
Inflammation in Chronic Wounds




Abstract
:
{kind=link}
{kind=link}
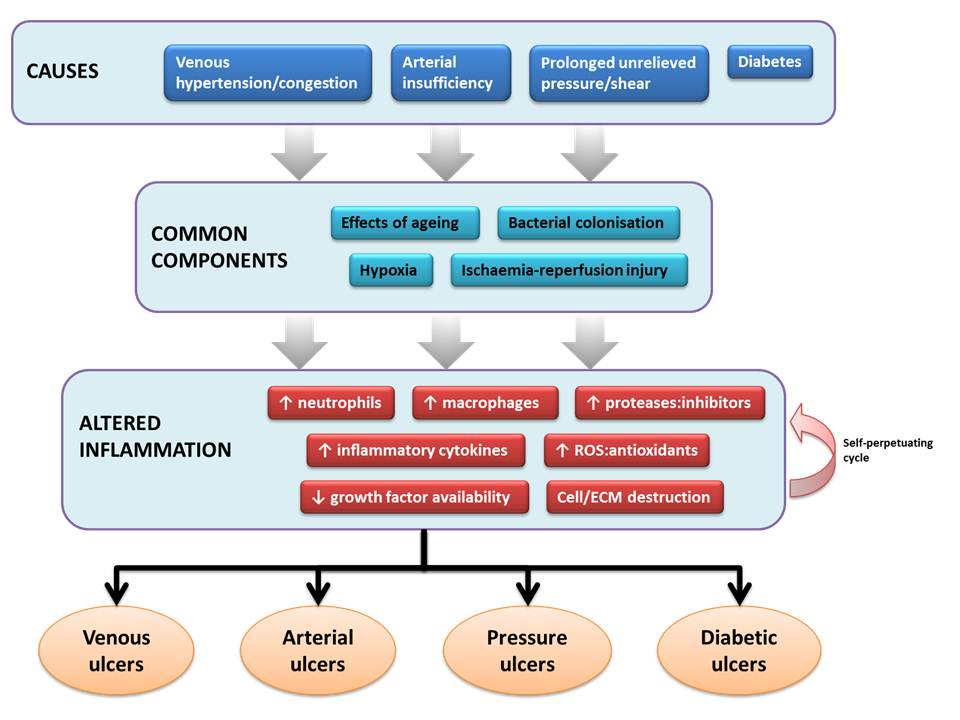
1. Overview
2. Causes
2.1. Venous Ulcers
2.2. Arterial Ulcers
2.3. Pressure Ulcers
2.4. Diabetic Ulcers
3. Normal Cutaneous Healing
3.1. Haemostasis
3.2. Inflammation
3.3. Proliferation
3.4. Remodelling
3.5. Necessity of Inflammation
4. Pathophysiology
4.1. Ageing
4.2. Hypoxia
4.3. Ischaemia-Reperfusion Injury
4.4. Bacterial Colonisation
5. Current Treatments
6. Future Prospects
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AGE | advanced glycation end-products |
| APC | activated protein C |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| FDA | Food and Drug Administration |
| FGF | fibroblast growth factor |
| IL | interleukin |
| MMP | matrix metalloproteinase |
| NF | nuclear factor |
| NO | nitric oxide |
| PDGF | platelet-derived growth factor |
| ROS | reactive oxygen species |
| TGF | transforming growth factor |
| TIMP | tissue inhibitor of matrix metalloproteinase |
| TNF | tumour necrosis factor |
| VEGF | vascular endothelial growth factor |
References
- Li, J.; Chen, J.; Kirsner, R. Pathophysiology of acute wound healing. Clin. Dermatol. 2007, 25, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Reinke, J.M.; Sorg, H. Wound repair and regeneration. Eur. Surg. Res. 2012, 49, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Nunan, R.; Harding, K.G.; Martin, P. Clinical challenges of chronic wounds: Searching for an optimal animal model to recapitulate their complexity. Dis. Model. Mech. 2014, 7, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Fonder, M.A.; Lazarus, G.S.; Cowan, D.A.; Aronson-Cook, B.; Kohli, A.R.; Mamelak, A.J. Treating the chronic wound: A practical approach to the care of nonhealing wounds and wound care dressings. J. Am. Acad. Dermatol. 2008, 58, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Diegelmann, R.F.; Evans, M.C. Wound healing: An overview of acute, fibrotic and delayed healing. Front. Biosci. 2004, 9, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R. Growth factors and chronic wound healing: Past, present, and future. Adv. Skin Wound Care 2004, 17, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Gordillo, G.M.; Roy, S.; Kirsner, R.; Lambert, L.; Hunt, T.K.; Gottrup, F.; Gurtner, G.C.; Longaker, M.T. Human skin wounds: A major and snowballing threat to public health and the economy. Wound Repair Regen. 2009, 17, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T. Understanding the financial benefits of optimising wellbeing in patients living with a wound. Wounds Int. 2013, 4, 13–17. [Google Scholar]
- Driscoll, P. Wound prevalence and wound management, 2012–2020. 2013. Available online: http://blog.mediligence.com/2013/01/29/wound-prevalence-and-wound-management-2012-2020/ (accessed on 14 September 2015).
- Mustoe, T. Understanding chronic wounds: A unifying hypothesis on their pathogenesis and implications for therapy. Am. J. Surg. 2004, 187, S65–S70. [Google Scholar] [CrossRef]
- Rudolph, R. Location of the force of wound contraction. Surg. Gynecol. Obstet. 1979, 148, 547–551. [Google Scholar] [PubMed]
- McGrath, M.H.; Simon, R.H. Wound geometry and the kinetics of wound contraction. Plast Reconstr. Surg. 1983, 72, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Volk, S.W.; Bohling, M.W. Comparative wound healing—Are the small animal veterinarian‘s clinical patients an improved translational model for human wound healing research? Wound Repair Regen. 2013, 21, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Demidova-Rice, T.N.; Hamblin, M.R.; Herman, I.M. Acute and impaired wound healing: Pathophysiology and current methods for drug delivery, part 1: Normal and chronic wounds: Biology, causes, and approaches to care. Adv. Skin Wound Care 2012, 25, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Word, R. Medical and surgical therapy for advanced chronic venous insufficiency. Surg. Clin. North Am. 2010, 90, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Trent, J.T.; Falabella, A.; Eaglstein, W.H.; Kirsner, R.S. Venous ulcers: Pathophysiology and treatment options. Ostomy Wound Manage. 2005, 51, 38–54. [Google Scholar] [PubMed]
- Bonham, P.A. Assessment and management of patients with venous, arterial, and diabetic/neuropathic lower extremity wounds. AACN Clin. Issues 2003, 14, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Fowkes, F.G.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.; Williams, L.J.; Mensah, G.A.; Criqui, M.H. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340. [Google Scholar] [CrossRef]
- Grey, J.E.; Harding, K.G.; Enoch, S. Venous and arterial leg ulcers. BMJ 2006, 332, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Bansal, C.; Scott, R.; Stewart, D.; Cockerell, C.J. Decubitus ulcers: A review of the literature. Int. J. Dermatol. 2005, 44, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Garwood, C.S.; Steinberg, J.S.; Kim, P.J. Bioengineered alternative tissues in diabetic wound healing. Clin. Podiatr. Med. Surg. 2015, 32, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Baltzis, D.; Eleftheriadou, I.; Veves, A. Pathogenesis and treatment of impaired wound healing in diabetes mellitus: New insights. Adv. Ther. 2014, 31, 817–836. [Google Scholar] [CrossRef] [PubMed]
- Falanga, V. Wound healing and its impairment in the diabetic foot. Lancet 2005, 366, 1736–1743. [Google Scholar] [CrossRef]
- Blakytny, R.; Jude, E. The molecular biology of chronic wounds and delayed healing in diabetes. Diabet. Med. 2006, 23, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Peppa, M.; Stavroulakis, P.; Raptis, S.A. Advanced glycoxidation products and impaired diabetic wound healing. Wound Repair Regen. 2009, 17, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Gary Sibbald, R.; Woo, K.Y. The biology of chronic foot ulcers in persons with diabetes. Diabetes Metab. Res. Rev. 2008, 24, S25–S30. [Google Scholar] [CrossRef] [PubMed]
- Berlanga-Acosta, J.; Schultz, G.S.; Lopez-Mola, E.; Guillen-Nieto, G.; Garcia-Siverio, M.; Herrera-Martinez, L. Glucose toxic effects on granulation tissue productive cells: The diabetics‘ impaired healing. Biomed. Res. Int. 2013, 2013, 256043. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, S.; Wallqvist, A.; Reifman, J.; Mitrophanov, A.Y. Computational approach to characterize causative factors and molecular indicators of chronic wound inflammation. J. Immunol. 2014, 192, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Noli, C.; Miolo, A. The mast cell in wound healing. Vet. Dermatol. 2001, 12, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Jackson, C.J. Extracellular matrix reorganization during wound healing and its impact on abnormal scarring. Adv. Wound Care 2013, 4, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Grose, R. Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 2003, 83, 835–870. [Google Scholar] [PubMed]
- Martin, P. Wound healing—Aiming for perfect skin regeneration. Science 1997, 276, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Barker, T.H. The role of ECM proteins and protein fragments in guiding cell behavior in regenerative medicine. Biomaterials 2011, 32, 4211–4214. [Google Scholar] [CrossRef] [PubMed]
- Eckes, B.; Nischt, R.; Krieg, T. Cell-matrix interactions in dermal repair and scarring. Fibrogenesis Tissue Repair 2010, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Darby, I.; Skalli, O.; Gabbiani, G. α-Smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab. Invest. 1990, 63, 21–29. [Google Scholar] [PubMed]
- Simon, P.E.; outran, H.A.; Romo III, T.; Pafford, W.; Pearson, J.M.; Yalamanchili, H.; Zoumalan, R.A. Skin Wound Healing. 2014. Available online: http://emedicine.medscape.com/article/884594-overview (accessed on 24 December 2014).
- Desmouliere, A.; Redard, M.; Darby, I.; Gabbiani, G. Apoptosis mediates the decrease in cellularity during the transition between granulation-tissue and scar. Am. J. Pathol. 1995, 146, 56–66. [Google Scholar] [PubMed]
- Ferguson, M.W.; O‘Kane, S. Scar-free healing: From embryonic mechanisms to adult therapeutic intervention. Philos. Trans. R Soc. Lond. B Biol. Sci. 2004, 359, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Szpaderska, A.M.; DiPietro, L.A. Inflammation in surgical wound healing: Friend or foe? Surgery 2005, 137, 571–573. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef] [PubMed]
- You, H.-J.; Han, S.-K. Cell therapy for wound healing. J. Korean Med. Sci. 2014, 29, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Brockes, J.P.; Kumar, A.; Velloso, C.P. Regeneration as an evolutionary variable. J Anat. 2001, 199, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Colwell, A.S.; Krummel, T.M.; Longaker, M.T.; Lorenz, H.P. An in vivo mouse excisional wound model of scarless healing. Plast. Reconstr. Surg. 2006, 117, 2292–2296. [Google Scholar] [CrossRef] [PubMed]
- Rowlatt, U. Intrauterine wound healing in a 20 week human fetus. Virchows Arch. A Pathol. Anat. Histol. 1979, 381, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Adzick, N.S.; Lorenz, H.P. Cells, matrix, growth factors, and the surgeon. The biology of scarless fetal wound repair. Ann. Surg. 1994, 220, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Stojadinovic, A.; Carlson, J.W.; Schultz, G.S.; Davis, T.A.; Elster, E.A. Topical advances in wound care. Gynecol. Oncol. 2008, 111, S70–S80. [Google Scholar] [CrossRef] [PubMed]
- Schultz, G.S.; Mast, B.A. Molecular analysis of the environment of healing and chronic wounds: Cytokines, proteases, and growth factors. Wounds Compend. Clin. Res. Pract. 1998, 10, 1f–9f. [Google Scholar]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in wound repair: Molecular and cellular mechanisms. J. Invest. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Van der Veer, W.M.; Bloemen, M.C.; Ulrich, M.M.; Molema, G.; van Zuijlen, P.P.; Middelkoop, E.; Niessen, F.B. Potential cellular and molecular causes of hypertrophic scar formation. Burns 2009, 35, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Mast, B.A.; Schultz, G.S. Interactions of cytokines, growth factors, and proteases in acute and chronic wounds. Wound Repair Regen. 1996, 4, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Schreml, S.; Szeimies, R.M.; Prantl, L.; Karrer, S.; Landthaler, M.; Babilas, P. Oxygen in acute and chronic wound healing. Br. J. Dermatol. 2010, 163, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Gosain, A.; DiPietro, L.A. Aging and wound healing. World J. Surg. 2004, 28, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Engeland, C.G.; Bosch, J.A.; Cacioppo, J.T.; Marucha, P.T. Mucosal wound healing: The roles of age and sex. Arch. Surg. 2006, 141, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Mills, S.J.; Ashworth, J.J. Ageing and wound healing. Biogerontology 2002, 3, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Greenwell-Wild, T.; Horan, M.A.; Wahl, S.M.; Ferguson, M.W. Topical estrogen accelerates cutaneous wound healing in aged humans associated with an altered inflammatory response. Am. J. Pathol. 1999, 155, 1137–1146. [Google Scholar] [CrossRef]
- Ashcroft, G.S.; Horan, M.A.; Ferguson, M.W. The effects of ageing on cutaneous wound healing in mammals. J. Anat. 1995, 187, 1–26. [Google Scholar] [PubMed]
- Ashcroft, G.S.; Horan, M.A.; Ferguson, M.W.J. Aging is associated with reduced deposition of specific extracellular matrix components, upregulation of angiogenesis, and an altered inflammatory response in a murine incisional wound healing model. J. Invest. Dermatol. 1997, 108, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Gouin, J.P.; Hantsoo, L.; Kiecolt-Glaser, J.K. Immune Dysregulation and chronic stress among older adults: A review. Neuroimmunomodulation 2008, 15, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Pereyra, L.H.; Lopez-Neblina, F.; Toledo, A.H. Reactive oxygen species and molecular biology of ischemia/reperfusion. Ann. Transplant. 2004, 9, 81–83. [Google Scholar] [PubMed]
- Peschen, M.; Lahaye, T.; Hennig, B.; Weyl, A.; Simon, J.C.; Vanscheidt, W. Expression of the adhesion molecules ICAM-1, VCAM-1, LFA-1 and VLA-4 in the skin is modulated in progressing stages of chronic venous insufficiency. Acta Derm. Venereol. 1999, 79, 27–32. [Google Scholar] [PubMed]
- Soneja, A.; Drews, M.; Malinski, T. Role of nitric oxide, nitroxidative and oxidative stress in wound healing. Pharmacol. Rep. 2005, 57, 108–119. [Google Scholar] [PubMed]
- Connelly, L.; Palacios-Callender, M.; Ameixa, C.; Moncada, S.; Hobbs, A.J. Biphasic regulation of NF-κB activity underlies the pro- and anti-inflammatory actions of nitric oxide. J. Immunol. 2001, 166, 3873–3881. [Google Scholar] [CrossRef] [PubMed]
- Anaya-Prado, R.; Toledo-Pereyra, L.H.; Lentsch, A.B.; Ward, P.A. Ischemia/reperfusion injury. J. Surg. Res. 2002, 105, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Mudge, E.J. Recent accomplishments in wound healing. Int. Wound J. 2015, 12, 4–9. [Google Scholar] [CrossRef] [PubMed]
- James, G.A.; Swogger, E.; Wolcott, R.; Pulcini, E.; Secor, P.; Sestrich, J.; Costerton, J.W.; Stewart, P.S. Biofilms in chronic wounds. Wound Repair Regen. 2008, 16, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Usui, M.L.; Lippman, S.I.; James, G.A.; Stewart, P.S.; Fleckman, P.; Olerud, J.E. Biofilms and inflammation in chronic wounds. Adv. Wound Care 2013, 2, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Wolcott, R.D.; Rhoads, D.D.; Dowd, S.E. Biofilms and chronic wound inflammation. J. Wound Care 2008, 17, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Schultz, G.S.; Sibbald, R.G.; Falanga, V.; Ayello, E.A.; Dowsett, C.; Harding, K.; Romanelli, M.; Stacey, M.C.; Teot, L.; Vanscheidt, W. Wound bed preparation: A systematic approach to wound management. Wound Repair Regen. 2003, 11, S1–S28. [Google Scholar] [CrossRef] [PubMed]
- Hershcovitch, M.D.; Hom, D.B. Update in wound healing in facial plastic surgery. Arch. Facial Plast. Surg. 2012, 14, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Meier, K.; Nanney, L.B. Emerging new drugs for wound repair. Expert. Opin. Emerg. Drugs 2006, 11, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Perspective article: Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.E.; Bolton, L.L.; Verco, S.; di Zerega, G.S. NorLeu-angiotensin (1–7) [DSC127] as a therapy for the healing of diabetic foot ulcers. Adv. Wound Care 2015, 4, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Sogabe, Y.; Abe, M.; Yokoyama, Y.; Ishikawa, O. Basic fibroblast growth factor stimulates human keratinocyte motility by Rac activation. Wound Repair Regen. 2006, 14, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.C.; Phillips, L.G.; Lawrence, W.T.; Bishop, J.B.; Youngerman, J.S.; Hayward, P.G.; Broemeling, L.D.; Heggers, J.P. The safety and effect of topically applied recombinant basic fibroblast growth factor on the healing of chronic pressure sores. Ann. Surg. 1992, 216, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.L.; Parer-Richard, C.; Daures, J.P.; Clouet, S.; Vannereau, D.; Bringer, J.; Rodier, M.; Jacob, C.; Comte-Bardonnet, M. Effect of topical basic fibroblast growth factor on the healing of chronic diabetic neuropathic ulcer of the foot. A pilot, randomized, double-blind, placebo-controlled study. Diabetes Care 1995, 18, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Uchi, H.; Igarashi, A.; Urabe, K.; Koga, T.; Nakayama, J.; Kawamori, R.; Tamaki, K.; Hirakata, H.; Ohura, T.; Furue, M. Clinical efficacy of basic fibroblast growth factor (bFGF) for diabetic ulcer. Eur. J. Dermatol. 2009, 19, 461–468. [Google Scholar] [PubMed]
- Kusumanto, Y.H.; van Weel, V.; Mulder, N.H.; Smit, A.J.; van den Dungen, J.J.; Hooymans, J.M.; Sluiter, W.J.; Tio, R.A.; Quax, P.H.; Gans, R.O.; et al. Treatment with intramuscular vascular endothelial growth factor gene compared with placebo for patients with diabetes mellitus and critical limb ischemia: A double-blind randomized trial. Hum. Gene Ther. 2006, 17, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Saaristo, A.; Tammela, T.; Farkkila, A.; Karkkainen, M.; Suominen, E.; Yla-Herttuala, S.; Alitalo, K. Vascular endothelial growth factor-C accelerates diabetic wound healing. Am. J. Pathol. 2006, 169, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Genentech Announces Full Year and Fourth Quarter 2007 Results. 2008. Available online: https://www.gene.com/media/press-releases/10967/2008-01-14/genentech-announces-full-year-and-fourth (accessed on 30 November 2016).
- Dinh, T.; Braunagel, S.; Rosenblum, B.I. Growth factors in wound healing: The present and the future? Clin. Podiatr. Med. Surg. 2015, 32, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Sorg, H.; Harder, Y.; Krueger, C.; Reimers, K.; Vogt, P.M. The nonhematopoietic effects of erythropoietin in skin regeneration and repair: From basic research to clinical use. Med. Res. Rev. 2013, 33, 637–664. [Google Scholar] [CrossRef] [PubMed]
- Mosnier, L.O.; Zlokovic, B.V.; Griffin, J.H. The cytoprotective protein C pathway. Blood 2006, 109, 3161–3172. [Google Scholar] [CrossRef] [PubMed]
- Uchiba, M.; Okajima, K.; Oike, Y.; Ito, Y.; Fukudome, K.; Isobe, H.; Suda, T. Activated protein C induces endothelial cell proliferation by mitogen-activated protein kinase activation in vitro and angiogenesis in vivo. Circ. Res. 2004, 95, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.J.; Xue, M.; Thompson, P.; Davey, R.A.; Whitmont, K.; Smith, S.; Buisson-Legendre, N.; Sztynda, T.; Furphy, L.J.; Cooper, A.; et al. Activated protein C prevents inflammation yet stimulates angiogenesis to promote cutaneous wound healing. Wound Repair Regen. 2005, 13, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Julovi, S.M.; Xue, M.; Dervish, S.; Sambrook, P.N.; March, L.; Jackson, C.J. Protease activated receptor-2 mediates activated protein C-induced cutaneous wound healing via inhibition of p38. Am. J. Pathol. 2011, 179, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Whitmont, K.; Reid, I.; Tritton, S.; March, L.; Xue, M.; Lee, M.; Fulcher, G.; Sambrook, P.; Slobedman, E.; Cooper, A.; et al. Treatment of chronic leg ulcers with topical activated protein C. Arch. Dermatol. 2008, 144, 1479–1483. [Google Scholar] [CrossRef] [PubMed]
- Wijewardena, A.; Vandervord, E.; Lajevardi, S.S.; Vandervord, J.; Jackson, C.J. Combination of activated protein C and topical negative pressure rapidly regenerates granulation tissue over exposed bone to heal recalcitrant orthopedic wounds. Int. J. Low. Extrem. Wounds 2011, 10, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Whitmont, K.; McKelvey, K.J.; Fulcher, G.; Reid, I.; March, L.; Xue, M.; Cooper, A.; Jackson, C.J. Treatment of chronic diabetic lower leg ulcers with activated protein C: A randomised placebo-controlled, double-blind pilot clinical trial. Int. Wound J. 2013, 12, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Whitmont, K.; Fulcher, G.; Reid, I.; Xue, M.; McKelvey, K.; Xie, Y.; Aboud, M.; Ward, C.; Smith, M.M.; Cooper, A.; et al. Low circulating protein C levels are associated with lower leg ulcers in patients with diabetes. Biomed. Res. Int. 2013, 2013, 719570. [Google Scholar] [CrossRef] [PubMed]
- Kapila, S.; Reid, I.; Dixit, S.; Fulcher, G.; March, L.; Jackson, C.; Cooper, A. Use of dermal injection of activated protein C for treatment of large chronic wounds secondary to pyoderma gangrenosum. Clin. Exper. Dermatol. 2014, 39, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Marti-Carvajal, A.J.; Sola, I.; Lathyris, D.; Cardona, A.F. Human recombinant activated protein C for severe sepsis. Cochrane Database Syst. Rev. 2012, 3, CD004388. [Google Scholar]
- Sarangi, P.P.; Lee, H.-W.; Kim, M. Activated protein C action in inflammation. Br. J. Haematol. 2010, 148, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Mosnier, L.O.; Sinha, R.K.; Burnier, L.; Bouwens, E.A.; Griffin, J.H. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood 2012, 120, 5237–5246. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, R.; Liang, H.; Clarke, E.; Jackson, C.; Xue, M. Inflammation in Chronic Wounds. Int. J. Mol. Sci. 2016, 17, 2085. https://doi.org/10.3390/ijms17122085
Zhao R, Liang H, Clarke E, Jackson C, Xue M. Inflammation in Chronic Wounds. International Journal of Molecular Sciences. 2016; 17(12):2085. https://doi.org/10.3390/ijms17122085
Chicago/Turabian StyleZhao, Ruilong, Helena Liang, Elizabeth Clarke, Christopher Jackson, and Meilang Xue. 2016. "Inflammation in Chronic Wounds" International Journal of Molecular Sciences 17, no. 12: 2085. https://doi.org/10.3390/ijms17122085
APA StyleZhao, R., Liang, H., Clarke, E., Jackson, C., & Xue, M. (2016). Inflammation in Chronic Wounds. International Journal of Molecular Sciences, 17(12), 2085. https://doi.org/10.3390/ijms17122085