
Abnormal Mitochondrial cAMP/PKA Signaling Is Involved in Sepsis-Induced Mitochondrial and Myocardial Dysfunction

Abstract
:1. Introduction
2. Results
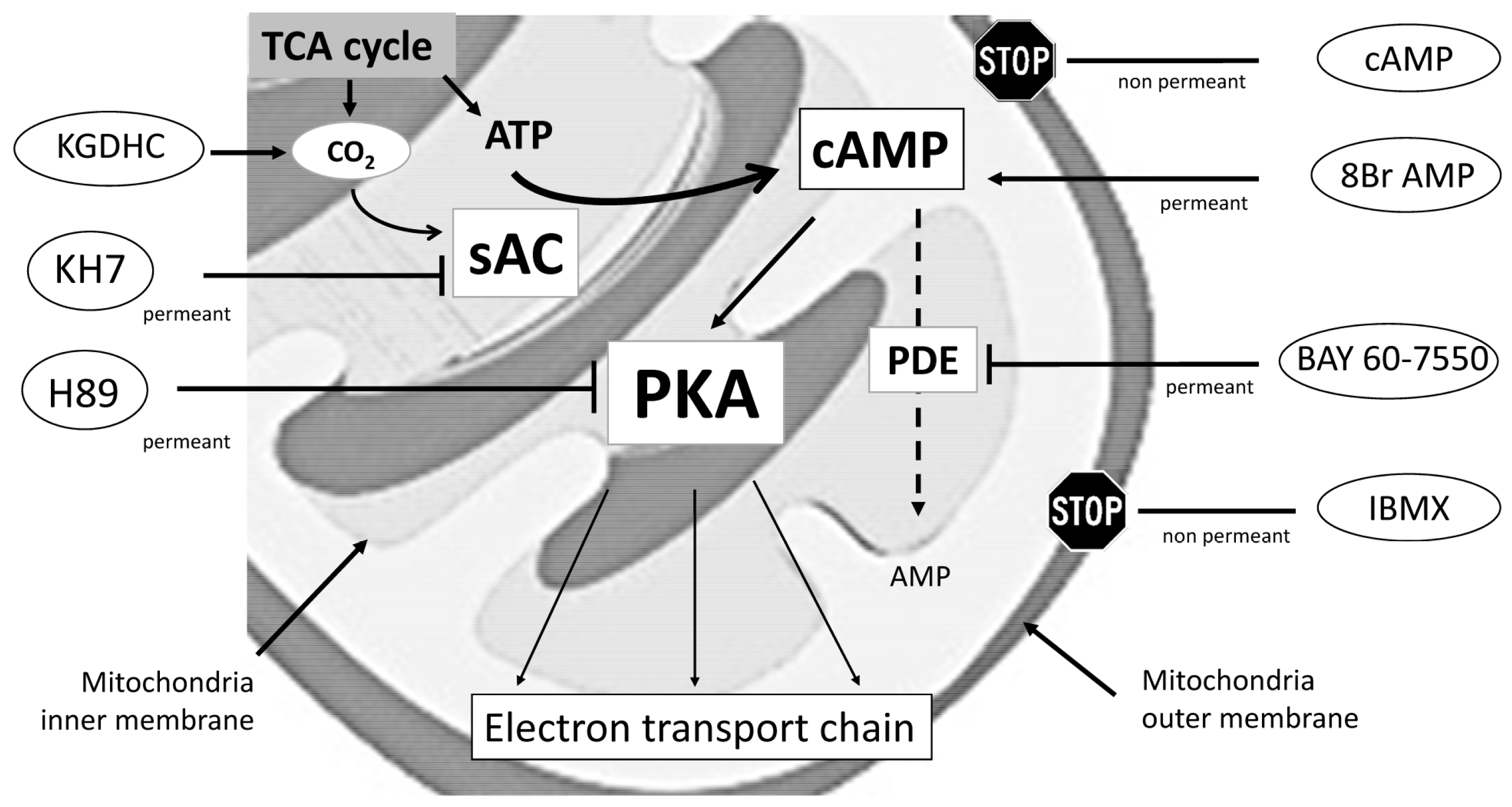
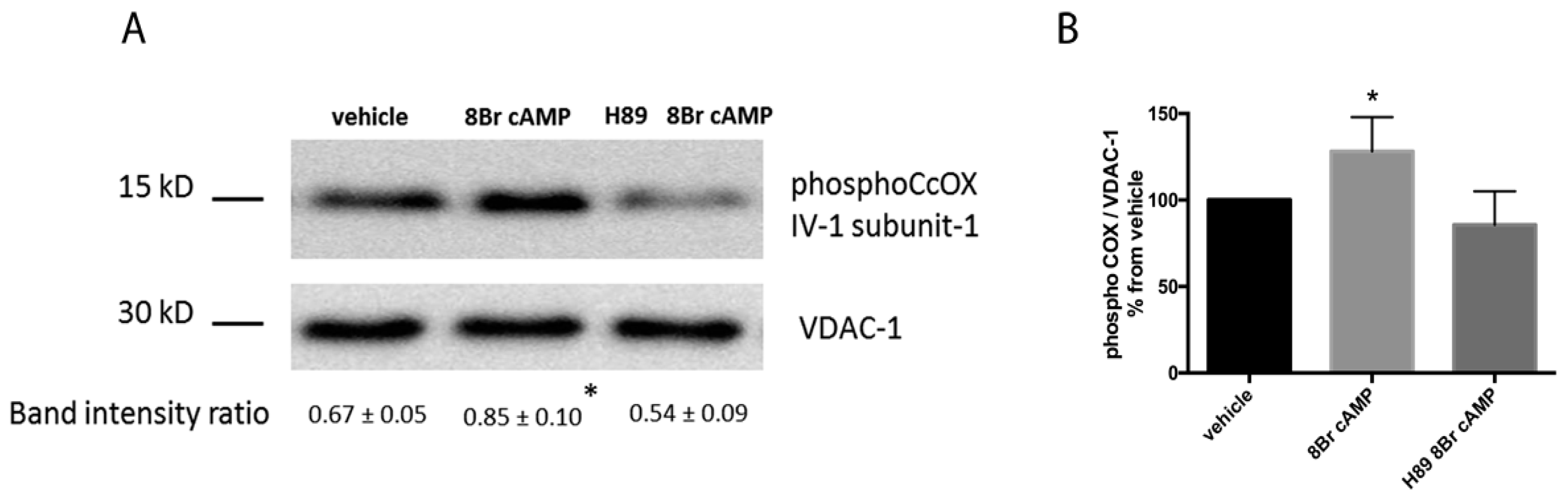
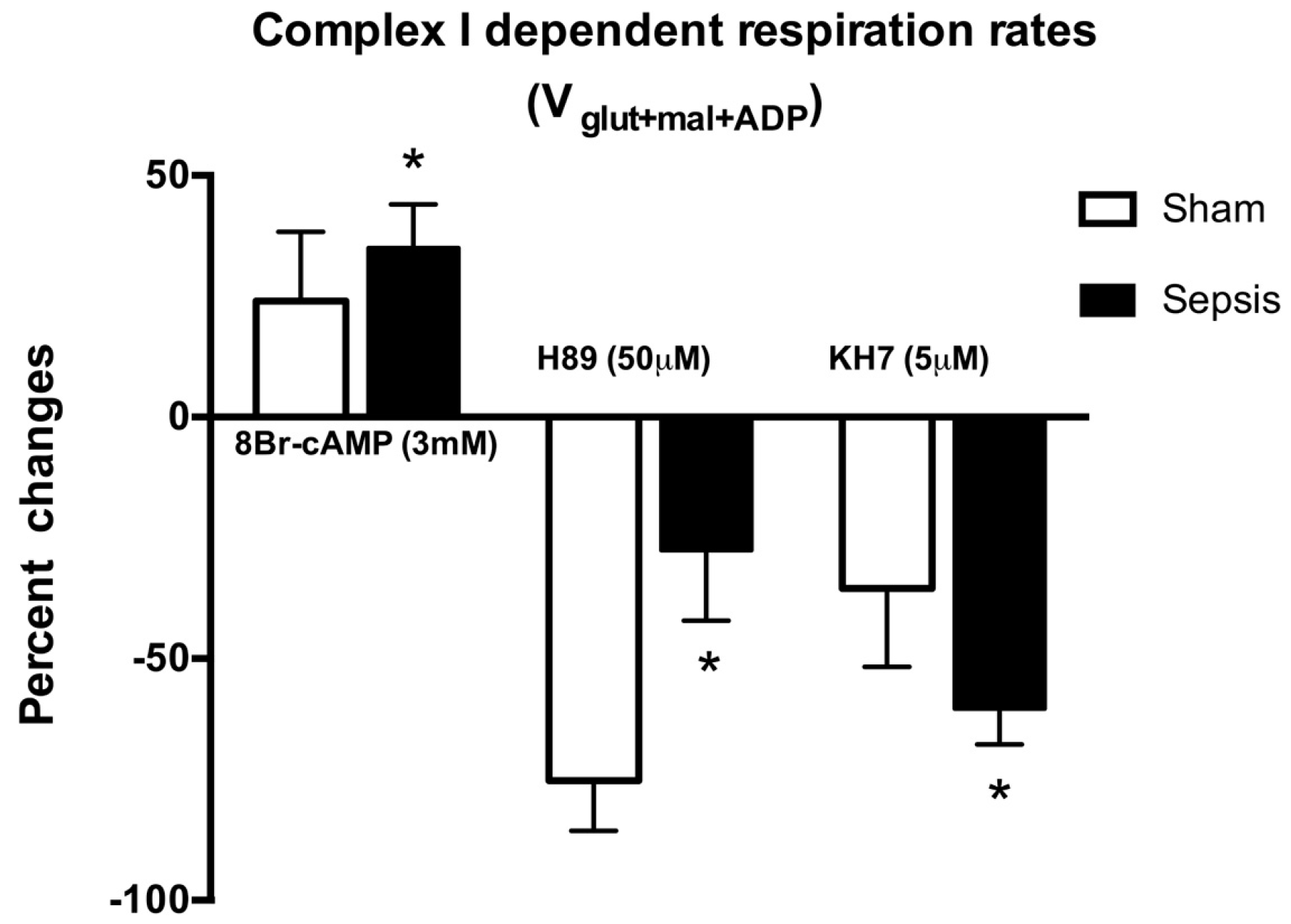
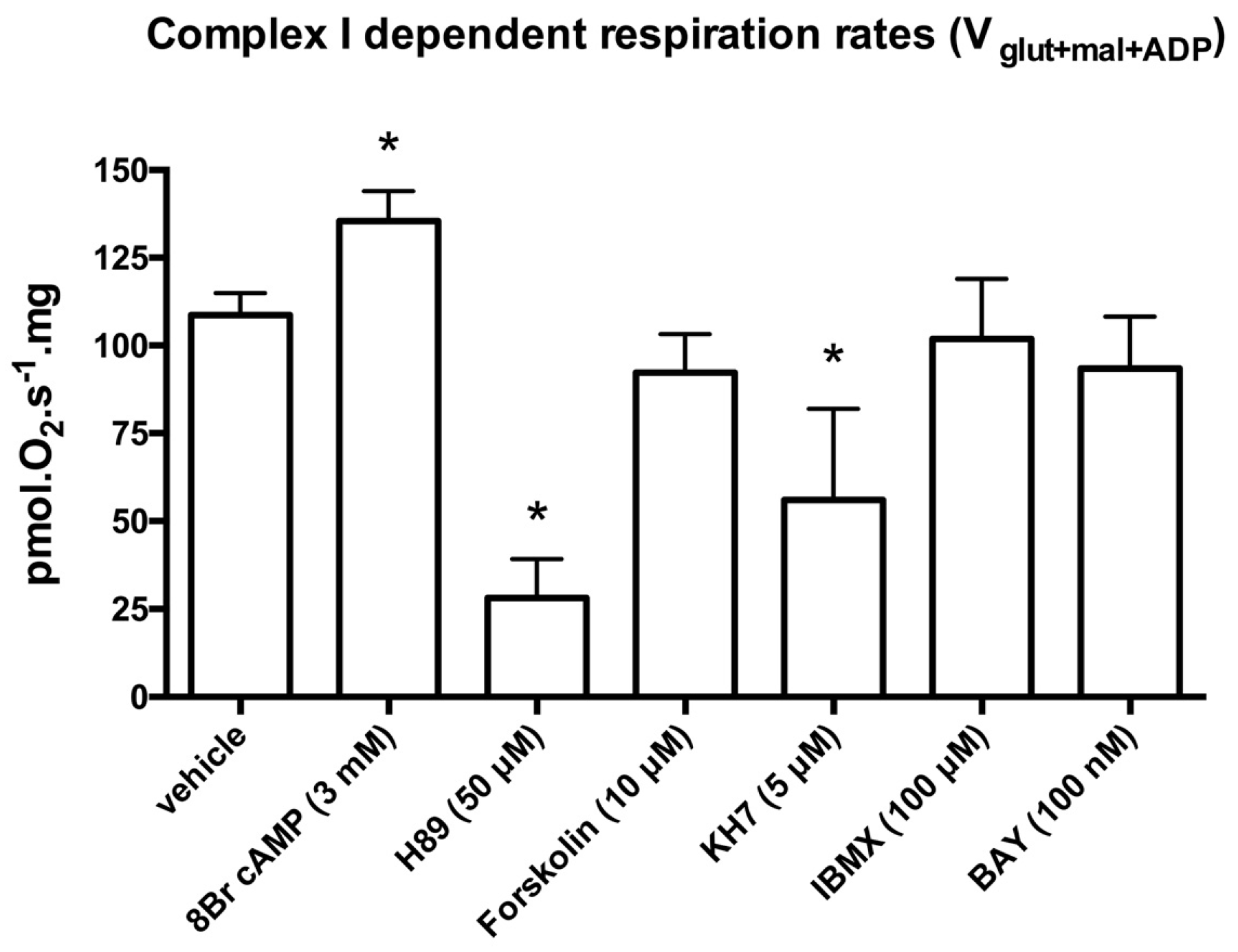
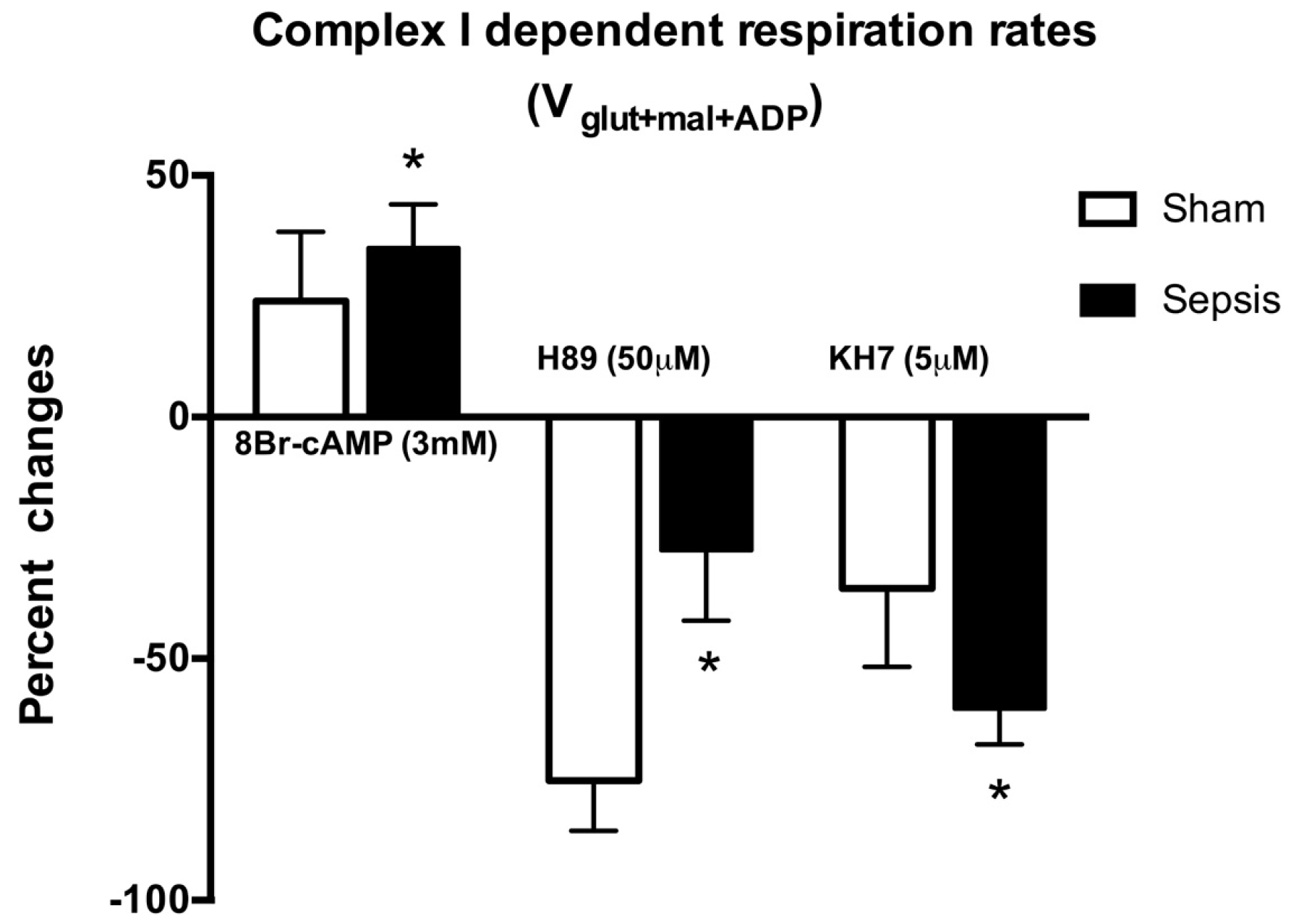
2.1. Locally Produced cAMP Stimulates Mitochondria Respiration and Increases Ser-58 CcOX Phosphorylation in Control Cardiac Fibers
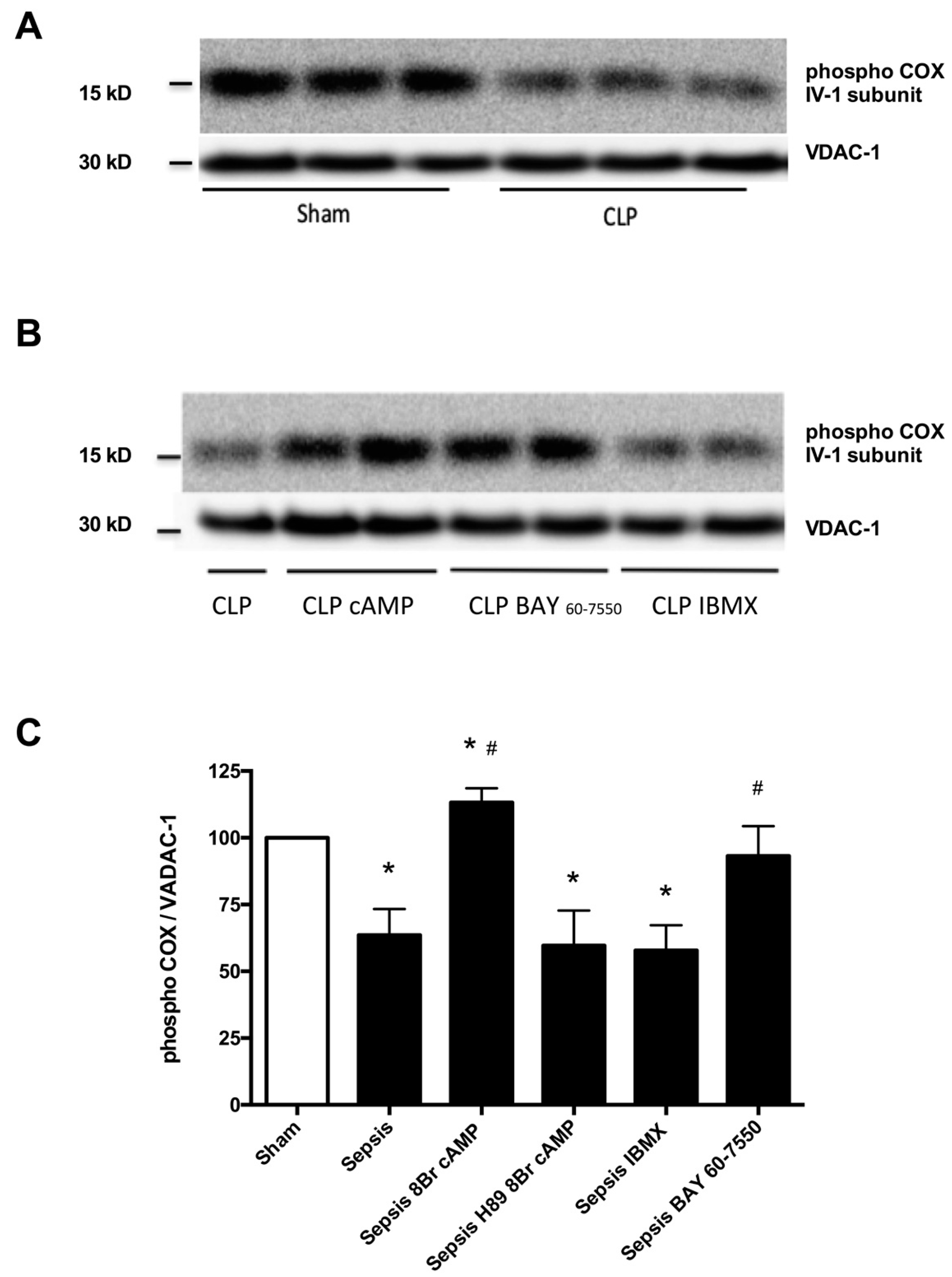
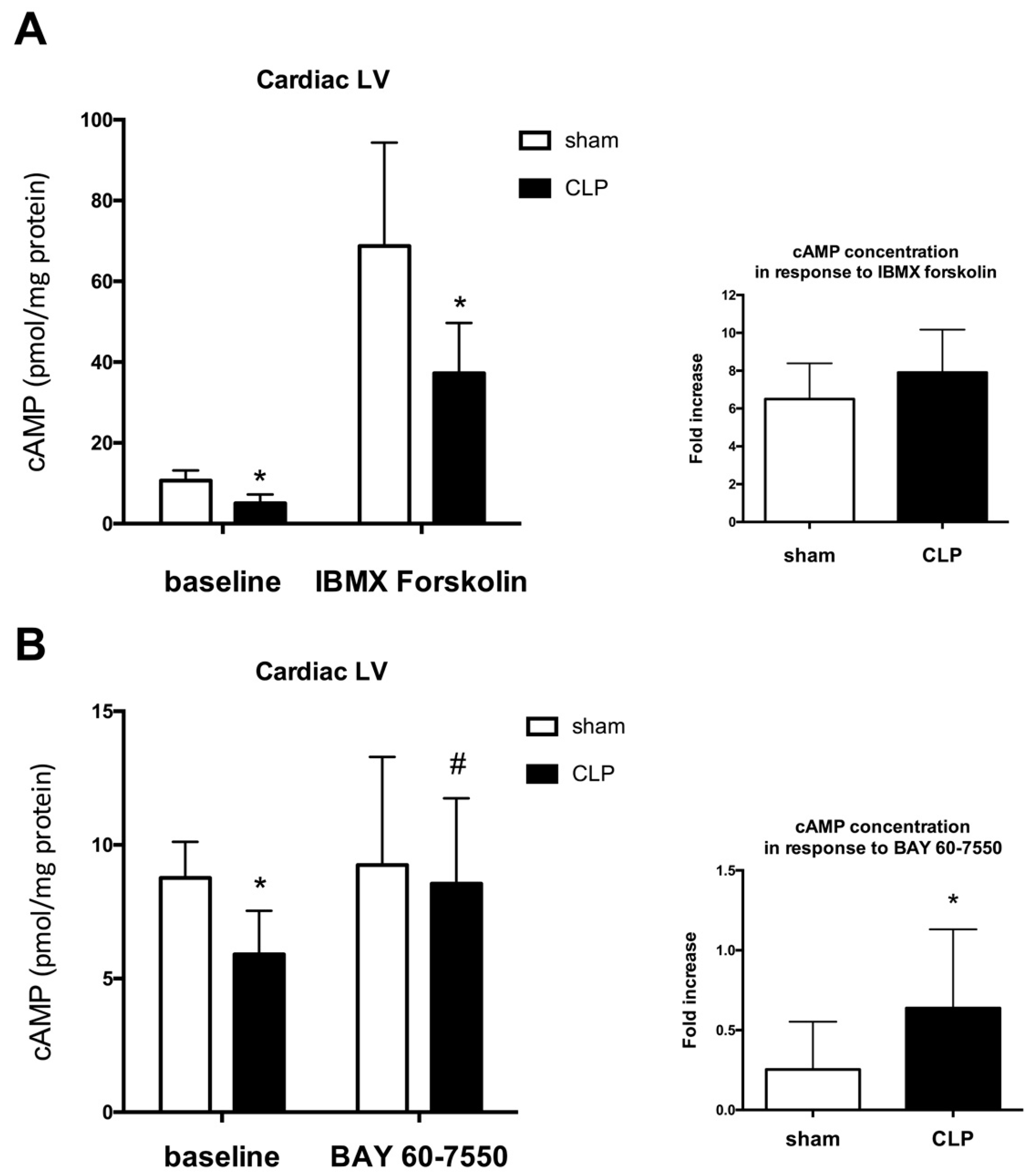
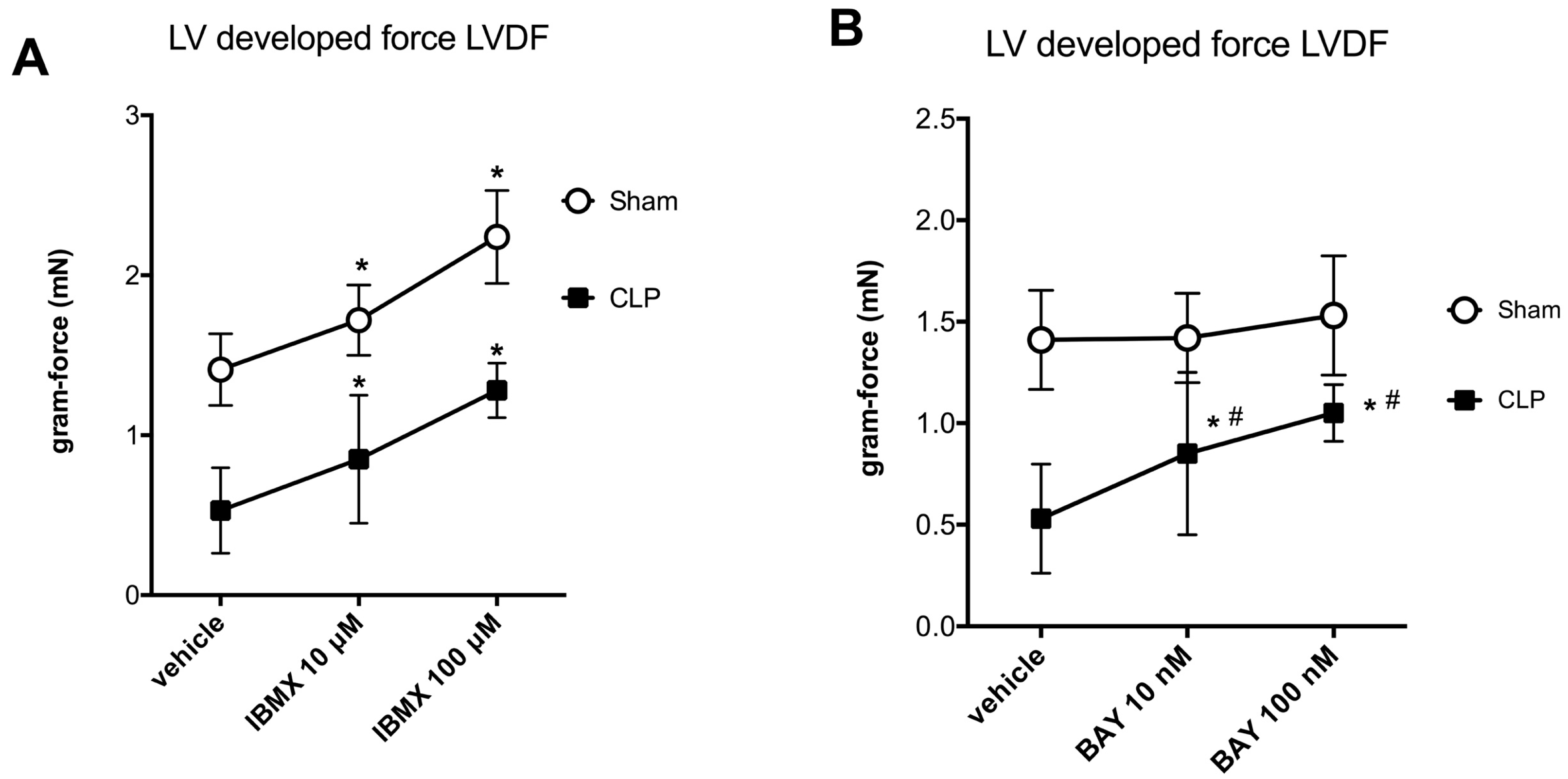
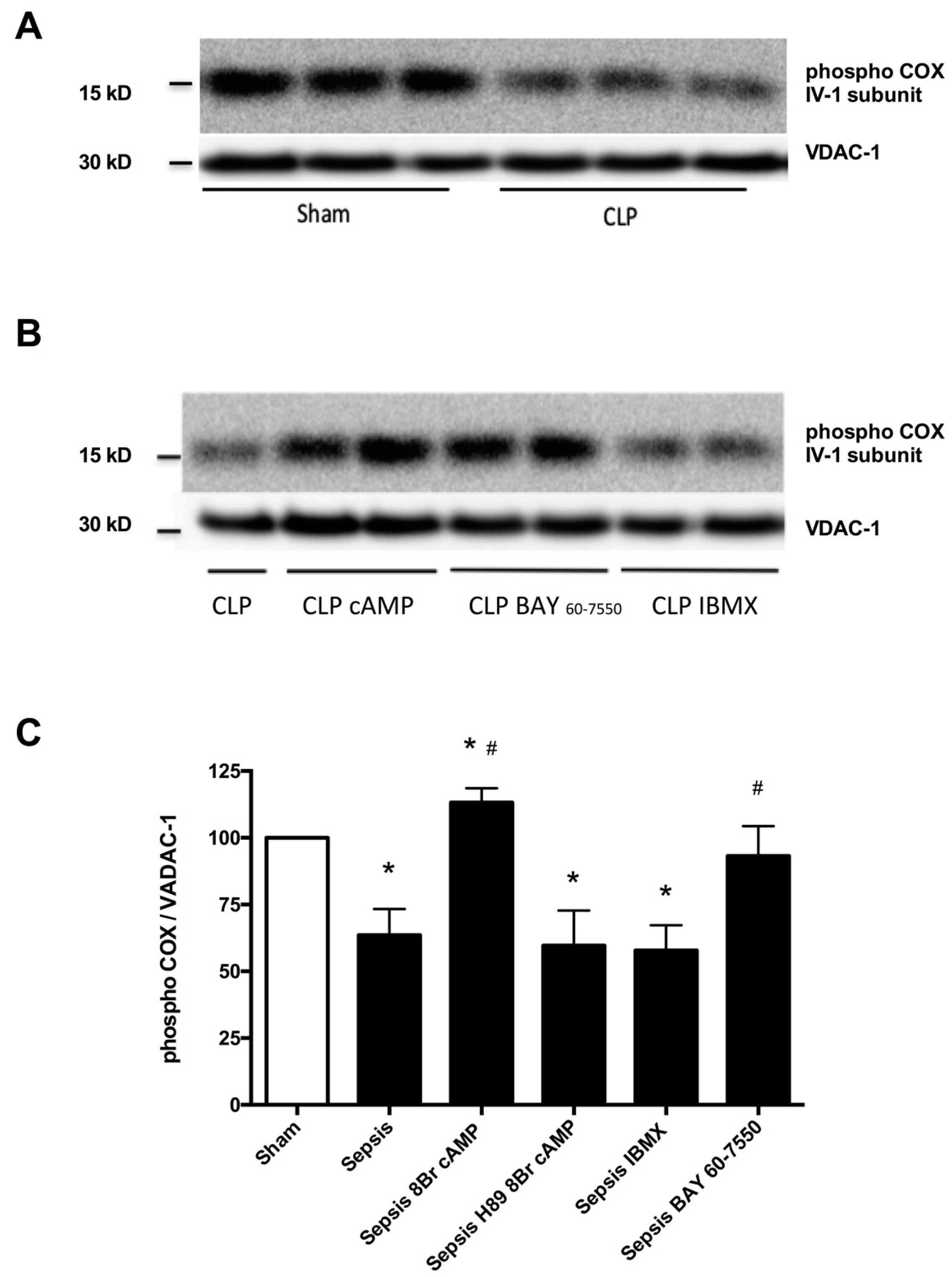
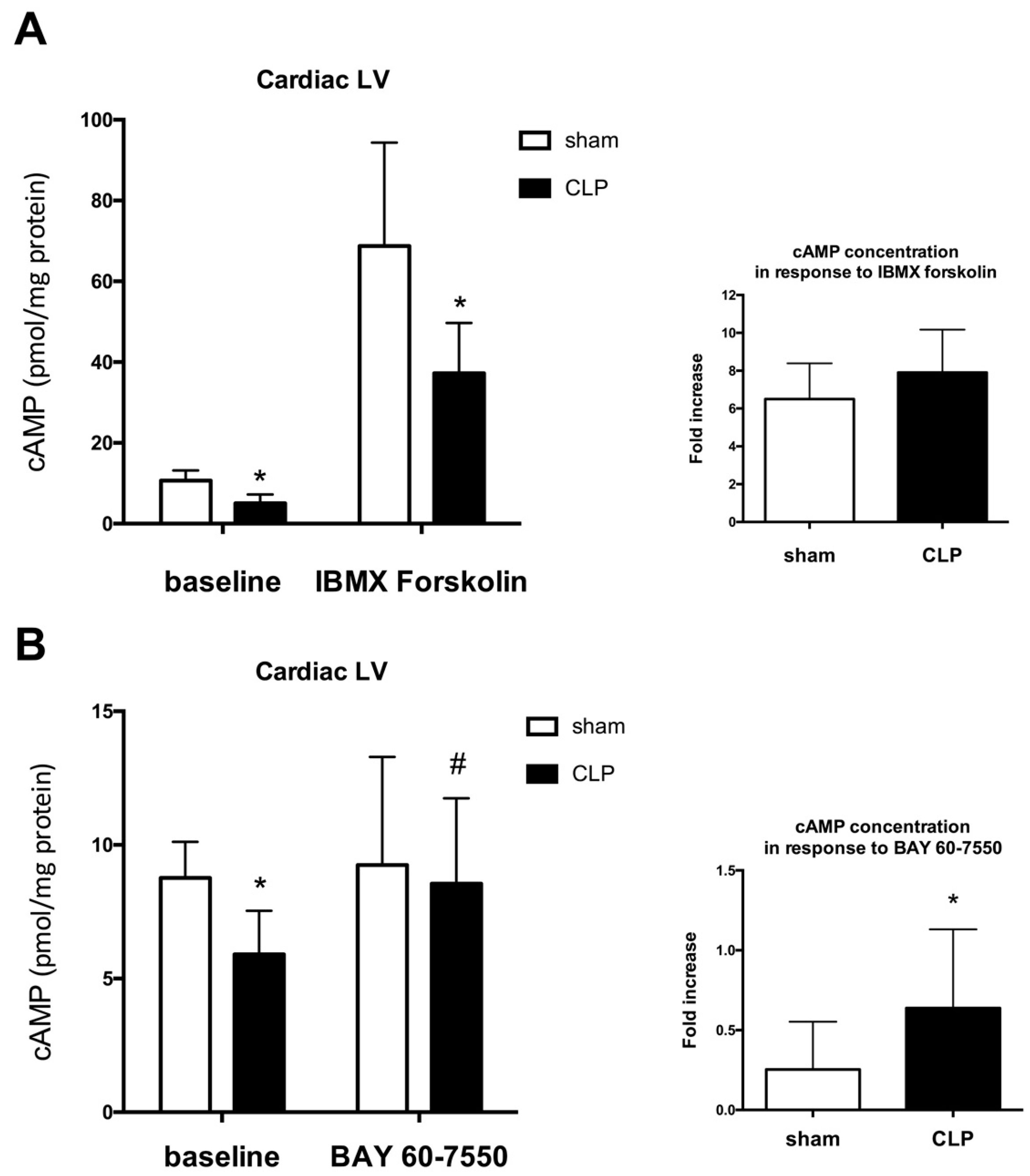
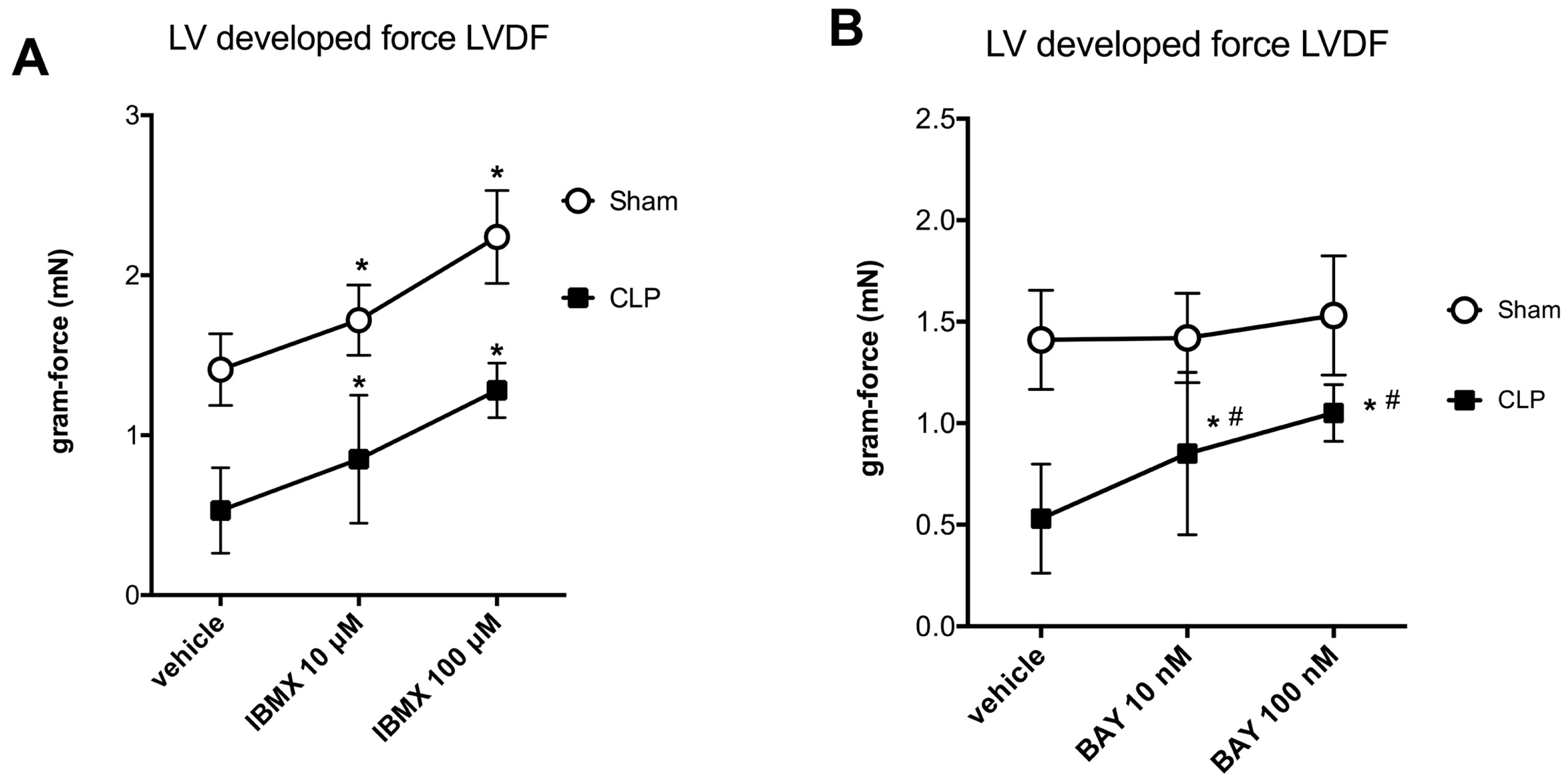
2.2. Blunted Mitochondrial cAMP-PKA Signaling in the Septic Heart Is Improved by PDE2 Inhibition
3. Discussion
Study Limitation
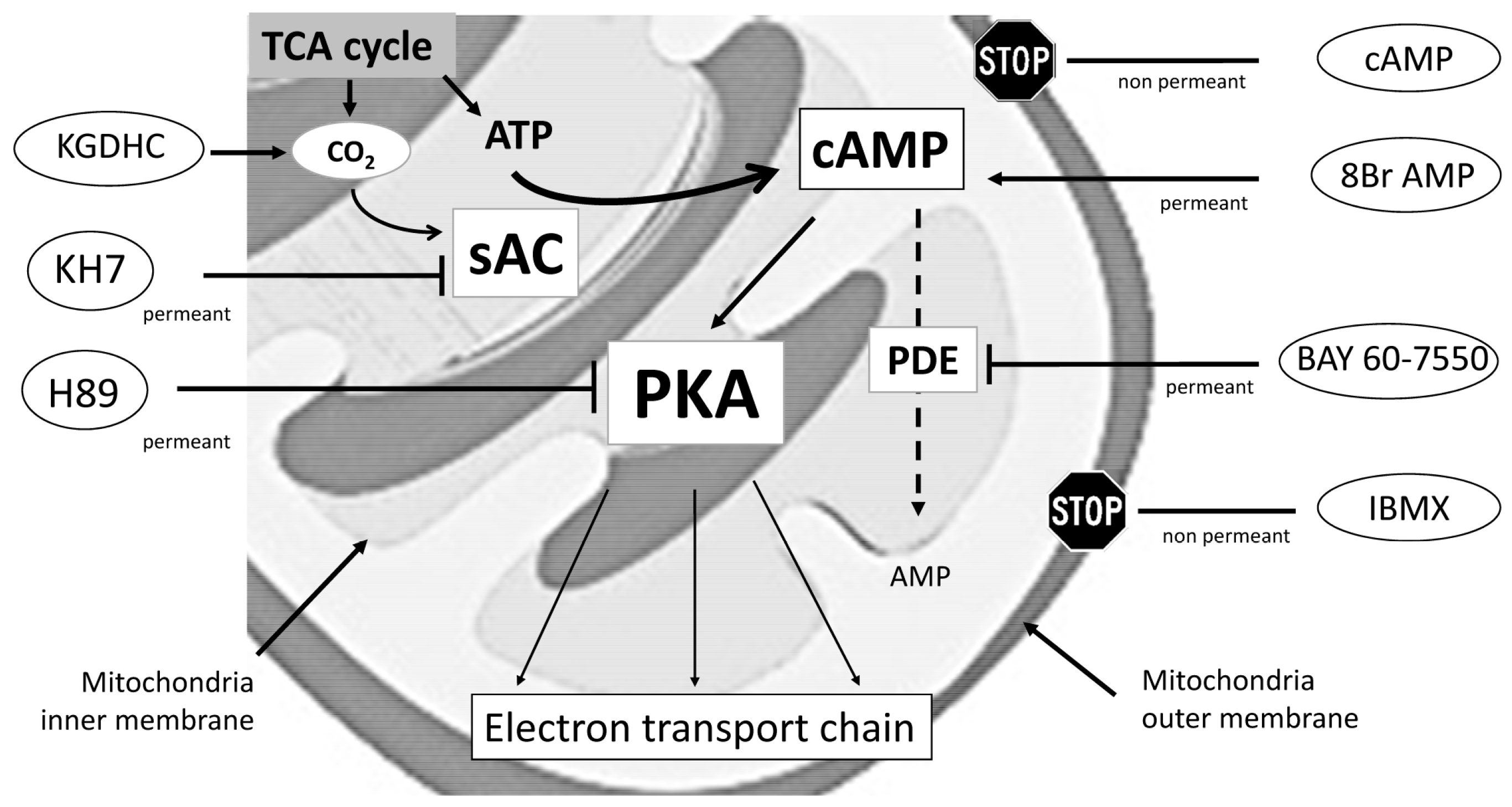
4. Materials and Methods
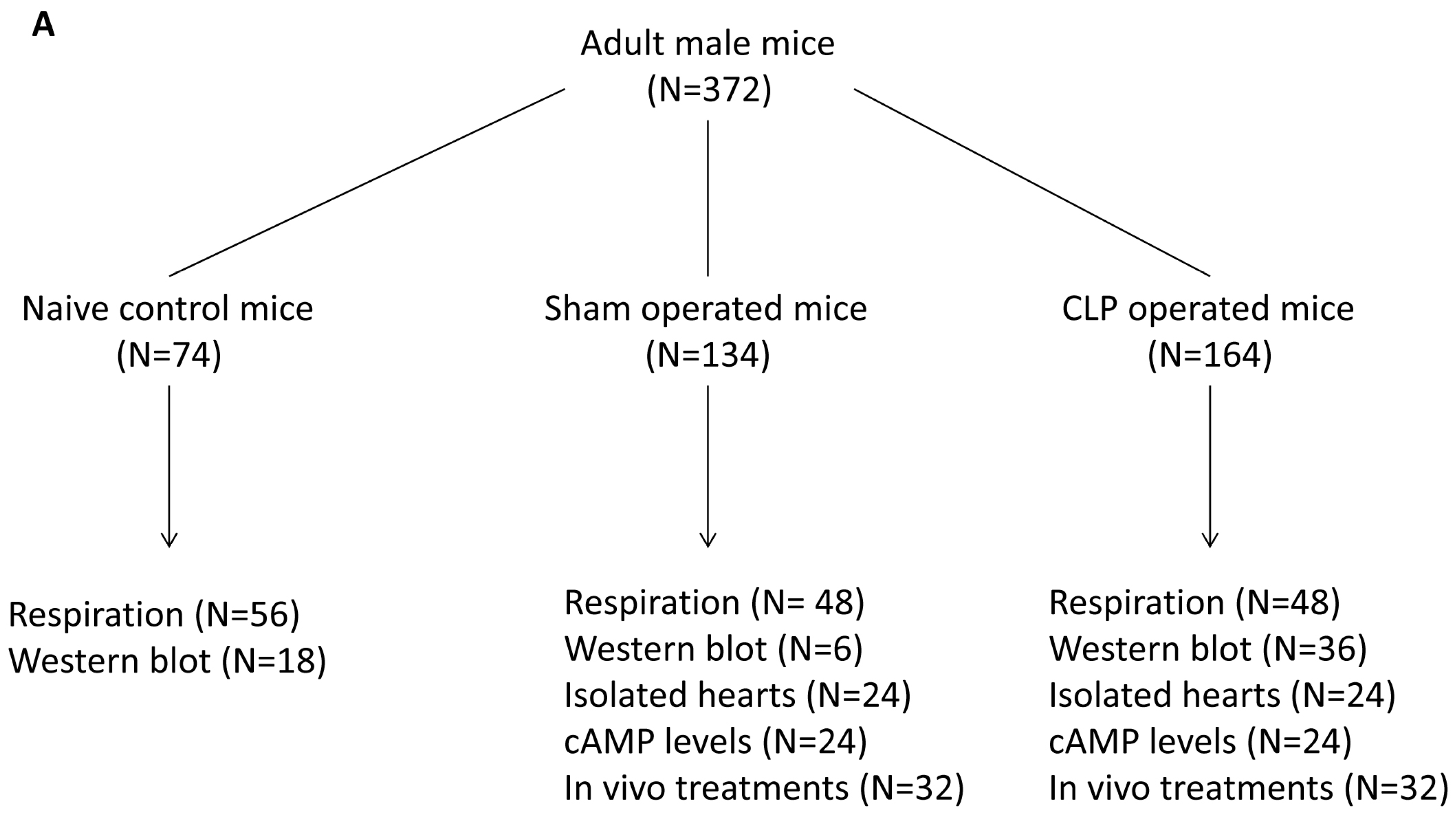
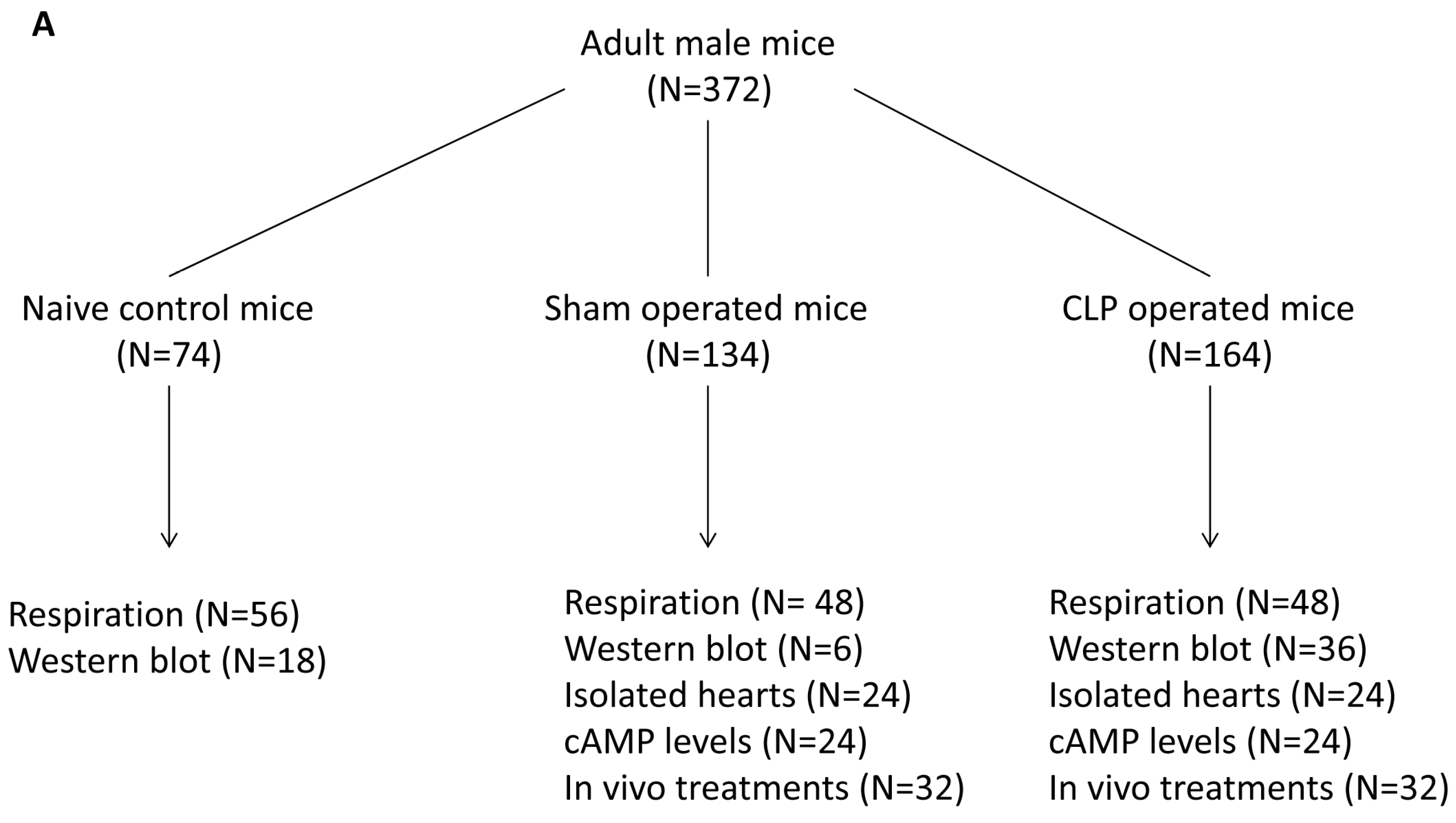
4.1. Animal Care and Model of Sepsis
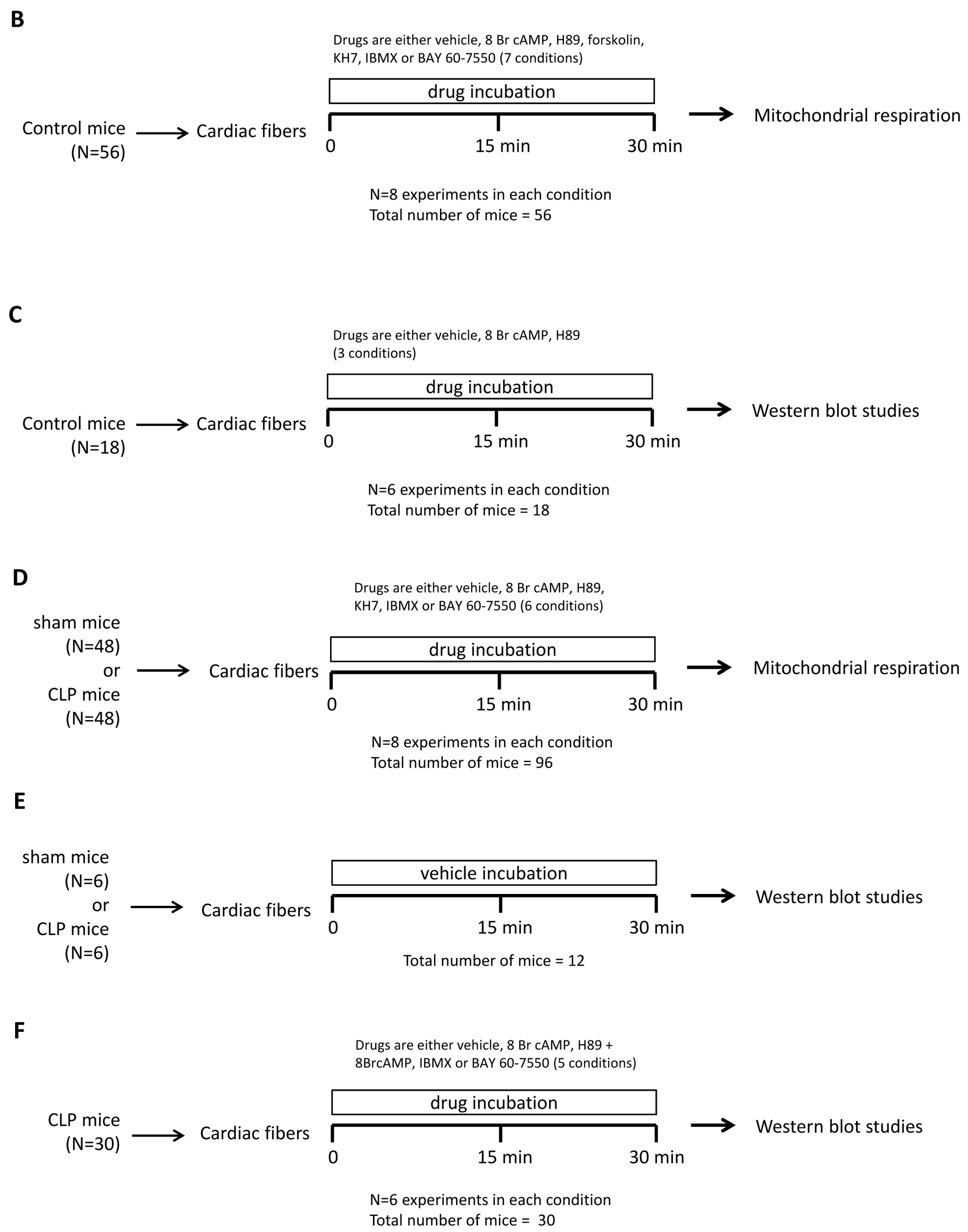
4.2. Mitochondrial Respiration in Permeabilized Cardiac Fibers
4.3. CO2-Generating α-Ketoglutarate Dehydrogenase Complex
4.4. cAMP Measurement
4.5. Isolated Perfused Heart Preparation
4.6. Western Blotting
4.7. Titration of Agonists and Inhibitors
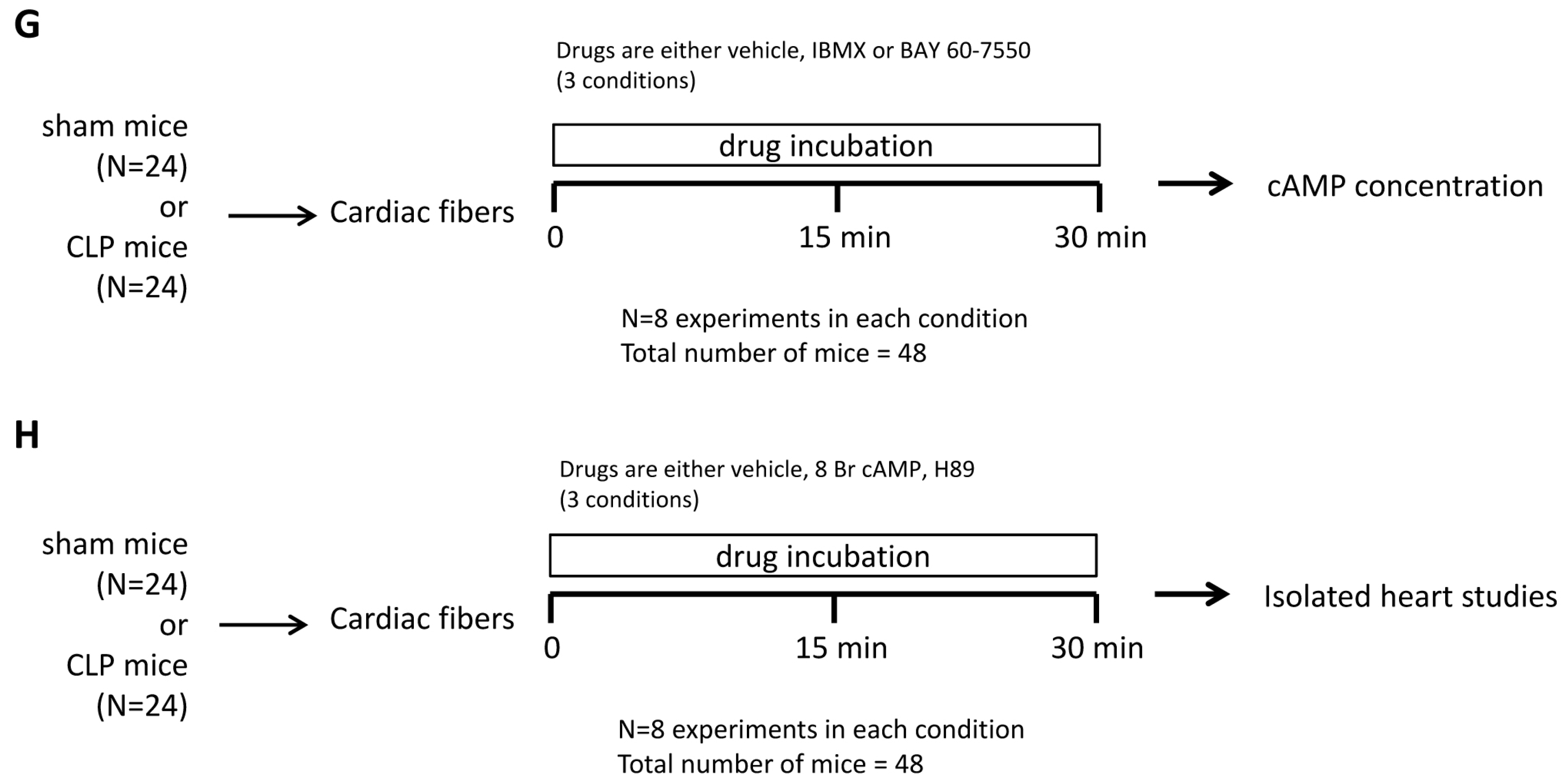
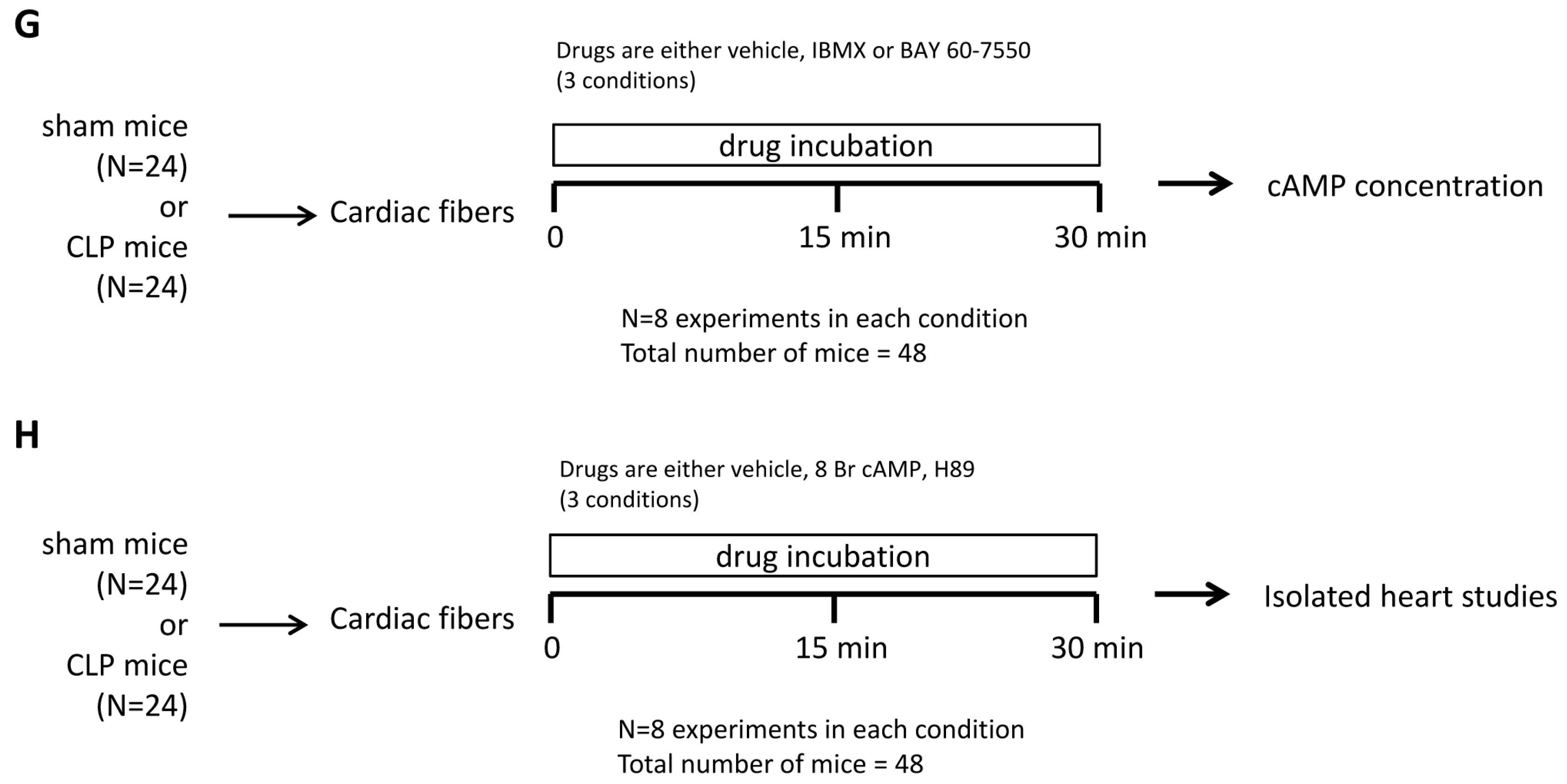
4.8. Study Design
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Guellich, A.; Mehel, H.; Fischmeister, R. Cyclic AMP synthesis and hydrolysis in the normal and failing heart. Pflug. Arch. 2014, 466, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.J. Adrenergic signaling in heart failure: A balance of toxic and protective effects. Pflug. Arch. 2014, 466, 1139–1150. [Google Scholar] [CrossRef] [PubMed]
- Skroblin, P.; Grossmann, S.; Schafer, G.; Rosenthal, W.; Klussmann, E. Mechanisms of protein kinase A anchoring. Int. Rev. Cell Mol. Biol. 2010, 283, 235–330. [Google Scholar] [PubMed]
- Najafi, A.; Sequeira, V.; Kuster, D.W.; van der Velden, J. β-adrenergic receptor signalling and its functional consequences in the diseased heart. Eur. J. Clin. Investig. 2016, 46, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Hancox, J.C. Regulation of cardiac Na+-Ca2+ exchanger activity by protein kinase phosphorylation—Still a paradox? Cell Calcium 2009, 45, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dutta, K.; Carmody, M.W.; Cala, S.E.; Davidoff, A.J. Depressed PKA activity contributes to impaired SERCA function and is linked to the pathogenesis of glucose-induced cardiomyopathy. J. Mol. Cell. Cardiol. 2002, 34, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Rababa’h, A.; Singh, S.; Suryavanshi, S.V.; Altarabsheh, S.E.; Deo, S.V.; McConnell, B.K. Compartmentalization role of A-kinase anchoring proteins (AKAPs) in mediating protein kinase A (PKA) signaling and cardiomyocyte hypertrophy. Int. J. Mol. Sci. 2015, 16, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Hill, G.E.; Anderson, J.L.; Lyden, E.R. Ketamine inhibits the proinflammatory cytokine-induced reduction of cardiac intracellular cAMP accumulation. Anesth. Analg. 1998, 87, 1015–1019. [Google Scholar] [PubMed]
- Matsuda, N.; Hattori, Y.; Akaishi, Y. Impairment of cardiac β-adrenoceptor cellular signaling by decreased expression of G(s α) in septic rabbits. Anesthesiology 2000, 93, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Reithmann, C.; Hallstrom, S.; Pilz, G.; Kapsner, T.; Schlag, G.; Werdan, K. Desensitization of rat cardiomyocyte adenylyl cyclase stimulation by plasma of noradrenaline-treated patients with septic shock. Circ. Shock 1993, 41, 48–59. [Google Scholar] [PubMed]
- Carre, J.E.; Singer, M. Cellular energetic metabolism in sepsis: The need for a systems approach. Biochim. Biophys. Acta 2008, 1777, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Lancel, S.; Hassoun, S.M.; Favory, R.; Decoster, B.; Motterlini, R.; Neviere, R. Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J. Pharmacol. Exp. Ther. 2009, 329, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Larche, J.; Lancel, S.; Hassoun, S.M.; Favory, R.; Decoster, B.; Marchetti, P.; Chopin, C.; Neviere, R. Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardial dysfunction and mortality. J. Am. Coll. Cardiol. 2006, 48, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Valsecchi, F.; Konrad, C.; Manfredi, G. Role of soluble adenylyl cyclase in mitochondria. Biochim. Biophys. Acta 2014, 1842, 2555–2560. [Google Scholar] [CrossRef] [PubMed]
- Valsecchi, F.; Ramos-Espiritu, L.S.; Buck, J.; Levin, L.R.; Manfredi, G. cAMP and mitochondria. Physiology 2013, 28, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab. 2009, 9, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Lefkimmiatis, K.; Leronni, D.; Hofer, A.M. The inner and outer compartments of mitochondria are sites of distinct cAMP/PKA signaling dynamics. J. Cell Biol. 2013, 202, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Huttemann, M.; Lee, I.; Samavati, L.; Yu, H.; Doan, J.W. Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochim. Biophys. Acta 2007, 1773, 1701–1720. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Signorile, A.; Larizza, M.; Pacelli, C.; Cocco, T.; Papa, S. Activation of the cAMP cascade in human fibroblast cultures rescues the activity of oxidatively damaged complex I. Free Radic. Biol. Med. 2012, 52, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Huttemann, M.; Helling, S.; Sanderson, T.H.; Sinkler, C.; Samavati, L.; Mahapatra, G.; Varughese, A.; Lu, G.; Liu, J.; Ramzan, R.; et al. Regulation of mitochondrial respiration and apoptosis through cell signaling: Cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim. Biophys. Acta 2012, 1817, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Huttemann, M. Energy crisis: The role of oxidative phosphorylation in acute inflammation and sepsis. Biochim. Biophys. Acta 2014, 1842, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Veksler, V.; Gellerich, F.N.; Saks, V.; Margreiter, R.; Kunz, W.S. Analysis of mitochondrial function in situ in permeabilized muscle fibers; tissues and cells. Nat. Protoc. 2008, 3, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Gatti, D.L.; Bai, Y.; Manfredi, G. Protein phosphorylation and prevention of cytochrome oxidase inhibition by ATP: Coupled mechanisms of energy metabolism regulation. Cell Metab. 2011, 13, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Russwurm, M.; Gunnewig, K.; Gertz, M.; Zoidl, G.; Ramos, L.; Buck, J.; Levin, L.R.; Rassow, J.; Manfredi, G.; et al. A phosphodiesterase 2A isoform localized to mitochondria regulates respiration. J. Biol. Chem. 2011, 286, 30423–30432. [Google Scholar] [CrossRef] [PubMed]
- Rudiger, A.; Singer, M. Mechanisms of sepsis-induced cardiac dysfunction. Crit. Care Med. 2007, 35, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Risoe, P.K.; Ryg, U.; Wang, Y.Y.; Rutkovskiy, A.; Smedsrød, B.; Valen, G.; Dahle, M.K. Cecal ligation and puncture sepsis is associated with attenuated expression of adenylyl cyclase 9 and increased miR142-3p. Shock 2011, 36, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Risoe, P.K.; Wang, Y.; Stuestol, J.F.; Aasen, A.O.; Wang, J.E.; Dahle, M.K. Lipopolysaccharide attenuates mRNA levels of several adenylyl cyclase isoforms in vivo. Biochim. Biophys. Acta 2007, 1772, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Thangamalai, R.; Kandasamy, K.; Sukumarn, S.V.; Reddy, N.; Singh, V.; Choudhury, S.; Parida, S.; Singh, T.U.; Boobalan, R.; Mishra, S.K. Atorvastatin prevents sepsis-induced downregulation of myocardial beta1-adrenoceptors and decreased cAMP response in mice. Shock 2014, 41, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Rudiger, A.; Dyson, A.; Felsmann, K.; Carré, J.E.; Taylor, V.; Hughes, S.; Clatworthy, I.; Protti, A.; Pellerin, D.; Lemm, J.; et al. Early functional and transcriptomic changes in the myocardium predict outcome in a long-term rat model of sepsis. Clin. Sci. 2013, 124, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Lochner, A.; Moolman, J.A. The many faces of H89: A review. Cardiovasc. Drug Rev. 2006, 24, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Huang, Z.; Deutschman, C.S.; Levy, R.J. Caffeine restores myocardial cytochrome oxidase activity and improves cardiac function during sepsis. Crit. Care Med. 2009, 37, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Park, W.S.; Jung, W.K.; Lee, D.Y.; Moon, C.; Yea, S.S.; Park, S.G.; Seo, S.K.; Park, C.; Choi, Y.H.; Kim, G.Y.; et al. Cilostazol protects mice against endotoxin shock and attenuates LPS-induced cytokine expression in RAW 264.7 macrophages via MAPK inhibition and NF-κB inactivation: Not involved in cAMP mechanisms. Int. Immunopharmacol. 2010, 10, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Flemming, S.; Schlegel, N.; Wunder, C.; Meir, M.; Baar, W.; Wollborn, J.; Roewer, N.; Germer, C.T.; Schick, M.A. Phosphodiesterase 4 inhibition dose dependently stabilizes microvascular barrier functions and microcirculation in a rodent model of polymicrobial sepsis. Shock 2014, 41, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, R.; Melbostad, H.; Loomis, W.; Porcides, R.D.; Wolf, P.; Tobar, M.; Hoyt, D.B. LPS-induced acute lung injury is attenuated by phosphodiesterase inhibition: Effects on proinflammatory mediators; metallo proteinases; NF-κB; and ICAM-1 expression. J. Trauma 2006, 60, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.A.; Marques, R.J.; Vicente, J.A.; Santos, M.S.; Monteiro, P.; Moreno, A.J.; Custódio, J.B. Sildenafil citrate concentrations not affecting oxidative phosphorylation depress H2O2 generation by rat heart mitochondria. Mol. Cell. Biochem. 2008, 309, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Rentsendorj, O.; D’Alessio, F.R.; Pearse, D.B. Phosphodiesterase 2A is a major negative regulator of iNOS expression in lipopolysaccharide-treated mouse alveolar macrophages. J. Leukoc. Biol. 2014, 96, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Hoover, D.B.; Ozment, T.R.; Wondergem, R.; Li, C.; Williams, D.L. Impaired heart rate regulation and depression of cardiac chronotropic and dromotropic function in polymicrobial sepsis. Shock 2015, 43, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Shrum, B.; Anantha, R.V.; Xu, S.X.; Donnelly, M.; Haeryfar, S.M.; McCormick, J.K.; Mele, T. A robust scoring system to evaluate sepsis severity in an animal model. BMC Res. Notes 2014. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Respiration Parameters | Sham | Sepsis |
|---|---|---|
| Vglut+mal respiration rate | ||
| vehicle | 14.5 ± 1.7 | 16.3 ± 2.3 |
| IBMX | 15.8 ± 4.2 | 14.9 ± 4.2 |
| BAY 60 7550 | 15.1 ± 3.7 | 10.3 ± 5.1 # |
| Vglut+mal+ADP respiration rate | ||
| vehicle | 103.6 ± 14.7 | 87.7 ± 4.5 * |
| IBMX | 101.9 ± 28.0 | 79.1 ± 30.3 * |
| BAY 60 7550 | 93.4 ± 20.9 | 84.4 ± 29.1 * |
| Respiratory control ratio | ||
| vehicle | 4.67 ± 0.91 | 3.18 ± 0.40 * |
| IBMX | 4.46 ± 0.59 | 3.11 ± 0.28 * |
| BAY 60 7550 | 4.35 ± 0.82 | 3.88 ± 0.31 # |
| Cardiac Parameters | Sham | Sepsis |
|---|---|---|
| LVDF (gram-force) | ||
| (1) Ex vivo perfusion | ||
| ● vehicle | 1.41 ± 0.28 | 0.53 ± 0.34 * |
| ● IBMX | 2.24 ± 0.37 # | 1.28 ± 0.20 *,# |
| ● BAY 60 7550 | 1.53 ± 0.82 | 1.05 ± 0.25 *,# |
| (2) In vivo BAY 60 7550 | 1.51 ± 0.28 | 0.62 ± 0.31 * |
| Coronary pressure (mmHg) | ||
| (1) Ex vivo perfusion | ||
| ● vehicle | 88 ± 14 | 86 ± 17 |
| ● IBMX | 85 ± 3 | 88 ± 6 |
| ● BAY 60 7550 | 84 ± 23 | 84 ± 25 |
| (2) In vivo BAY 60 7550 | 85 ± 14 | 52 ± 23 *,# |
| MVO2 (μL·min−1·g−1) | ||
| (1) Ex vivo perfusion | ||
| ● vehicle | 185 ± 14 | 95 ± 23 * |
| ● IBMX | 275 ± 42 # | 205 ± 25 *,# |
| ● BAY 60 7550 | 192 ± 23 | 142 ± 23 *,# |
| (2) In vivo BAY 60 7550 | 189 ± 14 | 97 ± 14 * |
| Efficiency | ||
| (1) Ex vivo perfusion | ||
| ● vehicle | 4.1 ± 0.85 | 3.0 ± 0.85 * |
| ● IBMX | 4.3 ± 0.57 | 3.3 ± 0.57 * |
| ● BAY 60 7550 | 4.3 ± 0.28 | 4.0 ± 0.57 # |
| (2) In vivo BAY 60 7550 | 4.4 ± 0.85 | 3.8 ± 0.85 # |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neviere, R.; Delguste, F.; Durand, A.; Inamo, J.; Boulanger, E.; Preau, S. Abnormal Mitochondrial cAMP/PKA Signaling Is Involved in Sepsis-Induced Mitochondrial and Myocardial Dysfunction. Int. J. Mol. Sci. 2016, 17, 2075. https://doi.org/10.3390/ijms17122075
Neviere R, Delguste F, Durand A, Inamo J, Boulanger E, Preau S. Abnormal Mitochondrial cAMP/PKA Signaling Is Involved in Sepsis-Induced Mitochondrial and Myocardial Dysfunction. International Journal of Molecular Sciences. 2016; 17(12):2075. https://doi.org/10.3390/ijms17122075
Chicago/Turabian StyleNeviere, Remi, Florian Delguste, Arthur Durand, Jocelyn Inamo, Eric Boulanger, and Sebastien Preau. 2016. "Abnormal Mitochondrial cAMP/PKA Signaling Is Involved in Sepsis-Induced Mitochondrial and Myocardial Dysfunction" International Journal of Molecular Sciences 17, no. 12: 2075. https://doi.org/10.3390/ijms17122075
APA StyleNeviere, R., Delguste, F., Durand, A., Inamo, J., Boulanger, E., & Preau, S. (2016). Abnormal Mitochondrial cAMP/PKA Signaling Is Involved in Sepsis-Induced Mitochondrial and Myocardial Dysfunction. International Journal of Molecular Sciences, 17(12), 2075. https://doi.org/10.3390/ijms17122075