
Epigenetic Modifications in Essential Hypertension


Abstract
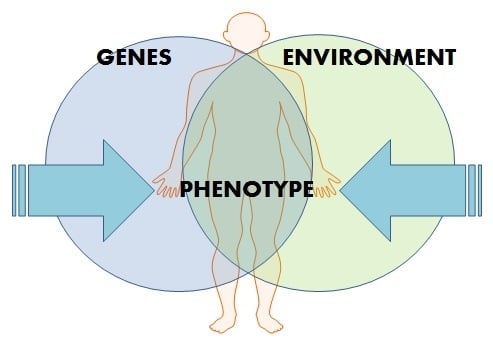
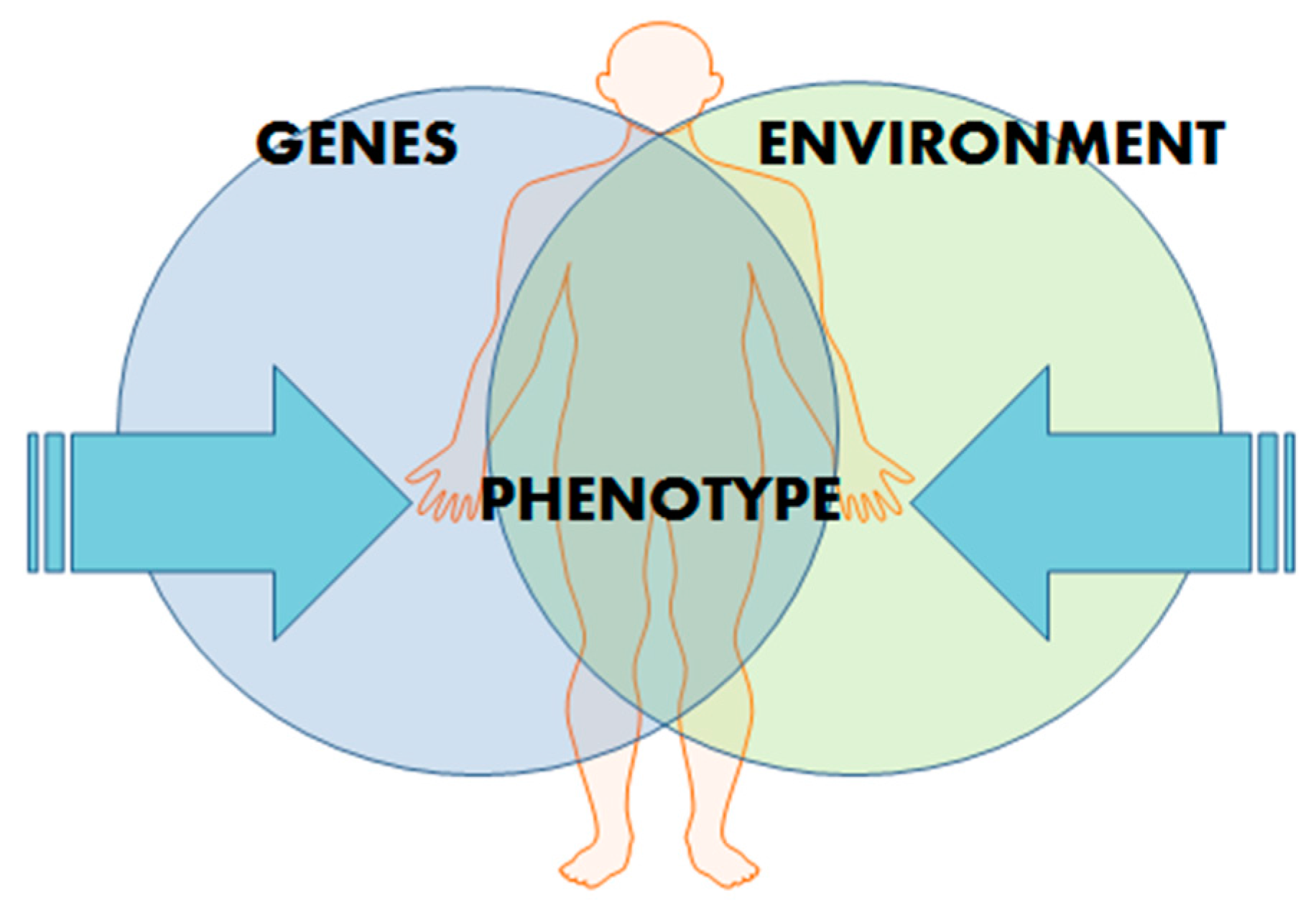
:
1. Introduction
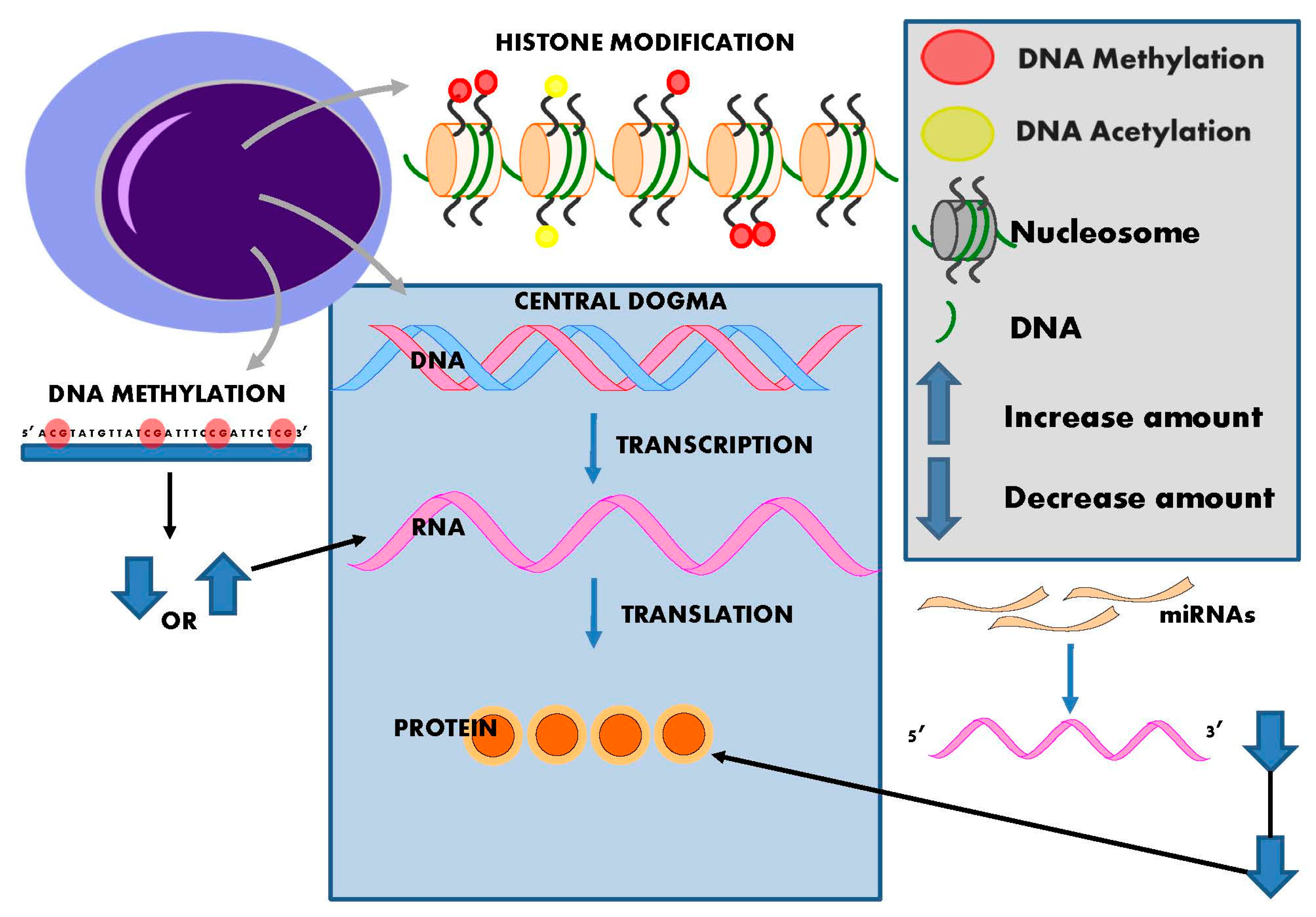
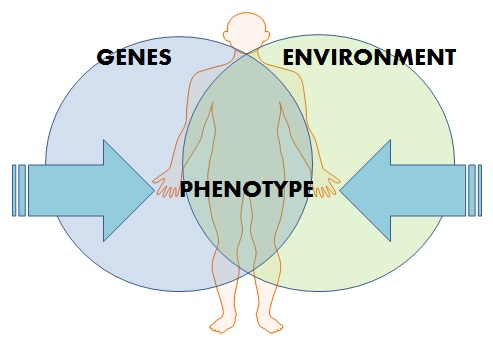
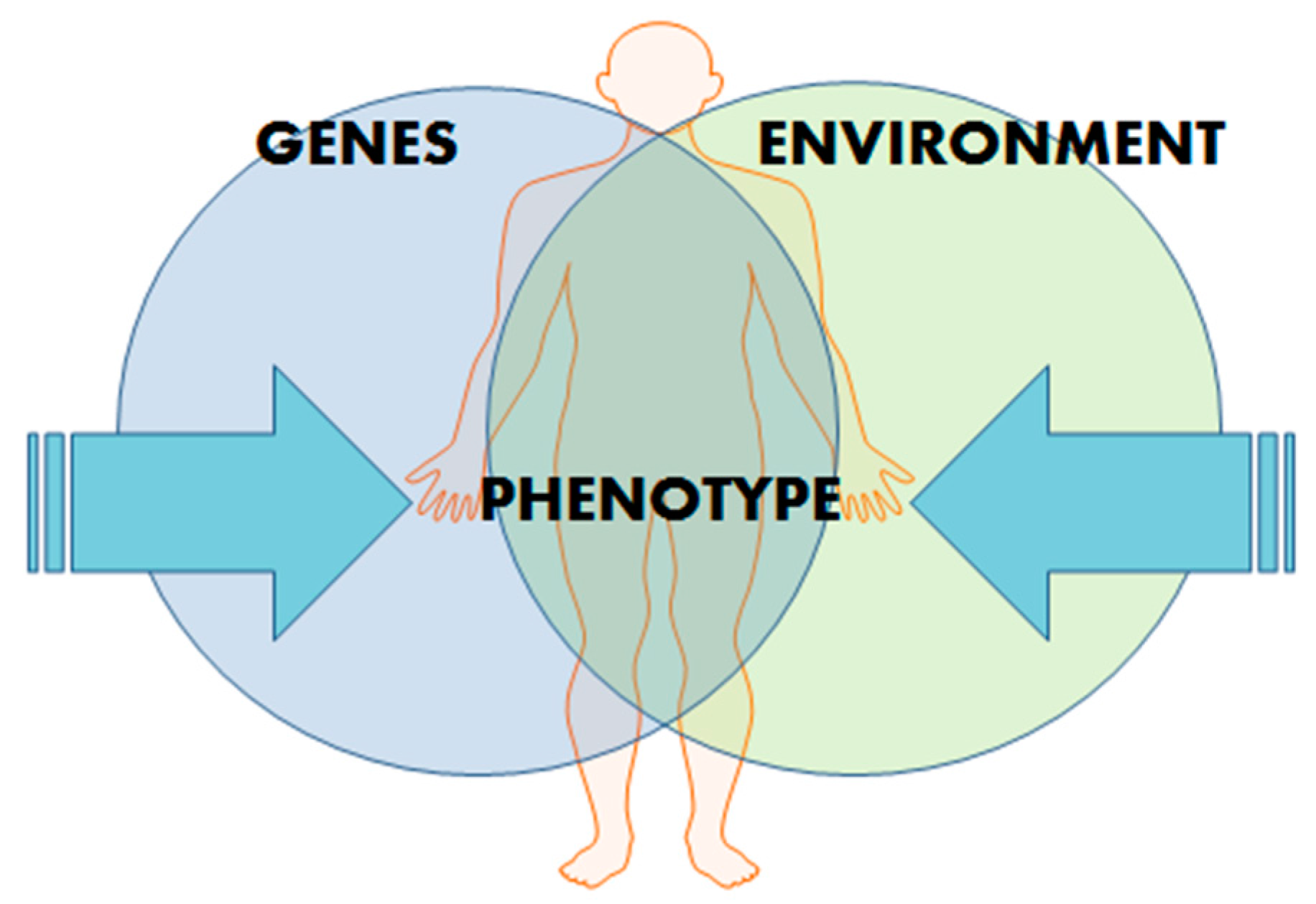
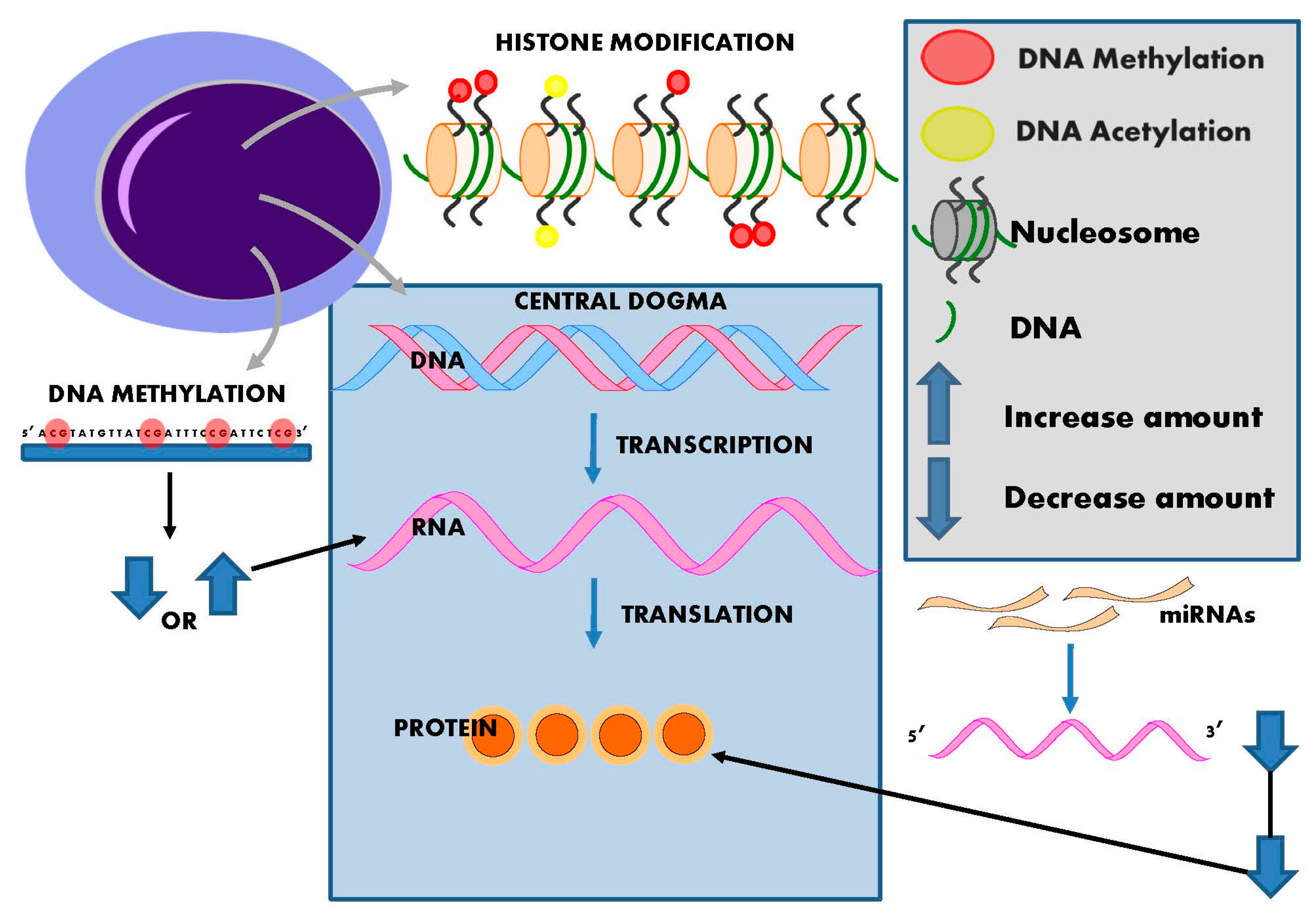
2. Epigenetics
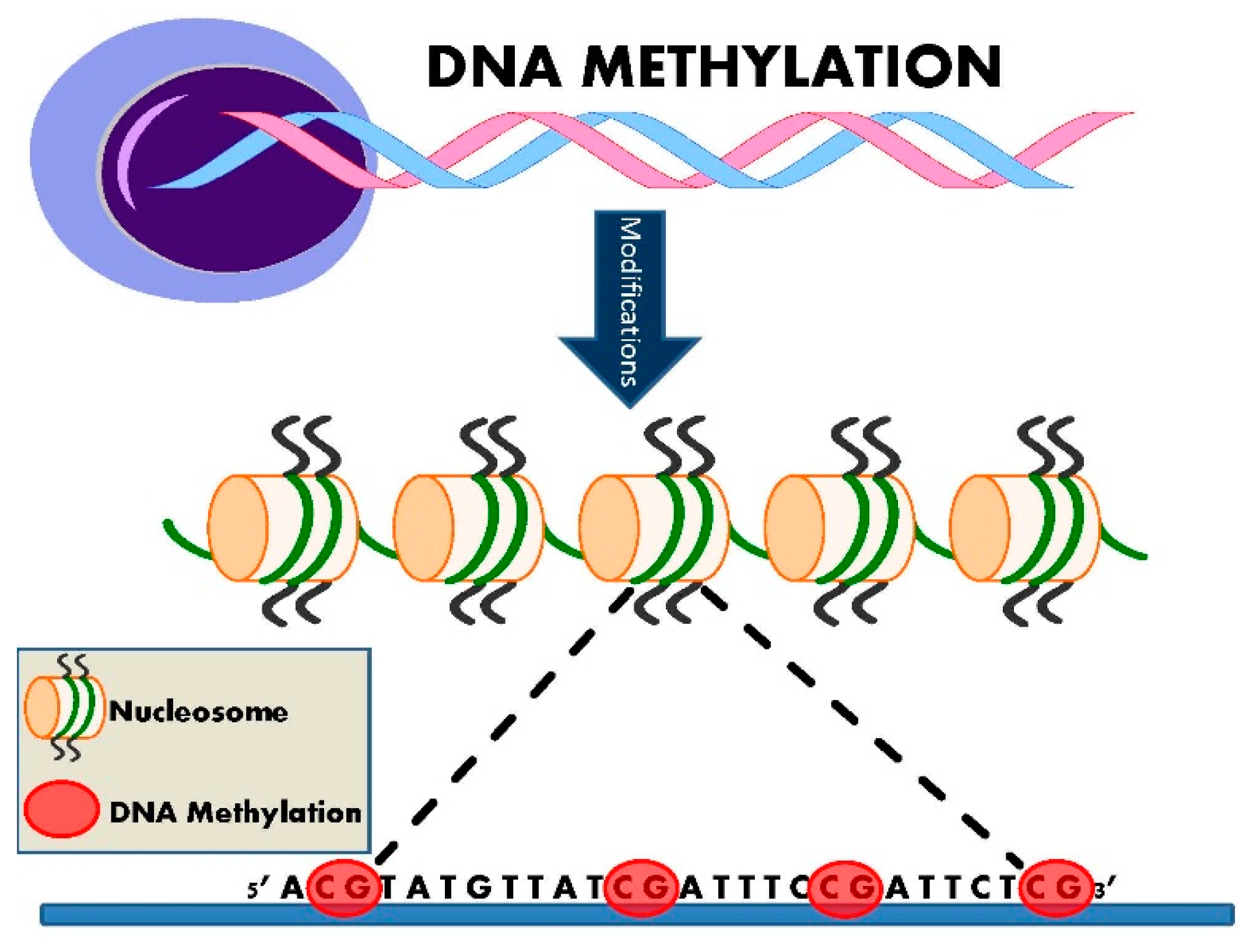
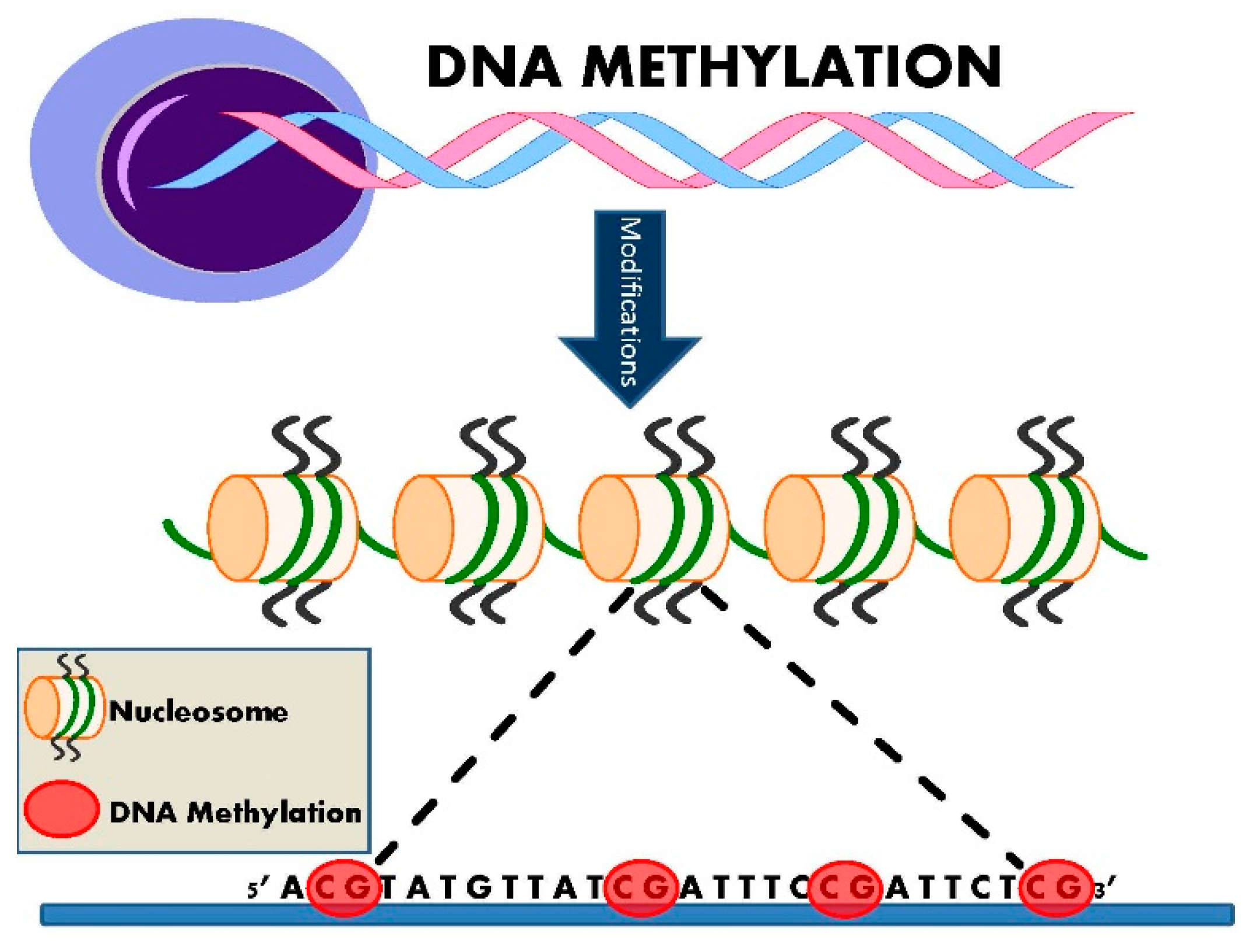
3. DNA Methylation
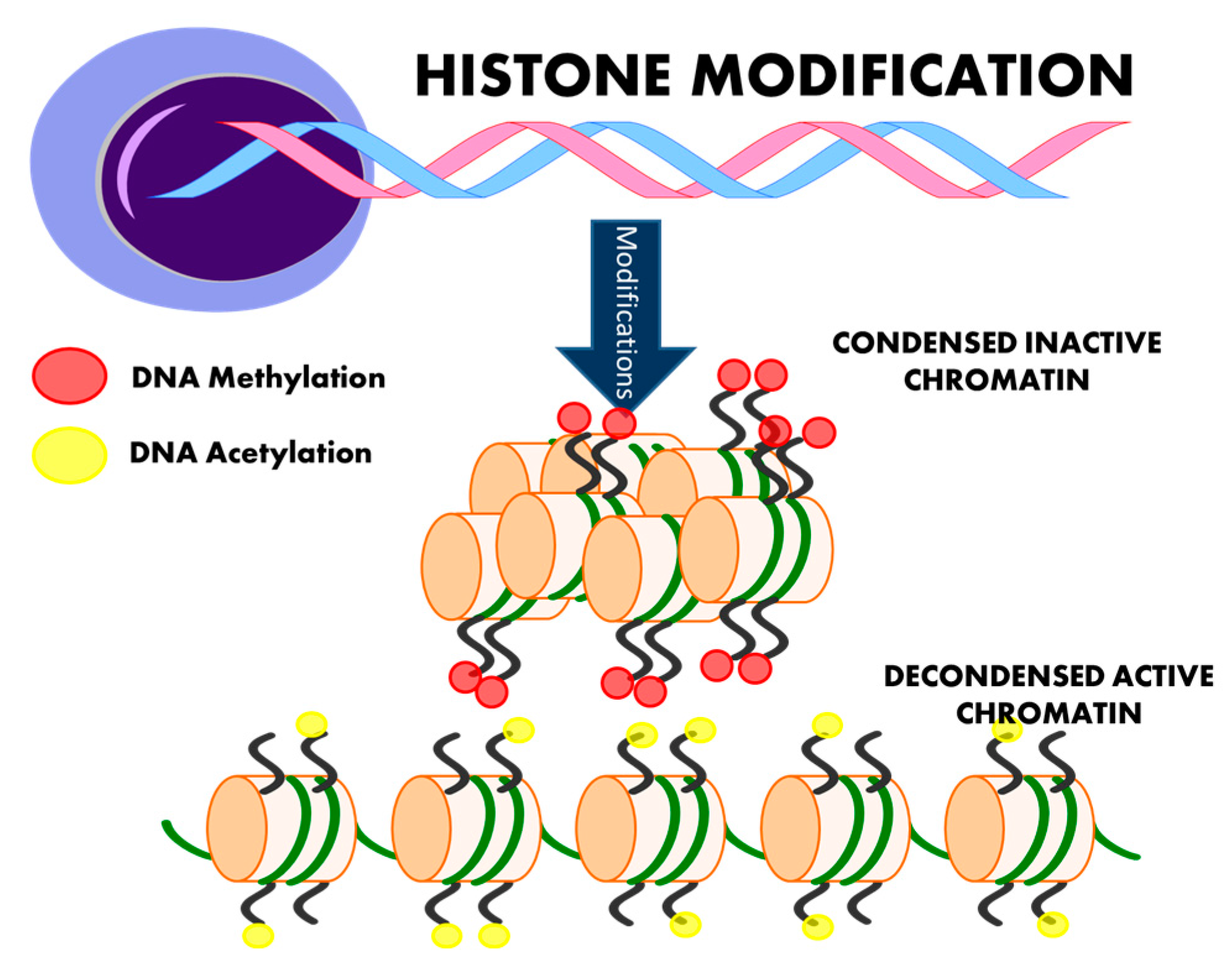
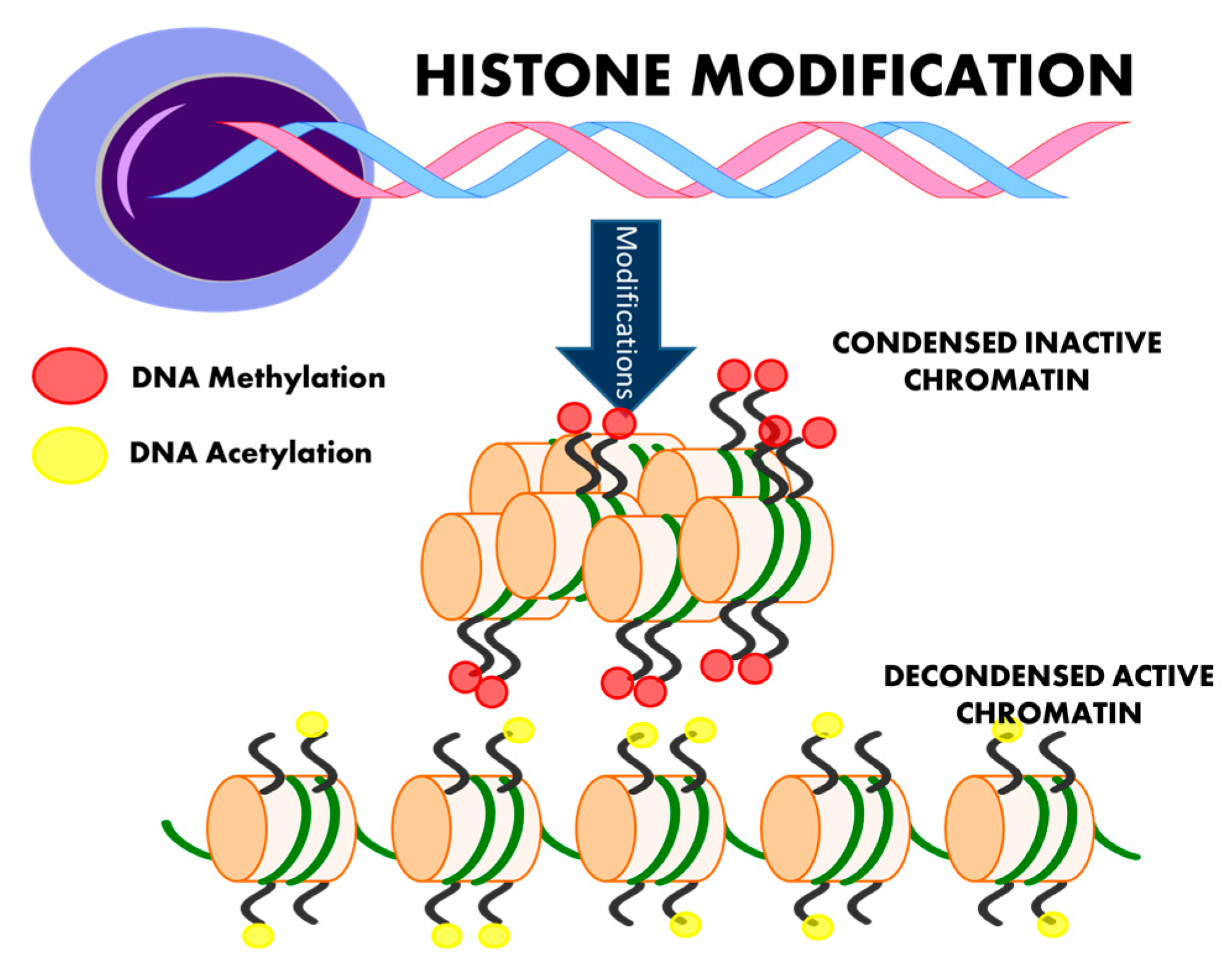
4. Histone Modification and Hypertension
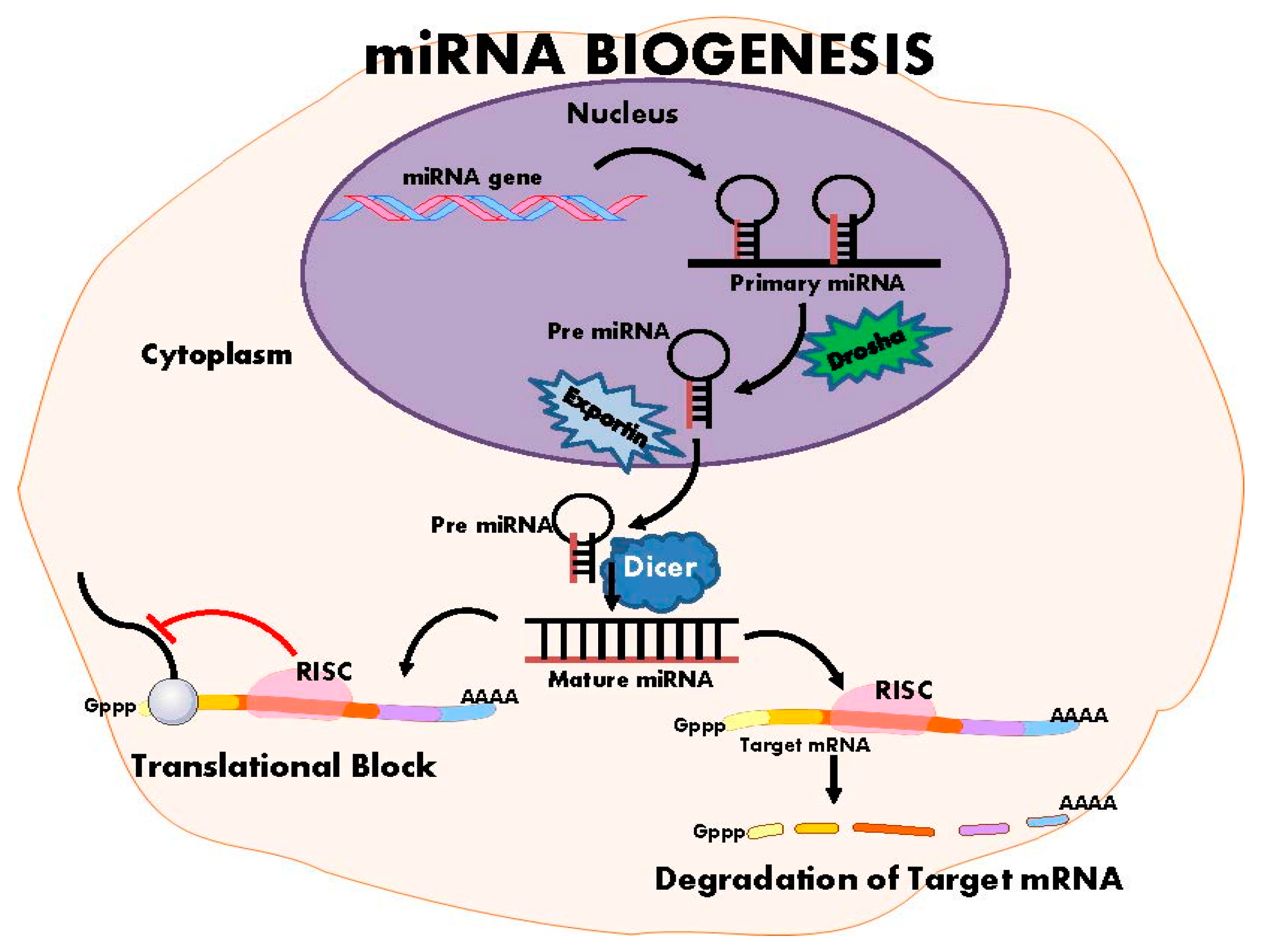
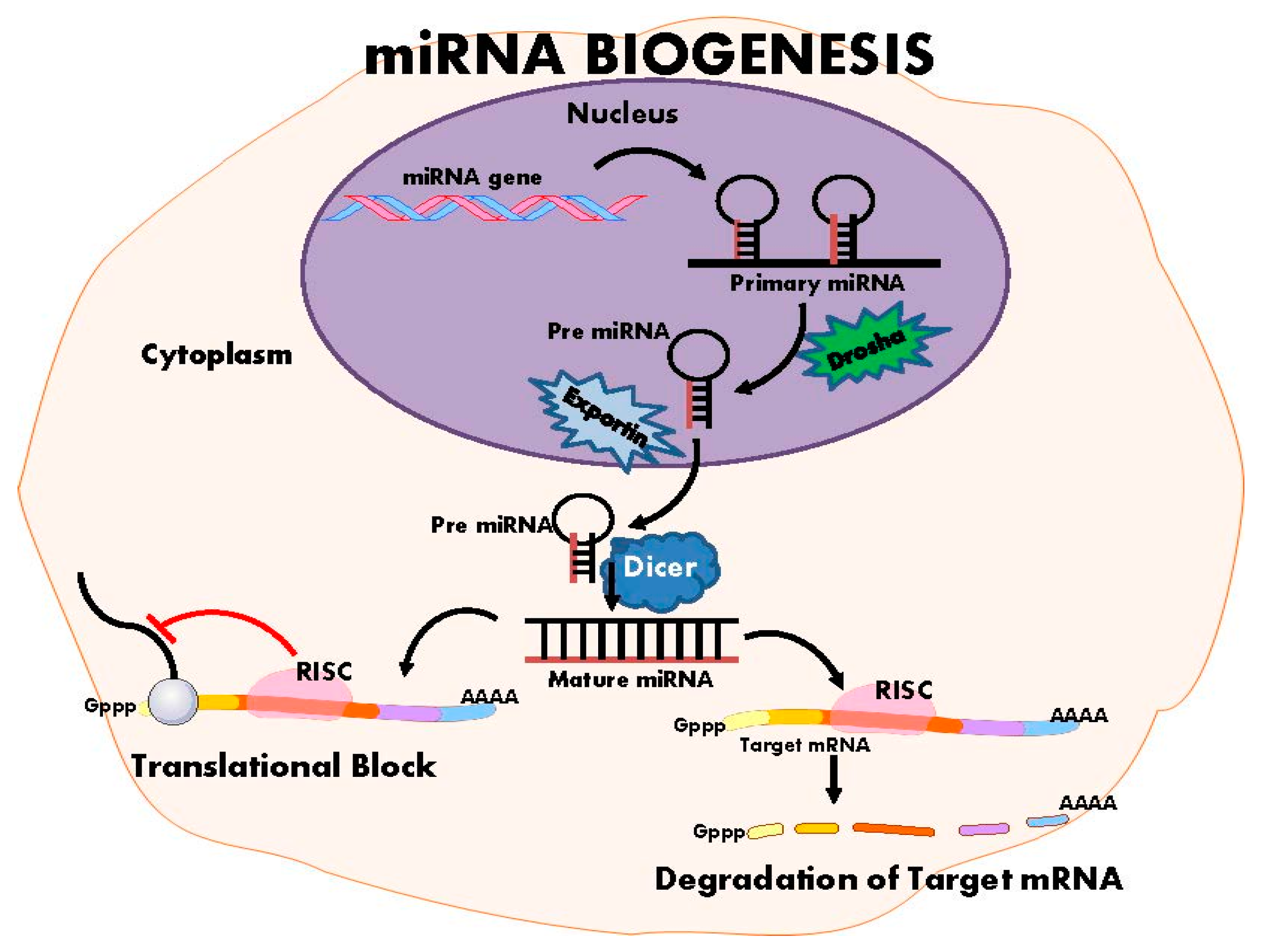
5. Non-Coding RNAs and Hypertension
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; AlMazroa, M.A.; Amann, M.; Anderson, H.R.; Andrews, K.G.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the global burden of disease study 2010. Lancet 2013, 380, 2224–2260. [Google Scholar] [CrossRef]
- Poulter, N.R.; Prabhakaran, D.; Caulfield, M. Hypertension. Lancet 2015, 386, 801–812. [Google Scholar] [CrossRef]
- Kunes, J.; Zicha, J. The interaction of genetic and environmental factors in the etiology of hypertension. Physiol. Res. 2009, 58 (Suppl. 2), S33–S41. [Google Scholar] [PubMed]
- Loscalzo, J.; Handy, D.E. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease (2013 grover conference series). Pulm. Circ. 2014, 4, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, J.M.; Martino, F.; Thum, T. Epigenetic modifications in cardiovascular disease. Basic Res. Cardiol. 2012, 107, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Khalil, C.A. The emerging role of epigenetics in cardiovascular disease. Ther. Adv. Chronic Dis. 2014, 5, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gong, L.; Tan, Y.; Hui, R.; Wang, Y. Hypertensive epigenetics: From DNA methylation to microRNAs. J. Hum. Hypertens. 2015, 29, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Bátkai, S.; Thum, T. MicroRNAs in hypertension: Mechanisms and therapeutic targets. Curr. Hypertens. Rep. 2012, 14, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Millis, R.M. Epigenetics and hypertension. Curr. Hypertens. Rep. 2011, 13, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Raftopoulos, L.; Katsi, V.; Makris, T.; Tousoulis, D.; Stefanadis, C.; Kallikazaros, I. Epigenetics, the missing link in hypertension. Life Sci. 2015, 129, 22–26. [Google Scholar] [CrossRef] [PubMed]
- López-Jaramillo, P.; Camacho, P.A.; Forero-Naranjo, L. The role of environment and epigenetics in hypertension. Expert Rev. Cardiovasc. Ther. 2013, 11, 1455–1457. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Hildmann, T.; Novik, K.L.; Lewin, J.; Tost, J.; Cox, A.V.; Andrews, T.D.; Howe, K.L.; Otto, T.; Olek, A.; et al. DNA methylation profiling of the human major histocompatibility complex: A pilot study for the human epigenome project. PLoS Biol. 2004, 2. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A. Project set to map marks on genome. Nature 2010, 463, 596–597. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, G.; Feil, R. New insights into establishment and maintenance of DNA methylation imprints in mammals. Philos. Trans. R. Soc. Biol. Sci. 2013, 368. [Google Scholar] [CrossRef]
- Kunes, J.; Kadlecova, M.; Vaneckova, I.; Zicha, J. Critical developmental periods in the pathogenesis of hypertension. Physiol. Res. 2012, 61 (Suppl. 1), S9–S17. [Google Scholar] [PubMed]
- Wilson, A.G. Epigenetic regulation of gene expression in the inflammatory response and relevance to common diseases. J. Periodontol. 2008, 79, 1514–1519. [Google Scholar] [PubMed]
- Miranda, T.B.; Jones, P.A. DNA methylation: The nuts and bolts of repression. J. Cell. Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Fouse, S.D.; Nagarajan, R.P.; Costello, J.F. Genome-scale DNA methylation analysis. Epigenomics 2010, 2, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Smolarek, I.; Wyszko, E.; Barciszewska, A.M.; Nowak, S.; Gawronska, I.; Jablecka, A.; Barciszewska, M.Z. Global DNA methylation changes in blood of patients with essential hypertension. Med. Sci. Monit. Basic Res. 2010, 16, CR149–CR155. [Google Scholar]
- Kato, N.; Loh, M.; Takeuchi, F.; Verweij, N.; Wang, X.; Zhang, W.; Kelly, T.N.; Saleheen, D.; Lehne, B.; Leach, I.M.; et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat. Genet. 2015, 47, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Friso, S.; Pizzolo, F.; Choi, S.-W.; Guarini, P.; Castagna, A.; Ravagnani, V.; Carletto, A.; Pattini, P.; Corrocher, R.; Olivieri, O. Epigenetic control of 11 β-hydroxysteroid dehydrogenase 2 gene promoter is related to human hypertension. Atherosclerosis 2008, 199, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.; Goyal, D.; Leitzke, A.; Gheorghe, C.P.; Longo, L.D. Brain renin-angiotensin system: Fetal epigenetic programming by maternal protein restriction during pregnancy. Reprod. Sci. 2010, 17, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Falkner, B.; Zhu, H.; Shi, H.; Su, S.; Xu, X.; Sharma, A.K.; Dong, Y.; Treiber, F.; Gutin, B.; et al. A genome-wide methylation study on essential hypertension in young african american males. PLoS ONE 2013, 8, e53938. [Google Scholar] [CrossRef] [PubMed]
- Pei, F.; Wang, X.; Yue, R.; Chen, C.; Huang, J.; Huang, J.; Li, X.; Zeng, C. Differential expression and DNA methylation of angiotensin type 1A receptors in vascular tissues during genetic hypertension development. Mol. Cell. Biochem. 2015, 402, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rivière, G.; Lienhard, D.; Andrieu, T.; Vieau, D.; Frey, B.M.; Frey, F.J. Epigenetic regulation of somatic angiotensin-converting enzyme by DNA methylation and histone acetylation. Epigenetics 2011, 6, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-A.; Baek, I.; Seok, Y.M.; Yang, E.; Cho, H.-M.; Lee, D.-Y.; Hong, S.H.; Kim, I.K. Promoter hypomethylation upregulates Na+-K+-2Cl− cotransporter 1 in spontaneously hypertensive rats. Biochem. Biophys. Res. Commun. 2010, 396, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, P.; Sansonnens, A.; Dick, B.; Frey, F.J. In vivo 11β-HSD-2 activity variability, salt-sensitivity, and effect of licorice. Hypertension 2001, 38, 1330–1336. [Google Scholar] [CrossRef] [PubMed]
- Udali, S.; Guarini, P.; Moruzzi, S.; Choi, S.-W.; Friso, S. Cardiovascular epigenetics: From DNA methylation to microRNAs. Mol. Asp. Med. 2013, 34, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Bogdarina, I.; Welham, S.; King, P.J.; Burns, S.P.; Clark, A.J. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ. Res. 2007, 100, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hong, X.-M.; Xing, H.-X.; Li, J.-P.; Huo, Y.; Xu, X.-P. E112D polymorphism in the prolylcarboxypeptidase gene is associated with blood pressure response to benazepril in chinese hypertensive patients. Chin. Med. J. 2009, 122, 2461–2465. [Google Scholar] [PubMed]
- Esler, M.; Eikelis, N.; Schlaich, M.; Lambert, G.; Alvarenga, M.; Kaye, D.; El-Osta, A.; Guo, L.; Barton, D.; Pier, C.; et al. Human sympathetic nerve biology: Parallel influences of stress and epigenetics in essential hypertension and panic disorder. Ann. N. Y. Acad. Sci. 2008, 1148, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Uludag, M.O.; Usanmaz, S.E.; Ayaloglu-Butun, F.; Akcali, K.C.; Demirel-Yilmaz, E. Resveratrol affects histone 3 lysine 27 methylation of vessels and blood biomarkers in DOCA salt-induced hypertension. Mol. Biol. Rep. 2015, 42, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.E.; Matouk, C.C.; Rachlis, A.; Lin, S.; Tai, S.C.; D’Abreo, C.; Marsden, P.A. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J. Biol. Chem. 2005, 280, 24824–24838. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-A.; Cho, H.-M.; Lee, D.-Y.; Kim, K.-C.; Han, H.S.; Kim, I.K. Tissue-specific upregulation of angiotensin-converting enzyme 1 in spontaneously hypertensive rats through histone code modifications. Hypertension 2012, 59, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-M.; Lee, D.-Y.; Kim, H.Y.; Lee, H.-A.; Seok, Y.M.; Kim, I.K. Upregulation of the Na+-K+-2Cl− cotransporter 1 via histone modification in the aortas of angiotensin II-induced hypertensive rats. Hypertens. Res. 2012, 35, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.D.; Zineh, I.; Burkley, B.; Gong, Y.; Langaee, T.Y.; Turner, S.T.; Chapman, A.B.; Boerwinkle, E.; Gums, J.G.; Cooper-DeHoff, R.M.; et al. Effects of genetic variation in H3K79 methylation regulatory genes on clinical blood pressure and blood pressure response to hydrochlorothiazide. J. Transl. Med. 2012, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Mu, S.; Shimosawa, T.; Ogura, S.; Wang, H.; Uetake, Y.; Kawakami-Mori, F.; Marumo, T.; Yatomi, Y.; Geller, D.S.; Tanaka, H.; et al. Epigenetic modulation of the renal β-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat. Med. 2011, 17, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.R.; Lokhandwala, M.F.; Banday, A.A. Resveratrol prevents endothelial nitric oxide synthase uncoupling and attenuates development of hypertension in spontaneously hypertensive rats. Eur. J. Pharmacol. 2011, 667, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Iturbe, B. Arteriolar remodeling in essential hypertension: Are connective tissue growth factor and transforming growth factor involved? Kidney Int. 2006, 69, 1104–1105. [Google Scholar] [CrossRef] [PubMed]
- Katholi, R.E.; Naftilan, A.J.; Oparil, S. Importance of renal sympathetic tone in the development of DOCA-salt hypertension in the rat. Hypertension 1980, 2, 266–273. [Google Scholar] [CrossRef] [PubMed]
- DiBona, G.F. Physiology in perspective: The wisdom of the body. Neural control of the kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R633–R641. [Google Scholar] [CrossRef] [PubMed]
- Guild, S.-J.; Eppel, G.A.; Malpas, S.C.; Rajapakse, N.W.; Stewart, A.; Evans, R.G. Regional responsiveness of renal perfusion to activation of the renal nerves. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R1177–R1186. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Ring, A.M.; Lifton, R.P. Molecular physiology of the WNK kinases. Annu. Rev. Physiol. 2008, 70, 329–355. [Google Scholar] [CrossRef] [PubMed]
- Chiga, M.; Rai, T.; Yang, S.-S.; Ohta, A.; Takizawa, T.; Sasaki, S.; Uchida, S. Dietary salt regulates the phosphorylation of OSR1/SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int. 2008, 74, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.; Marshall, E.; MacGillivray, T.; Mittal, M.; Xue, W.; Kenyon, C.J.; Brown, R.W. Dietary electrolyte-driven responses in the renal WNK kinase pathway in vivo. J. Am. Soc. Nephrol. 2006, 17, 2402–2413. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Rezai-Zadeh, N.; Seto, E. Negative regulation of histone deacetylase 8 activity by cyclic AMP-dependent protein kinase A. Mol. Cell. Biol. 2004, 24, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, N.M.; Snyder, S.K.; Kolla, S.S.; Smith, C.L. Chromatin-dependent regulation of the MMTV promoter by cAMP signaling is mediated through distinct pathways. Exp. Cell Res. 2003, 287, 361–373. [Google Scholar] [CrossRef]
- Nguyen Dinh Cat, A.L.; Ouvrard-Pascaud, A.; Tronche, F.; Clemessy, M.; Gonzalez-Nunez, D.; Farman, N.; Jaisser, F. Conditional transgenic mice for studying the role of the glucocorticoid receptor in the renal collecting duct. Endocrinology 2009, 150, 2202–2210. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.F. Non-coding RNAs, epigenetics and complexity. Gene 2008, 410, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.Z.; Charchar, F.J. MicroRNAs in essential hypertension and blood pressure regulation. In MicroRNA: Medical Evidence; Springer: Berlin, Germany, 2015; pp. 215–235. [Google Scholar]
- Marques, F.Z.; Morris, B.J. Neurogenic hypertension: Revelations from genome-wide gene expression profiling. Curr. Hypertens. Rep. 2012, 14, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Sethupathy, P.; Borel, C.; Gagnebin, M.; Grant, G.R.; Deutsch, S.; Elton, T.S.; Hatzigeorgiou, A.G.; Antonarakis, S.E. Human microRNA-155 on chromosome 21 differentially interacts with its polymorphic target in the agtr1 3′ untranslated region: A mechanism for functional single-nucleotide polymorphisms related to phenotypes. Am. J. Hum. Genet. 2007, 81, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Liu, T.; Jiang, F.; Liu, C.; Zhao, X.; Gao, Y.; Wang, H.; Liu, Z. MicroRNA-155 regulates angiotensin II type 1 receptor expression in umbilical vein endothelial cells from severely pre-eclamptic pregnant women. Int. J. Mol. Med. 2011, 27, 393–399. [Google Scholar] [PubMed]
- Marques, F.Z.; Campain, A.E.; Tomaszewski, M.; Zukowska-Szczechowska, E.; Yang, Y.H.J.; Charchar, F.J.; Morris, B.J. Gene expression profiling reveals renin mRNA overexpression in human hypertensive kidneys and a role for microRNAs. Hypertension 2011, 58, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chabrashvili, T.; Borrego, L.; Aslam, S.; Umans, J.G. Angiotensin II infusion alters vascular function in mouse resistance vessels: Roles of O–·2 and endothelium. J. Vasc. Res. 2005, 43, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef]
- Annilo, T.; Kepp, K.; Laan, M. Natural antisense transcript of natriuretic peptide precursor A (NPPA): Structural organization and modulation of NPPA expression. BMC Mol. Biol. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Kong, Q.; Kone, B.C. CREB trans-activation of disruptor of telomeric silencing-1 mediates forskolin inhibition of ctgf transcription in mesangial cells. Am. J. Physiol. Ren. Physiol. 2010, 298, F617–F624. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybyłowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Johnson, R.; Stanton, L.W. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 2012, 31, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Paralkar, V.R.; Weiss, M.J. A new “Linc” between noncoding RNAs and blood development. Genes Dev. 2011, 25, 2555–2558. [Google Scholar] [CrossRef] [PubMed]
- Pauli, A.; Rinn, J.L.; Schier, A.F. Non-coding RNAs as regulators of embryogenesis. Nat. Rev. Genet. 2011, 12, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Niland, C.N.; Merry, C.R.; Khalil, A.M. Emerging roles for long non-coding RNAs in cancer and neurological disorders. Front. Genet. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Ørom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Tissue Type/Sample Size | Findings |
|---|---|---|
| Smolarek, et al. [19] | Human: Peripheral blood; 60 with EH; 30 controls | Mean 5mC amount in DNA significantly decreased as HT severity increased. |
| Kato, et al. [20] | Human: Peripheral blood, cord blood, muscle, liver, fat; 320,251 individuals of East Asian, European and South Asian ancestry | Multiple genetic variants involved with vascular smooth muscle (IGFBP3, KCNK3, PDE3A, PRDM6) and renal function (ARHGAP24, OSR1, SLC22A7, TBX2) discovered to be correlated with BP modulation. Two-fold enrichment discovered between DNA methylation and sentinel blood pressure SNPs, providing evidence for DNA methylation role in blood pressure regulation. |
| Friso, et al. [21] | Human: Peripheral blood; 25 with EH; 32 with prednisone therapy | HSD11B2 gene promoter methylation associated with EH onset via disruption to THF/The ratio. |
| Goyal, et al. [22] | Rat: Tissues: brain; 20 MLP pups, 17 control pups | Hypomethylation of RAAS system genes such as ACE resulting in HT in offspring. |
| Wang, et al. [23] | Human: Peripheral blood; 8 EH; 8 control | PRCP gene hypomethylated in EH, linked to disruption in cleavage of angiotensin II and III. |
| Pei, et al. [24] | Rat: Tissue: aorta; 6 Spontaneously HT; 6 WKY control | Atgr1a gene progressively hypomethylated as SHR age progressed. Indicating increased expression of Atgr1a in aging SHR. |
| Riviere, et al. [25] | Rat: Cultured endothelial cells from WKY | Hypermethylation associated with trasciptional repression of sACE, indicating a role for epigenetics in sACE modulation during HT. |
| Lee, et al. [26] | Rat: Tissues: aorta, heart; SHR and WKY | Hypomethylation of Sic2a2 gene lead to increased expression of NKCC1 which was positively correlated with HT. |
| Reference | Tissue Type/Sample Size | Findings |
|---|---|---|
| Han, et al. [33] | Rat: Tissue: Aorta, renal artery; SHR and WKY | Up-regulated histone modifier H3K27me3 in renal artery of SHR correlated with HT improvement after resveratrol intake. |
| Fish, et al. [34] | Human: Umbilical vein endothelial cells | Endothelial cell nucleosomes corresponding to eNOS enriched in various histones relevant to eNOS expression. |
| Lee, et al. [35] | Rat: Tissue: adrenal gland, aorta, heart, kidney, liver, and lung. SHR and WKY | Higher expression of Ace1 mRNA & protein in SHR. Ace1 promoter enriched with H3Ac and H3K4me3 in SHR. |
| Cho, et al. [36] | Rat: Tissue: Mesenteric artery, aorta; SD and Sham rat. | Nkcc1 up-regulated in SD rat. Acetylated histone H3 up-regulated, trimethylated histone H3 down-regulated. |
| Duarte, et al. [37] | Human: Peripheral blood; First sample: 206 mixed sex, normotensive; Second sample: 730 mixed sex, HT and normotensive. | DOT1L strongly associated with increased BP in Caucasians. Possibly via mediation of hypermethylation of H3. |
| Mu, et al. [38] | Mouse: Tissue: Kidney; norepinephrine infused-C57 BL/6j, Adrb1 knockout and Adrb2 knockout mice. | WNK4 down-regulation caused increased H3 & H4 acetylation, leading to overexpression of NCC and therefore promoting HT onset. |
| Reference | Tissue Type/Sample Size | Findings |
|---|---|---|
| Goyal, et al. [22] | Rat: Tissues: brain; 20 maternal low protein pups, 17 control pups | mmu-miR-27a and mmu-miR-27b regulate ACE1 and was upregulated (3.3- and 8.8-fold respectively) in MLP rat; mmu-mir-330 regulates angiotensin II type 2 receptor (A) and was downregulated 3.5-fold in MLP rat. |
| Sethupathy, et al. [55] | Human: Fibroblasts from monozygotic twin; n = 2 | Has-miR-155 binds to 3’UTR of AGR1 mRNA “A” allele causing a reduction in AGTR1 mRNA, reducing the pressor effect in response to angiotensin II. |
| Cheng, et al. [56] | Human: Endothelial cells from pre-eclamptic placentas | Has-miR-155 up-regulated in preeclampsia placentas, indicating involvement in regulation of AGTR1. |
| Marques, et al. [57] | Human: Tissue: kidney; Sample 1: 42 mixed sex, Polish individuals of mixed HT status; Sample 2: 22 male only, mixed HT status. All samples untreated for HT | Has-miR-181a & has-miR-663 is able to bind 3’UTR of renin mRNA, found to be underexpressed in EH. These miRNA able to regulate renin mRNA directly, explaining overexpression of renin in EH kidney. |
| Wang, et al. [58] | Mouse: Tissue: Mesenteric arterioles; 16 male C57Bl/5 mice; 16 sham mouse control | siRNA targeting p22phox mRNA demonstrated inhibition of contractile response from angiotensin II, consequently lowering BP. |
| Cabili, et al. [59] | Human: Tissue: 24 various and cells lines; 24 human samples | lincRNAs may promote the transcription of their neighbouring coding genes, including those implicated in EH and BP regulation. |
| Annilo, et al. [60] | Human and Mouse: Various tissues and cell lines; n = not disclosed | Seven blood pressure candidate genes ADD3, NPPA, ATP1A1, NPR2, CYP17A1, ACSM3 and SLC14A2 were connected with cis-lncRNA transcripts. |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wise, I.A.; Charchar, F.J. Epigenetic Modifications in Essential Hypertension. Int. J. Mol. Sci. 2016, 17, 451. https://doi.org/10.3390/ijms17040451
Wise IA, Charchar FJ. Epigenetic Modifications in Essential Hypertension. International Journal of Molecular Sciences. 2016; 17(4):451. https://doi.org/10.3390/ijms17040451
Chicago/Turabian StyleWise, Ingrid A., and Fadi J. Charchar. 2016. "Epigenetic Modifications in Essential Hypertension" International Journal of Molecular Sciences 17, no. 4: 451. https://doi.org/10.3390/ijms17040451
APA StyleWise, I. A., & Charchar, F. J. (2016). Epigenetic Modifications in Essential Hypertension. International Journal of Molecular Sciences, 17(4), 451. https://doi.org/10.3390/ijms17040451