
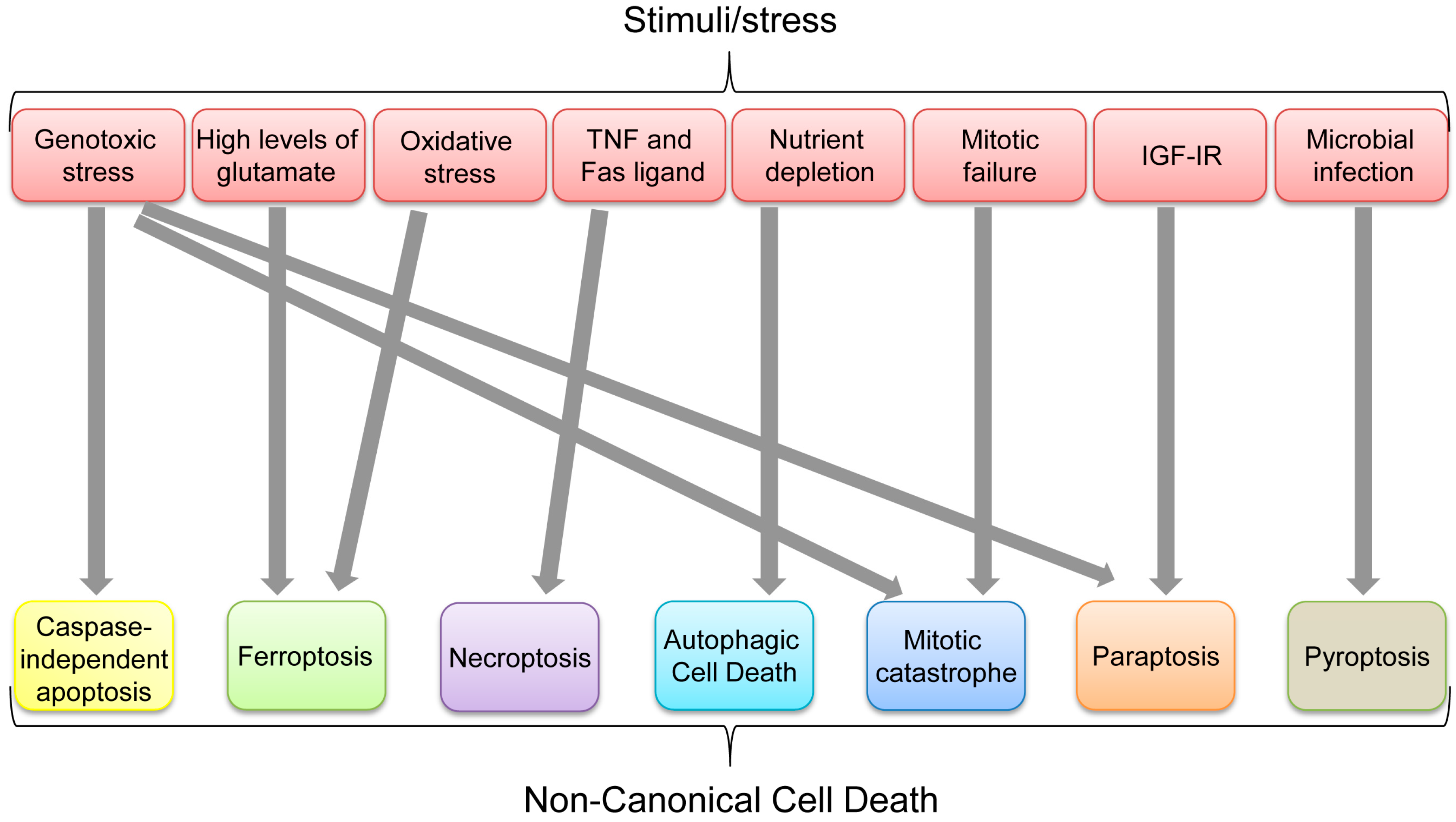
Non-Canonical Cell Death Induced by p53

Abstract
:
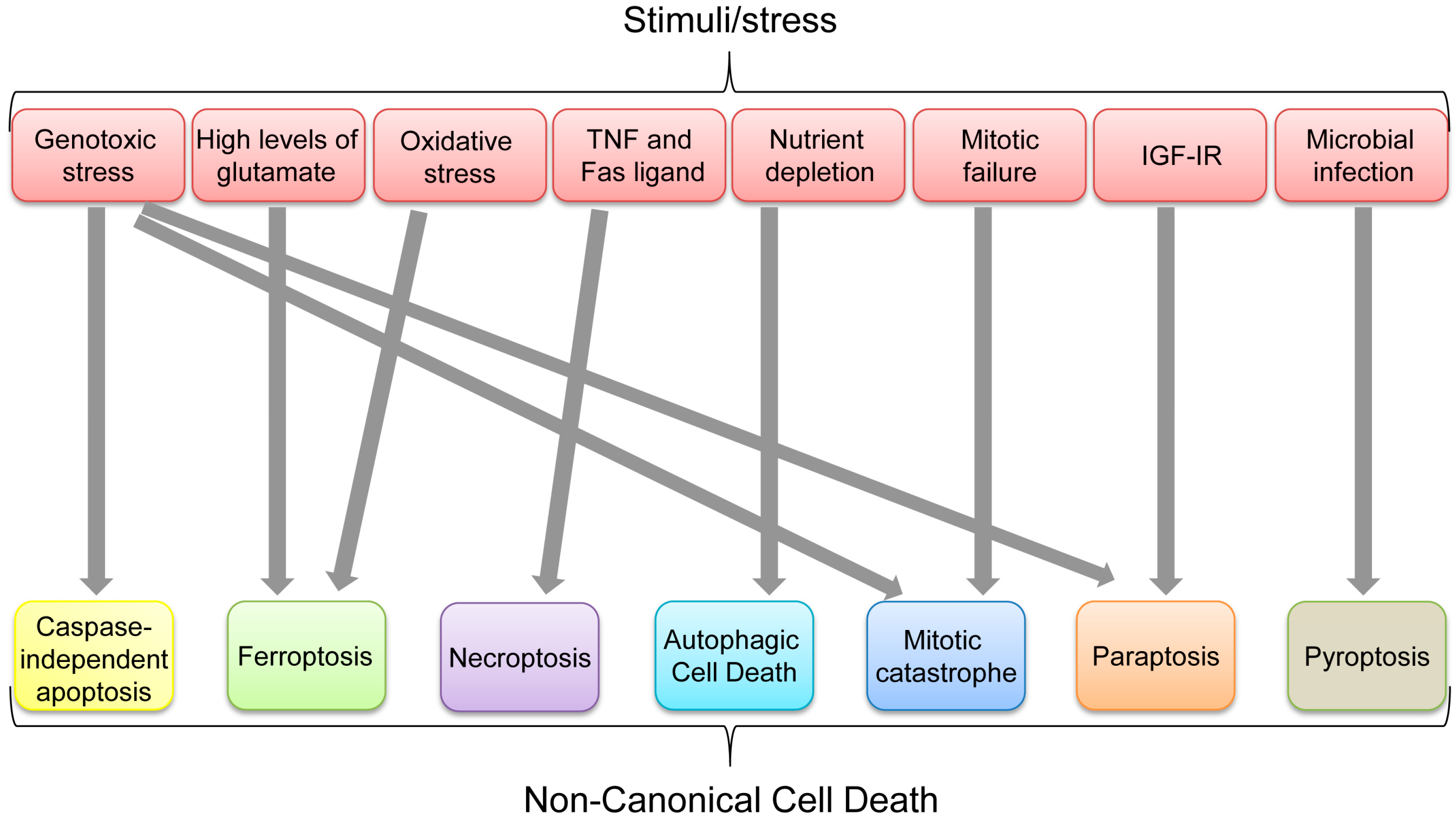
1. Introduction
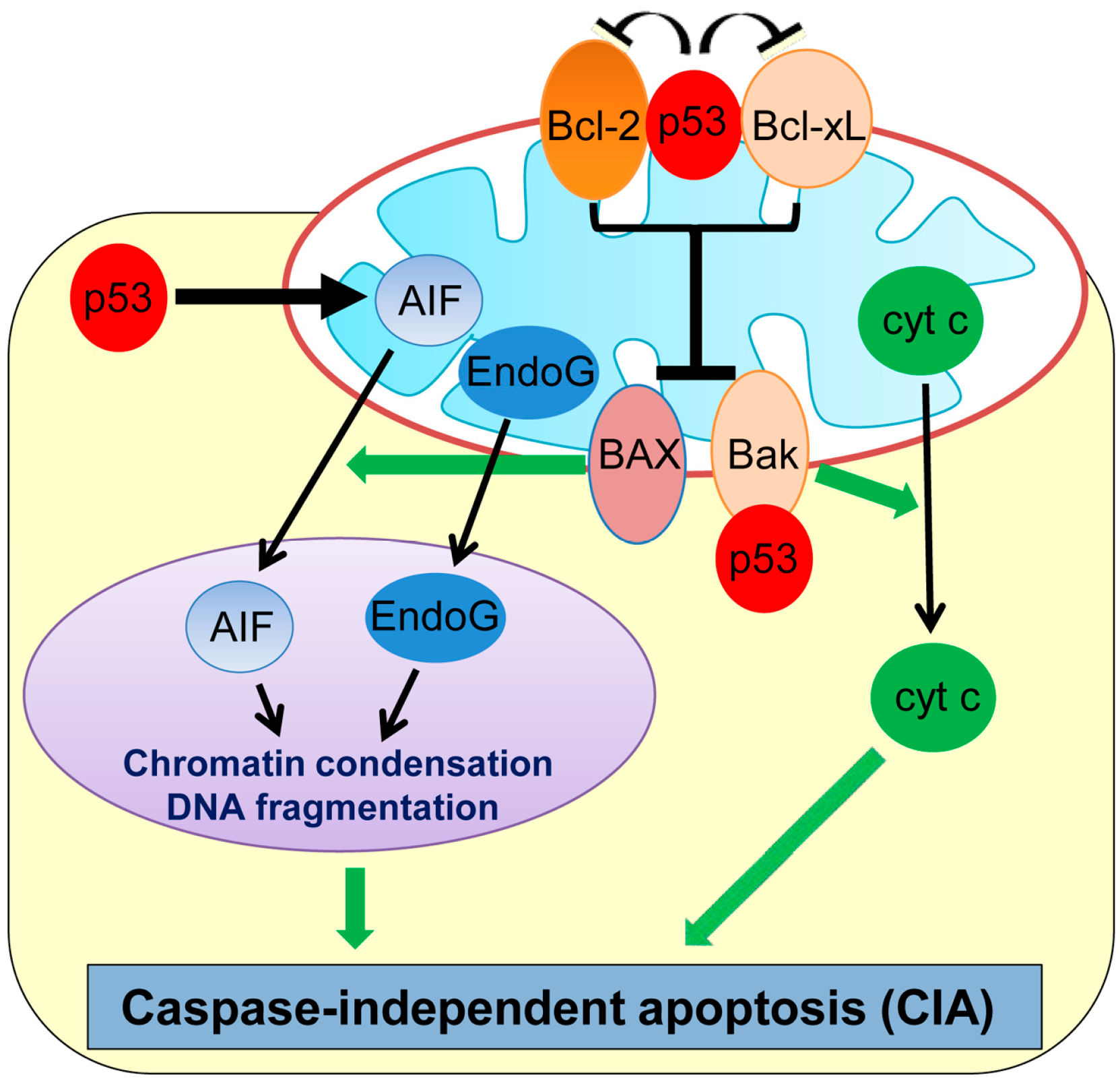
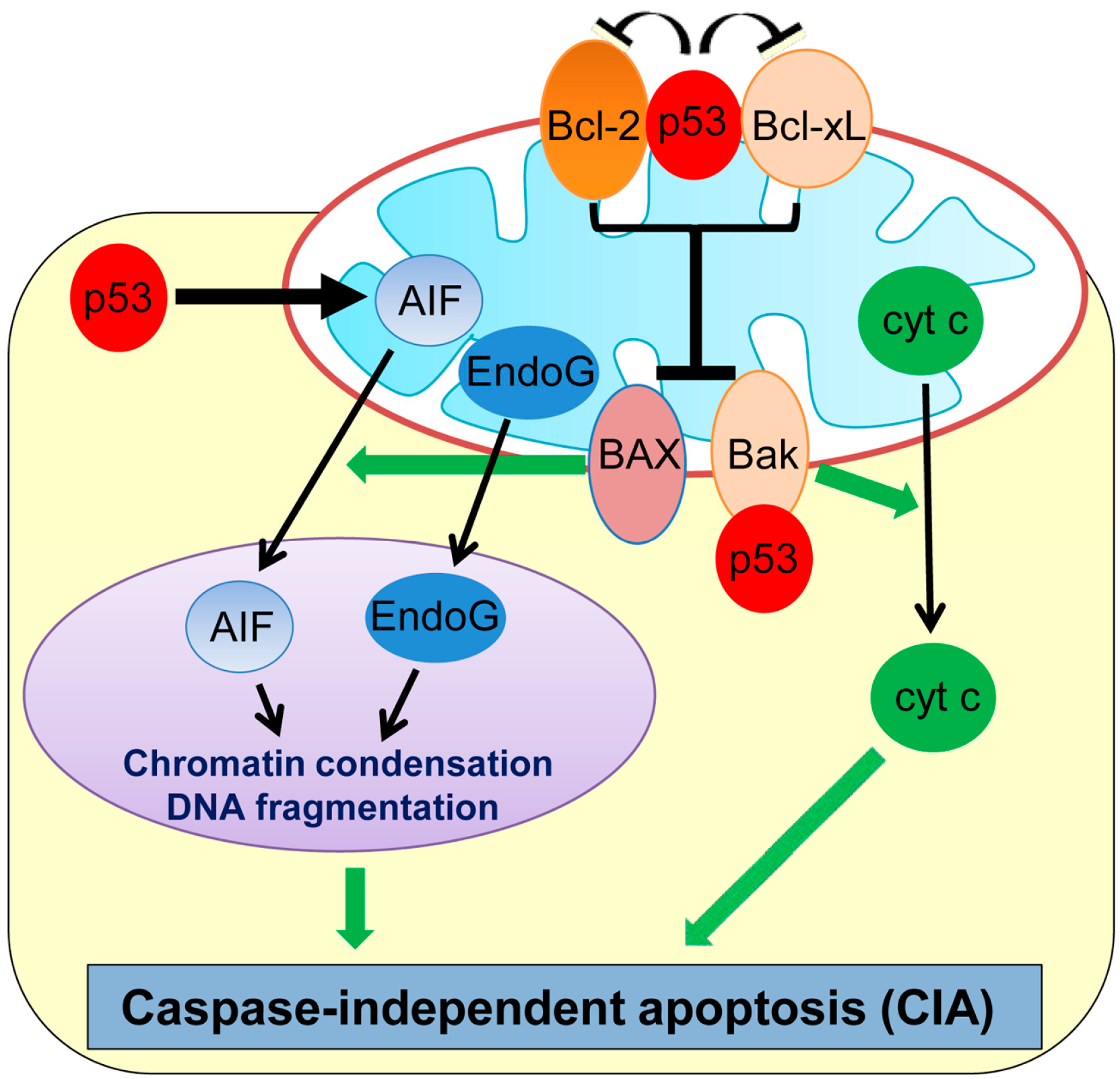
2. Caspase-Independent Apoptosis (CIA)
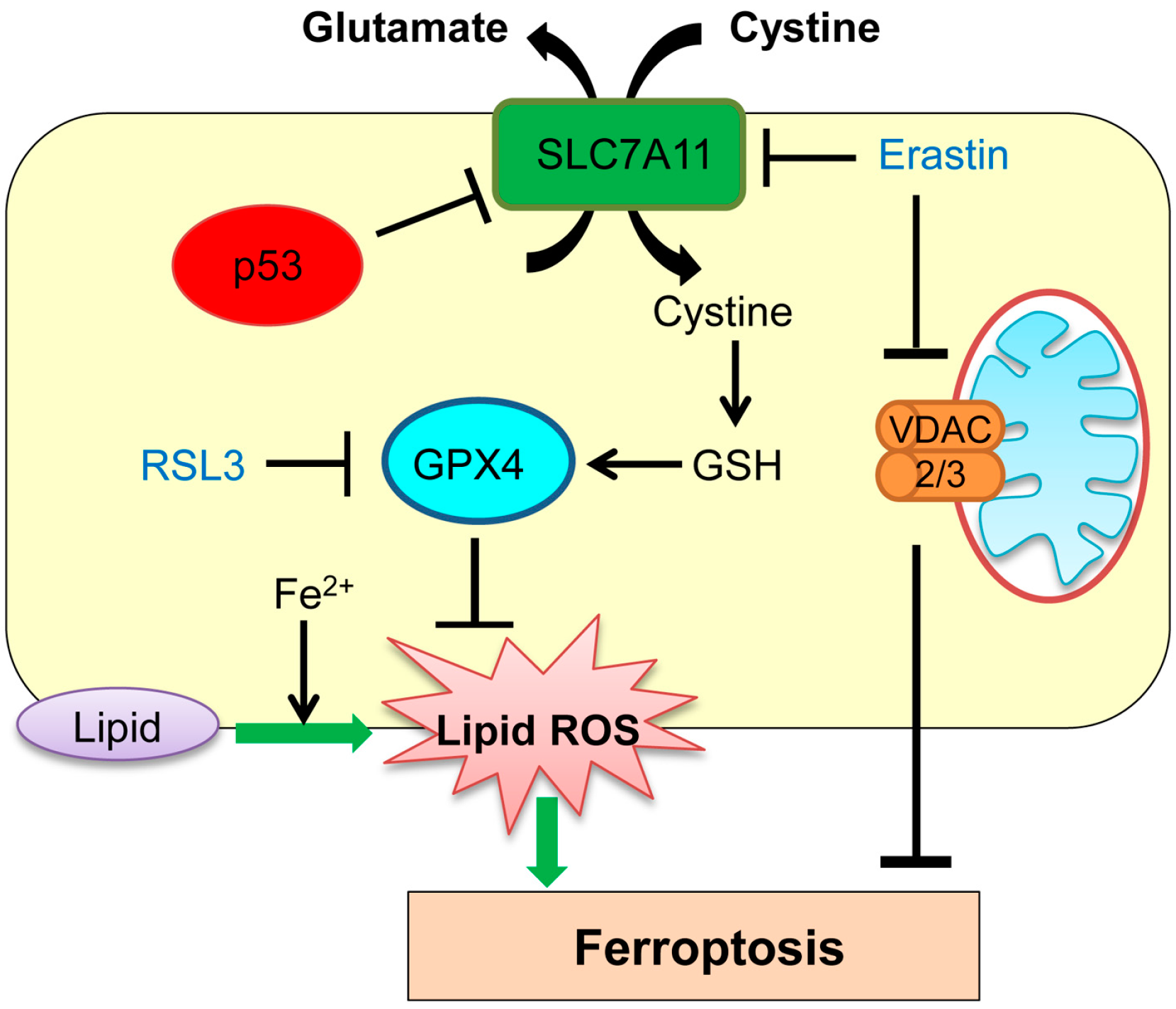
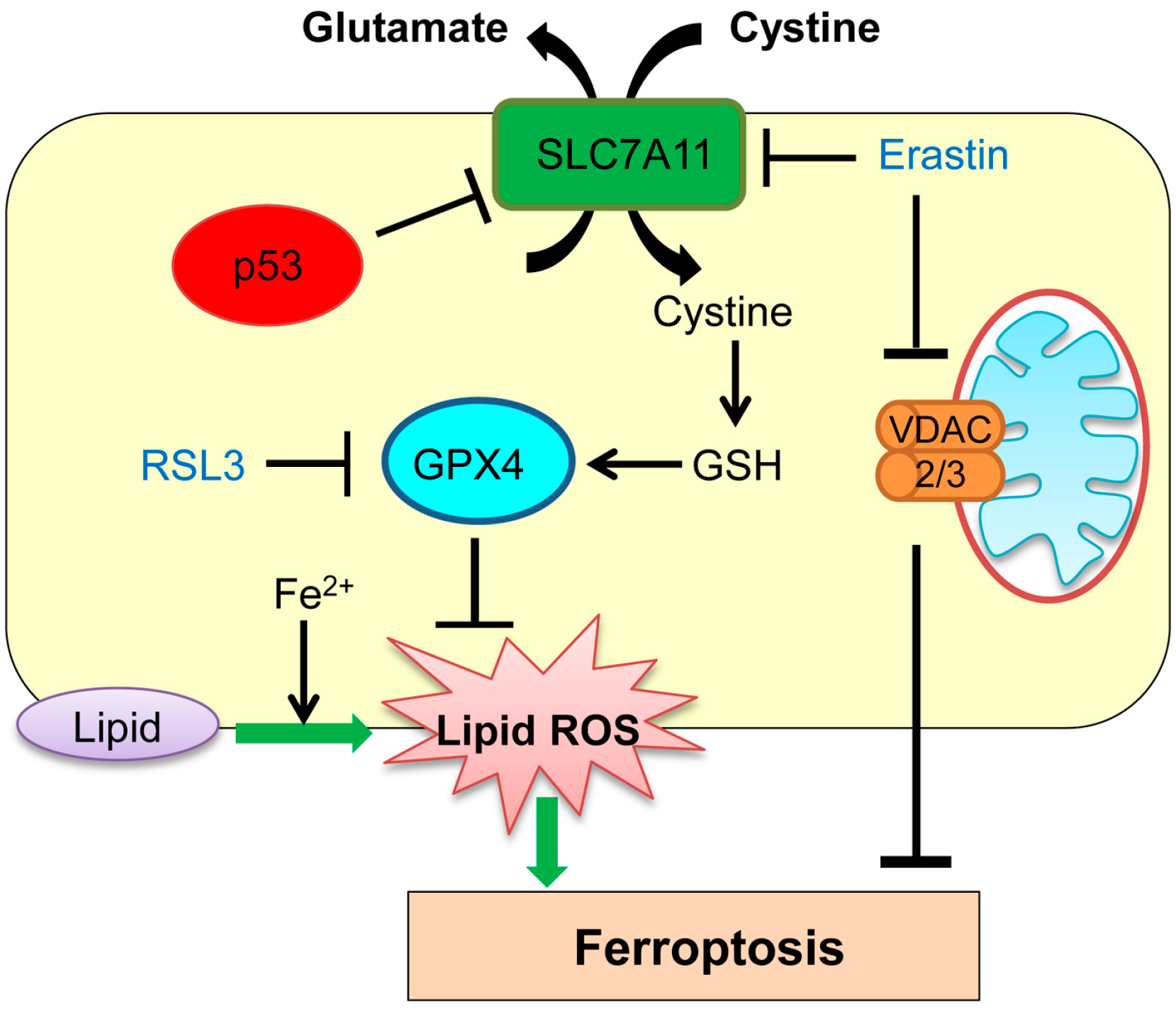
3. Ferroptosis
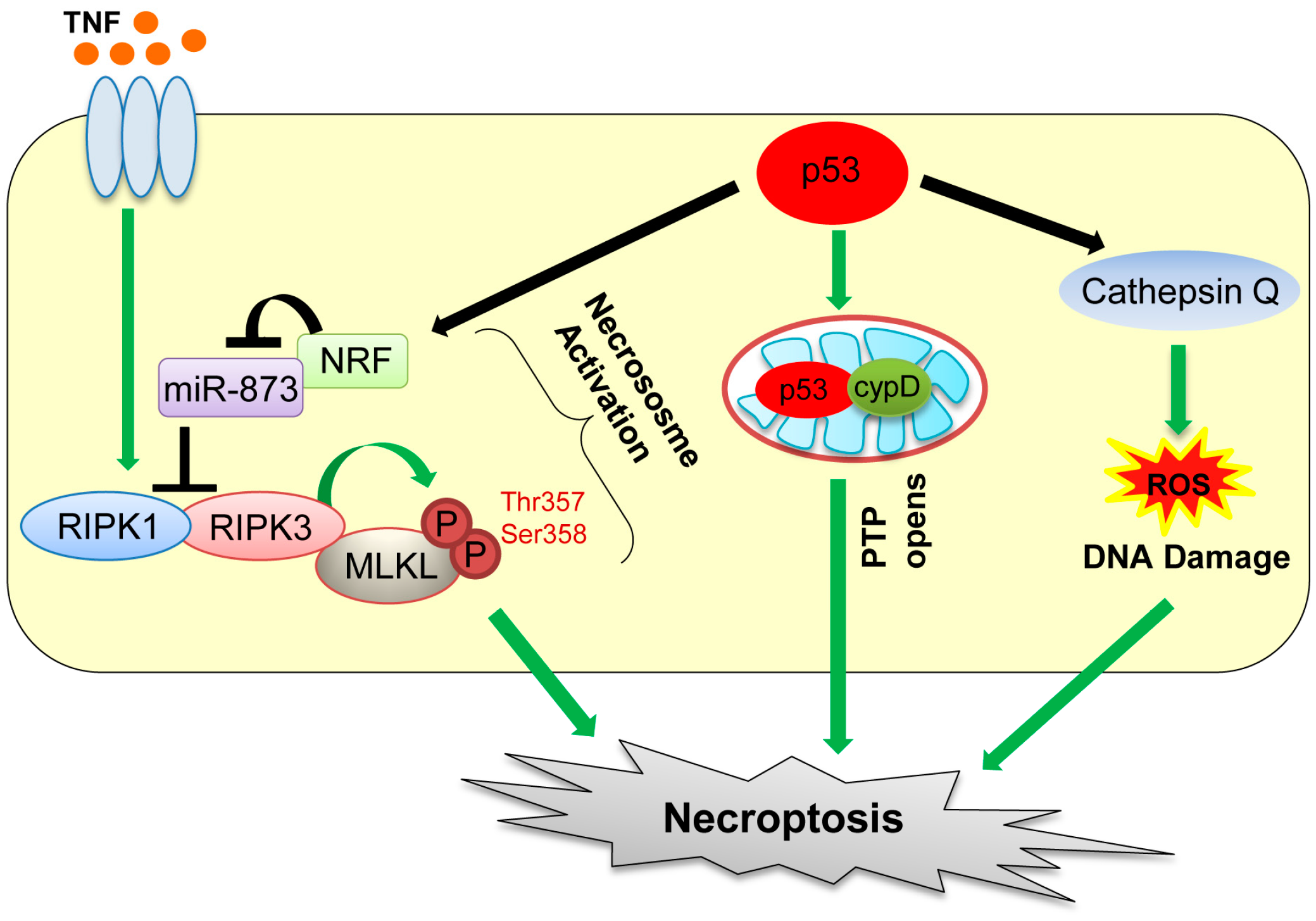
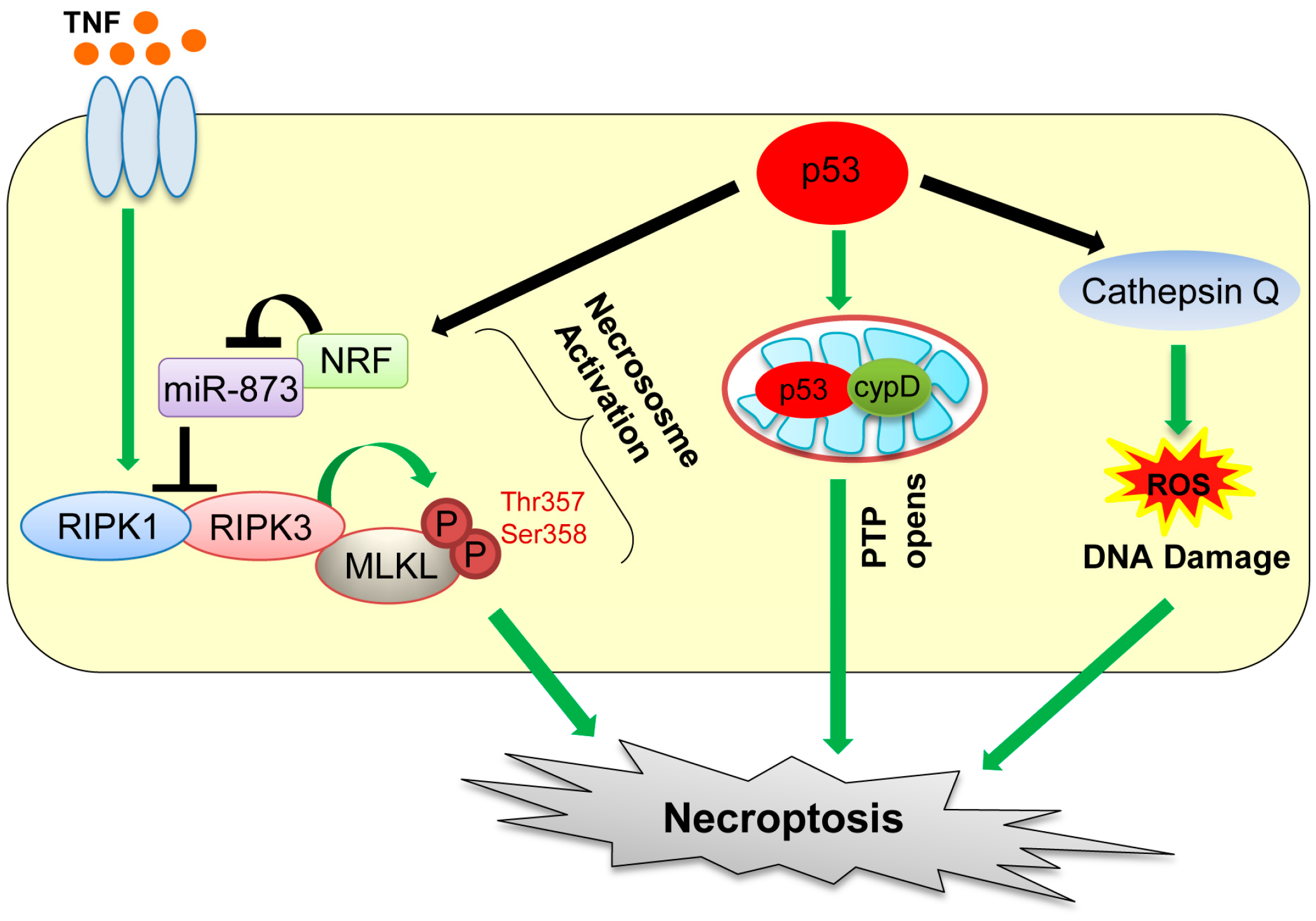
4. Necroptosis
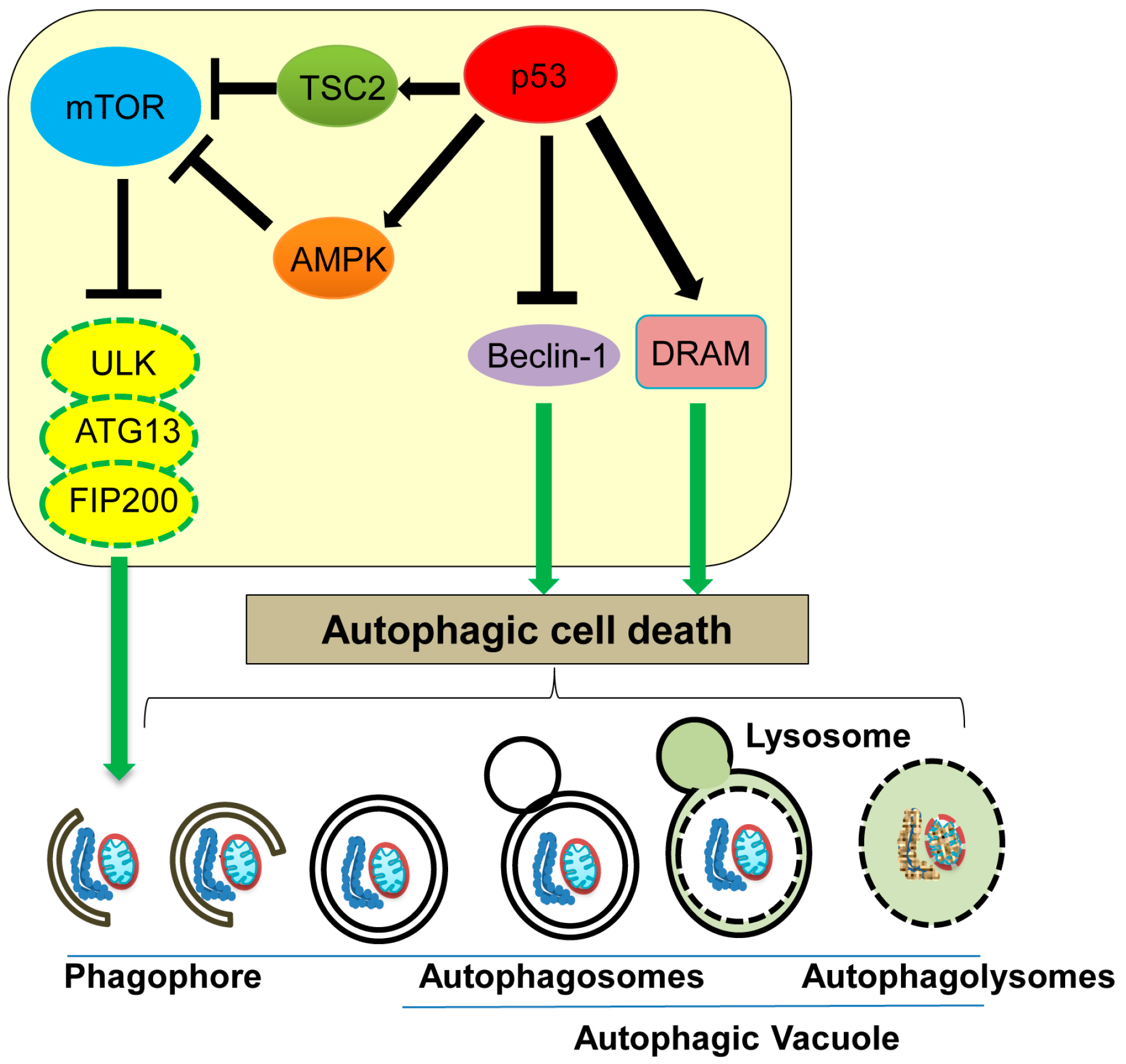
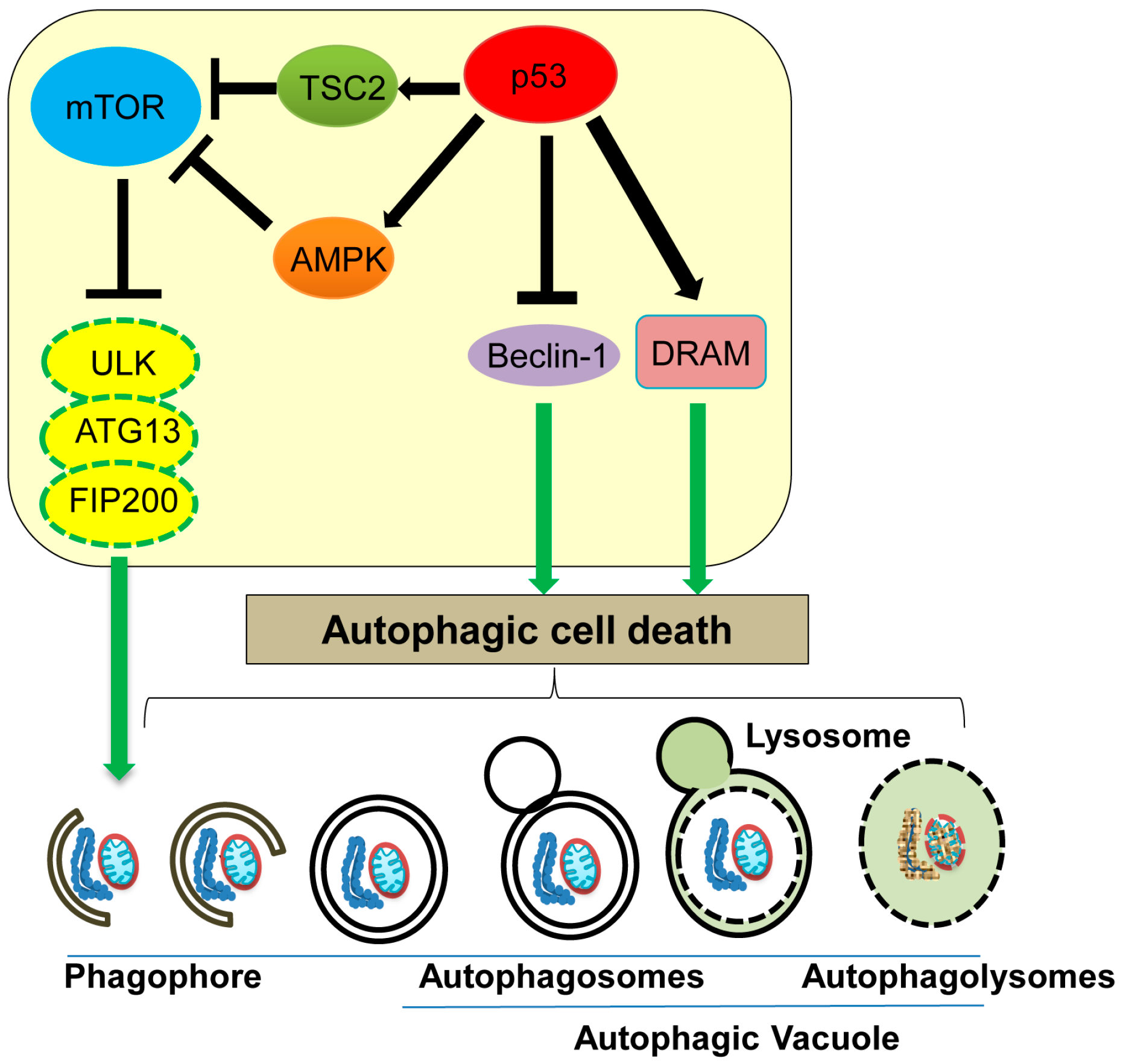
5. Autophagic Cell Death
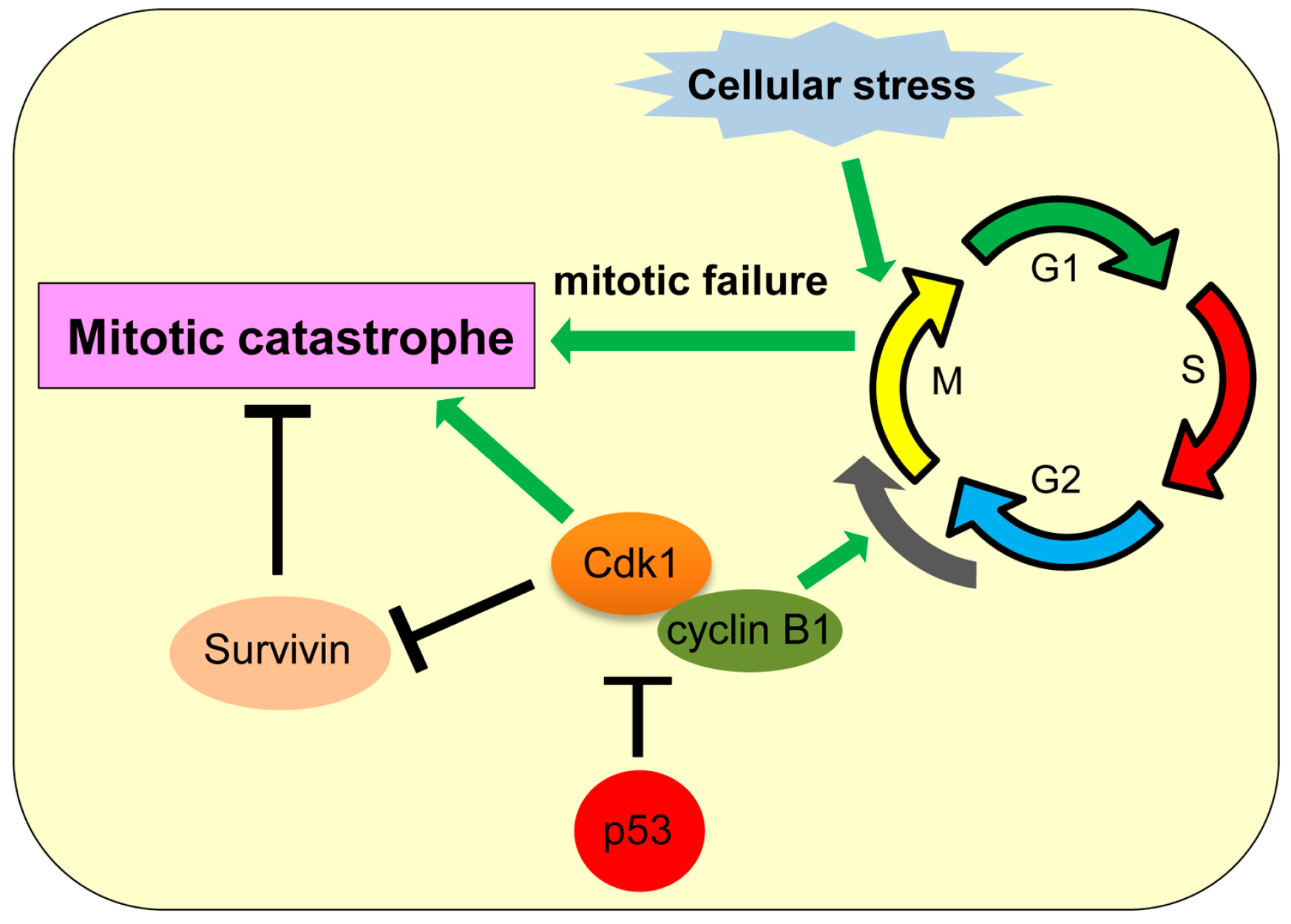
6. Mitotic Catastrophe
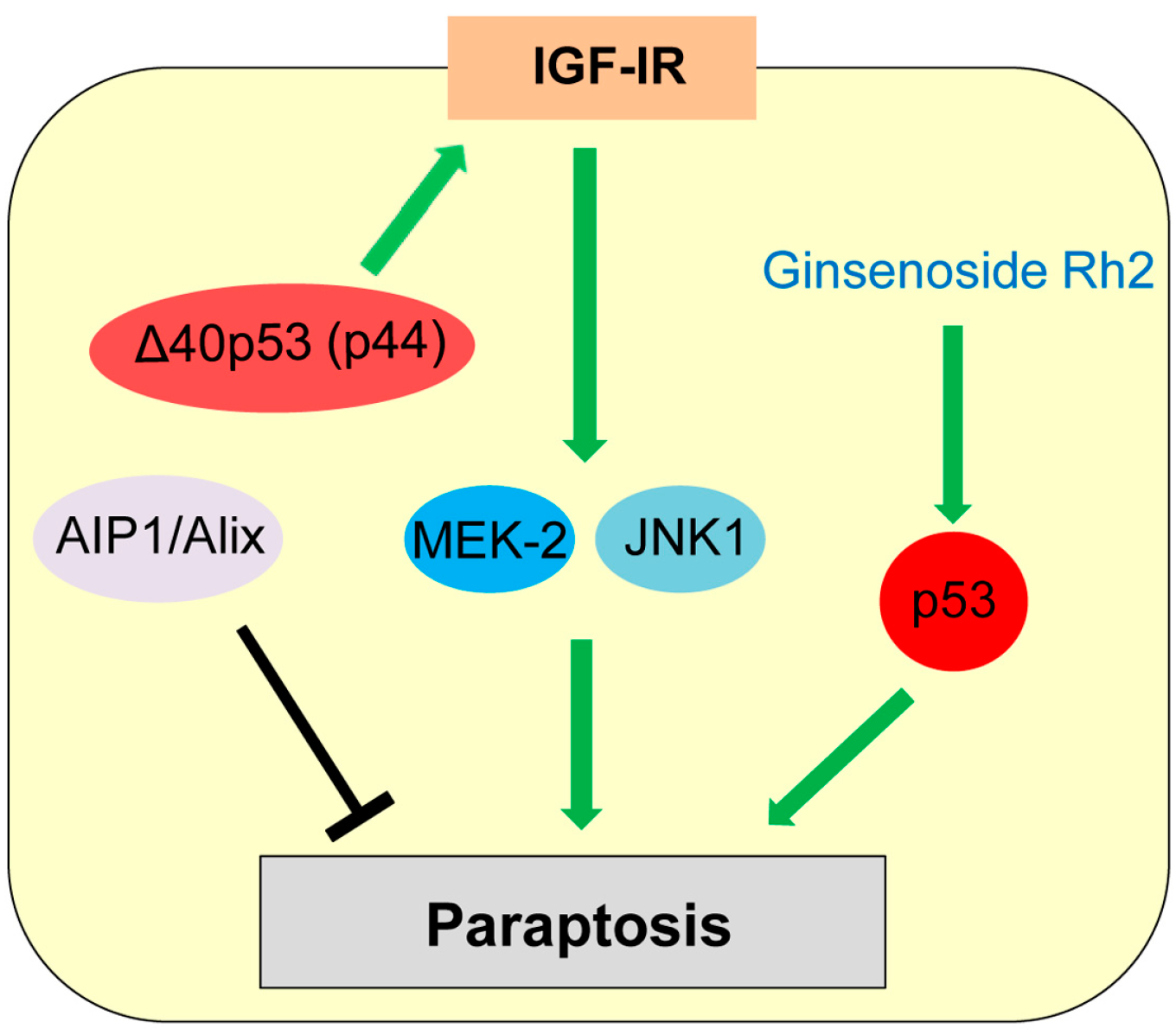
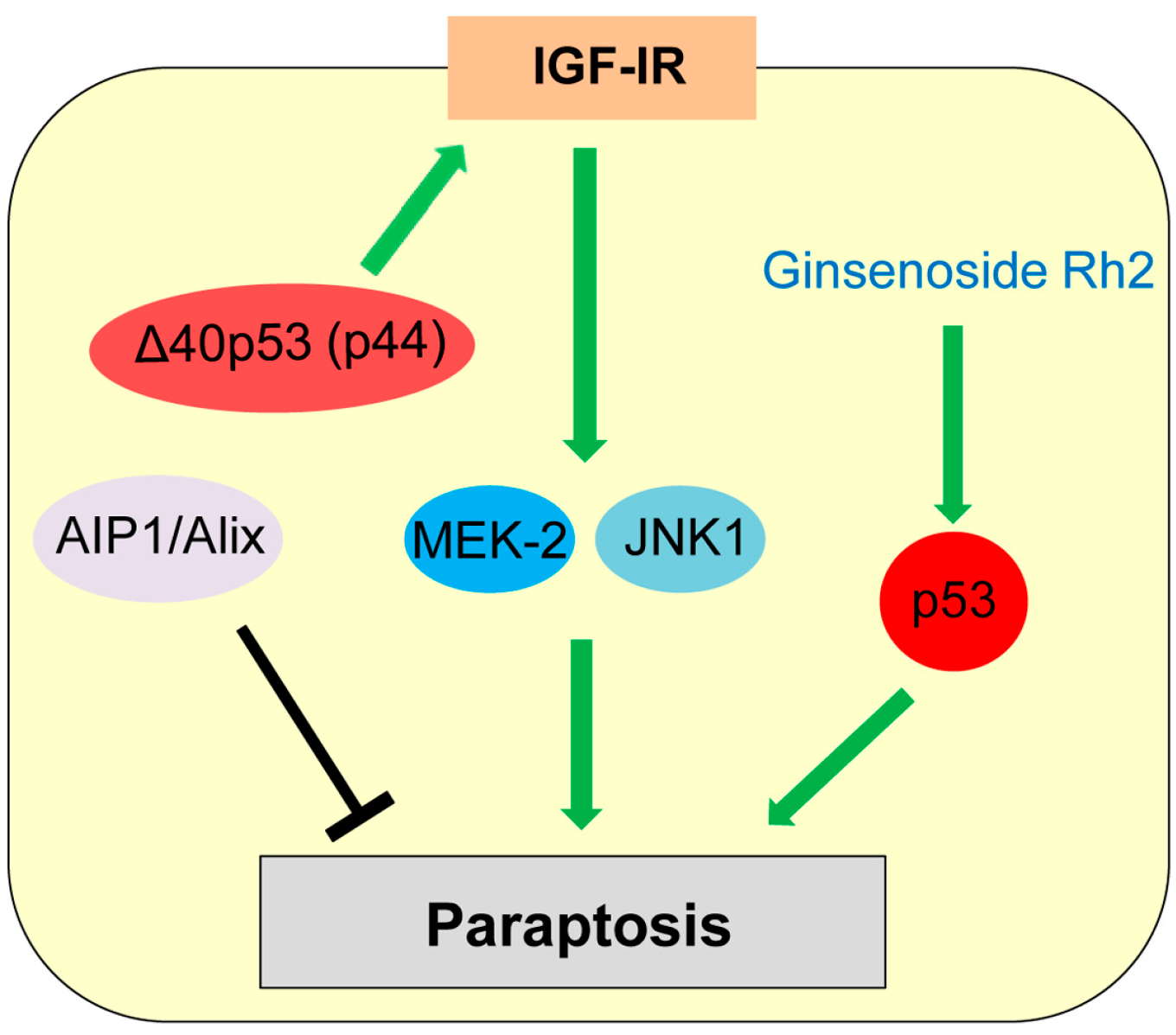
7. Paraptosis
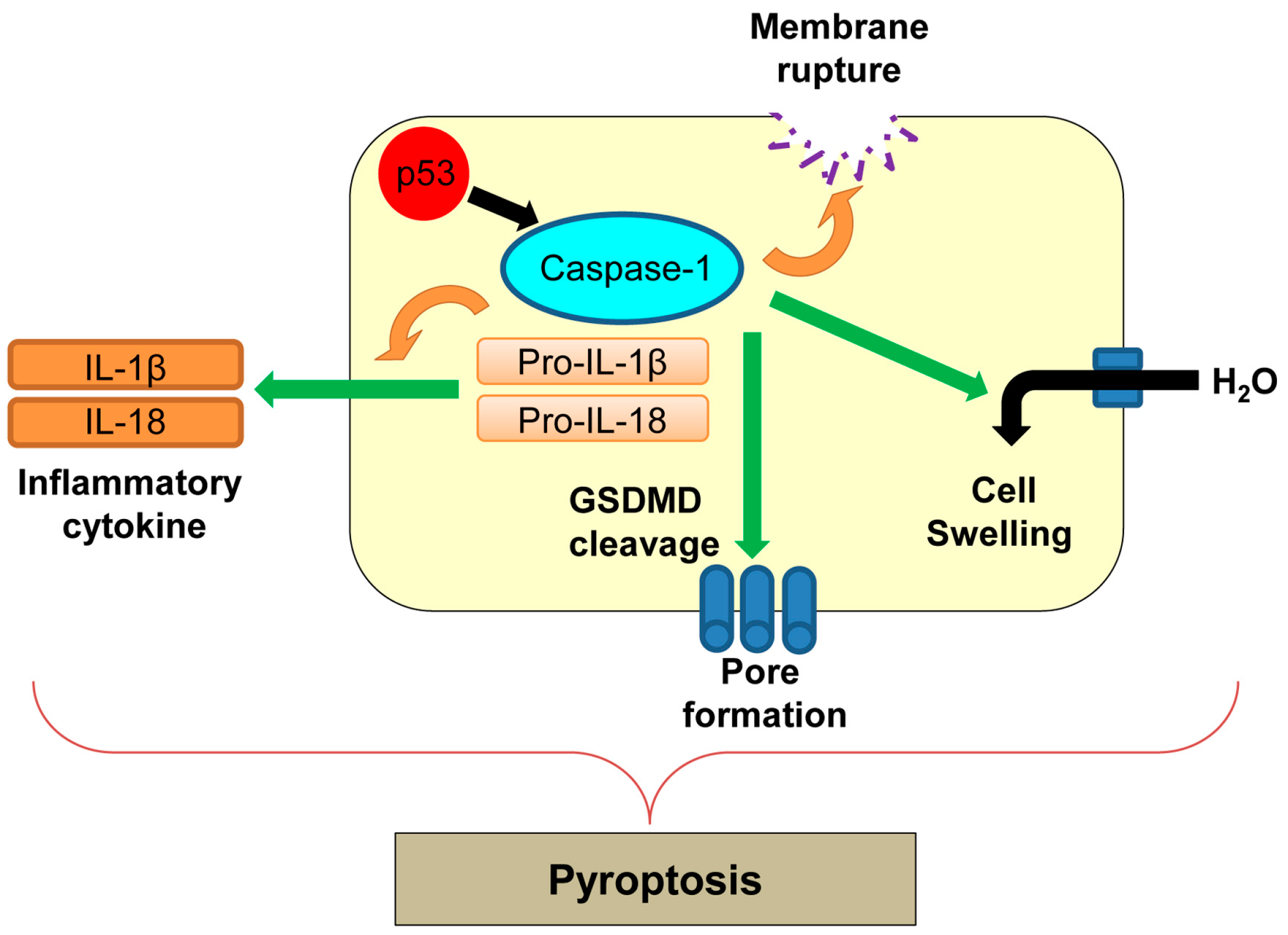
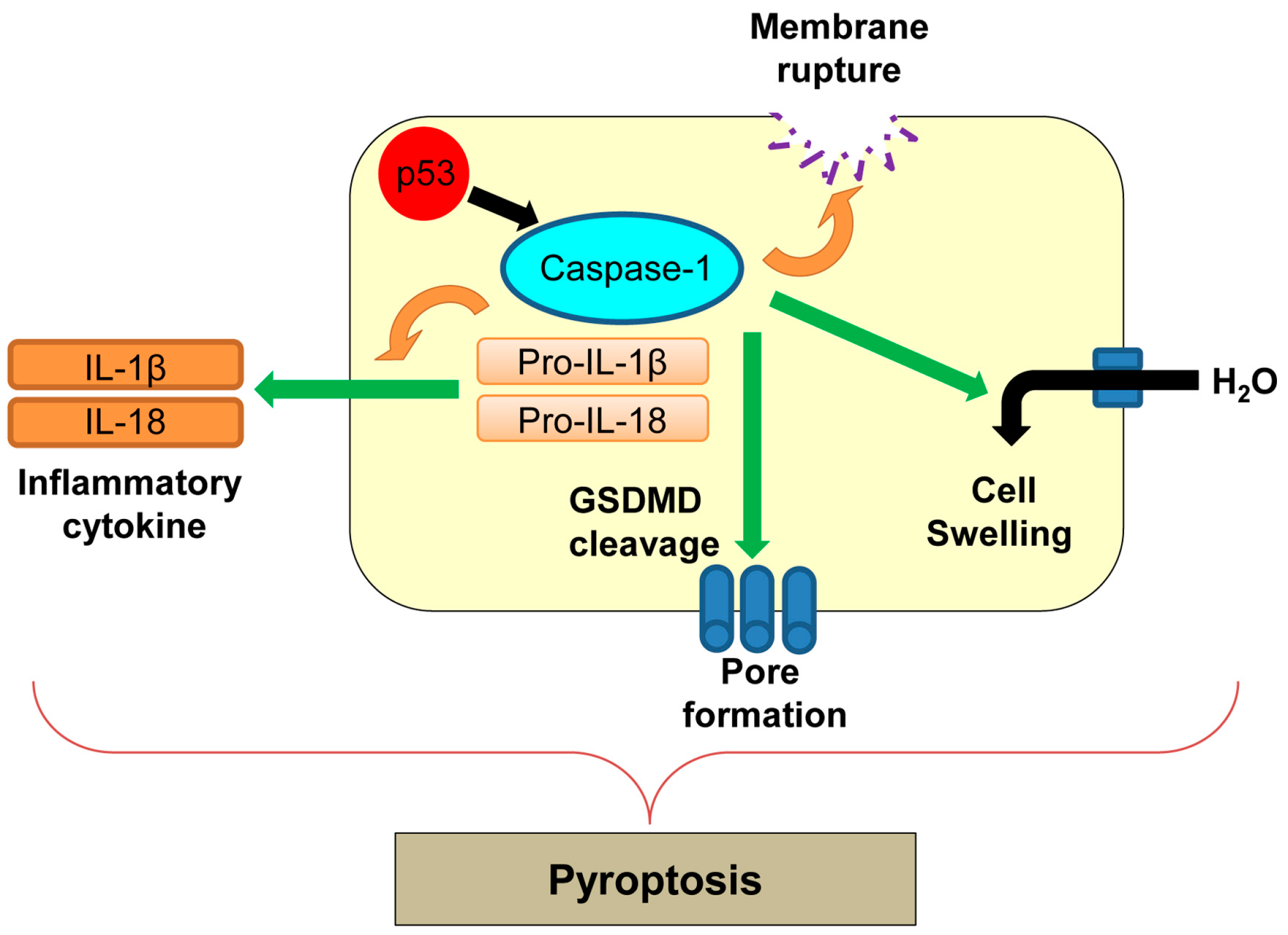
8. Pyroptosis
9. Efferocytosis
10. Conclusions and Discussion
Acknowledgments
Conflicts of Interest
References
- Vogt, C. Untersuchungen Uber Die Entwicklungsgeschichte der Geburtshelferkröte (Alytes Obstetricans); Jent und Gassmann: Solothurn, Switzerland, 1984. [Google Scholar]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the nomenclature committee on cell death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.E.; Yuan, J.Y.; Horvitz, H.R. Mechanisms and functions of cell death. Annu. Rev. Cell Biol. 1991, 7, 663–698. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.D.; Weil, M.; Raff, M.C. Programmed cell death in animal development. Cell 1997, 88, 347–354. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; El-Deiry, W.S. Trail and apoptosis induction by TNF-family death receptors. Oncogene 2003, 22, 8628–8633. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H. The Fas signaling pathway: More than a paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Gaidano, G. Messengers of cell death: Apoptotic signaling in health and disease. Haematologica 2003, 88, 212–218. [Google Scholar] [PubMed]
- Evans-Storms, R.B.; Cidlowski, J.A. Regulation of apoptosis by steroid hormones. J. Steroid Biochem. Mol. Biol. 1995, 53, 1–8. [Google Scholar] [CrossRef]
- Herbst, S.; Schaible, U.E.; Schneider, B.E. Interferon gamma activated macrophages kill mycobacteria by nitric oxide induced apoptosis. PLoS ONE 2011, 6, e19105. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Zavaleta, N.; Carmody, A.; Messer, R.; Whitmire, W.M.; Caldwell, H.D. Chlamydia pneumoniae inhibits activated human T lymphocyte proliferation by the induction of apoptotic and pyroptotic pathways. J. Immunol. 2011, 186, 7120–7126. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.Y.; Chen, X.Y.; Liu, Y.J.; Zhong, H.M.; Jiang, F.L.; Liu, Y. Oxidative stress-mediated intrinsic apoptosis in human promyelocytic leukemia HL-60 cells induced by organic arsenicals. Sci. Rep. 2016, 6, 29865. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [PubMed]
- Duprez, L.; Wirawan, E.; Vanden Berghe, T.; Vandenabeele, P. Major cell death pathways at a glance. Microbes Infect. 2009, 11, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Mak, T.W. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Diederich, M.; Cerella, C. Non-canonical programmed cell death mechanisms triggered by natural compounds. Semin. Cancer Biol. 2016, 40–41, 4–34. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Iwakuma, T. Targeting oncogenic mutant p53 for cancer therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef] [PubMed]
- Iwakuma, T.; Lozano, G. Crippling p53 activities via knock-in mutations in mouse models. Oncogene 2007, 26, 2177–2184. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.; Levine, A. p53 research: The past thirty years and the next thirty years. Cold Spring Harb. Perspect. Biol. 2010, 2, a000893. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.C.; Strasser, A. BH3-only proteins-essential initiators of apoptotic cell death. Cell 2000, 103, 839–842. [Google Scholar] [CrossRef]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Tajeddine, N.; Kroemer, G. Targeting p53 to mitochondria for cancer therapy. Cell Cycle 2008, 7, 1949–1955. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S. The role of p53 in chemosensitivity and radiosensitivity. Oncogene 2003, 22, 7486–7495. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Rothman, J.H. Cell death: Hook, line and linker. Curr. Biol. 2007, 17, R286–R289. [Google Scholar] [CrossRef] [PubMed]
- Ekert, P.G.; Read, S.H.; Silke, J.; Marsden, V.S.; Kaufmann, H.; Hawkins, C.J.; Gerl, R.; Kumar, S.; Vaux, D.L. Apaf-1 and caspase-9 accelerate apoptosis, but do not determine whether factor-deprived or drug-treated cells die. J. Cell Biol. 2004, 165, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Padhye, S.; Dandawate, P.; Yusufi, M.; Ahmad, A.; Sarkar, F.H. Perspectives on medicinal properties of plumbagin and its analogs. Med. Res. Rev. 2012, 32, 1131–1158. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, W.; Zhou, H.; Luo, N.; Nie, C.; Zhao, X.; Yuan, Z.; Liu, X.; Wei, Y. Mitochondrial p53 phosphorylation induces Bak-mediated and caspase-independent cell death. Oncotarget 2015, 6, 17192–17205. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.J.; Kim, Y.S.; Kim, J.U.; Han, S.M.; Shin, J.W.; Yang, H.O. Mechanism of taxol-induced apoptosis in human SKOV3 ovarian carcinoma cells. J. Cell Biochem. 2004, 91, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Broker, L.E.; Huisman, C.; Span, S.W.; Rodriguez, J.A.; Kruyt, F.A.; Giaccone, G. Cathepsin B mediates caspase-independent cell death induced by microtubule stabilizing agents in non-small cell lung cancer cells. Cancer Res. 2004, 64, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [PubMed]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic dnase when released from mitochondria. Nature 2001, 412, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, G.; Schotte, P.; van Gurp, M.; Demol, H.; Hoorelbeke, B.; Gevaert, K.; Rodriguez, I.; Ruiz-Carrillo, A.; Vandekerckhove, J.; Declercq, W.; et al. Endonuclease G: A mitochondrial protein released in apoptosis and involved in caspase-independent DNA degradation. Cell Death Differ. 2001, 8, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Miramar, M.D.; Costantini, P.; Ravagnan, L.; Saraiva, L.M.; Haouzi, D.; Brothers, G.; Penninger, J.M.; Peleato, M.L.; Kroemer, G.; Susin, S.A. NADH oxidase activity of mitochondrial apoptosis-inducing factor. J. Biol. Chem. 2001, 276, 16391–16398. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Cande, C.; Stephanou, N.C.; Jiang, S.; Gurbuxani, S.; Larochette, N.; Daugas, E.; Garrido, C.; Kroemer, G.; Wu, H. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat. Struct. Biol. 2002, 9, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, M.; Daugas, E.; Susin, S.A.; Zamzami, N.; Metivier, D.; Nieminen, A.L.; Brothers, G.; Penninger, J.M.; Kroemer, G. Dominant cell death induction by extramitochondrially targeted apoptosis-inducing factor. FASEB J. 2001, 15, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Cregan, S.P.; Fortin, A.; MacLaurin, J.G.; Callaghan, S.M.; Cecconi, F.; Yu, S.W.; Dawson, T.M.; Dawson, V.L.; Park, D.S.; Kroemer, G.; et al. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J. Cell Biol. 2002, 158, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.S.; Novak, R.; Murray, P.J.; Eischen, C.M.; Susin, S.A.; Kroemer, G.; Halle, A.; Weber, J.R.; Tuomanen, E.I.; Cleveland, J.L. Apoptosis-inducing factor mediates microglial and neuronal apoptosis caused by pneumococcus. J. Infect. Dis. 2001, 184, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 2009, 1787, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human Bax gene. Cell 1995, 80, 293–299. [Google Scholar] [PubMed]
- Wu, Y.; Mehew, J.W.; Heckman, C.A.; Arcinas, M.; Boxer, L.M. Negative regulation of Bcl-2 expression by p53 in hematopoietic cells. Oncogene 2001, 20, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Stambolsky, P.; Weisz, L.; Shats, I.; Klein, Y.; Goldfinger, N.; Oren, M.; Rotter, V. Regulation of AIF expression by p53. Cell Death Differ. 2006, 13, 2140–2149. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; de Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by Gpx4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-Ras-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Ou, Y.; Jiang, L.; Gu, W. Ferroptosis: A missing puzzle piece in the p53 blueprint? Mol. Cell. Oncol. 2016, 3, e1046581. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kroemer, G. Ferroptosis in p53-dependent oncosuppression and organismal homeostasis. Cell Death Differ. 2015, 22, 1237–1238. [Google Scholar] [CrossRef] [PubMed]
- Proskuryakov, S.Y.; Konoplyannikov, A.G.; Gabai, V.L. Necrosis: A specific form of programmed cell death? Exp. Cell Res. 2003, 283, 1–16. [Google Scholar] [CrossRef]
- Edinger, A.L.; Thompson, C.B. Death by design: Apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 2004, 16, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Majno, G.; Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 1995, 146, 3–15. [Google Scholar] [PubMed]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kroemer, G. Necroptosis: A specialized pathway of programmed necrosis. Cell 2008, 135, 1161–1163. [Google Scholar] [CrossRef] [PubMed]
- Laster, S.M.; Wood, J.G.; Gooding, L.R. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J. Immunol. 1988, 141, 2629–2634. [Google Scholar] [PubMed]
- Matsumura, H.; Shimizu, Y.; Ohsawa, Y.; Kawahara, A.; Uchiyama, Y.; Nagata, S. Necrotic death pathway in Fas receptor signaling. J. Cell Biol. 2000, 151, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase rip as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dugger, D.L.; Wickliffe, K.E.; Kapoor, N.; de Almagro, M.C.; Vucic, D.; Komuves, L.; Ferrando, R.E.; French, D.M.; Webster, J.; et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 2014, 343, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Chan, F.K. Necrosis-dependent and independent signaling of the RIP kinases in inflammation. Cytokine Growth Factor Rev. 2014, 25, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.M.; Cook, W.D.; Okamoto, T.; Murphy, J.; Lawlor, K.E.; Vince, J.E.; Vaux, D.L. TNF can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis. 2013, 4, e465. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.C.; Ren, D.; Wang, G.X.; Chen, D.Y.; Westergard, T.D.; Kim, H.; Sasagawa, S.; Hsieh, J.J.; Cheng, E.H. The p53-cathepsin axis cooperates with Ros to activate programmed necrotic death upon DNA damage. Proc. Natl. Acad. Sci. USA 2009, 106, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, F.; Liu, C.Y.; An, T.; Zhang, J.; Zhou, L.Y.; Wang, M.; Dong, Y.H.; Li, N.; Gao, J.N.; et al. The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873. Cell Death Differ. 2016, 23, 1394–1405. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Mortimore, G.E.; Poso, A.R.; Kadowaki, M.; Wert, J.J., Jr. Multiphasic control of hepatic protein degradation by regulatory amino acids. General features and hormonal modulation. J. Biol. Chem. 1987, 262, 16322–16327. [Google Scholar] [PubMed]
- Cao, Y.; Klionsky, D.J. Physiological functions of ATG6/Beclin 1: A unique autophagy-related protein. Cell Res. 2007, 17, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-ATG13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Sebti, S.; Titone, R.; Zhou, Y.; Isidoro, C.; Ross, T.S.; Hibshoosh, H.; Xiao, G.; Packer, M.; Xie, Y.; et al. Decreased BECN1 mRNA expression in human breast cancer is associated with estrogen receptor-negative subtypes and poor prognosis. EBioMedicine 2015, 2, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by Beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [PubMed]
- Shi, Y.H.; Ding, Z.B.; Zhou, J.; Qiu, S.J.; Fan, J. Prognostic significance of Beclin 1-dependent apoptotic activity in hepatocellular carcinoma. Autophagy 2009, 5, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Galluzzi, L.; Morselli, E.; Kepp, O.; Malik, S.A.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010, 22, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK β1, TSC2, and pten expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Criollo, A.; Dessen, P.; Kroemer, G. Dram: A phylogenetically ancient regulator of autophagy. Cell Cycle 2009, 8, 2319–2320. [Google Scholar] [CrossRef] [PubMed]
- Mrschtik, M.; O’Prey, J.; Lao, L.Y.; Long, J.S.; Beaumatin, F.; Strachan, D.; O’Prey, M.; Skommer, J.; Ryan, K.M. Dram-3 modulates autophagy and promotes cell survival in the absence of glucose. Cell Death Differ 2015, 22, 1714–1726. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. Dram, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.Y.; Gang, H.; Aviv, Y.; Dhingra, R.; Margulets, V.; Kirshenbaum, L.A. p53 mediates autophagy and cell death by a mechanism contingent on Bnip3. Hypertension 2013, 62, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.S.; Wilkinson, S.; James, J.; Ryan, K.M.; Vousden, K.H. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 2009, 16, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, A.B. BNIP3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010, 6, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Criollo, A.; Tasdemir, E.; Vicencio, J.M.; Tajeddine, N.; Hickman, J.A.; Geneste, O.; Kroemer, G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-xl. Autophagy 2007, 3, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Ash, D.; Shaha, C. Beclin-1-p53 interaction is crucial for cell fate determination in embryonal carcinoma cells. J. Cell. Mol. Med. 2014, 18, 2275–2286. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Nishimura, T.; Muramatsu, K.; Kodera, H.; Kumada, S.; Sugai, K.; Kasai-Yoshida, E.; Sawaura, N.; Nishida, H.; Hoshino, A.; et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 2013, 45, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Cullup, T.; Kho, A.L.; Dionisi-Vici, C.; Brandmeier, B.; Smith, F.; Urry, Z.; Simpson, M.A.; Yau, S.; Bertini, E.; McClelland, V.; et al. Recessive mutations in EPG5 cause vici syndrome, a multisystem disorder with defective autophagy. Nat. Genet. 2013, 45, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Oz-Levi, D.; Ben-Zeev, B.; Ruzzo, E.K.; Hitomi, Y.; Gelman, A.; Pelak, K.; Anikster, Y.; Reznik-Wolf, H.; Bar-Joseph, I.; Olender, T.; et al. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am. J. Hum. Genet. 2012, 91, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Klionsky, D.J. Autophagy contributes to lysosomal storage disorders. Autophagy 2012, 8, 715–716. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Valent, A.; Raslova, H.; Yakushijin, K.; Horne, D.; Feunteun, J.; Lenoir, G.; Medema, R.; et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene 2004, 23, 4362–4370. [Google Scholar] [CrossRef] [PubMed]
- Denisenko, T.V.; Sorokina, I.V.; Gogvadze, V.; Zhivotovsky, B. Mitotic catastrophe and cancer drug resistance: A link that must to be broken. Drug Resist. Updates 2016, 24, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Russell, P.; Nurse, P. CDC25+ functions as an inducer in the mitotic control of fission yeast. Cell 1986, 45, 145–153. [Google Scholar] [CrossRef]
- Molz, L.; Booher, R.; Young, P.; Beach, D. CDC2 and the regulation of mitosis: Six interacting mcs genes. Genetics 1989, 122, 773–782. [Google Scholar] [CrossRef]
- Ayscough, K.; Hayles, J.; MacNeill, S.A.; Nurse, P. Cold-sensitive mutants of p34CDC2 that suppress a mitotic catastrophe phenotype in fission yeast. Mol. Gen. Genet. 1992, 232, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Allan, L.A.; Clarke, P.R. Phosphorylation of caspase-9 by CDK1/cyclin B1 protects mitotic cells against apoptosis. Mol. Cell 2007, 26, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.L.; Johnson, C.E.; Freel, C.D.; Parrish, A.B.; Day, J.L.; Buchakjian, M.R.; Nutt, L.K.; Thompson, J.W.; Moseley, M.A.; Kornbluth, S. Restraint of apoptosis during mitosis through interdomain phosphorylation of caspase-2. EMBO J. 2009, 28, 3216–3227. [Google Scholar] [CrossRef] [PubMed]
- Smits, V.A.; Medema, R.H. Checking out the G2/M transition. Biochim. Biophys. Acta 2001, 1519, 1–12. [Google Scholar] [CrossRef]
- Margottin-Goguet, F.; Hsu, J.Y.; Loktev, A.; Hsieh, H.M.; Reimann, J.D.; Jackson, P.K. Prophase destruction of Emi1 by the SCF(βTrCP/Slimb) ubiquitin ligase activates the anaphase promoting complex to allow progression beyond prometaphase. Dev. Cell 2003, 4, 813–826. [Google Scholar] [CrossRef]
- Yoshikawa, R.; Kusunoki, M.; Yanagi, H.; Noda, M.; Furuyama, J.I.; Yamamura, T.; Hashimoto-Tamaoki, T. Dual antitumor effects of 5-fluorouracil on the cell cycle in colorectal carcinoma cells: A novel target mechanism concept for pharmacokinetic modulating chemotherapy. Cancer Res. 2001, 61, 1029–1037. [Google Scholar] [PubMed]
- Chan, T.A.; Hermeking, H.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. 14-3-3σ is required to prevent mitotic catastrophe after DNA damage. Nature 1999, 401, 616–620. [Google Scholar] [PubMed]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.M.; Osborne, T.P.; Wheatley, S.P. Phosphorylation of survivin at threonine 34 inhibits its mitotic function and enhances its cytoprotective activity. Cell Cycle 2009, 8, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xiao, Z.; Chen, J.; Ng, S.C.; Sowin, T.; Sham, H.; Rosenberg, S.; Fesik, S.; Zhang, H. Human Chk1 expression is dispensable for somatic cell death and critical for sustaining G2 DNA damage checkpoint. Mol. Cancer Ther. 2003, 2, 543–548. [Google Scholar] [PubMed]
- Yun, J.; Chae, H.D.; Choy, H.E.; Chung, J.; Yoo, H.S.; Han, M.H.; Shin, D.Y. p53 negatively regulates CDC2 transcription via the CCAAT-binding NF-Y transcription factor. J. Biol. Chem. 1999, 274, 29677–29682. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.R.; DePrimo, S.E.; Agarwal, A.; Agarwal, M.L.; Schonthal, A.H.; Katula, K.S.; Stark, G.R. Mechanisms of G2 arrest in response to overexpression of p53. Mol. Biol. Cell 1999, 10, 3607–3622. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, A.P.; Maximo, V.; Lima, J.; Singh, K.K.; Soares, P.; Videira, A. Involvement of p53 in cell death following cell cycle arrest and mitotic catastrophe induced by rotenone. Biochim. Biophys. Acta 2011, 1813, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Ianzini, F.; Bertoldo, A.; Kosmacek, E.A.; Phillips, S.L.; Mackey, M.A. Lack of p53 function promotes radiation-induced mitotic catastrophe in mouse embryonic fibroblast cells. Cancer Cell Int. 2006, 6, 11. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taylor, B.F.; McNeely, S.C.; Miller, H.L.; Lehmann, G.M.; McCabe, M.J., Jr.; States, J.C. p53 suppression of arsenite-induced mitotic catastrophe is mediated by p21CIP1/WAF1. J. Pharmacol. Exp. Ther. 2006, 318, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Fragkos, M.; Beard, P. Mitotic catastrophe occurs in the absence of apoptosis in p53-null cells with a defective G1 checkpoint. PLoS ONE 2011, 6, e22946. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.; McGuirk, M.; Hockenberry, T.N.; Wu, Q.; Ashar, H.; Black, S.; Wen, S.F.; Wang, L.; Kirschmeier, P.; Bishop, W.R.; et al. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene 2002, 21, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Salmela, A.L.; Kallio, M.J. Mitosis as an anti-cancer drug target. Chromosoma 2013, 122, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Mc Gee, M.M. Targeting the mitotic catastrophe signaling pathway in cancer. Mediat. Inflamm. 2015, 2015, 146282. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, S.; de Belle, I.; Bredesen, D.E. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 14376–14381. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, S.; Poksay, K.; de Belle, I.; Lafuente, M.J.; Liu, B.; Nasir, J.; Bredesen, D.E. Paraptosis: Mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ. 2004, 11, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Kim, I.Y.; Saha, S.; Choi, K.S. Paraptosis in the anti-cancer arsenal of natural products. Pharmacol. Ther. 2016, 162, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Sugimori, N.; Espinoza, J.L.; Trung, L.Q.; Takami, A.; Kondo, Y.; An, D.T.; Sasaki, M.; Wakayama, T.; Nakao, S. Paraptosis cell death induction by the thiamine analog benfotiamine in leukemia cells. PLoS ONE 2015, 10, e0120709. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhao, J.; Wang, C.Z.; Searle, J.; He, T.C.; Yuan, C.S.; Du, W. Ginsenoside RH2 induces apoptosis and paraptosis-like cell death in colorectal cancer cells through activation of p53. Cancer Lett. 2011, 301, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; O’Riordan, K.J.; Burns-Cusato, M.; Andrzejewski, M.E.; del Alcazar, C.G.; Burger, C.; Scrable, H.; Puglielli, L. Altered longevity-assurance activity of p53:p44 in the mouse causes memory loss, neurodegeneration and premature death. Aging Cell 2010, 9, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Neuberg, M.; Buckbinder, L.; Seizinger, B.; Kley, N. The p53/IGF-1 receptor axis in the regulation of programmed cell death. Endocrine 1997, 7, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, C.; Kley, N.; Werner, H.; LeRoith, D. p53 regulates insulin-like growth factor-I (IGF-I) receptor expression and IGF-I-induced tyrosine phosphorylation in an osteosarcoma cell line: Interaction between p53 and Sp1. Endocrinology 1998, 139, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Allen, H.; Banerjee, S.; Franklin, S.; Herzog, L.; Johnston, C.; McDowell, J.; Paskind, M.; Rodman, L.; Salfeld, J.; et al. Mice deficient in IL-1β-converting enzyme are defective in production of mature IL-1β and resistant to endotoxic shock. Cell 1995, 80, 401–411. [Google Scholar] [CrossRef]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Hilbi, H.; Moss, J.E.; Hersh, D.; Chen, Y.; Arondel, J.; Banerjee, S.; Flavell, R.A.; Yuan, J.; Sansonetti, P.J.; Zychlinsky, A. Shigella-induced apoptosis is dependent on caspase-1 which binds to IPAB. J. Biol. Chem. 1998, 273, 32895–32900. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Radha, V.; Furukawa, Y.; Swarup, G. Direct transcriptional activation of human caspase-1 by tumor suppressor p53. J. Biol. Chem. 2001, 276, 10585–10588. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Birge, R.B. Efferocytosis. Curr. Biol. 2016, 26, R558–R559. [Google Scholar] [CrossRef] [PubMed]
- Sarode, G.S. Efferocytosis in oral squamous cell carcinoma. J. Oral Maxillofac. Pathol. 2016, 20, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Stanford, J.C.; Young, C.; Hicks, D.; Owens, P.; Williams, A.; Vaught, D.B.; Morrison, M.M.; Lim, J.; Williams, M.; Brantley-Sieders, D.M.; et al. Efferocytosis produces a prometastatic landscape during postpartum mammary gland involution. J. Clin. Investig. 2014, 124, 4737–4752. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.E.; Sun, M.; Zhang, R.; Febbraio, M.; Silverstein, R.; Hazen, S.L. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J. Exp. Med. 2006, 203, 2613–2625. [Google Scholar] [CrossRef] [PubMed]
- Parks, B.W.; Black, L.L.; Zimmerman, K.A.; Metz, A.E.; Steele, C.; Murphy-Ullrich, J.E.; Kabarowski, J.H. CD36, but not G2A, modulates efferocytosis, inflammation, and fibrosis following bleomycin-induced lung injury. J. Lipid Res. 2013, 54, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- McCubbrey, A.L.; Nelson, J.D.; Stolberg, V.R.; Blakely, P.K.; McCloskey, L.; Janssen, W.J.; Freeman, C.M.; Curtis, J.L. MicroRNA-34a negatively regulates efferocytosis by tissue macrophages in part via SIRT1. J. Immunol. 2016, 196, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Komarova, E.A.; Krivokrysenko, V.; Wang, K.; Neznanov, N.; Chernov, M.V.; Komarov, P.G.; Brennan, M.L.; Golovkina, T.V.; Rokhlin, O.W.; Kuprash, D.V.; et al. p53 is a suppressor of inflammatory response in mice. FASEB J. 2005, 19, 1030–1032. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode of Cell Death | Definition | Major Mediators | Role of p53 | Reference |
|---|---|---|---|---|
| Caspase-independent apoptosis (CIA) | Cell death occurring independently of caspases | AIF, EndoG | p53 transcriptionally upregulates AIF and also induces cytoplasmic translocation of AIF from mitochondria. | [33,34,35,45] |
| Ferroptosis | Iron-dependent regulated cell death | GPX4, SLC7A11 | p53 transcriptionally represses SLC7A11. | [57] |
| Necroptosis (programmed necrotic cell death) | The regulated form of necrotic cell death | RIPK1/RIPK3, cypD, Cathepsin Q | p53 transactivates cathepsin Q, indirectly increases RIPK1/RIPK3 via the NRF-miR-873 axis, and directly interacts with cypD in mitochondria. | [64,67,68,73] |
| Autophagic cell death | Non-apoptotic, non-necrotic cell death resulting from the process of autophagy | ATG proteins, Beclin-1, DRAM | Nuclear p53 increases TSC2 and AMPK levels to inhibit mTOR activity, as well as increases DRAM levels, promoting the autophagic process. Cytoplasmic p53 binds with Beclin-1 and promotes its degradation to inhibit autophagy. | [87,88,89,91,92,93,98] |
| Mitotic catastrophe | Cell death caused by impaired mitosis | Cdk1 | p53 inhibits transcription of cdk1. | [109,110,111] |
| Paraptosis | Programmed cell death induced by IGF-IR with swelling of mitochondria or endoplasmic reticulum (ER) and cytoplasmic vacuolization | IGF-IR, ALG-2-interacting protein (AIP1, also known as ALG-2 interacting protein-X, Alix) | Δ40p53 (p44) upregulates IGF-IR. | [135,139,141] |
| Pyroptosis | Inflammatory form of regulated cell death which is triggered by microbial infection | Caspase-1 | p53 transactivates caspase-1, but its direct involvement in pyroptosis remains unclear. | [143,149] |
| Efferocytosis | The process by which phagocytes engulf and digest dead or dying cells | miR-34a | p53 transcriptionally upregulates miR-34a. | [155,156] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranjan, A.; Iwakuma, T. Non-Canonical Cell Death Induced by p53. Int. J. Mol. Sci. 2016, 17, 2068. https://doi.org/10.3390/ijms17122068
Ranjan A, Iwakuma T. Non-Canonical Cell Death Induced by p53. International Journal of Molecular Sciences. 2016; 17(12):2068. https://doi.org/10.3390/ijms17122068
Chicago/Turabian StyleRanjan, Atul, and Tomoo Iwakuma. 2016. "Non-Canonical Cell Death Induced by p53" International Journal of Molecular Sciences 17, no. 12: 2068. https://doi.org/10.3390/ijms17122068
APA StyleRanjan, A., & Iwakuma, T. (2016). Non-Canonical Cell Death Induced by p53. International Journal of Molecular Sciences, 17(12), 2068. https://doi.org/10.3390/ijms17122068