2H-chromen-2-one | 2.217 |
| 11 | 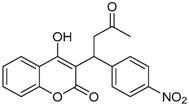 | 4-hydroxy-3-(1-(4-nitrophenyl)-3-oxobutyl)-2H-chromen-2-one | 2.288 |
| 12 | 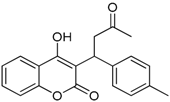 | 4-hydroxy-3-(3-oxo-1-p-tolylbutyl)-2H-chromen-2-one | 2.318 |
| 13 |  | 3-(1-(4-fluorophenyl)-3-oxobutyl)-4-hydroxy-2H-chromen-2-one | 2.341 |
| 14 |  | 3-(1-(4-tert-butylphenyl)-3-oxobutyl)-4-hydroxy-2H-chromen-2-one | 2.179 |
| 15 | 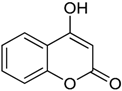 | 4-hydroxy-2H-chromen-2-one | 3.421 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goya Jorge, E.; Rayar, A.M.; Barigye, S.J.; Jorge Rodríguez, M.E.; Sylla-Iyarreta Veitía, M. Development of an in Silico Model of DPPH• Free Radical Scavenging Capacity: Prediction of Antioxidant Activity of Coumarin Type Compounds. Int. J. Mol. Sci. 2016, 17, 881. https://doi.org/10.3390/ijms17060881
Goya Jorge E, Rayar AM, Barigye SJ, Jorge Rodríguez ME, Sylla-Iyarreta Veitía M. Development of an in Silico Model of DPPH• Free Radical Scavenging Capacity: Prediction of Antioxidant Activity of Coumarin Type Compounds. International Journal of Molecular Sciences. 2016; 17(6):881. https://doi.org/10.3390/ijms17060881
Chicago/Turabian StyleGoya Jorge, Elizabeth, Anita Maria Rayar, Stephen J. Barigye, María Elisa Jorge Rodríguez, and Maité Sylla-Iyarreta Veitía. 2016. "Development of an in Silico Model of DPPH• Free Radical Scavenging Capacity: Prediction of Antioxidant Activity of Coumarin Type Compounds" International Journal of Molecular Sciences 17, no. 6: 881. https://doi.org/10.3390/ijms17060881
APA StyleGoya Jorge, E., Rayar, A. M., Barigye, S. J., Jorge Rodríguez, M. E., & Sylla-Iyarreta Veitía, M. (2016). Development of an in Silico Model of DPPH• Free Radical Scavenging Capacity: Prediction of Antioxidant Activity of Coumarin Type Compounds. International Journal of Molecular Sciences, 17(6), 881. https://doi.org/10.3390/ijms17060881