
MicroRNA in Metabolic Re-Programming and Their Role in Tumorigenesis


Abstract
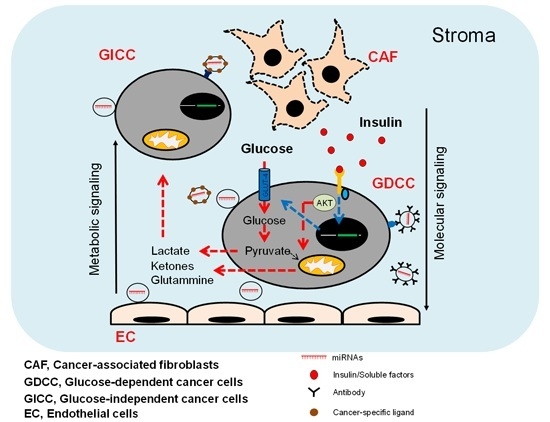
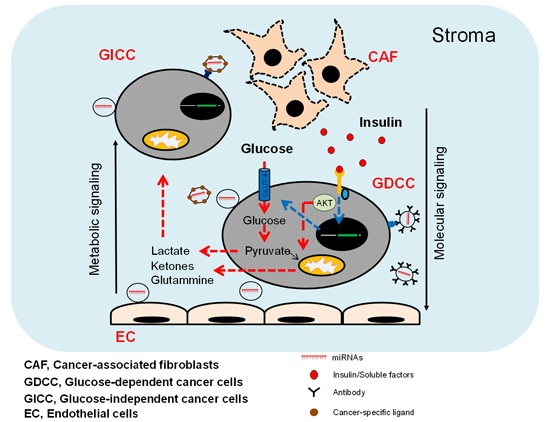
:
1. Introduction
2. Tumour Environment and Cancer Metabolic Reprogramming
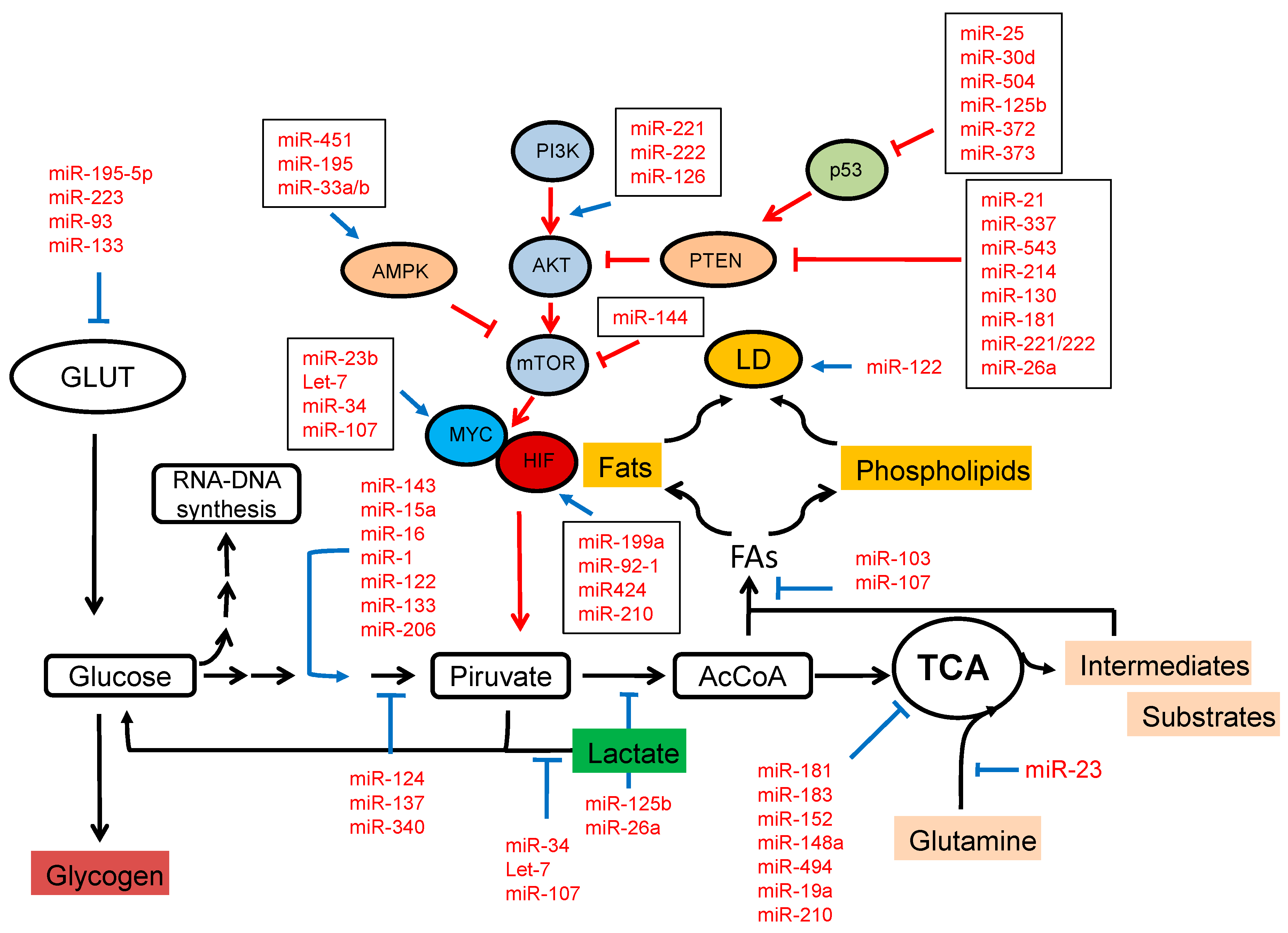
2.1. miRNA Regulate Glucose Metabolism
2.2. miRNAs Regulate the Tricarboxylic Acid Cycle
2.3. miRNAs and Lipid Metabolism
2.4. Regulation of Signalling Pathways by miRNAs
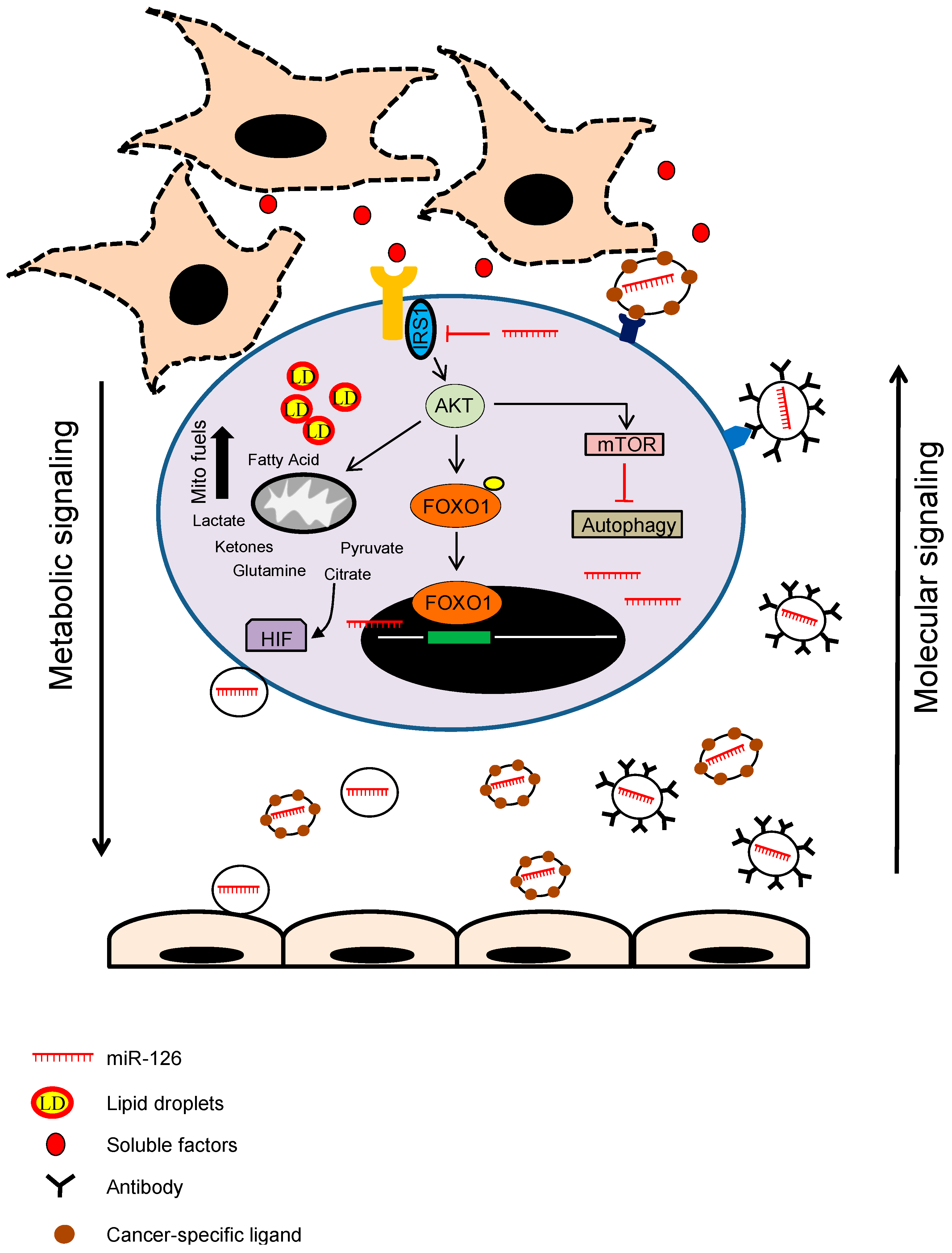
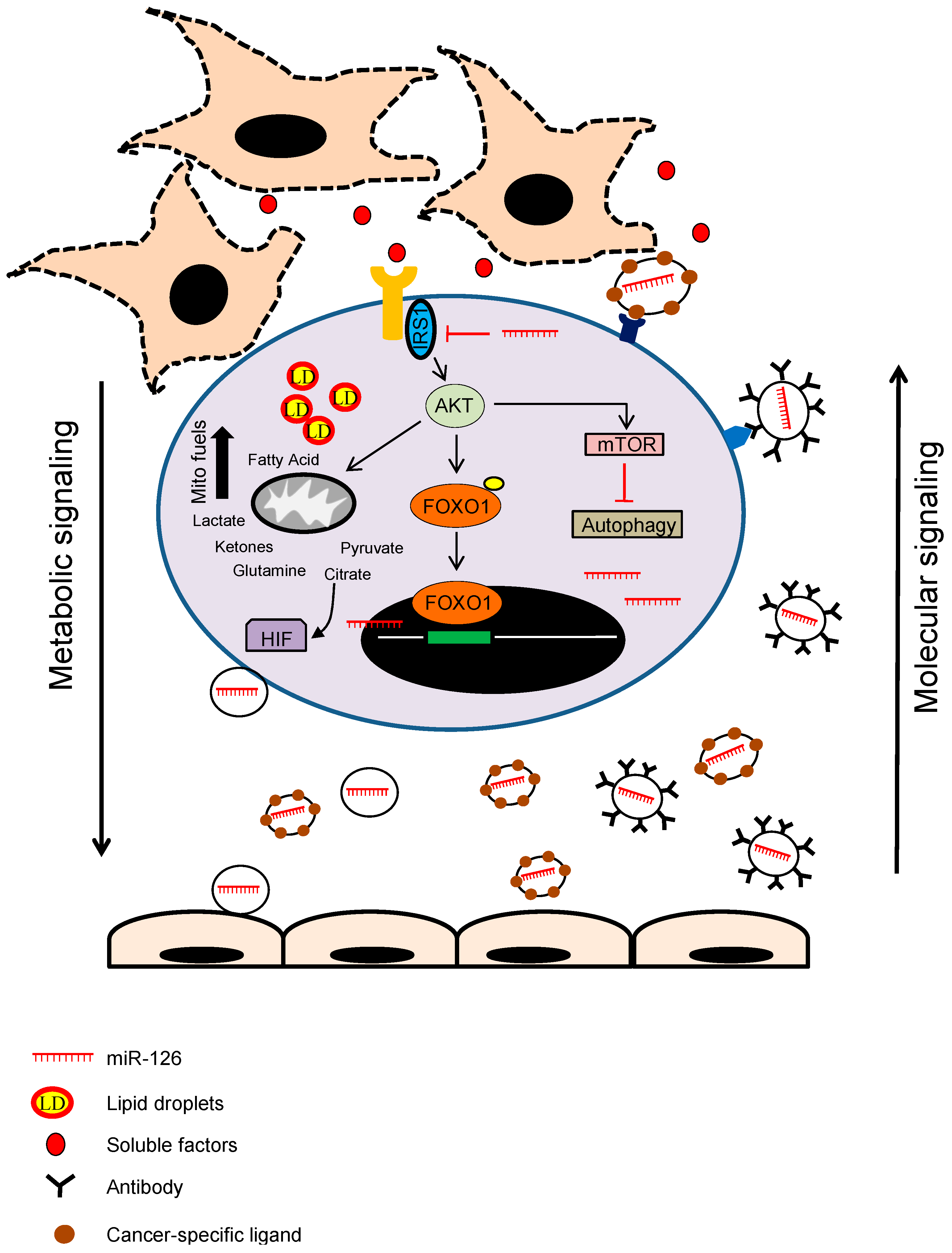
3. miRNAs as Cancer Stroma Messengers Regulating Cancer Metabolism and Tumorigenesis
4. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
References
- Xing, Y.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of the tumour microenvironment. FEBS J. 2015, 282, 3892–3898. [Google Scholar] [CrossRef] [PubMed]
- Salani, B.; Ravera, S.; Amaro, A.; Salis, A.; Passalacqua, M.; Millo, E.; Damonte, G.; Marini, C.; Pfeffer, U.; Sambuceti, G.; et al. IGF1 regulates PKM2 function through Akt phosphorylation. Cell Cycle 2015, 14, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Buzzai, M.; Bauer, D.E.; Jones, R.G.; Deberardinis, R.J.; Hatzivassiliou, G.; Elstrom, R.L.; Thompson, C.B. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid β-oxidation. Oncogene 2005, 24, 4165–4173. [Google Scholar] [CrossRef] [PubMed]
- Hanai, J.I.; Doro, N.; Seth, P.; Sukhatme, V.P. ATP citrate lyase knockdown impacts cancer stem cells in vitro. Cell Death Dis. 2013, 4, e696. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Cirri, P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Challagundla, K.B.; Fanini, F.; Vannini, I.; Wise, P.; Murtadha, M.; Malinconico, L.; Cimmino, A.; Fabbri, M. MicroRNAs in the tumor microenvironment: Solving the riddle for a better diagnostics. Expert Rev. Mol. Diagn. 2014, 14, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. MicroRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007, 302, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Calin, G.A.; Croce, C.M. MicroRNAs in Cancer. Annu. Rev. Med. 2009, 60, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.; Qi, M.; Wu, B.; Song, Y.; Wang, Y.; Li, T. MicroRNA-195-5p suppresses glucose uptake and proliferation of human bladder cancer T24 cells by regulating GLUT3 expression. FEBS Lett. 2012, 586, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Heneidi, S.; Lee, J.M.; Layman, L.C.; Stepp, D.W.; Gamboa, G.M.; Chen, B.S.; Chazenbalk, G.; Azziz, R. miRNA-93 inhibits GLUT4 and is overexpressed in adipose tissue of polycystic ovary syndrome patients and women with insulin resistance. Diabetes 2013, 62, 2278–2286. [Google Scholar] [CrossRef] [PubMed]
- Peschiaroli, A.; Giacobbe, A.; Formosa, A.; Markert, E.K.; Bongiorno-Borbone, L.; Levine, A.J.; Candi, E.; D’Alessandro, A.; Zolla, L.; Finazzi Agrò, A.; et al. miR-143 regulates hexokinase 2 expression in cancer cells. Oncogene 2013, 32, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhang, L.F.; Zhang, H.W.; Hu, S.; Lu, M.H.; Liang, S.; Li, B.; Li, Y.; Li, D.; Wang, E.D.; et al. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. 2012, 31, 1985–1998. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Chen, W.G.; Li, X.W. MicroRNA-143 acts as a tumor suppressor by targeting hexokinase 2 in human prostate cancer. Am. J. Cancer Res. 2015, 5, 2056–2063. [Google Scholar] [PubMed]
- Calin, G.A.; Cimmino, A.; Fabbri, M.; Ferracin, M.; Wojcik, S.E.; Shimizu, M.; Taccioli, C.; Zanesi, N.; Garzon, R.; Aqeilan, R.I.; et al. miR-15a and miR-16-1 cluster functions in human leukemia. Proc. Natl. Acad. Sci. USA 2008, 105, 5166–5171. [Google Scholar] [CrossRef] [PubMed]
- Fabani, M.M.; Gait, M.J. miR-122 targeting with LNA/2′-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA-peptide conjugates. RNA 2008, 14, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Happel, C.; Manna, S.K.; Acquaah-Mensah, G.; Carrerero, J.; Kumar, S.; Nasipuri, P.; Krausz, K.W.; Wakabayashi, N.; Dewi, R.; et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Rathore, M.G.; Saumet, A.; Rossi, J.F.; de Bettignies, C.; Tempé, D.; Lecellier, C.H.; Villalba, M. The NF-κB member p65 controls glutamine metabolism through miR-23a. Int. J. Biochem. Cell Biol. 2012, 44, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Cheng, T. Q's next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, K.M.; Dewhirst, M.W. Tumor metabolism of lactate: The influence and therapeutic potential for MCT and CD147 regulation. Future Oncol. 2010, 6, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Wilfred, B.R.; Wang, W.X.; Nelson, P.T. Energizing miRNA research: A review of the role of miRNAs in lipid metabolism, with a prediction that miR-103/107 regulates human metabolic pathways. Mol. Genet. Metab. 2007, 91, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Wu, T.; Miao, L.; Mei, Y.; Wu, M. miR-181a regulates lipid metabolism via IDH1. Sci. Rep. 2015, 5, 8801. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Sasayama, T.; Tanaka, K.; Nakamizo, S.; Nishihara, M.; Mizukawa, K.; Kohta, M.; Koyama, J.; Miyake, S.; Taniguchi, M.; et al. MicroRNA-183 upregulates HIF-1α by targeting isocitrate dehydrogenase 2 (IDH2) in glioma cells. J. Neurooncol. 2013, 111, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, Y.; Zhang, H.; Huang, P.; Luthra, R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 2010, 29, 4362–4368. [Google Scholar] [CrossRef] [PubMed]
- Eichner, L.J.; Perry, M.C.; Dufour, C.R.; Bertos, N.; Park, M.; St-Pierre, J.; Giguère, V. miR-378* mediates metabolic shift in breast cancer cells via the PGC-1β/ERRγ transcriptional pathway. Cell Metab. 2010, 12, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Abadi, A.; Akhtar, M.; Hettinga, B.P.; Tarnopolsky, M.A. miRNA in the regulation of skeletal muscle adaptation to acute endurance exercise in C57Bl/6J male mice. PLoS ONE 2009, 4, e5610. [Google Scholar] [CrossRef] [PubMed]
- Tili, E.; Michaille, J.J.; Luo, Z.; Volinia, S.; Rassenti, L.Z.; Kipps, T.J.; Croce, C.M. The down-regulation of miR-125b in chronic lymphocytic leukemias leads to metabolic adaptation of cells to a transformed state. Blood 2012, 120, 2631–2638. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Liu, Y.; Jin, X.; Lu, W.; Liu, J.; Xia, Z.; Yuan, Q.; Zhao, X.; Xu, N.; Liang, S. MicroRNA-26a regulates glucose metabolism by direct targeting PDHX in colorectal cancer cells. BMC Cancer 2014, 14, 443. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhao, X.; Zhou, Y.; Hu, Y. miR-124, miR-137 and miR-340 regulate colorectal cancer growth via inhibition of the Warburg effect. Oncol. Rep. 2012, 28, 1346–1352. [Google Scholar] [PubMed]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Ferlito, M.; Kent, O.A.; Fox-Talbot, K.; Wang, R.; Liu, D.; Raghavachari, N.; Yang, Y.; Wheelan, S.J.; Murphy, E.; et al. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ. Res. 2012, 110, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Aschrafi, A.; Schwechter, A.D.; Mameza, M.G.; Natera-Naranjo, O.; Gioio, A.E.; Kaplan, B.B. MicroRNA-338 regulates local cytochrome c oxidase IV mRNA levels and oxidative posphorylation in the axons of sympathetic neurons. J. Neurosci. 2008, 28, 12581–12590. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Dávalos, A.; Goedeke, L.; Smibert, P.; Ramírez, C.M.; Warrier, N.P.; Andreo, U.; Cirera-Salinas, D.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef] [PubMed]
- Lynn, F.C. Meta-regulation: MicroRNA regulation of glucose and lipid metabolism. Trends Endocrinol. Metab. 2009, 20, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Hsu, S.D.; Hsu, C.S.; Lai, T.C.; Chen, S.J.; Shen, R.; Huang, Y.; Chen, H.C.; Lee, C.H.; Tsai, T.F.; et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Investig. 2012, 122, 2884–2897. [Google Scholar] [CrossRef] [PubMed]
- Pol, A.; Gross, S.P.; Parton, R.G. Review: Biogenesis of the multifunctional lipid droplet: Lipids, proteins, and sites. J. Cell Biol. 2014, 204, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Herms, A.; Bosch, M.; Reddy, B.J.; Schieber, N.L.; Fajardo, A.; Rupérez, C.; Fernández-Vidal, A.; Ferguson, C.; Rentero, C.; Tebar, F.; et al. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat. Commun. 2015, 6, 7176. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A. Drivers of the Warburg phenotype. Cancer J. 2015, 21, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Rane, S.; He, M.; Sayed, D.; Vashistha, H.; Malhotra, A.; Sadoshima, J.; Vatner, D.E.; Vatner, S.F.; Abdellatif, M. Downregulation of miR-199a derepresses hypoxia-inducible factor-1α and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res. 2009, 104, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Shanafelt, T.D.; Cimmino, A.; Taccioli, C.; Volinia, S.; Liu, C.G.; Calin, G.A.; Croce, C.M.; Chan, D.A.; Giaccia, A.J.; et al. Aberrant regulation of pVHL levels by microRNA promotes the HIF/VEGF axis in CLL B cells. Blood 2009, 113, 5568–5574. [Google Scholar] [CrossRef] [PubMed]
- Chow, T.F.; Youssef, Y.M.; Lianidou, E.; Romaschin, A.D.; Honey, R.J.; Stewart, R.; Pace, K.T.; Yousef, G.M. Differential expression profiling of microRNAs and their potential involvement in renal cell carcinoma pathogenesis. Clin. Biochem. 2010, 43, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.; Subramanian, I.V.; Adhikari, N.; Zhang, X.; Joshi, H.P.; Basi, D.; Chandrashekhar, Y.S.; Hall, J.L.; Roy, S.; Zeng, Y.; et al. Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-α isoforms and promotes angiogenesis. J. Clin. Investig. 2010, 120, 4141–4154. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.J.; Souza, A.L.; Clish, C.B.; Puigserver, P. A hypoxia-induced positive feedback loop promotes hypoxia-inducible factor 1α stability through miR-210 suppression of glycerol-3-phosphate dehydrogenase 1-like. Mol. Cell. Biol. 2011, 31, 2696–2706. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- He, X.Y.; Chen, J.X.; Zhang, Z.; Li, C.L.; Peng, Q.L.; Peng, H.M. The let-7a microRNA protects from growth of lung carcinoma by suppression of k-Ras and c-Myc in nude mice. J. Cancer Res. Clin. Oncol. 2010, 136, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shyh-Chang, N.; Segrè, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.P.; Kharas, M.G.; et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 2011, 147, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Braun, C.J.; Zhang, X.; Savelyeva, I.; Wolff, S.; Moll, U.M.; Schepeler, T.; Ørntoft, T.F.; Andersen, C.L.; Dobbelstein, M. p53-Responsive microRNAs 192 and 215 are capable of inducing cell cycle arrest. Cancer Res. 2008, 68, 10094–10104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.G.; Zheng, J.N.; Pei, D.S. P53/microRNA-34-induced metabolic regulation: New opportunities in anticancer therapy. Mol. Cancer 2014, 13, 115. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.L.; Xue, G.; Mei, Q.; Wang, Y.Z.; Ding, F.X.; Liu, M.F.; Lu, M.H.; Tang, Y.; Yu, H.Y.; Sun, S.H. Repression of the miR-17–92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J. 2009, 28, 2719–2732. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Miyazono, K. p53 actions on microRNA expression and maturation pathway. Methods Mol. Biol. 2013, 962, 165–181. [Google Scholar] [PubMed]
- Kumar, M.; Lu, Z.; Takwi, A.A.; Chen, W.; Callander, N.S.; Ramos, K.S.; Young, K.H.; Li, Y. Negative regulation of the tumor suppressor p53 gene by microRNAs. Oncogene 2011, 30, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.; Stoop, H.; Nagel, R.; Liu, Y.P.; van Duijse, J.; Drost, J.; et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 2006, 124, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Kefas, B.; Comeau, L.; Erdle, N.; Montgomery, E.; Amos, S.; Purow, B. Pyruvate kinase M2 is a target of the tumor-suppressive microRNA-326 and regulates the survival of glioma cells. Neuro Oncol. 2010, 12, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Nuovo, G.; Palatini, J.; de Lay, M.; Van Brocklyn, J.; Ostrowski, M.C.; Chiocca, E.A.; Lawler, S.E. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol. Cell 2010, 37, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Hatziapostolou, M.; Polytarchou, C.; Iliopoulos, D. miRNAs link metabolic reprogramming to oncogenesis. Trends Endocrinol. Metab. 2013, 24, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Z.; Li, C.; Chen, Q.; Jing, Y.; Carpenter, R.; Jiang, Y.; Kung, H.F.; Lai, L.; Jiang, B.H. miR-21 induced angiogenesis through AKT and ERK activation and HIF-1α expression. PLoS ONE 2011, 6, e19139. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; He, T.; Liang, L.; Zhang, X.; Yuan, H. Upregulation of microRNA‑337 promotes the proliferation of endometrial carcinoma cells via targeting PTEN. Mol. Med. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhou, H.X.; Yang, M.X.; Wang, Y.; Cao, W.M.; Lu, K.F.; Wu, R.Q. miR-543 promotes proliferation and invasion of non-small cell lung cancer cells by inhibiting PTEN. Biochem. Biophys. Res. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, L.; Chen, Z.; Zhu, M.; Gu, Y. MicroRNA-214 acts as a potential oncogene in breast cancer by targeting the PTEN-PI3K/Akt signaling pathway. Int. J. Mol. Med. 2016, 37, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yan, D.; Wu, W.; Zhu, J.; Ye, W.; Shu, Q. MicroRNA-130a promotes the metastasis and epithelial-mesenchymal transition of osteosarcoma by targeting PTEN. Oncol. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Cui, L.; Mei, Z.; Liu, M.; Zhang, D. miR-181a mediates metabolic shift in colon cancer cells via the PTEN/AKT pathway. FEBS Lett. 2014, 588, 1773–1779. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wu, X.; Liu, B.; Wang, C.; Liu, Y.; Zhou, Q.; Xu, K. miR-26a enhances metastasis potential of lung cancer cells via AKT pathway by targeting PTEN. Biochim. Biophys. Acta 2012, 1822, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Sudarsanam, S.; Johnson, D.E. Functional consequences of mTOR inhibition. Curr. Opin. Drug Discov. Dev. 2010, 13, 31–40. [Google Scholar]
- Xiang, C.; Cui, S.P.; Ke, Y. miR-144 inhibits cell proliferation of renal cell carcinoma by targeting MTOR. J. Huazhong Univ. Sci. Technol. Med. Sci. 2016, 36, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, L.; Ge, Y.; Zhou, X.; Zhang, A.; Zhang, C.; Zhong, Y.; You, Y.; Pu, P.; Kang, C. miR-221/222 promote malignant progression of glioma through activation of the Akt pathway. Int. J. Oncol. 2010, 36, 913–920. [Google Scholar] [PubMed]
- Chen, H.; Li, L.; Wang, S.; Lei, Y.; Ge, Q.; Lv, N.; Zhou, X.; Chen, C. Reduced miR-126 expression facilitates angiogenesis of gastric cancer through its regulation on VEGF-A. Oncotarget 2014, 5, 11873–11885. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, M.; Nocchi, L.; Staffolani, S.; Manzella, N.; Amati, M.; Goodwin, J.; Kluckova, K.; Nguyen, M.; Strafella, E.; Bajzikova, M.; et al. MicroRNA-126 suppresses mesothelioma malignancy by targeting IRS1 and interfering with the mitochondrial function. Antioxid. Redox Signal. 2014, 21, 2109–2125. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.H.; Chu, T.Y. Repression of miR-126 and upregulation of adrenomedullin in the stromal endothelium by cancer-stromal cross talks confers angiogenesis of cervical cancer. Oncogene 2014, 33, 3636–3647. [Google Scholar] [CrossRef] [PubMed]
- Guay, C.; Regazzi, R. MicroRNAs and the functional β cell mass: For better or worse. Diabetes Metab. 2015, 41, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Hong, J.; Cao, Y.; Shi, J.; Gu, W.; Ning, G.; Zhang, Y.; Wang, W. Elevated circulating microRNA-122 is associated with obesity and insulin resistance in young adults. Eur. J. Endocrinol. 2015, 172, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE 2011, 6, e23937. [Google Scholar] [CrossRef] [PubMed]
- Van der Poorten, D.; George, J. Disease-specific mechanisms of fibrosis: Hepatitis C virus and nonalcoholic steatohepatitis. Clin. Liver Dis. 2008, 12, 805–824. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Bhattacharyya, S.N. Insulin-like growth factor-1 prevents miR-122 production in neighbouring cells to curtail its intercellular transfer to ensure proliferation of human hepatoma cells. Nucleic Acids Res. 2014, 42, 7170–7185. [Google Scholar] [CrossRef] [PubMed]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Sun, F.; Dong, N.; Sun, Z.; Diao, Y.; Zheng, C.; Sun, J.; Yang, Y.; Jiang, D. MicroRNA-7 directly targets insulin-like growth factor 1 receptor to inhibit cellular growth and glucose metabolism in gliomas. Diagn. Pathol. 2014, 9, 211. [Google Scholar] [CrossRef] [PubMed]
- Sayed, D.; Abdellatif, M. AKT-ing via microRNA. Cell Cycle 2010, 9, 3213–3217. [Google Scholar] [CrossRef] [PubMed]
- Popper, H.H. Progression and metastasis of lung cancer. Cancer Metastasis Rev. 2016, 35, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Sha, J.; Jiang, C.; Gao, X.; Qu, L.; Yan, H.; Xu, T.; Jiang, Q.; Gao, H. miR-122 inhibits metastasis and epithelial-mesenchymal transition of non-small-cell lung cancer cells. Oncotargets Ther. 2015, 8, 3175–3184. [Google Scholar] [PubMed]
- Zhang, T.; Guo, P.; Zhang, Y.; Xiong, H.; Yu, X.; Xu, S.; Wang, X.; He, D.; Jin, X. The antidiabetic drug metformin inhibits the proliferation of bladder cancer cells in vitro and in vivo. Int. J. Mol. Sci. 2013, 14, 24603–24618. [Google Scholar] [CrossRef] [PubMed]
- Pulito, C.; Donzelli, S.; Muti, P.; Puzzo, L.; Strano, S.; Blandino, G. microRNAs and cancer metabolism reprogramming: The paradigm of metformin. Ann. Transl. Med. 2014, 2, 58. [Google Scholar] [PubMed]
- Kato, K.; Iwama, H.; Yamashita, T.; Kobayashi, K.; Fujihara, S.; Fujimori, T.; Kamada, H.; Kobara, H.; Masaki, T. The anti-diabetic drug metformin inhibits pancreatic cancer cell proliferation in vitro and in vivo: Study of the microRNAs associated with the antitumor effect of metformin. Oncol. Rep. 2016, 35, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yuan, Y.; Huang, L.; Qiao, M.; Zhang, Y. Metformin alters the expression profiles of microRNAs in human pancreatic cancer cells. Diabetes Res. Clin. Pract. 2012, 96, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Cufí, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Quirantes, R.; Segura-Carretero, A.; Micol, V.; Joven, J.; Bosch-Barrera, J.; Del Barco, S.; Martin-Castillo, B.; et al. Metformin lowers the threshold for stress-induced senescence: A role for the microRNA-200 family and miR-205. Cell Cycle 2012, 11, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Blandino, G.; Valerio, M.; Cioce, M.; Mori, F.; Casadei, L.; Pulito, C.; Sacconi, A.; Biagioni, F.; Cortese, G.; Galanti, S.; et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat. Commun. 2012, 3, 865. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Y.; Xu, B.; Li, L. A New Role for an Old Drug: Metformin Targets MicroRNAs in Treating Diabetes and Cancer. Drug Dev. Res. 2015, 76, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Maida, Y.; Takakura, M.; Nishiuchi, T.; Yoshimoto, T.; Kyo, S. Exosomal transfer of functional small RNAs mediates cancer-stroma communication in human endometrium. Cancer Med. 2016, 5, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Kohlhapp, F.J.; Mitra, A.K.; Lengyel, E.; Peter, M.E. MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene 2015, 34, 5857–5868. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; de Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of cancer-associated fibroblasts by IDH3α downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; de Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Shi, H.; Lin, F.; Dasari, S.; Bednash, J.; Thorne, S.; Watkins, S.; Joshi, R.; Thomas, S.M. Enhancement of head and neck squamous cell carcinoma proliferation, invasion, and metastasis by tumor-associated fibroblasts in preclinical models. Head Neck 2014, 36, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Hatziapostolou, M.; Polytarchou, C.; Aggelidou, E.; Drakaki, A.; Poultsides, G.A.; Jaeger, S.A.; Ogata, H.; Karin, M.; Struhl, K.; Hadzopoulou-Cladaras, M.; et al. An HNF4α-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 2011, 147, 1233–1247. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Yang, H.; He, L.; Zhao, J.J.; Coppola, D.; Dalton, W.S.; Cheng, J.Q. MicroRNA-155 is regulated by the transforming growth factor β/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol. Cell. Biol. 2008, 28, 6773–6784. [Google Scholar] [CrossRef] [PubMed]
- Higgs, G.; Slack, F. The multiple roles of microRNA-155 in oncogenesis. J. Clin. Bioinform. 2013, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Pochampally, R.; Watabe, K.; Lu, Z.; Mo, Y.Y. Exosome-mediated transfer of miR-10b promotes cell invasion in breast cancer. Mol. Cancer 2014, 13, 256. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, L.; Li, D.; Yin, Y.; Zhang, C.Y.; Li, J.; Zhang, Y. Microvesicle-delivery miR-150 promotes tumorigenesis by up-regulating VEGF, and the neutralization of miR-150 attenuate tumor development. Protein Cell 2013, 4, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Pan, J.S.; Jin, L.X.; Wu, J.; Ren, Y.D.; Chen, P.; Xiao, C.; Han, J. MicroRNA-17~92 inhibits colorectal cancer progression by targeting angiogenesis. Cancer Lett. 2016, 376, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Rupaimoole, R.; Shen, F.; Pradeep, S.; Pecot, C.V.; Ivan, C.; Nagaraja, A.S.; Gharpure, K.M.; Pham, E.; Hatakeyama, H.; McGuire, M.H.; et al. A miR-192-EGR1-HOXB9 regulatory network controls the angiogenic switch in cancer. Nat. Commun. 2016, 7, 11169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bai, M.; Deng, T.; Liu, R.; Wang, X.; Qu, Y.; Duan, J.; Zhang, L.; Ning, T.; Ge, S.; et al. Cell-derived microvesicles mediate the delivery of miR-29a/c to suppress angiogenesis in gastric carcinoma. Cancer Lett. 2016, 375, 331–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liu, M.; Wang, C.; Lin, C.; Sun, Y.; Jin, D. Down-regulation of miR-206 promotes proliferation and invasion of laryngeal cancer by regulating VEGF expression. Anticancer Res. 2011, 31, 3859–3863. [Google Scholar] [PubMed]
- Hulsmans, M.; Holvoet, P. MicroRNA-containing microvesicles regulating inflammation in association with atherosclerotic disease. Cardiovasc Res. 2013, 100, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Krause, B.J.; Carrasco-Wong, I.; Dominguez, A.; Arnaiz, P.; Farías, M.; Barja, S.; Mardones, F.; Casanello, P. Micro-RNAs Let7e and 126 in Plasma as Markers of Metabolic Dysfunction in 10 to 12 Years Old Children. PLoS ONE 2015, 10, e0128140. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.S.; Park, S.Y.; Ma, D.; Zhang, J.; Lee, W. The induction of microRNA targeting IRS-1 is involved in the development of insulin resistance under conditions of mitochondrial dysfunction in hepatocytes. PLoS ONE 2011, 6, e17343. [Google Scholar] [CrossRef]
- Zhao, X.; Zhu, D.; Lu, C.; Yan, D.; Li, L.; Chen, Z. MicroRNA-126 inhibits the migration and invasion of endometrial cancer cells by targeting insulin receptor substrate 1. Oncol. Lett. 2016, 11, 1207–1212. [Google Scholar] [PubMed]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, C.; Cheng, J.; Chen, B.; Ke, Q.; Lv, Z.; Wu, J.; Zhou, Y. MicroRNA-145 suppresses hepatocellular carcinoma by targeting IRS1 and its downstream Akt signaling. Biochem. Biophys. Res. Commun. 2014, 446, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Shi, B.; Huang, K.; Fan, G. MicroRNA-128 suppresses cell growth and metastasis in colorectal carcinoma by targeting IRS1. Oncol. Rep. 2015, 34, 2797–2805. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Xu, M.; Chen, X.; Nie, L.; Chen, N.; Gong, J.; Zhang, M.; Su, Z.; Huang, L.; Zhou, Q. miR200c targets IRS1 and suppresses prostate cancer cell growth. Prostate 2015, 75, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhang, F.; Lin, X.; Huang, C.; Zhang, Y.; Li, Y.; Lin, J.; Chen, W.; Lin, X. MicroRNA-1225-5p inhibits proliferation and metastasis of gastric carcinoma through repressing insulin receptor substrate-1 and activation of β-catenin signaling. Oncotarget 2016, 7, 4647–4663. [Google Scholar] [PubMed]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gan, B.; Liu, D.; Paik, J.H. FoxO family members in cancer. Cancer Biol. Ther. 2011, 12, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Seton-Rogers, S. Hypoxia: HIF switch. Nat. Rev. Cancer 2011, 11, 391. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, M.; Monaco, F.; Manzella, N.; Rohlena, J.; Rohlenova, K.; Staffolani, S.; Gaetani, S.; Ciarapica, V.; Amati, M.; Bracci, M.; et al. MicroRNA-126 induces autophagy by altering cell metabolism in malignant mesothelioma. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, H.M.; Holloway, G.P.; Steinberg, G.R. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: Implications for obesity. Mol. Cell. Endocrinol. 2013, 366, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Clark, G.O.; Scherer, P.E.; Orci, L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta 2010, 1801, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Miranda, D.A.; Koves, T.R.; Gross, D.A.; Chadt, A.; Al-Hasani, H.; Cline, G.W.; Schwartz, G.J.; Muoio, D.M.; Silver, D.L. Re-patterning of skeletal muscle energy metabolism by fat storage-inducing transmembrane protein 2. J. Biol. Chem. 2011, 286, 42188–42199. [Google Scholar] [CrossRef] [PubMed]
- Ivashov, V.A.; Zellnig, G.; Grillitsch, K.; Daum, G. Identification of triacylglycerol and steryl ester synthases of the methylotrophic yeast Pichia pastoris. Biochim. Biophys. Acta 2013, 1831, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Suzuki, J.; Zenimaru, Y.; Takahashi, S.; Koizumi, T.; Noriki, S.; Yamaguchi, O.; Otsu, K.; Shen, W.J.; Kraemer, F.B.; et al. Cardiac overexpression of hormone-sensitive lipase inhibits myocardial steatosis and fibrosis in streptozotocin diabetic mice. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E1109–E1118. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Zhang, J.; Choi, A.M.; Kim, H.P. Mitochondrial dysfunction induces formation of lipid droplets as a generalized response to stress. Oxid. Med. Cell. Longev. 2013, 2013, 327167. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| miRNA | Target Gene | Function | Cancer Hallmarks | Ref. |
|---|---|---|---|---|
| miR-199a | HIF1α | HIF inhibition | Hypoxia | [47] |
| miR-92-1 | VHL | HIF stabilization | Hypoxia | [48] |
| miR-20a/b | VHL | HIF stabilization | Hypoxia | [49] |
| miR-21 | VHL | HIF stabilization | Hypoxia | [49] |
| miR-22 | VHL | HIF stabilization | Hypoxia | [49] |
| miR-101 | VHL | HIF stabilization | Hypoxia | [49] |
| miR-106a/b | VHL | HIF stabilization | Hypoxia | [49] |
| miR-150 | VHL | HIF stabilization | Hypoxia | [49] |
| miR-200b | VHL | HIF stabilization | Hypoxia | [49] |
| miR-424 | Cullin-2 | HIF stabilization | Hypoxia | [50] |
| miR-210 | GPD1L | HIF stabilization | Hypoxia | [51] |
| miR-23a/b | GLS | Glutaminolysis inhibition | Metabolism | [52] |
| Let-7a | c-MYC | MYC pathway | Insulin pathway | [53] |
| RAS | PI3K/Akt signaling | [54] | ||
| p53 | p53 signaling | [22] | ||
| IGF-IR | Insulin/IGF-IR | [80] | ||
| miR-9 | E-cadherin | Induced by c-MYC | Insulin pathway | [55] |
| miR-34 | LDHA, MYC, SIRT1 | p53 signaling | Apoptosis | [57] |
| miR-194/miR-215 | p53 signaling | Apoptosis/cell cycle arrest | [56] | |
| miR-107 | CDKN1A/p21, HIF | p53 signaling | Hypoxia | [22] |
| miR-17-92 | KRAS | Suppressed by p53 | Proliferation | [58] |
| miR-16-1 | CDK6 | Suppressed by p53 | Cell cycle arrest | [59] |
| miR-143 | Suppressed by p53 | [59] | ||
| miR-145 | Suppressed by p53 | [59] | ||
| miR-25 | p53 | Suppress p53 signaling | Proliferation/cell cycle arrest | [60] |
| miR-30d | p53 | Suppress p53 signaling | [60] | |
| miR-504 | p53 | Suppress p53 signaling | [60] | |
| miR-125b | p53 | Suppress p53 signaling | [60] | |
| miR-372 | p53 | Suppress p53 signaling | [61] | |
| miR-373 | p53 | Suppress p53 signaling | [61] | |
| miR-326 | PKM2 | AMPK signaling | Metabolism | [62] |
| miR-451 | CAB9/miR-195 | LKB1/AMPK signaling | Proliferation | [63] |
| miR-33a/b | AMPK, IRS2 | AMPK/FOXO1 signaling | Glucose metabolism | [38] |
| miR-21 | PTEN, HIF, VEGF | PI3K/Akt activation | Proliferation/angiogenesis | [65] |
| miR-337 | PTEN | PI3K/Akt signaling | [66] | |
| miR-543 | PTEN | PI3K/Akt signaling | [67] | |
| miR-214 | PTEN | PI3K/Akt signaling | [68] | |
| miR-130 | PTEN | PI3K/Akt signaling | [69] | |
| miR-181 | PTEN | PI3K/Akt signaling | [70] | |
| miR-221/222 | PTEN | PI3K/Akt signaling | [75] | |
| miR-26a | PTEN | PI3K/Akt signaling | Metastasis/angiogenesis | [71] |
| miR-144 | mTOR | PI3K/Akt/mTOR signaling | Proliferation/cell cycle arrest | [73] |
| miR-126 | IRS1, VEGF | PI3K/Akt/mTOR signaling | Proliferation/angiogenesis | [76,77,78] |
| miR-7 | IGF-IR | Insulin/IGF-IR signaling | Insulin secretion | [79,85] |
| miR-375 | IGF-IR | Insulin/IGF-IR signaling | [79] | |
| let-7 | IGF-IR | Insulin/IGF-IR signaling | [79] | |
| miR-122 | IGF-IR | IGF-IR/PI3K/Akt | metastasis | [41,80] |
| miR-320 | IGF-IR | Insulin/IGF-IR signaling | [86] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomasetti, M.; Amati, M.; Santarelli, L.; Neuzil, J. MicroRNA in Metabolic Re-Programming and Their Role in Tumorigenesis. Int. J. Mol. Sci. 2016, 17, 754. https://doi.org/10.3390/ijms17050754
Tomasetti M, Amati M, Santarelli L, Neuzil J. MicroRNA in Metabolic Re-Programming and Their Role in Tumorigenesis. International Journal of Molecular Sciences. 2016; 17(5):754. https://doi.org/10.3390/ijms17050754
Chicago/Turabian StyleTomasetti, Marco, Monica Amati, Lory Santarelli, and Jiri Neuzil. 2016. "MicroRNA in Metabolic Re-Programming and Their Role in Tumorigenesis" International Journal of Molecular Sciences 17, no. 5: 754. https://doi.org/10.3390/ijms17050754
APA StyleTomasetti, M., Amati, M., Santarelli, L., & Neuzil, J. (2016). MicroRNA in Metabolic Re-Programming and Their Role in Tumorigenesis. International Journal of Molecular Sciences, 17(5), 754. https://doi.org/10.3390/ijms17050754