
Case Characterization, Clinical Features and Risk Factors in Drug-Induced Liver Injury



Abstract
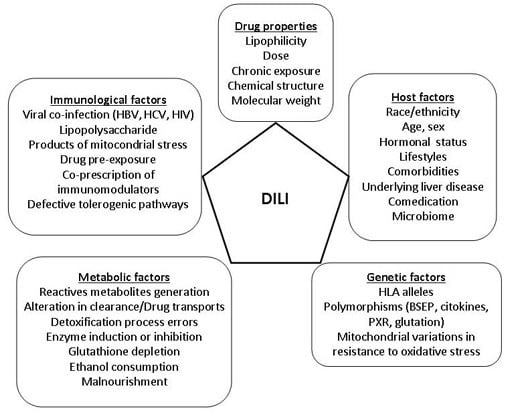
:
1. Introduction
2. Case Characterization
3. Clinical Features
4. Risk Factors
4.1. Drug Factors
4.1.1. Drug Dose and Lipophilicity
4.1.2. Reactive Metabolites and Oxidative Stress
4.1.3. Mitochondrial Hazards
4.1.4. Hepatobiliary Transporter Inhibition
4.2. Host Factors
4.2.1. Age
4.2.2. Gender
4.2.3. Race
4.2.4. Underlying Liver Disease
4.2.5. Comorbidities
4.2.6. Drug-Drug Interactions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DILI | Drug-Induced Liver Injury |
| GWAs | Genome-wide association study |
| HDS | Herbal and Diet Supplements |
| ALF | Acute liver failure |
| DILIN | Drug Induced Liver Injury Network |
| ALT | Alanin-aminotransferase |
| ULN | Upper limit of normal |
| ALP | Alkaline Phosphatase |
| TB | Total Bilirrubin |
| AST | Aspartate-aminotransferase |
| OLT | Orthotopic liver transplantation |
| GGT | G-glutamyl transpeptidase |
| AIH | Autoimmune hepatitis |
| RUCAM | Roussel Uclaf Causality Assessment Method |
| CTLA-4 | Cytotoxic-T lymphocyte A-4 |
| PD-1 | Programed death-1 |
| SOS | Sinusoidal obstruction syndrome |
| HLA | Human Leukocyte Antigen |
| DAMPS | Damage Associated Molecular Patterns |
| FDA | Food and Drug Administration |
| ROS | Reactive oxygen species |
| mtDNA | Mitochondrial deoxyribonucleic acid |
| BSEP | Bile salt export pump |
| MRP | Multidrug resistance-associated protein |
| HIV | Human immunodeficiency virus |
| HBV | Hepatitis B virus |
| HCV | Hepatitis C virus |
| NAFLD | Non-alcoholic fatty liver disease |
| CYP | Cytochrome |
| GPRD | General Practice Research Database |
References
- Andrade, R.J.; Lucena, M.I.; Kaplowitz, N.; García-Muñoz, B.; Borraz, Y.; Pachkoria, K.; García-Cortés, M.; Fernández, M.C.; Pelaez, G.; Rodrigo, L.; et al. Outcome of acute idiosyncratic drug-induced liver injury: Long-term follow-up in a hepatotoxicity registry. Hepatology 2006, 44, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Andrade, R.J.; Lucena, M.I.; Fernández, M.C.; Pelaez, G.; Pachkoria, K.; García-Ruiz, E.; García-Muñoz, B.; González-Grande, R.; Pizarro, A.; Durán, J.A.; et al. Drug-induced liver injury: An analysis of 461 incidences submitted to the Spanish registry over a 10-year period. Gastroenterology 2005, 129, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Navarro, V.J.; Seeff, L.B. Liver injury induced by herbal complementary and alternative medicine. Clin. Liver Dis. 2013, 17, 715–735. [Google Scholar] [CrossRef] [PubMed]
- Sgro, C.; Clinard, F.; Ouazir, K.; Chanay, H.; Allard, C.; Guilleminet, C.; Lenoir, C.; Lemoine, A.; Hillon, P. Incidence of drug-induced hepatic injuries: A French population-based study. Hepatology 2002, 36, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, E.S.; Bergmann, O.M.; Bjornsson, H.K.; Kvaran, R.B.; Olafsson, S. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology 2013, 144, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Bonkovsky, H.L.; Fontana, R.; Lee, W.; Stolz, A.; Talwalkar, J.; Reddy, K.R.; Watkins, P.B.; Navarro, V.; Barnhart, H.; et al. Features and Outcomes of 899 Patients With Drug-Induced Liver Injury: The DILIN Prospective Study. Gastroenterology 2015, 148, 1340–1352. [Google Scholar] [CrossRef] [PubMed]
- Suk, K.T.; Kim, D.J.; Kim, C.H.; Park, S.H.; Yoon, J.H.; Kim, Y.S.; Baik, G.H.; Kim, J.B.; Kweon, Y.O.; Kim, B.I.; et al. A prospective nationwide study of drug-induced liver injury in Korea. Am. J. Gastroenterol. 2012, 107, 1380–1387. [Google Scholar] [PubMed]
- Andrade, R.J.; López-Ortega, S.; López-Vega, M.C.; Robles, M.; Cueto, I.; Lucena, M.I. Idiosyncratic drug hepatotoxicity: A 2008 update. Expert Rev. Clin. Pharmacol. 2008, 1, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Ostapowicz, G.; Fontana, R.J.; Schiødt, F.V.; Larson, A.; Davern, T.J.; Han, S.H.; McCashland, T.M.; Shakil, A.O.; Hay, H.E.; Hynan, L.; et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann. Intern. Med. 2002, 137, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Reuben, A.; Koch, D.G.; Lee, W.M. Group ALFS. Drug-induced acute liver failure: Results of a U.S. multicenter, prospective study. Hepatology 2010, 52, 2065–2076. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, P.H.; Fontana, R.J. Clinical features, diagnosis, and natural history of drug-induced liver injury. Semin. Liver Dis. 2014, 34, 134–144. [Google Scholar] [PubMed]
- Aithal, G.P.; Watkins, P.B.; Andrade, R.J.; Larrey, D.; Molokhia, M.; Takikawa, H.; Hunt, C.M.; Wilke, R.A.; Avigan, M.; Kaplowitz, N.; et al. Case definition and phenotype standardization in drug-induced liver injury. Clin. Pharmacol. Ther. 2011, 89, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Robles-Diaz, M.; Garcia-Cortes, M.; Medina-Caliz, I.; González-Jiménez, A.; González-Grande, R.; Navarro, J.M.; Castiella, A.; Zapata, E.M.; Romero-Gómez, M.; Blanco, S.; et al. The value of serum aspartate aminotransferase and gamma-glutamyl transpetidase as biomarkers in hepatotoxicity. Liver Int. 2015, 35, 2474–2482. [Google Scholar] [CrossRef] [PubMed]
- Robles-Diaz, M.; Lucena, M.I.; Kaplowitz, N.; Stephens, C.; Medina-Cáliz, I.; González-Jiménez, A.; Ulzurrun, E.; González, A.F.; Fernández, M.C.; Romero-Gómez, M.; et al. Use of Hy’s Law and a New Composite Algorithm to Predict Acute Liver Failure in Patients With Drug-Induced Liver Injury. Gastroenterology 2014, 147, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Chalasani, N.P.; Lee, W.M.; Fontana, R.J.; Bonkovsky, H.L.; Watkins, P.B.; Hayashi, P.H.; Davern, T.J.; Navarro, V.; Reddy, R.; et al. Hepatic histological findings in suspected drug-induced liver injury: Systematic evaluation and clinical associations. Hepatology 2014, 59, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.J.; Watkins, P.B.; Bonkovsky, H.L.; Chalasani, N.; Davern, T.; Serrano, J.; Rochon, J.; DILIN Study Group. Drug-Induced Liver Injury Network (DILIN) prospective study: Rationale, design and conduct. Drug Saf. 2009, 32, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, H.J. The spectrum of hepatotoxicity. Perspect. Biol. Med. 1968, 12, 135–161. [Google Scholar] [CrossRef] [PubMed]
- FDA. Draft Guidance for Industry. Drug-Induced Liver Injury: Premarketing Clinical Evaluation. Available online: https://www.federalregister.gov/articles/2009/07/30/E9-18135/guidance-for-industry-on-drug-induced-liver-injury-premarketing-clinical-evaluation-availability (accessed on 16 February 2016).
- Pachkoria, K.; Lucena, M.I.; Crespo, E.; Ruiz-Cabello, F.; López-Ortega, S.; Fernández, M.A.; Romero-Gómez, M.; Madrazo, A.; Durán, J.A.; de Dios, A.M.; et al. Analysis of IL-10, IL-4 and TNF-α polymorphisms in drug-induced liver injury (DILI) and its outcome. J. Hepatol. 2008, 49, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Andrade, R.J.; Robles, M.; Fernández-Castañer, A.; López-Ortega, S.; López-Vega, M.C.; Lucena, M.I. Assessment of drug-induced hepatotoxicity in clinical practice: A challenge for gastroenterologists. World J. Gastroenterol. 2007, 13, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, H.J. Hepatotoxicity: The Adverse Effects of Drugs and Other Chemicals on the Liver, 2nd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 1999. [Google Scholar]
- Fisher, K.; Vuppalanchi, R.; Saxena, R. Drug-Induced Liver Injury. Arch. Pathol. Lab. Med. 2015, 139, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Kakar, S. Histological patterns in drug-induced liver disease. J. Clin. Pathol. 2009, 62, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.J. Pathogenesis of idiosyncratic drug-induced liver injury and clinical perspectives. Gastroenterology 2014, 146, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Goodman, Z.D. Drug hepatotoxicity. Clin. Liver Dis. 2002, 6, 381–397. [Google Scholar] [CrossRef]
- Larrey, D. Drug-induced liver diseases. J. Hepatol. 2000, 32, 77–88. [Google Scholar] [CrossRef]
- Lucena, M.I.; Andrade, R.J.; Kaplowitz, N.; García-Cortés, M.; Fernández, M.C.; Romero-Gómez, M.; Bruguera, M.; Hallal, H.; Robles-Díaz, M.; Rodríguez-González, J.F.; et al. Phenotypic characterization of idiosyncratic drug-induced liver injury: The influence of age and sex. Hepatology 2009, 49, 2001–2009. [Google Scholar] [CrossRef] [PubMed]
- Robles-Diaz, M.; Gonzalez-Jimenez, A.; Medina-Caliz, I.; Stephens, C.; García-Cortés, M.; García-Muñoz, B.; Ortega-Alonso, A.; Blanco-Reina, E.; González-Grande, R.; Jiménez-Pérez, M.; et al. Distinct phenotype of hepatotoxicity associated with illicit use of anabolic androgenic steroids. Aliment. Pharmacol. Ther. 2015, 41, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.A.; Vuppalanchi, R.; Fontana, R.J.; Stolz, A.; Kleiner, D.E.; Hayashi, P.H.; Gu, J.; Hoofnagle, J.H.; Chalasani, N. Clinical and histologic features of azithromycin-induced liver injury. Clin. Gastroenterol. Hepatol. 2015, 13, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Orman, E.S.; Conjeevaram, H.S.; Vuppalanchi, R.; Freston, J.W.; Rochon, J.; Kleiner, D.E.; Hayashi, P.H.; DILIN Research Group. Clinical and histopathologic features of fluoroquinolone-induced liver injury. Clin. Gastroenterol. Hepatol. 2011, 9, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, E.S.; Jonasson, J.G. Drug-induced cholestasis. Clin. Liver Dis. 2013, 17, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Benichou, C. Criteria of drug-induced liver disorders. Report of an international consensus meeting. J. Hepatol. 1990, 11, 272–276. [Google Scholar] [PubMed]
- Medina-Cáliz, I.; Robles-Díaz, M.; García-Muñoz, B.; Stephens, C.; Ortega-Alonso, A.; García-Cortés, M.; González-Jiménez, A.; Sanabria-Cabrera, J.; Moreno, I.; Fernández, C.; et al. Definition, incidence and risk factors for chronicity following acute idiosyncratic drug-induced licer injury. J. Hepatol. 2016, in press. [Google Scholar]
- Delemos, A.S.; Foureau, D.M.; Jacobs, C.; Ahrens, W.; Russo, M.W.; Bonkovsky, H.L. Drug-induced liver injury with autoimmune features. Semin. Liver Dis. 2014, 34, 194–204. [Google Scholar] [PubMed]
- Castiella, A.; Zapata, E.; Lucena, M.I.; Andrade, R.J. Drug-induced autoimmune liver disease: A diagnostic dilemma of an increasingly reported disease. World J. Hepatol. 2014, 6, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Danan, G.; Teschke, R. RUCAM in Drug and Herb Induced Liver Injury: The Update. Int. J. Mol. Sci. 2015, 17. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, E.; Talwalkar, J.; Treeprasertsuk, S.; Kamath, P.S.; Takahashi, N.; Sanderson, S.; Neuhauser, M.; Lindor, K. Drug-induced autoimmune hepatitis: Clinical characteristics and prognosis. Hepatology 2010, 51, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Foureau, D.M.; Walling, T.L.; Maddukuri, V.; Anderson, W.; Cullbreath, K.; Kleiner, D.E.; Ahrens, W.A.; Jacobs, C.; Watkins, P.B.; Chalasani, N.; et al. Comparative analysis of portal hepatic infiltrating leucocytes in acute drug-induced liver injury, idiopathic autoimmune and viral hepatitis. Clin. Exp. Immunol. 2015, 180, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, S.; Lopes, S.; Magro, F.; Cardoso, H.; Horta e Vale, A.M.; Marques, M.; Mariz, E.; Bernardes, M.; Lopes, J.; Carneiro, F.; et al. Autoimmune hepatitis and anti-tumor necrosis factor α therapy: A single center report of 8 cases. World J. Gastroenterol. 2015, 21, 7584–7588. [Google Scholar] [CrossRef] [PubMed]
- Perdices, E.V.; Medina-Cáliz, I.; Hernando, S.; Ortega, A.; Martín-Ocaña, F.; Navarro, J.M.; Peláez, G.; Castiella, A.; Hallal, H.; Romero-Gómez, M.; et al. Hepatotoxicity associated with statin use: Analysis of the cases included in the Spanish Hepatotoxicity Registry. Rev. Esp. Enferm. Dig. 2014, 106, 246–254. [Google Scholar] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Voskens, C.J.; Goldinger, S.M.; Loquai, C.; Robert, C.; Kaehler, K.C.; Berking, C.; Bergmann, T.; Bockmeyer, C.L.; Eigentler, T.; Fluck, M.; et al. The price of tumor control: An analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS ONE 2013, 8, e53745. [Google Scholar] [CrossRef] [PubMed]
- Goekkurt, E.; Stoehlmacher, J.; Stueber, C.; Wolschke, C.; Eiermann, T.; Iacobelli, S.; Zander, A.R.; Ehninger, G.; Kröger, N. Pharmacogenetic analysis of liver toxicity after busulfan/cyclophosphamide-based allogeneic hematopoietic stem cell transplantation. Anticancer Res. 2007, 27, 4377–4380. [Google Scholar] [PubMed]
- Andrade, R.J.; Robles, M.; Ulzurrun, E.; Lucena, M.I. Drug-induced liver injury: Insights from genetic studies. Pharmacogenomics 2009, 10, 1467–1487. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.K.; Donaldson, P.T.; Bhatnagar, P.; Shen, Y.; Pe’er, I.; Floratos, A.; Daly, M.J.; Goldstein, D.B.; John, S.; Nelson, M.R.; et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 2009, 41, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Molokhia, M.; Shen, Y.; Urban, T.J.; Aithal, G.P.; Andrade, R.J.; Day, C.P.; Ruiz-Cabello, F.; Donaldson, P.T.; Stephens, C.; et al. Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology 2011, 141, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.B.; Lewitzky, S.; Leroy, E.; Yang, F.; Zhao, X.; Klickstein, L.; Wright, T.M.; Meyer, J.; Paulding, C.A. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat. Genet. 2010, 42, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Alfirevic, A.; Gonzalez-Galarza, F.; Bell, C.; Martinsson, K.; Platt, V.; Bretland, G.; Evely, J.; Lichtenfels, M.; Cederbrant, K.; French, N.; et al. In silico analysis of HLA associations with drug-induced liver injury: Use of a HLA-genotyped DNA archive from healthy volunteers. Genome Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Schaid, D.J.; Spraggs, C.F.; McDonnell, S.K.; Parham, L.R.; Cox, C.J.; Ejlertsen, B.; Finkelstein, D.M.; Rappold, E.; Curran, J.; Cardon, L.R.; et al. Prospective validation of HLA-DRB1*07:01 allele carriage as a predictive risk factor for lapatinib-induced liver injury. J. Clin. Oncol. 2014, 32, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.; Nicoletti, P.; Bjornsson, E.; Lucena, M.I.; Andrade, R.J.; Grove, J.; Stephens, C.; Hallberg, P.; Maitland-van der Zee, A.H.; Martin, J.H.; et al. HLA-A*33:01 is strongly associated with drug-induced liver injury (DILI) due to terbinafine and several other unrelated compounds. Hepatology 2015, 65 (Suppl. 1), 325A–326A. [Google Scholar]
- Stephens, C.; Andrade, R.J.; Lucena, M.I. Mechanisms of drug-induced liver injury. Curr. Opin. Allergy Clin. Immunol. 2014, 14, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Suzuki, A.; Borlak, J.; Andrade, R.J.; Lucena, M.I. Drug-induced liver injury: Interactions between drug properties and host factors. J. Hepatol. 2015, 63, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Dara, L.; Liu, Z.; Kaplowitz, N. Mechanisms of Adaptation and Progression in Idiosyncratic Drug Induced Liver Injury, Clinical Implications. Liver Int. 2016, 32, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.H.; Ju, C.; Ramaiah, S.K.; Uetrecht, J.; Jaeschke, H. Mechanisms of immune-mediated liver injury. Toxicol. Sci. 2010, 115, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Uetrecht, J.P. New concepts in immunology relevant to idiosyncratic drug reactions: The “danger hypothesis” and innate immune system. Chem. Res. Toxicol. 1999, 12, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Lammert, C.; Einarsson, S.; Saha, C.; Niklasson, A.; Bjornsson, E.; Chalasani, N. Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: Search for signals. Hepatology 2008, 47, 2003–2009. [Google Scholar] [CrossRef] [PubMed]
- Lammert, C.; Bjornsson, E.; Niklasson, A.; Chalasani, N. Oral medications with significant hepatic metabolism at higher risk for hepatic adverse events. Hepatology 2010, 51, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Carrascosa, M.F.; Salcines-Caviedes, J.R.; Lucena, M.I.; Andrade, R.J. Acute liver failure following atorvastatin dose escalation: Is there a threshold dose for idiosyncratic hepatotoxicity? J. Hepatol. 2015, 62, 751–752. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; Decrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Borlak, J.; Tong, W. High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology 2013, 58, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Wang, K.; Li, H.; Shi, Q. A comprehensive study of the association between drug hepatotoxicity and daily dose, liver metabolism, and lipophilicity using 975 oral medications. Oncotarget 2015, 6, 17031–17038. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Laverty, H.; Srivastava, A.; Antoine, D.J.; Naisbitt, D.; Williams, D.P. Drug bioactivation and protein adduct formation in the pathogenesis of drug-induced toxicity. Chem. Biol. Interact. 2011, 192, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Stepan, A.F.; Walker, D.P.; Bauman, J.; Price, D.A.; Baillie, T.A.; Kalgutkar, A.S.; Aleo, M.D. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: A perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 2011, 24, 1345–1410. [Google Scholar] [CrossRef] [PubMed]
- Obach, R.S.; Kalgutkar, A.S.; Soglia, J.R.; Zhao, S.X. Can in vitro metabolism-dependent covalent binding data in liver microsomes distinguish hepatotoxic from nonhepatotoxic drugs? An analysis of 18 drugs with consideration of intrinsic clearance and daily dose. Chem. Res. Toxicol. 2008, 21, 1814–1822. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.N.; Kelly, J.M.; Tripathy, S.; Zhao, S.X.; Lam, W.W.; Kalgutkar, A.S.; Obach, R.S. Can in vitro metabolism-dependent covalent binding data distinguish hepatotoxic from nonhepatotoxic drugs? An analysis using human hepatocytes and liver S-9 fraction. Chem. Res. Toxicol. 2009, 22, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.J.; Henstock, P.V.; Dunn, M.C.; Smith, A.R.; Chabot, J.R.; de Graaf, D. Cellular imaging predictions of clinical drug-induced liver injury. Toxicol. Sci. 2008, 105, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.; Nie, A.; Brandon Parker, J.; Sawant, S.; Piechta, L.A.; Kelley, M.F.; Maark Kao, L.; Jim Proctor, S.; Verheyen, G.; Johnson, M.D.; et al. Oxidative stress/reactive metabolite gene expression signature in rat liver detects idiosyncratic hepatotoxicants. Toxicol. Appl. Pharmacol. 2014, 275, 189–97. [Google Scholar] [CrossRef] [PubMed]
- Pulkes, T.; Hanna, M.G. Human mitochondrial DNA diseases. Adv. Drug Deliv. Rev. 2001, 49, 27–43. [Google Scholar] [CrossRef]
- Boelsterli, U.A.; Lim, P.L. Mitochondrial abnormalities—A link to idiosyncratic drug hepatotoxicity? Toxicol. Appl. Pharmacol. 2007, 220, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Porceddu, M.; Buron, N.; Roussel, C.; Labbe, G.; Fromenty, B.; Borgne-Sanchez, A. Prediction of liver injury induced by chemicals in human with a multiparametric assay on isolated mouse liver mitochondria. Toxicol. Sci. 2012, 129, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Dykens, J.A.; Will, Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov. Today 2007, 12, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Labbe, G.; Pessayre, D.; Fromenty, B. Drug-induced liver injury through mitochondrial dysfunction: Mechanisms and detection during preclinical safety studies. Fundam. Clin. Pharmacol. 2008, 22, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Tung, C.W.; Shi, Q.; Guo, L.; Shi, L.; Fang, H.; Borlak, J.; Tong, W. A testing strategy to predict risk for drug-induced liver injury in humans using high-content screen assays and the “rule-of-two” model. Arch. Toxicol. 2014, 88, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; García-Martín, E.; Andrade, R.J.; Martínez, C.; Stephens, C.; Ruiz, J.D.; Ulzurrun, E.; Fernández, M.C.; Romero-Gómez, M.; Castiella, A.; et al. Mitochondrial superoxide dismutase and glutathione peroxidase in idiosyncratic drug-induced liver injury. Hepatology 2010, 52, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Dara, L.; Win, S.; Than, T.A.; Yuan, L.; Abbasi, S.Q.; Liu, Z.X.; Kaplowitz, N. Regulation of drug-induced liver injury by signal transduction pathways: Critical role of mitochondria. Trends Pharmacol. Sci. 2013, 34, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Ulzurrun, E.; Stephens, C.; Crespo, E.; Ruiz-Cabello, F.; Ruiz-Nuñez, J.; Saenz-López, P.; Moreno-Herrera, I.; Robles-Diaz, M.; Hallal, M.; Moreno-Planas, J.M.; et al. Role of chemical structures and the 1331T>C bile salt export pump polymorphism in idiosyncratic drug-induced liver injury. Liver Int. 2013, 33, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.E.; Trauner, M.; van Staden, C.J.; Lee, P.H.; Ramachandran, B.; Eschenberg, M.; Afshari, C.A.; Qualls, C.W., Jr.; Lightfoot-Dunn, R.; Hamadeh, H.K. Interference with bile salt export pump function is a susceptibility factor for human liver injury in drug development. Toxicol. Sci. 2010, 118, 485–500. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.; Stahl, S.; Paul, N.; Barber, J.; Kenna, J.G. In vitro inhibition of the bile salt export pump correlates with risk of cholestatic drug-induced liver injury in humans. Drug Metab. Dispos. 2012, 40, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Pérez, L.M.; Milkiewicz, P.; Elias, E.; Coleman, R.; Sánchez Pozzi, E.J.; Roma, M.G. Oxidative stress induces internalization of the bile salt export pump, Bsep, and bile salt secretory failure in isolated rat hepatocyte couplets: A role for protein kinase C and prevention by protein kinase A. Toxicol. Sci. 2006, 91, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Aleo, M.D.; Luo, Y.; Swiss, R.; Bonin, P.D.; Potter, D.M.; Will, Y. Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology 2014, 60, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Geier, A.; Wagner, M.; Dietrich, C.G.; Trauner, M. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regeneration. Biochim. Biophys. Acta 2007, 1773, 283–308. [Google Scholar] [CrossRef] [PubMed]
- Köck, K.; Ferslew, B.C.; Netterberg, I.; Yang, K.; Urban, T.J.; Swaan, P.W.; Stewart, P.W.; Brouwer, K.L. Risk factors for development of cholestatic drug-induced liver injury: Inhibition of hepatic basolateral bile acid transporters multidrug resistance-associated proteins 3 and 4. Drug Metab. Dispos. 2014, 42, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.E.; van Staden, C.J.; Chen, Y.; Kalyanaraman, N.; Kalanzi, J.; Dunn, R.T., 2nd.; Afshari, C.A.; Hamadeh, H.K. A multifactorial approach to hepatobiliary transporter assessment enables improved therapeutic compound development. Toxicol. Sci. 2013, 136, 216–241. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Hilmer, S.N. Drug-induced liver injury in older adults. Ther. Adv. Drug Saf. 2010, 1, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Felker, D.; Lynn, A.; Wang, S.; Johnson, D.E. Evidence for a potential protective effect of carnitine-pantothenic acid co-treatment on valproic acid-induced hepatotoxicity. Expert Rev. Clin. Pharmacol. 2014, 7, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Fountain, F.F.; Tolley, E.; Chrisman, C.R.; Self, T.H. Isoniazid hepatotoxicity associated with treatment of latent tuberculosis infection: A 7-year evaluation from a public health tuberculosis clinic. Chest 2005, 128, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Heubi, J.E.; Partin, J.C.; Partin, J.S.; Schubert, W.K. Reye’s syndrome: Current concepts. Hepatology 1987, 7, 155–164. [Google Scholar] [CrossRef] [PubMed]
- De Abajo, F.J.; Montero, D.; Madurga, M.; García Rodríguez, L.A. Acute and clinically relevant drug-induced liver injury: A population based case-control study. Br. J. Clin. Pharmacol. 2004, 58, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Larrey, D. Epidemiology and individual susceptibility to adverse drug reactions affecting the liver. Semin. Liver Dis. 2002, 22, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.J.; Hayashi, P.H.; Gu, J.; Reddy, K.R.; Barnhart, H.; Watkins, P.B.; Serrano, J.; Lee, W.M.; Chalasani, N.; Stolz, A.; et al. Idiosyncratic drug-induced liver injury is associated with substantial morbidity and mortality within 6 months from onset. Gastroenterology 2014, 147, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Aceti, A.; Pasquazzi, C.; Zechini, B.; de Bac, C.; Group, L. Hepatotoxicity development during antiretroviral therapy containing protease inhibitors in patients with HIV: The role of hepatitis B and C virus infection. J. Acquir. Immune Defic. Syndr. 2002, 29, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Servoss, J.C.; Kitch, D.W.; Andersen, J.W.; Reisler, R.B.; Chung, R.T.; Robbins, G.K. Predictors of antiretroviral-related hepatotoxicity in the adult AIDS Clinical Trial Group (1989–1999). J. Acquir. Immune Defic. Syndr. 2006, 43, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, M.S.; Thomas, D.L.; Mehta, S.H.; Chaisson, R.E.; Moore, R.D. Hepatotoxicity associated with nevirapine or efavirenz-containing antiretroviral therapy: Role of hepatitis C and B infections. Hepatology 2002, 35, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.C.; Lee, C.H.; Lee, M.C.; Wang, J.Y.; Yu, C.J.; Lee, L.N. Hepatotoxicity due to first-line anti-tuberculosis drugs: A five-year experience in a Taiwan medical centre. Int. J. Tuberc. Lung Dis 2013, 17, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.M.; Cheng, Y.J.; Li, Y.L.; Liu, C.E.; Hsu, W.H. Antituberculosis treatment and hepatotoxicity in patients with chronic viral hepatitis. Lung 2014, 192, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Liu, C.H.; Hu, F.C.; Chang, H.C.; Liu, J.L.; Chen, J.M.; Yu, C.J.; Lee, L.N.; Kao, J.H.; Yang, P.C. Risk factors of hepatitis during anti-tuberculous treatment and implications of hepatitis virus load. J. Infect. 2011, 62, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Lomtadze, N.; Kupreishvili, L.; Salakaia, A.; Vashakidze, S.; Sharvadze, L.; Kempker, R.R.; Magee, M.J.; del Rio, C.; Blumberg, H.M. Hepatitis C virus co-infection increases the risk of anti-tuberculosis drug-induced hepatotoxicity among patients with pulmonary tuberculosis. PLoS ONE 2013, 8, e83892. [Google Scholar] [CrossRef] [PubMed]
- Ungo, J.R.; Jones, D.; Ashkin, D.; Hollender, E.S.; Bernstein, D.; Albanese, A.P.; Pitchenik, A.E. Antituberculosis drug-induced hepatotoxicity. The role of hepatitis C virus and the human immunodeficiency virus. Am. J. Respir. Crit. Care Med. 1998, 157, 1871–1876. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Andrade, R.J. Drug-induced liver injury: Expanding our knowledge by enlarging population analysis with prospective and scoring causality assessment. Gastroenterology 2015, 148, 1271–1273. [Google Scholar] [CrossRef] [PubMed]
- Tilling, L.; Townsend, S.; David, J. Methotrexate and hepatic toxicity in rheumatoid arthritis and psoriatic arthritis. Clin. Drug Investig. 2006, 26, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Amital, H.; Arnson, Y.; Chodick, G.; Shalev, V. Hepatotoxicity rates do not differ in patients with rheumatoid arthritis and psoriasis treated with methotrexate. Rheumatology (Oxford) 2009, 48, 1107–1110. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P. Hepatotoxicity related to antirheumatic drugs. Nat. Rev. Rheumatol. 2011, 7, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Medina-Cáliz, I.; Garcia-Muñoz, B.; Robles Diaz, M.; Stephens, C.; González-Jiménez, A.; García-Cortés, M.; Ortega, A.; Hidalgo, R.; Fernández, M.C.; Romero-Gómez, M.; et al. Risk factors for chronicity in idiosyncratic drug-induced liver injury (DILI). Hepatology 2014, 60 (Suppl. 4), 139A. [Google Scholar]
- Lucena, M.I.; Andrade, R.J.; Vicioso, L.; González, F.J.; Pachkoria, K.; García-Muñoz, B. Prolonged cholestasis after raloxifene and fenofibrate interaction: A case report. World J. Gastroenterol. 2006, 12, 5244–5246. [Google Scholar] [PubMed]
- Björnsson, E.; Jacobsen, E.I.; Kalaitzakis, E. Hepatotoxicity associated with statins: Reports of idiosyncratic liver injury post-marketing. J. Hepatol. 2012, 56, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P. Pharmacogenetic testing in idiosyncratic drug-induced liver injury: Current role in clinical practice. Liver Int. 2015, 35, 1801–1808. [Google Scholar] [CrossRef] [PubMed]
- Andrade, R.J. Reducing Risk of Severe Liver Injury in Patients Treated With Isoniazid. Clin. Gastroenterol. Hepatol. 2015, 13, 1683–1685. [Google Scholar] [CrossRef] [PubMed]
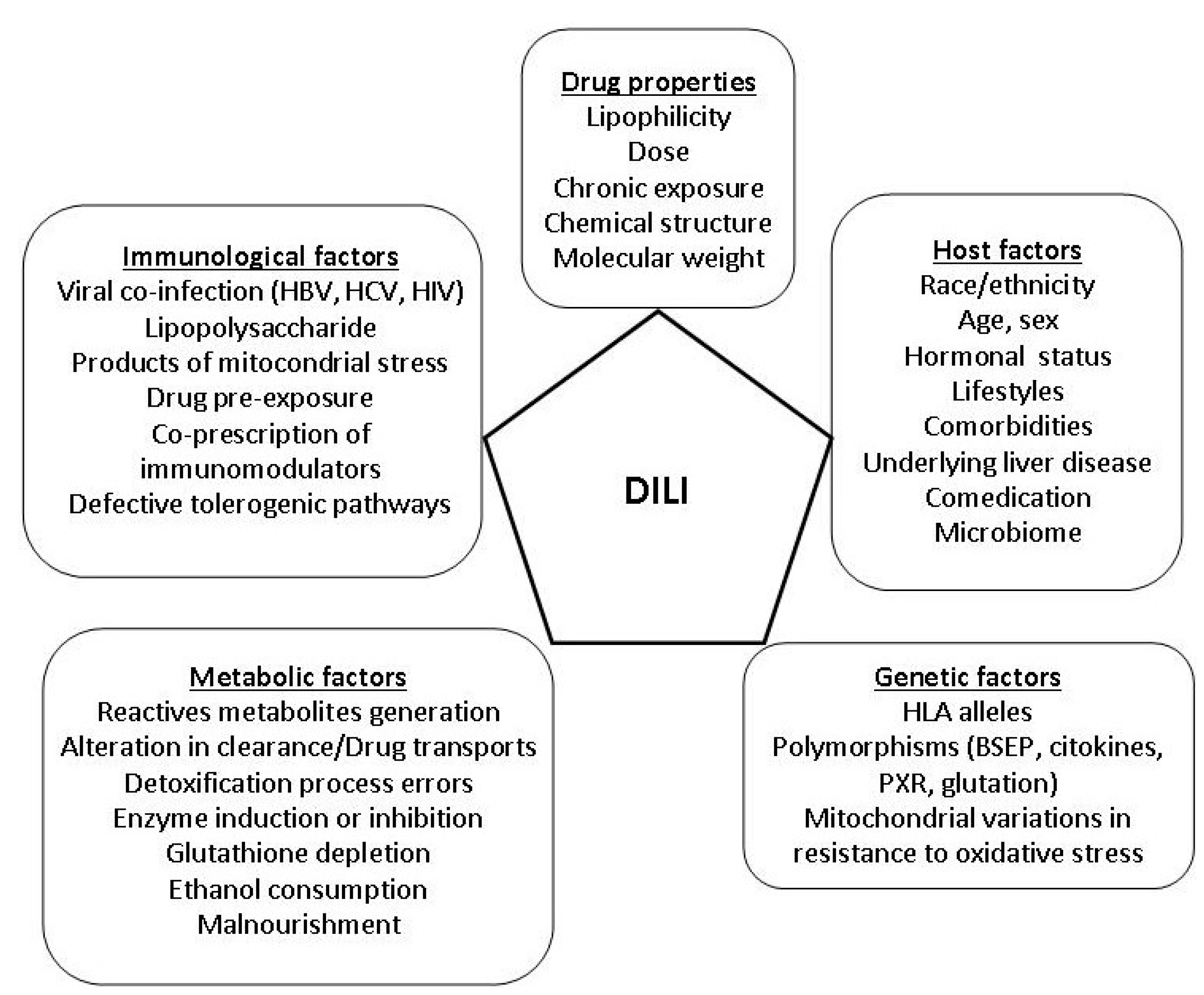

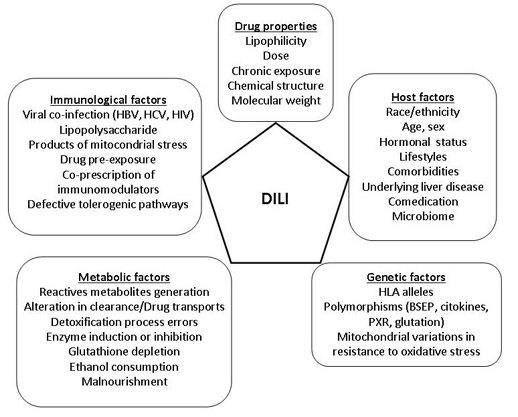{kind=link}
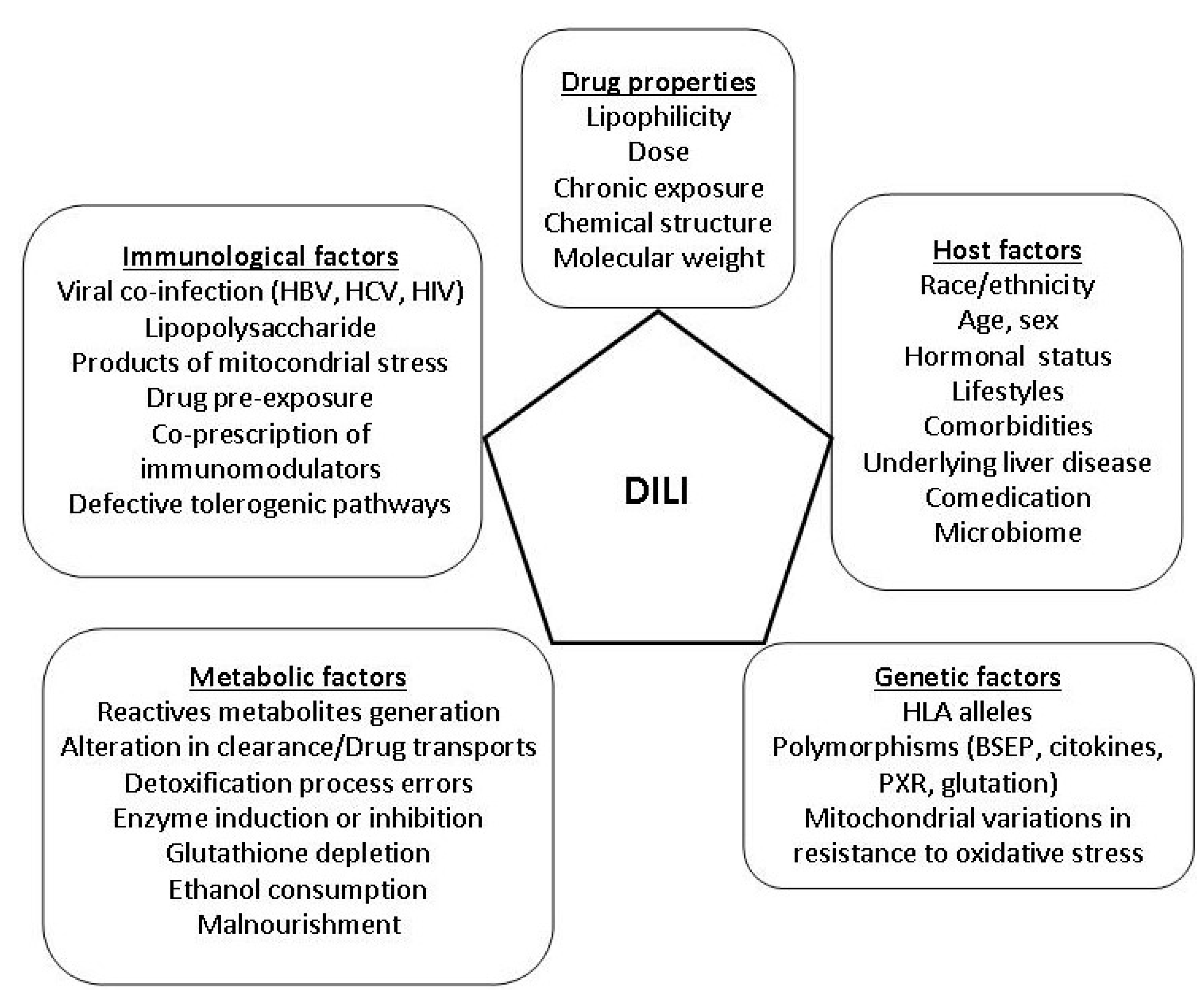{kind=link}
| R = (ALT Patient/ULN)/(ALP Patient/ULN) | |
|---|---|
| Hepatocellular | R ≥ 5 |
| Cholestatic | R ≤ 2 |
| Mixed | R > 2 and <5 |
| Mild | Elevated ALT or ALP Values Reaching Criteria for DILI, but TB < 2 × ULN |
|---|---|
| Moderate | Elevated ALT/ALP values reaching criteria for DILI and TB ≥ 2 × ULN, or symptomatic hepatitis |
| Severe | Elevated ALT/ALP values reaching criteria for DILI, T ≥ 2 × ULN, and one of the following: (1) International normalized ratio (INR) ≥ 1.5, (2) ascites and/or encephalopathy, disease duration <26 weeks, and absence of underlying cirrhosis or (3) other organ failure considered to be due to DILI |
| Fatal | Death or transplantation due to DILI |
| Drug | Type of Liver Damage | Time to Onset | Immunoallergic Features (Rash. Fever, Esonophilia) | Cases of Acute Liver Failure | Cases of Chronic Liver Injury |
|---|---|---|---|---|---|
| Acarbose | HC | 2–8 months | Not typical | No | No |
| Albendazole | HC, Mx | Few days–2 months | Maybe present | No | No |
| Allopurinol | HC, Mx | 2–6 weeks | Yes (DRESS syndrome) | Yes | Yes |
| Amiodarone | HC | Few days–several years | Cases of Reye syndrome | Yes | Yes |
| Amitryptiline | HC, Chol | 1–14 months | Frequent | Yes | Yes |
| Amoxicillin | HC, Chol | Few days–2 weeks | Yes (Stevens-Johnson syndrome) | Yes | Yes (rare) |
| Amoxicillin-clavulanic acid | Chol | Few days–8 weeks After antibiotic is completed (few days–6 weeks) | Not prominent | Yes | Yes (rare) |
| Ampicillin | HC, Chol | Few days–2 weeks | Yes (Stevens-Johnson syndrome) | Yes | Yes |
| Androgenic steroids | Chol | 1–4 months | No | No | No |
| Asparaginase | HC | 2–3 weeks | Rare | Yes | No |
| Atorvastatin | Chol, Mx, HC | 1 month-several years | Yes (autoimmune hepatitis) | Yes | Yes |
| Azathioprine | Chol | 2–12 months | Uncommon | Yes | No |
| Bupropion | Chol, HC | 1–3 months | Uncommon | Yes | No |
| Captopril | Chol | 2–12 weeks | Infrequent | Yes | Yes |
| Carbamazepine | Mx, Chol, HC | 1–8 weeks | Yes (DRESS syndrome) | Yes | Yes |
| Celecoxib | Chol, HC | Few days-few weeks | Yes (Stevens-Johnson syndrome) | No | Yes |
| Chlorpromazine | Chol | 1–5 weeks | Some cases (mild) | Yes | Yes |
| Chlorpropamide | Chol, HC, Mx | 2–12 weeks | Yes | No | No |
| Ciprofloxacin | Chol, HC | 2 days–2 weeks | Many cases | Yes | No |
| Clarithromycin | Chol, HC | 1–3 weeks | No | Yes (HC cases) | Yes |
| Clindamycin | HC, Mx | 1–3 weeks | Typical | No | Yes |
| Clopidogrel | HC | 2–24 weeks | Mild, not prominent | Yes | No |
| Cloxacillin | Chol | 1–6 weeks | No | No | No |
| Contraceptives | Chol | Few cycles | No | No | No |
| Cyproheptadine | Chol, Mx | 1–6 weeks | No | No | No |
| Diazepam | Chol, Mx | 1–6 months | No | No | No |
| Diclofenac | HC | 2–6 months | Yes | Yes | Yes (rare) |
| Dicloxacillin | Chol | 1–6 weeks | Yes, not prominent | No | No |
| Didanosine | HC | Few weeks | Not prominent | Yes | Yes |
| Disulfiram | HC | 2–12 weeks | Not uncommon | Yes | No |
| Enalapril | Chol | 2–12 weeks | Infrequent | Yes | Yes |
| Erythromycin | Chol | 1–3 weeks | Common | Yes | Yes |
| Fluoxetine | HC | 2–12 weeks | No | No | No |
| Flutamide | HC | 1–10 months | Rare | Yes | No |
| Fluvastatin | Chol, Mx | 1–4 months | Uncommon | Yes (rare) | No |
| Fosinopril | Chol | 2–12 weeks | Infrequent | No | Yes |
| Glibenclamide | Chol, Mx | 3–12 weeks | Not typical | Yes | Yes |
| Gold preparations (iv) | Chol | 1–8 weeks | No | Yes | No |
| Halothane | HC | 2–14 days | Yes | Yes | Yes (if repeated exposure) |
| Ibuprofen | HC, Chol | Few days–3 weeks | Prominent (Stevens-Johnson syndrome) | Yes | Yes |
| Imipramine | Chol, HC | 1–8 weeks | Not prominent | Yes (rare) | Yes (rare) |
| Indomethacin | HC | 1–8 weeks | Not common | Yes (rare) | No |
| Irbesartan | HC | 1–4 weeks | No | No | No |
| Isoniazid | HC | 2 weeks–6 months | Uncommon (mild) | Yes | Yes (rare) |
| Ketoconazole | HC, Chol | 1–6 months | Rare | Yes | Yes (rare) |
| Leflunomide | Chol, HC | 1–6 months | Not prominent | Yes | No |
| Lovastatin | Chol | Few weeks–several years | No | Yes (rare) | Yes |
| Mebendazole | HC | Few days | Typical | No | No |
| Mesalazine | Chol, HC | 1–6 months | No | No | No |
| Methimazole | Chol, Mx | 2–12 weeks | Uncommon | Rare | Yes (rare) |
| Methotrexate | 5–10 years | No | No | Yes (cases of cirrhosis) | |
| Minocycline | HC | 1–3 months | Common (autoimmune markers) | Yes | Yes |
| Mirtazapine | HC | Several months–several years | Uncommon | No | No |
| Nitrofurantoin | HC | 1–2 weeks | Typically | Yes | Yes (autoimmune hepatitis) |
| Nefazodone | HC | 6 weeks–8 months | Uncommon | Yes | No |
| Norfloxacin | HC, Chol | 1 day–3 weeks | Many cases | Yes | No |
| Omeprazole | HC | 1–4 weeks | Rare | Yes (rare) | No |
| Paroxetine | HC, Mx | 2–16 weeks | Uncommon | Yes | No |
| Penicillamine | Chol | 1–6 weeks | Common | Yes | Yes |
| Pentamidine | HC | Few days | No | No | No |
| Phenytoin | HC | 2–8 weeks | Common (DRESS) | Yes | Rare |
| Pioglitazone | HC, Chol | 1–6 months | Rare | Yes (HC cases) | No |
| Pravastatin | Chol, HC | 2–9 months | Uncommon | No | No |
| Pyrazinamide | HC | 4–8 weeks | Uncommon | Yes | No |
| Risperidone | Chol | Few days (even years) | Rare | No | No |
| Rofecoxib | Chol, Mx | 1–12 weeks | Uncommon | No | No |
| Rosiglitazone | HC, Chol | 1–12 weeks | Rare | Yes (HC cases) | No |
| Simvastatin | HC, Chol | 1–6 months | Uncommon | Yes (rare) | No |
| Sulfasalazine | Mx | Few days–weeks | Common (DRESS) | Yes | Yes |
| Sulindac | HC, Mx | Few days–weeks | Prominent | Yes | Yes |
| Tamoxifen | Chol, Mx, HC | 6 months | Uncommon | Yes | Yes (cases of fatty liver) |
| Telithromycin | HC | Few days–1 week | Uncommon | Yes | No |
| Terbinafine | HC, Chol | 6 weeks | Uncommon (Stevens-Johnson) | Yes | Yes |
| Thiabendazole | Chol | 1–2 weeks | Rare | Yes | Yes |
| Ticlopidine | Chol | 6 weeks | Not common (mild) | Yes | Yes |
| Tetracycline | HC | Few days | No | Yes (pregnancy) | No |
| Tolcapone | HC | 1–5 months | No | Yes | No |
| Trazodone | HC | Few days–6 months | Not prominent | Yes (rare) | Yes (rare) |
| Trimethoprin-sulfamethoxazol | Chol, Mx | Few days–weeks | Common (DRESS syndrome) | Yes | Yes |
| Troglitazone | HC | 1–6 months | Uncommon | Yes | Yes |
| Valproic acid | HC | 1–6 months | Rare | Yes (Reye like-syndrome) | Yes (cases of cirrhosis) |
| Venlafaxine | Chol, HC | 1–3 months | Uncommon | No | No |
| Verapamil | Mx, Chol | 2–8 weeks | Rare | No | No |
| Zidovudine | Chol | 1–4 weeks | Not common | Yes | No |
| Type of Damage | Drug | Histological Features |
|---|---|---|
| Acute hepatocellular injury | Isoniazid, aspirin, sulfamides | Lobular predominant lymphocytic-plasmacytic infiltration +/− hepatocellular degeneration, lobular disarray, no cholestasis |
| Autoimmune-like hepatitis | Nitrofurantoin, minocycline, Ipilimumab | Plasma cells and interface hepatitis |
| Pure cholestasis | Anabolic steroids, estrogens | Hepatocyte cholestasis and dilated biliary canaliculi with bile plugs, without evidence of necrosis or inflammation |
| Cholestasis hepatitis | Phenytoin, amoxicillin-clavulanate, fluorquinolones, macrolides, azithromycin | Portal and ductal inflammation as well as hepatocyte necrosis with marked predominance centrilobular cholestasis |
| Granulomatous hepatitis | Isoniazid, interferon, phenytoin, allopurinol | Nonnecrotizing epithelioid granulomas |
| Chronic hepatitis | Diclofenac, Methyldopa, Bentazepam | Portal predominant, interface hepatitis, portal-based fibrosis |
| Macrovesicular steatosis | Tetracycline, steroids, gold, 5-fluorouracil, methotrexate, tamoxifen | Variable degrees of accumulation of large fat droplets with peripheral displacement of the nucleus without significant inflammation or cholestasis or alternate pattern |
| Microvesicular steatosis | Valproic acid, tetracycline, zidovudine | Diffuse hepatocyte accumulation of small fat droplets maintaining a central placement of the nucleus without significant inflammation or cholestasis or alternate pattern |
| Non-alcoholic fatty liver | Tamoxifen, amiodarone | Macrosteatosis and microsteatosis, hepatocyte ballooning and periportal inflammation |
| Vanishing bile duct syndrome | Amoxicillin-clavulanate, sulfonamides | Paucity of interlobular bile ducts |
| Fibrosis/cirrhosis | Methotrexate, amiodarone | Hepatic collagenization with minimal inflammation |
| Sinusoidal obstruction syndrome | Busulfan, oxaliplatin | Sinusoidal dilatation and congestion, central venule occlusions, perisinusoidal fibrosis |
| Liver adenoma | Oral contraceptives | Normal appearance of the hepatocytes. These are arranged in sheets and have no malignant features. These cells tend to be larger than normal hepatocytes, and their cytoplasm often contains fat or glycogen |
| Compounds | Number of Cases | HLA Allele | Odds Ratio (95% CI) | p Value |
|---|---|---|---|---|
| Flucloxacillin | 51 | B*57:01 | 80.6 (22.8–284.9) | 9 × 10−19 |
| Amoxicillin-clavulanate | 201 | A*02:01 | 2.3 (1.8–2.9) | 1.8 × 10−10 |
| DRB1*15:01-DQB1*06:02 | 2.8 (2.1–3.8) | 3.5 × 10−11 | ||
| Lumiracoxib | 41 | DRB1*15:01-DQB1*06:02 | 5.0 (3.6–7.0) | 6.8 × 10−25 |
| Lapatinib | 35 | DRB1*07:01-DQA1*02:01 | 2.9 (1.3–6.6) | 0.007 |
| Ximegalatran | 74 | DRB1*07:01-DQA1*02:01 | 4.4 (2.2–8.9) | 6 × 10−6 |
| Ticlopidine | 22 | A*33:03 | 13.0 (4.4–38.6) | 1.2 × 10−5 |
| Terbinafine | 14 | A*33:01 | 40.53 (12.51–288.9) | 6.7 × 10−10 |
| Fenofibrate | 7 | A*33:01 | 58.7 (12.31–279.8) | 3.2 × 10−7 |
| Ticlopidine | 5 | A*33:01 | 163.1 (16.2–1642) | 0.00002 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega-Alonso, A.; Stephens, C.; Lucena, M.I.; Andrade, R.J. Case Characterization, Clinical Features and Risk Factors in Drug-Induced Liver Injury. Int. J. Mol. Sci. 2016, 17, 714. https://doi.org/10.3390/ijms17050714
Ortega-Alonso A, Stephens C, Lucena MI, Andrade RJ. Case Characterization, Clinical Features and Risk Factors in Drug-Induced Liver Injury. International Journal of Molecular Sciences. 2016; 17(5):714. https://doi.org/10.3390/ijms17050714
Chicago/Turabian StyleOrtega-Alonso, Aida, Camilla Stephens, M. Isabel Lucena, and Raúl J. Andrade. 2016. "Case Characterization, Clinical Features and Risk Factors in Drug-Induced Liver Injury" International Journal of Molecular Sciences 17, no. 5: 714. https://doi.org/10.3390/ijms17050714
APA StyleOrtega-Alonso, A., Stephens, C., Lucena, M. I., & Andrade, R. J. (2016). Case Characterization, Clinical Features and Risk Factors in Drug-Induced Liver Injury. International Journal of Molecular Sciences, 17(5), 714. https://doi.org/10.3390/ijms17050714