
In Search of New Therapeutic Targets in Obesity Treatment: Sirtuins


Abstract
:1. Introduction
2. Short Review of the Silent Information Regulators (SIRTs) System
3. SIRTs in Control of Lipid and Glucose Metabolism
3.1. SIRTs in Lipid Metabolism
3.2. SIRTs in Insulin Secretion and Glucose Metabolism
4. SIRTs and Adipogenesis
5. Abnormal Activity of SIRTs System in Obesity
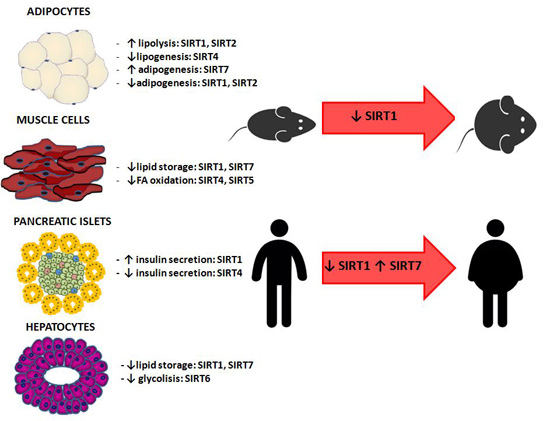
5.1. Animal Studies
5.2. Human Studies
5.3. Human Obesity Associated Changes in SIRTs Expression
5.4. Genetic Studies
6. Sirtuins as Targets for Obesity Treatment
7. Final Remarks and Conclusions
Acknowledgments
Conflicts of Interest
References
- Klaus, J.R.; Hurwitz, B.E.; Llabre, M.M.; Skyler, J.S.; Goldberg, R.B.; Marks, J.B.; Bilsker, M.S.; Schneiderman, N. Central obesity and insulin resistance in the cardiometabolic syndrome: Pathways to preclinical cardiovascular structure and function. J. Cardiometab. Syndr. 2009, 4, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Sanders, B.D.; Jackson, B.; Marmorstein, R. Structural basis for sirtuin function: What we know and what we don’t. Biochim. Biophys. Acta 2010, 1804, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins—Emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Nguyen, M.; Qin, F.X.; Tong, Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 2007, 6, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Parihar, P.; Solanki, I.; Mansuri, M.L.; Parihar, M.S. Mitochondrial sirtuins: Emerging roles in metabolic regulations, energy homeostasis and diseases. Exp. Gerontol. 2015, 61, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Zakhary, S.M.; Ayubcha, D.; Dileo, J.N.; Jose, R.; Leheste, J.R.; Horowitz, J.M.; Torres, G. Distribution analysis of deacetylase SIRT1 in rodent and human nervous systems. Anat. Rec. 2010, 293, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Wieser, V.; Gerner, R.R.; Bichler, A.; Enrich, B.; Moser, P.; Ebenbichler, C.F.; Kaser, S.; Tilg, H. Adipose tissue and liver expression of SIRT1, 3, and 6 increase after extensive weight loss in morbid obesity. J. Hepatol. 2013, 59, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Caton, P.W.; Richardson, S.J.; Kieswich, J.; Bugliani, M.; Holland, M.L.; Marchetti, P.; Morgan, N.G.; Yaqoob, M.M.; Holness, M.J.; Sugden, M.C. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia 2013, 56, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Acs, Z.; Bori, Z.; Takeda, M.; Osvath, P.; Berkes, I.; Taylor, A.W.; Yang, H.; Radak, Z. High altitude exposure alters gene expression levels of DNA repair enzymes, and modulates fatty acid metabolism by SIRT4 induction in human skeletal muscle. Respir. Physiol. Neurobiol. 2014, 196, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Chalkiadaki, A.; Guarente, L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat. Rev. Endocrinol. 2012, 8, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. SIRT1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Bordone, L.; Cohen, D.; Robinson, A.; Motta, M.C.; van Veen, E.; Czopik, A.; Steele, A.D.; Crowe, H.; Marmor, S.; Luo, J.; et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell 2007, 6, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; Kon, N.; Knight, C.; Matsumoto, M.; Gutiérrez-Juárez, R.; Rossetti, L.; Gu, W.; Accili, D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008, 8, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Tong, Q. SIRT2 suppresses adipocyte differentiation by deacetylating FOXO1 and enhancing FOXO1’s repressive interaction with PPARgamma. Mol. Biol. Cell 2009, 20, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Tao, R.; Gao, X.; Li, T.; Zhou, X.; Guan, K.L.; Xiong, Y.; Lei, Q.Y. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell 2013, 51, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Galonek, H.; Israelian, K.; Choy, W.; Morrison, M.; Xia, Y.; Wang, X.; Xu, Y.; Yang, Y.; Smith, J.J.; et al. Biochemical characterization, localization, and tissue distribution of the longer form of mouse SIRT3. Protein Sci. 2009, 18, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Pfluger, P.T.; Herranz, D.; Velasco-Miguel, S.; Serrano, M.; Tschöp, M.H. SIRT1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA 2008, 105, 9793–9798. [Google Scholar] [CrossRef] [PubMed]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Bruno, J.; Easlon, E.; Lin, S.J.; Cheng, H.L.; Alt, F.W.; Guarente, L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008, 22, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Fu, Y.C.; Xu, W.C.; Feng, Y.Q.; Fang, S.R.; Zhou, X.H. Resveratrol inhibits the expression of SREBP1 in cell model of steatosis via SIRT1-FOXO1 signaling pathway. Biochem. Biophys. Res. Commun. 2009, 380, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.; Lodish, H.; Sun, L. MicroRNAs in adipogenesis and as therapeutic targets for obesity. Expert Opin. Ther. Targets 2011, 15, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, S.; Blander, G.; Tse, J.G.; Krieger, M.; Guarente, L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol. Cell 2007, 28, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Groen, A.K.; Oude Elferink, R.P.; Verkade, H.J.; Kuipers, F. The ins and outs of reverse cholesterol transport. Ann. Med. 2004, 36, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Lomb, D.J.; Laurent, G.; Haigis, M.C. Sirtuins regulate key aspects of lipid metabolism. Biochim. Biophys. Acta 2010, 1804, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Savastano, S.; di Somma, C.; Colao, A.; Barrea, L.; Orio, F.; Finelli, C.; Pasanisi, F.; Contaldo, F.; Tarantino, G. Preliminary data on the relationship between circulating levels of Sirtuin 4, anthropometric and metabolic parameters in obese subjects according to growth hormone/insulin-like growth factor-1 status. Growth Horm. IGF Res. 2015, 25, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.; German, N.J.; Saha, A.K.; de Boer, V.C.; Davies, M.; Koves, T.R.; Dephoure, N.; Fischer, F.; Boanca, G.; Vaitheesvaran, B.; et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol. Cell 2013, 50, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.; Vallespinos-Serrano, M.; Trabulo, S.M.; Fernandez-Marcos, P.J.; Firment, A.N.; Vazquez, B.N.; Vieira, C.R.; Mulero, F.; Camara, J.A.; Cronin, U.P.; et al. MiR-93 Controls Adiposity via Inhibition of Sirt7 and Tbx3. Cell Rep. 2015, 12, 1594–1605. [Google Scholar] [CrossRef] [PubMed]
- Bordone, L.; Motta, M.C.; Picard, F.; Robinson, A.; Jhala, U.S.; Apfeld, J.; McDonagh, T.; Lemieux, M.; McBurney, M.; Szilvasi, A.; et al. SIRT1 regulates insulin secretion by repressing UCP2 in pancreatic β cells. PLoS Biol. 2006, 4, e31. [Google Scholar] [CrossRef]
- Bordone, L.; Motta, M.C.; Picard, F.; Robinson, A.; Jhala, U.S.; Apfeld, J.; McDonagh, T.; Lemieux, M.; McBurney, M.; Szilvasi, A.; et al. Correction: SIRT1 regulates insulin secretion by repressing UCP2 in pancreatic β cells. PLoS Biol. 2015, 13, e1002346. [Google Scholar] [CrossRef] [PubMed]
- Moynihan, K.A.; Grimm, A.A.; Plueger, M.M.; Bernal-Mizrachi, E.; Ford, E.; Cras-Méneur, C.; Permutt, M.A.; Imai, S. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005, 2, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.I.; Kitamura, T.; Kruse, J.P.; Raum, J.C.; Stein, R.; Gu, W.; Accili, D. FOXO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005, 2, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.J.; Meares, G.P.; Hansen, P.A.; Corbett, J.A. FoxO1 and SIRT1 regulate β-cell responses to nitric oxide. J. Biol. Chem. 2011, 286, 8338–8348. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, N.; Schwer, B.; Carobbio, S.; Waltregny, D.; North, B.J.; Castronovo, V.; Maechler, P.; Verdin, E. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J. Biol. Chem. 2007, 282, 33583–33592. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.; Grether-Beck, S.; Singh, M.; Kuck, F.; Jakob, S.; Kefalas, A.; Altinoluk-Hambüchen, S.; Graffmann, N.; Schneider, M.; Lindecke, A.; et al. MicroRNA-15b regulates mitochondrial ROS production and the senescence-associated secretory phenotype through sirtuin 4/SIRT4. Aging 2016, 8, 534–559. [Google Scholar]
- Bordone, L.; Guarente, L. Sirtuins and β-cell function. Diabetes Obes. Metab. 2007, 9, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The histone deacetylase SIRT6 regulates glucose homeostasis via Hif1α. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Hernando, C.; Suárez, Y.; Rayner, K.J.; Moore, K.J. MicroRNAs in lipid metabolism. Curr. Opin. Lipidol. 2011, 22, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschöp, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, T.; Milne, J.C.; Imamura, T.; Schenk, S.; Sonoda, N.; Babendure, J.L.; Lu, J.C.; Smith, J.J.; Jirousek, M.R.; Olefsky, J.M. SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Mol. Cell. Biol. 2009, 29, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, T.; Schenk, S.; Imamura, T.; Babendure, J.L.; Sonoda, N.; Bae, E.J.; Oh, D.Y.; Lu, M.; Milne, J.C.; Westphal, C.; et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E419–E428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. The direct involvement of Sirt1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J. Biol. Chem. 2007, 282, 34356–34364. [Google Scholar] [CrossRef] [PubMed]
- Puri, N.; Sodhi, K.; Haarstad, M.; Kim, D.H.; Bohinc, S.; Foglio, E.; Favero, G.; Abraham, N.G. Heme induced oxidative stress attenuates sirtuin1 and enhances adipogenesis in mesenchymal stem cells and mouse pre-adipocytes. J. Cell. Biochem. 2012, 113, 1926–1935. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, Z.; Zhang, W.; Hu, X.; Wei, H.; Peng, J.; Jiang, S. SIRT1 inhibits adipogenesis and promotes myogenic differentiation in C3H10T1/2 pluripotent cells by regulating Wnt signaling. Cell Biosci. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wang, L.; Kon, N.; Zhao, W.; Lee, S.; Zhang, Y.; Rosenbaum, M.; Zhao, Y.; Gu, W.; Farmer, S.R.; et al. Brown remodeling of white adipose tissue by SIRT1-dependent deacetylation of PPAR-γ. Cell 2012, 150, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Abdesselem, H.; Madani, A.; Hani, A.; Al-Noubi, M.; Goswami, N.; Ben Hamidane, H.; Billing, A.M.; Pasquier, J.; Bonkowski, M.S.; Halabi, N.; et al. SIRT1 limits adipocyte hyperplasia through c-Myc inhibition. J. Biol. Chem. 2016, 291, 2119–2135. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Lee, H.; Jung, C.H.; Jeon, T.I.; Ha, T.Y. MicroRNA-146b promotes adipogenesis by suppressing the SIRT1-FOXO1 cascade. EMBO Mol. Med. 2013, 5, 1602–1612. [Google Scholar] [CrossRef] [PubMed]
- Jing, E.; Gesta, S.; Kahn, C.R. SIRT2 regulates adipocyte differentiation through FOXO1 acetylation/deacetylation. Cell Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; He, M.; Liu, Y.; Paredes, S.; Villanova, L.; Brown, K.; Qiu, X.; Nabavi, N.; Mohrin, M.; Wojnoonski, K.; et al. SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep. 2013, 5, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Gao, Z.; Zhang, J.; Rivera, C.A.; Yin, J.; Weng, J.; Ye, J. Lack of SIRT1 (Mammalian Sirtuin 1) activity leads to liver steatosis in the SIRT1+/− mice: A role of lipid mobilization and inflammation. Endocrinology 2010, 151, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Mayoral, R.; Osborn, O.; McNelis, J.; Johnson, A.M.; Oh da, Y.; Izquierdo, C.L.; Chung, H.; Li, P.; Traves, P.G.; Bandyopadhyay, G.; et al. Adipocyte SIRT1 knockout promotes PPAR-γ activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol. Metab. 2015, 4, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Lagouge, M.; Canto, C.; Strehle, A.; Houten, S.M.; Milne, J.C.; Lambert, P.D.; Mataki, C.; Elliott, P.J.; Auwerx, J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008, 8, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Kikuchi, O.; Shimpuku, M.; Susanti, V.Y.; Yokota-Hashimoto, H.; Taguchi, R.; Shibusawa, N.; Sato, T.; Tang, L.; Amano, K.; et al. Hypothalamic SIRT1 prevents age-associated weight gain by improving leptin sensitivity in mice. Diabetologia 2014, 57, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Sarruf, D.A.; Li, P.; Osborn, O.; Sanchez-Alavez, M.; Talukdar, S.; Chen, A.; Bandyopadhyay, G.; Xu, J.; Morinaga, H.; et al. Neuronal SIRT1 deficiency increases insulin sensitivity in both brain and peripheral tissues. J. Biol. Chem. 2013, 288, 10722–10735. [Google Scholar] [CrossRef] [PubMed]
- Cakir, I.; Perello, M.; Lansari, O.; Messier, N.J.; Vaslet, C.A.; Nillni, E.A. Hypothalamic SIRT1 regulates food intake in a rodent model system. PLoS ONE 2009, 4, e8322. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Pak, Y.K.; Jang, P.G.; Namkoong, C.; Choi, Y.S.; Won, J.C.; Kim, K.S.; Kim, S.W.; Kim, H.S.; Park, J.Y.; et al. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat. Neurosci. 2006, 9, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Toorie, A.M.; Nillni, E.A. Minireview: Central Sirt1 regulates energy balance via the melanocortin system and alternate pathways. Mol. Endocrinol. 2014, 28, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Rothgiesser, K.M.; Erener, S.; Waibel, S.; Lüscher, B.; Hottiger, M.O. SIRT2 regulates NF-κB dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 2010, 123, 4251–4258. [Google Scholar] [CrossRef] [PubMed]
- Nasrin, N.; Wu, X.; Fortier, E.; Feng, Y.; Bare’, O.C.; Chen, S.; Ren, X.; Wu, Z.; Streeper, R.S.; Bordone, L. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J. Biol. Chem. 2010, 285, 31995–32002. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Sadhukhan, S.; Noriega, L.G.; Moullan, N.; He, B.; Weiss, R.S.; Lin, H.; Schoonjans, K.; Auwerx, J. Metabolic characterization of a SIRT5 deficient mouse model. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Nakamura, Y.; Tanaka, D.; Zhuang, X.; Fujita, Y.; Obara, A.; Hamasaki, A.; Hosokawa, M.; Inagaki, N. Overexpression of SIRT5 confirms its involvement in deacetylation and activation of carbamoyl phosphate synthetase 1. Biochem. Biophys. Res. Commun. 2010, 393, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Schwer, B.; Schumacher, B.; Lombard, D.B.; Xiao, C.; Kurtev, M.V.; Gao, J.; Schneider, J.I.; Chai, H.; Bronson, R.T.; Tsai, L.H.; et al. Neural sirtuin 6 (SIRT6) ablation attenuates somatic growth and causes obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 21790–21794. [Google Scholar] [CrossRef] [PubMed]
- Kanfi, Y.; Peshti, V.; Gil, R.; Naiman, S.; Nahum, L.; Levin, E.; Kronfeld-Schor, N.; Cohen, H.Y. SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell 2010, 9, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.G.; Ramadori, G.; Ioris, R.M.; Galiè, M.; Berglund, E.D.; Coate, K.C.; Fujikawa, T.; Pucciarelli, S.; Moreschini, B.; Amici, A.; et al. Enhanced insulin sensitivity in skeletal muscle and liver by physiological overexpression of SIRT6. Mol. Metab. 2015, 4, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Vakhrusheva, O.; Smolka, C.; Gajawada, P.; Kostin, S.; Boettger, T.; Kubin, T.; Braun, T.; Bober, E. SIRT7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ. Res. 2008, 102, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, T.; Karim, M.F.; Sato, Y.; Senokuchi, T.; Miyata, K.; Fukuda, T.; Go, C.; Tasaki, M.; Uchimura, K.; Kadomatsu, T.; et al. SIRT7 controls hepatic lipid metabolism by regulating the ubiquitin-proteasome pathway. Cell Metab. 2014, 19, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Crujeiras, A.B.; Parra, D.; Goyenechea, E.; Martínez, J.A. Sirtuin gene expression in human mononuclear cells is modulated by caloric restriction. Eur. J. Clin. Investig. 2008, 38, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Falchi, M.; Olsson, B.; Jacobson, P.; Cauchi, S.; Balkau, B.; Marre, M.; Lantieri, O.; Andersson, J.C.; Jernås, M.; et al. Association of sirtuin 1 (SIRT1) gene SNPs and transcript expression levels with severe obesity. Obesity 2012, 20, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.B.; Ølholm, J.; Paulsen, S.K.; Bennetzen, M.F.; Richelsen, B. Low SIRT1 expression, which is upregulated by fasting, in human adipose tissue from obese women. Int. J. Obes. 2008, 32, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Guo, T.; Traurig, M.; Mason, C.C.; Kobes, S.; Perez, J.; Knowler, W.C.; Bogardus, C.; Hanson, R.L.; Baier, L.J. SIRT1 is associated with a decrease in acute insulin secretion and a sex specific increase in risk for type 2 diabetes in Pima Indians. Mol. Genet. Metab. 2011, 104, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.S.; Lee, S.K.; Jang, Y.J.; Park, H.S.; Kim, J.H.; Lee, Y.J.; Heo, Y.S. Association between low SIRT1 expression in visceral and subcutaneous adipose tissues and metabolic abnormalities in women with obesity and type 2 diabetes. Diabetes Res. Clin. Pract. 2013, 101, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Costa Cdos, S.; Hammes, T.O.; Rohden, F.; Margis, R.; Bortolotto, J.W.; Padoin, A.V.; Mottin, C.C.; Guaragna, R.M. SIRT1 transcription is decreased in visceral adipose tissue of morbidly obese patients with severe hepatic steatosis. Obes. Surg. 2010, 20, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Finelli, C.; Scopacasa, F.; Pasanisi, F.; Contaldo, F.; Capone, D.; Savastano, S. Circulating levels of sirtuin 4, a potential marker of oxidative metabolism, related to coronary artery disease in obese patients suffering from NAFLD, with normal or slightly increased liver enzymes. Oxid. Med. Cell. Longev. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Xu, W.; Chen, Y.; Li, Z.; Zeng, Y.; Fu, Y. The expression of Sirtuins 1 and 4 in peripheral blood leukocytes from patients with type 2 diabetes. Eur. J. Histochem. 2011, 55. [Google Scholar] [CrossRef] [PubMed]
- Bober, E.; Fang, J.; Smolka, C.; Ianni, A.; Vakhrusheva, O.; Krüger, M.; Braun, T. SIRT7 promotes adipogenesis by binding to and inhibiting SIRT1. BMC Proc. 2012, 6. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, D.; Yi, C.; Wang, Y.; Wang, H.; Wang, J. MicroRNA-22 functions as a tumor suppressor by targeting SIRT1 in renal cell carcinoma. Oncol. Rep. 2015, 35, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Li, C.; Qi, W.; Zhang, Y.; Zhang, F.; Wu, J.X.; Hu, Y.N.; Wu, D.M.; Liu, Y.; Yan, T.T.; et al. Downregulation of miR-181a upregulates sirtuin-1 (SIRT1) and improves hepatic insulin sensitivity. Diabetologia 2012, 55, 2032–2043. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Noh, J.H.; Jung, K.H.; Eun, J.W.; Bae, H.J.; Kim, M.G.; Chang, Y.G.; Shen, Q.; Park, W.S.; Lee, J.Y.; et al. Sirtuin7 oncogenic potential in human hepatocellular carcinoma and its regulation by the tumor suppressors MiR-125a-5p and MiR-125b. Hepatology 2013, 57, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.; Chen, H.; Roix, J.J.; Kathiresan, S.; Hirschhorn, J.N.; Daly, M.J.; et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316, 1331–1336. [Google Scholar] [PubMed]
- Van den Berg, S.W.; Dollé, M.E.; Imholz, S.; van der A, D.L.; van’t Slot, R.; Wijmenga, C.; Verschuren, W.M.; Strien, C.; Siezen, C.L.; Hoebee, B.; et al. Genetic variations in regulatory pathways of fatty acid and glucose metabolism are associated with obesity phenotypes: A population-based cohort study. Int. J. Obes. 2009, 33, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Peeters, A.V.; Beckers, S.; Verrijken, A.; Mertens, I.; Roevens, P.; Peeters, P.J.; van Hul, W.; van Gaal, L.F. Association of SIRT1 gene variation with visceral obesity. Hum. Genet. 2008, 124, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Zillikens, M.C.; van Meurs, J.B.; Rivadeneira, F.; Amin, N.; Hofman, A.; Oostra, B.A.; Sijbrands, E.J.; Witteman, J.C.; Pols, H.A.; van Duijn, C.M.; et al. SIRT1 genetic variation is related to BMI and risk of obesity. Diabetes 2009, 58, 2828–2834. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, P.; Machicao, F.; Reinhardt, J.; Machann, J.; Schick, F.; Tschritter, O.; Stefan, N.; Fritsche, A.; Häring, H.U. SIRT1 genetic variants associate with the metabolic response of Caucasians to a controlled lifestyle intervention-the TULIP Study. BMC Med. Genet. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, Y.; Suzuki, K.; Hamajima, N.; Niwa, T. SIRT1 gene polymorphisms are associated with body fat and blood pressure in Japanese. Transl. Res. 2011, 157, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Kilic, U.; Gok, O.; Elibol-Can, B.; Ozgen, I.T.; Erenberk, U.; Uysal, O.; Dundaroz, M.R. SIRT1 gene variants are related to risk of childhood obesity. Eur. J. Pediatr. 2015, 174, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Figarska, S.M.; Vonk, J.M.; Boezen, H.M. SIRT1 polymorphism, long-term survival and glucose tolerance in the general population. PLoS ONE 2013, 8, e58636. [Google Scholar] [CrossRef] [PubMed]
- Zarrabeitia, M.T.; Valero, C.; Martín-Escudero, J.C.; Olmos, J.M.; Bolado-Carrancio, A.; de Sande-Nacarino, E.L.; Rodríguez-Rey, J.C.; Sainz, J.; Riancho, J.A. Association study of sirtuin 1 polymorphisms with bone mineral density and body mass index. Arch. Med. Res. 2012, 43, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Kedenko, L.; Lamina, C.; Kedenko, I.; Kollerits, B.; Kiesslich, T.; Iglseder, B.; Kronenberg, F.; Paulweber, B. Genetic polymorphisms at SIRT1 and FOXO1 are associated with carotid atherosclerosis in the SAPHIR cohort. BMC Med. Genet. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Della-Morte, D.; Wang, L.; Cabral, D.; Beecham, A.; McClendon, M.S.; Luca, C.C.; Blanton, S.H.; Sacco, R.L.; Rundek, T. Association of the sirtuin and mitochondrial uncoupling protein genes with carotid plaque. PLoS ONE 2011, 6, e27157. [Google Scholar] [CrossRef] [PubMed]
- Della-Morte, D.; Dong, C.; Bartels, S.; Cabral, D.; Blanton, S.H.; Sacco, R.L.; Rundek, T. Association of the sirtuin and mitochondrial uncoupling protein genes with carotid intima-media thickness. Transl. Res. 2012, 160, 389–390. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Della-Morte, D.; Cabral, D.; Wang, L.; Blanton, S.H.; Seemant, C.; Sacco, R.L.; Rundek, T. Sirtuin/uncoupling protein gene variants and carotid plaque area and morphology. Int. J. Stroke 2015, 10, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Wang, H.; Chen, H.; Pang, S.; Wang, L.; Liu, D.; Yan, B. Genetic analysis of the SIRT1 gene promoter in myocardial infarction. Biochem. Biophys. Res. Commun. 2012, 426, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Kilic, U.; Gok, O.; Bacaksiz, A.; Izmirli, M.; Elibol-Can, B.; Uysal, O. SIRT1 gene polymorphisms affect the protein expression in cardiovascular diseases. PLoS ONE 2014, 9, e90428. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.; Valladares-Salgado, A.; Garcia-Mena, J.; Ross, K.; Edwards, M.; Angeles-Martinez, J.; Ortega-Camarillo, C.; de la Peña, J.E.; Burguete-Garcia, A.I.; Wacher-Rodarte, N.; et al. Candidate gene association study conditioning on individual ancestry in patients with type 2 diabetes and metabolic syndrome from Mexico city. Diabetes Metab. Res. Rev. 2010, 26, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Koya, D.; Araki, S.; Babazono, T.; Umezono, T.; Toyoda, M.; Kawai, K.; Imanishi, M.; Uzu, T.; Suzuki, D.; et al. Association between single nucleotide polymorphisms within genes encoding sirtuin families and diabetic nephropathy in Japanese subjects with type 2 diabetes. Clin. Exp. Nephrol. 2011, 15, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, Y.; Mitsuda, Y.; Tsuruta, Y.; Suzuki, K.; Hamajima, N.; Niwa, T. Sirtuin 1 gene polymorphisms are associated with cholesterol metabolism and coronary artery calcification in Japanese hemodialysis patients. J. Ren. Nutr. 2012, 22, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Elghobashy, Y.; Tayel, S.; ALrefai, A.; Khamis, S.; Elbarbary, H. Relation of sirtuin 1 gene polymorphism with lipid profile in hemodialysis patients. Br. Biotechnol. J. 2014, 4, 932–945. [Google Scholar] [CrossRef]
- Mellini, P.; Valente, S.; Mai, A. Sirtuin modulators: An updated patent review (2012–2014). Expert Opin. Ther. Pat. 2015, 25, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Rayalam, S.; Yang, J.Y.; Ambati, S.; Della-Fera, M.A.; Baile, C.A. Resveratrol induces apoptosis and inhibits adipogenesis in 3T3-L1 adipocytes. Phytother. Res. 2008, 22, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, L.; Fernández-Quintela, A.; Arias, N.; Portillo, M.P. Resveratrol: Anti-obesity mechanisms of action. Molecules 2014, 19, 18632–18655. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K.J.; Baur, J.A.; Lewis, K.N.; Peshkin, L.; Price, N.L.; Labinskyy, N.; Swindell, W.R.; Kamara, D.; Minor, R.K.; Perez, E.; et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008, 8, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jin, Y.; Choi, Y.; Park, T. Resveratrol exerts anti-obesity effects via mechanisms involving down-regulation of adipogenic and inflammatory processes in mice. Biochem. Pharmacol. 2011, 81, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- De Ligt, M.; Timmers, S.; Schrauwen, P. Resveratrol and obesity: Can resveratrol relieve metabolic disturbances? Biochim. Biophys. Acta 2015, 1852, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Konings, E.; Timmers, S.; Boekschoten, M.V.; Goossens, G.H.; Jocken, J.W.; Afman, L.A.; Müller, M.; Schrauwen, P.; Mariman, E.C.; Blaak, E.E. The effects of 30 days resveratrol supplementation on adipose tissue morphology and gene expression patterns in obese men. Int. J. Obes. 2014, 38, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Kenney, R.D.; Gagne, D.J.; Frushour, B.P.; Ladd, W.; Galonek, H.L.; Israelian, K.; Song, J.; Razvadauskaite, G.; Lynch, A.V.; et al. Small molecule activators of SIRT1 replicate signaling pathways triggered by calorie restriction in vivo. BMC Syst. Biol. 2009, 3. [Google Scholar] [CrossRef] [PubMed]
- Howells, L.M.; Berry, D.P.; Elliott, P.J.; Jacobson, E.W.; Hoffmann, E.; Hegarty, B.; Brown, K.; Steward, W.P.; Gescher, A.J. Phase I randomized, double-blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases—Safety, pharmacokinetics, and pharmacodynamics. Cancer Prev. Res. 2011, 4, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.P.; Lee, H.Y.; Lau, D.P.; Supaat, W.; Chan, Y.H.; Koh, A.F. Effects of resveratrol in patients with type 2 diabetes mellitus on skeletal muscle SIRT1 expression and energy expenditure. Int. J. Sport Nutr. Exerc. Metab. 2014, 24, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Faghihzadeh, F.; Adibi, P.; Hekmatdoost, A. The effects of resveratrol supplementation on cardiovascular risk factors in patients with non-alcoholic fatty liver disease: A randomised, double-blind, placebo-controlled study. Br. J. Nutr. 2015, 114, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.M.; Vestergaard, P.F.; Clasen, B.F.; Radko, Y.; Christensen, L.P.; Stødkilde-Jørgensen, H.; Møller, N.; Jessen, N.; Pedersen, S.B.; Jørgensen, J.O. High-dose resveratrol supplementation in obese men: An investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes 2013, 62, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Conte, C.; Fontana, L.; Mittendorfer, B.; Imai, S.; Schechtman, K.B.; Gu, C.; Kunz, I.; Rossi Fanelli, F.; Patterson, B.W.; et al. Resveratrol supplementation does not improve metabolic function in nonobese women with normal glucose tolerance. Cell Metab. 2012, 16, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Baksi, A.; Kraydashenko, O.; Zalevkaya, A.; Stets, R.; Elliott, P.; Haddad, J.; Hoffmann, E.; Vlasuk, G.P.; Jacobson, E.W. A phase II, randomized, placebo-controlled, double-blind, multi-dose study of SRT2104, a SIRT1 activator, in subjects with type 2 diabetes. Br. J. Clin. Pharmacol. 2014, 78, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007, 450, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Usui, I.; Kanatani, Y.; Matsuya, Y.; Tsuneyama, K.; Fujisaka, S.; Bukhari, A.; Suzuki, H.; Senda, S.; Imanishi, S.; et al. Treatment with SRT1720, a SIRT1 activator, ameliorates fatty liver with reduced expression of lipogenic enzymes in MSG mice. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1179–E1186. [Google Scholar] [CrossRef] [PubMed]
- Pacholec, M.; Bleasdale, J.E.; Chrunyk, B.; Cunningham, D.; Flynn, D.; Garofalo, R.S.; Griffith, D.; Griffor, M.; Loulakis, P.; Pabst, B.; et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 2010, 285, 8340–8351. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Villalba, J.M.; Alcaín, F.J. Sirtuin activators and inhibitors. Biofactors 2012, 38, 349–359. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Gene | Polymorphism | Allele/Genotype | Association | Population | References |
|---|---|---|---|---|---|
| SIRT1 | rs2273773 (T/C) | C | Lower intima-media thickness in men | 1770 Austrian Caucasians | [98] |
| C | Higher intima-media thickness in women | 1770 Austrian Caucasians | [98] | ||
| C | Higher TC and LDL-C levels in male hemodialysis patients | 219 Japanese hemodialysis patients, 803 control subjects | [106] | ||
| T | Associated with cardiovascular diseases | 278 Turkish patients with CVD 135 controls | [103] | ||
| CT | Higher BMI compared to TT homozygotes | 3575 Dutch Caucasians | [90] | ||
| CT | Higher insulin levels | 120 obese Turkish children 120 lean controls | [95] | ||
| CC | Higher systolic and diastolic blood pressure in men | 1279 Japanese | [94] | ||
| TT | Higher fat content and higher fasting glucose in men | 1279 Japanese | [94] | ||
| TT | Higher diastolic blood pressure and higher TC and LDL-C levels | 70 Egyptian | [107] | ||
| No association with obesity and susceptibility to lifestyle modification | 196 German Caucasians | [93] | |||
| No influence on mortality and on glucose tolerance in obese individuals | 1390 Dutch Caucasians | [96] | |||
| rs7069102 (G/C) | G | Associated with CVD | 278 Turkish patients with CVD 135 controls | [103] | |
| CC | Lower risk of obesity but higher visceral fat Content in men | 1068 obese subjects, 313 lean controls (Belgian Caucasians) | [91] | ||
| GG | Higher fat content and higher systolic blood pressure in men | 1279 Japanese | [94] | ||
| No association with obesity | 896 obese subjects, 532 lean controls (French Caucasians) 154 Swedish families (732 subjects) 120 obese Turkish children and 120 lean controls | [77,95] | |||
| No association with obesity and susceptibility to lifestyle modification | 196 German Caucasians | [93] | |||
| No influence on mortality and on glucose tolerance in obese individuals | 1390 Dutch Caucasians | [96] | |||
| rs7895833 (A/G) | G | Lower BMI | 8598 Dutch Caucasians | [92] | |
| G | Higher BMI | 120 obese Turkish children 120 lean controls | [95] | ||
| rs7895833 (A/G) | A | Increased mortality in diabetic patients (in a haplotype with rs1467568G/ rs497849G) | 8598 Dutch Caucasians | [92] | |
| AA | higher BMI and higher fat content in men | 1279 Japanese | [94] | ||
| AA | higher diastolic blood pressure in women | 1279 Japanese | [94] | ||
| AG | Higher BMI | 120 obese Turkish children 120 lean controls | [95] | ||
| GG | Higher diastolic blood pressure and higher TC and LDL-C levels | 70 Egyptians | [107] | ||
| No association with BMI and fat content | 3501 Pima Indians 3003 Native Americans | [79] | |||
| rs1467568 (A/G) | G | lower BMI | 8598 Dutch Caucasians | [92] | |
| G | Increased mortality in diabetic patients (in a haplotype with rs7895833A/rs497849G) | 8598 Dutch Caucasians | [92] | ||
| G | Lower intima-media thickness in men | 1770 Austrian Caucasians | [98] | ||
| G | Higher intima-media thickness in women | 1770 Austrian Caucasians | [98] | ||
| No association with obesity | 896 obese subjects, 532 lean controls (French Caucasians) 154 Swedish families (732 subjects) | [77] | |||
| rs12413112 (G/A) | A | Higher BMI | 896 obese subjects, 532 lean controls (French Caucasians) 154 Swedish families (732 subjects) | [77] | |
| A | Lower energy expenditure and resistance to lifestyle interventions | 196 German Caucasians | [93] | ||
| A | Higher mean common intima-media thickness | 1770 Austrian Caucasians | [98] | ||
| No association with BMI and weight | 1279 Japanese | [94] | |||
| rs33957861 (C/T) | T | Higher BMI | 896 obese subjects, 532 lean controls (French Caucasians) 154 Swedish families (732 subjects) | [77] | |
| rs11599176 (A/G) | G | Higher BMI | 896 obese subjects, 532 lean controls (French Caucasians) 154 Swedish families (732 subjects) | [77] | |
| rs35689145 (G/A) | A | Higher BMI | 896 obese subjects, 532 lean controls (French Caucasians) 154 Swedish families (732 subjects) | [77] | |
| rs730821 (A/G) | No association with BMI and weight | 1279 Japanese | [94] | ||
| rs12778366 (C/T) | C | Reduced mortality in obese/overweight individuals | 1390 Dutch Caucasians | [96] | |
| C | Better glucose tolerance in men | 1390 Dutch Caucasians | [96] | ||
| rs12049646 (C/T) | T | Higher BMI in men | 1802 Spanish Caucasians | [97] | |
| rs3740051 (A/G) | G | Lower intima-media thickness in men | 1770 Austrian Caucasians | [98] | |
| G | Higher intima-media thickness in women | 1770 Austrian Caucasians | [98] | ||
| rs2236319 (A/G) | G | Lower intima-media thickness in men | 1770 Austrian Caucasians | [98] | |
| G | Higher intima-media thickness in women | 1770 Austrian Caucasians | [98] | ||
| A | Associated with diabetic nephropathy | 1502 Japanese patients with T2DM 1740 controls | [105] | ||
| rs10823108 (G/A) | A | Lower intima-media thickness in men | 1770 Austrian Caucasians | [98] | |
| A | Higher intima-media thickness in women | 1770 Austrian Caucasians | [98] | ||
| G | Associated with diabetic nephropathy | 1502 Japanese patients with T2DM 1740 controls | [105] | ||
| rs4746720 (T/C) | T | Associated with diabetic nephropathy | 1502 Japanese patients with T2DM 1740 controls | [105] | |
| No association with BMI and fat content | 3501 Pima Indians 3003 Native Americans | [79] | |||
| rs497849 (G/A) | G | Increased mortality in diabetic patients (in a haplotype with rs1467568G/rs7895833A) | 8598 Dutch Caucasians | [92] | |
| rs10509291 (T/A) | T | Associated with type 2 diabetes | 3501 Pima Indians | [79] | |
| T | Not associated with type 2 diabetes | 3003 Native Americans | [79] | ||
| No association with BMI and fat content | 6504 North Americans | [79] | |||
| rs7896005 (G/A) | G | Associated with type 2 diabetes | 3501 Pima Indians | [79] | |
| G | Not associated with type 2 diabetes | 3003 Native Americans | [79] | ||
| – | No association with BMI and fat content | 6504 North Americans | [79] | ||
| rs3758391 (C/T) | C | Protects from type 2 diabetes | 519 Mexican patients with T2DM 389 Mexican patients with MS 547 Mexican controls | [104] | |
| rs3818292 (A/G) | A | Associated with diabetic nephropathy | 1502 Japanese patients with T2DM 1740 controls | [105] | |
| SIRT2 | rs4802998 (A/G) | G | Higher intima-media thickness | 1356 North Americans | [100] |
| SIRT3 | rs12363280 (C/G) | G | Higher grey scale median indicator of plaque morphology and a predictor of stroke | 1356 North Americans | [99] |
| C | Lower intima media thickness | 1356 North Americans | [100] | ||
| rs4980329 (T/C) | T | Higher grey scale median—an indicator of plaque morphology and a predictor of stroke | 1356 North Americans | [99] | |
| rs3825075 (T/C) | TT | Lower intima media thickness in women | 1356 North Americans | [100] | |
| SIRT5 | rs4712032 (A/G) | G | Increased number of carotid plaques | 1356 North Americans | [99] |
| rs12216101 (G /T) | G | Increased number of carotid plaques | 1356 North Americans | [99] | |
| SIRT6 | rs107251 (C/T) | T | Increased number of carotid plaques | 1356 North Americans | [99,101] |
| rs3760905 (G/T) | T | Increased number of carotid plaques | 1356 North Americans | [99,101] | |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurylowicz, A. In Search of New Therapeutic Targets in Obesity Treatment: Sirtuins. Int. J. Mol. Sci. 2016, 17, 572. https://doi.org/10.3390/ijms17040572
Kurylowicz A. In Search of New Therapeutic Targets in Obesity Treatment: Sirtuins. International Journal of Molecular Sciences. 2016; 17(4):572. https://doi.org/10.3390/ijms17040572
Chicago/Turabian StyleKurylowicz, Alina. 2016. "In Search of New Therapeutic Targets in Obesity Treatment: Sirtuins" International Journal of Molecular Sciences 17, no. 4: 572. https://doi.org/10.3390/ijms17040572
APA StyleKurylowicz, A. (2016). In Search of New Therapeutic Targets in Obesity Treatment: Sirtuins. International Journal of Molecular Sciences, 17(4), 572. https://doi.org/10.3390/ijms17040572