
d-Lysergic Acid Diethylamide (LSD) as a Model of Psychosis: Mechanism of Action and Pharmacology



Abstract
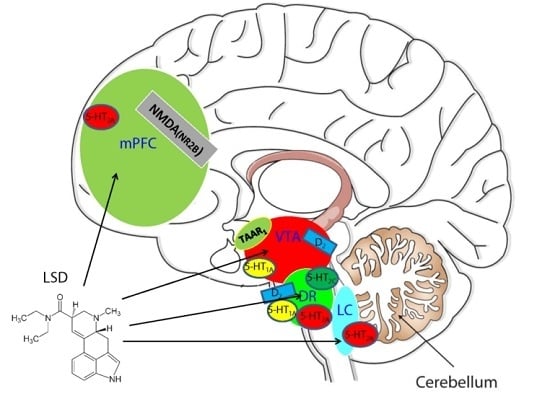
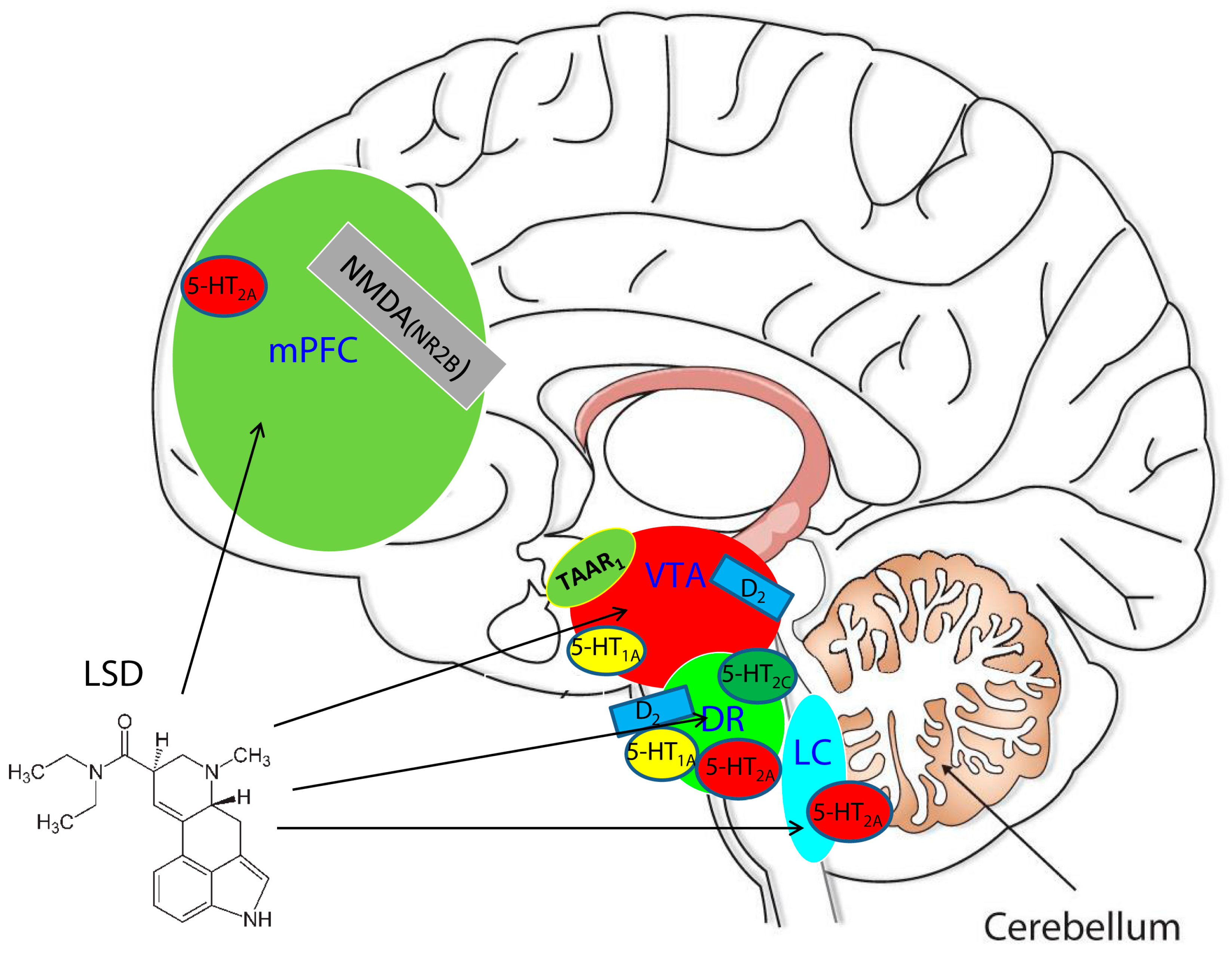
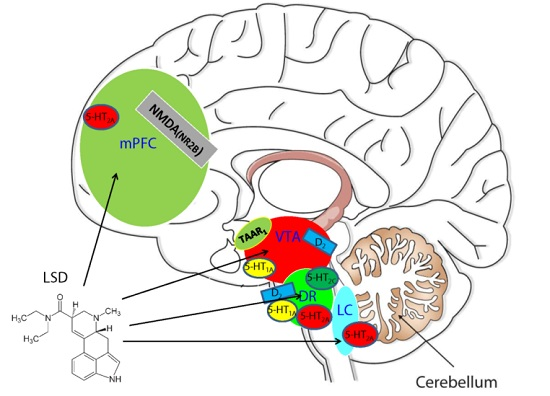
:
1. Introduction
1.1. LSD: “A Joyous Song of Being” (Albert Hoffman)
1.2. Psychosis and LSD: Human Studies
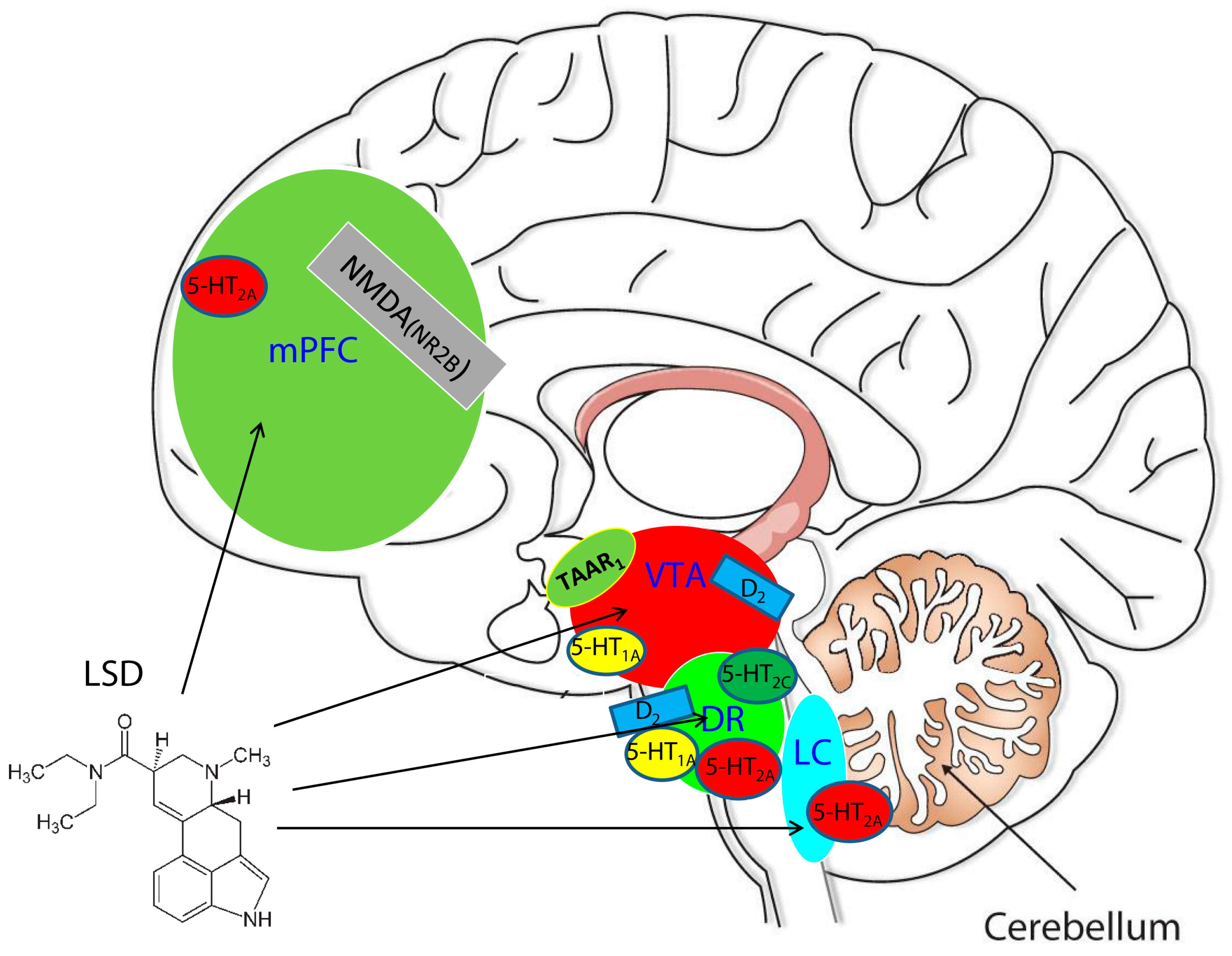
2. The Dopamine-Serotonin System and TAAR1 Receptor in the Pathogenesis of Psychosis and in the Mechanism of Action of Antipsychotic Drugs
3. In the Deep of the Mechanism: Who Are the Players?
3.1. The Serotonin System and LSD: The Playmaker or the Point Guard
3.2. The Dopamine System: The Power Forward
3.4. Glutamate: The Third Player or Small Forward
3.5. Trace Amine-Associated Receptor 1 (TAAR1) and LSD: The Shooting Guard
3.6. Prefrontal Cortex: The Center
4. LSD: An Animal Model of Psychosis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| p.o. | per os |
| i.v. | intra-venous |
| i.m. | intra-muscular |
| i.p. | intra-peritoneal |
References
- Hofmann, A.; Ott, J. LSD, My Problem Child; McGraw-Hill: New York, NY, USA, 1980; Volume 5. [Google Scholar]
- Savage, C. Lysergic acid diethylamide (LSD-25) A Clinical-Psychological Study. Am. J. Psychiatry 1952, 108, 896–900. [Google Scholar] [CrossRef] [PubMed]
- Pahnke, W.N.; Richards, W.A. Implications of LSD and experimental mysticism. J. Relig. Health 1966, 5, 175–208. [Google Scholar] [CrossRef] [PubMed]
- Hensala, J.D.; Epstein, L.J.; Blacker, K. LSD and psychiatric inpatients. Arch. Gen. Psychiatry 1967, 16, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Passie, T.; Halpern, J.H.; Stichtenoth, D.O.; Emrich, H.M.; Hintzen, A. The pharmacology of lysergic acid diethylamide: A review. CNS Neurosci. Ther. 2008, 14, 295–314. [Google Scholar] [CrossRef] [PubMed]
- Matthew, H. Lysergic acid diethylamide intoxication. Br. Med. J. 1968, 1, 380. [Google Scholar] [CrossRef] [PubMed]
- Goodman, N. The serotonergic system and mysticism: Could LSD and the nondrug-induced mystical experience share common neural mechanisms? J. Psychoact. Drugs 2002, 34, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.; Moffatt, J.D. Cardiovascular toxicity of novel psychoactive drugs: Lessons from the past. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 39, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Schmid, Y.; Enzler, F.; Gasser, P.; Grouzmann, E.; Preller, K.H.; Vollenweider, F.X.; Brenneisen, R.; Müller, F.; Borgwardt, S.; Liechti, M.E. Acute effects of lysergic acid diethylamide in healthy subjects. Biol. Psychiatry 2015, 78, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Pahnke, W.N. LSD and Religious Experience. LSD Man & Society; Wesleyan University Press: Middletown, CT, USA, 1967; pp. 60–85. [Google Scholar]
- Osmond, H.; Smythies, J. Schizophrenia: A new approach. Br. J. Psychiatry 1952, 98, 309–315. [Google Scholar] [CrossRef]
- Hartley, S.; Barrowclough, C.; Haddock, G. Anxiety and depression in psychosis: A systematic review of associations with positive psychotic symptoms. Acta Psychiatr. Scand. 2013, 128, 327–346. [Google Scholar] [CrossRef] [PubMed]
- Faerden, A.; Barrett, E.A.; Nesvåg, R.; Friis, S.; Finset, A.; Marder, S.R.; Ventura, J.; Andreassen, O.A.; Agartz, I.; Melle, I. Apathy, poor verbal memory and male gender predict lower psychosocial functioning one year after the first treatment of psychosis. Psychiatry Res. 2013, 210, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.; van Os, J. The schizophrenia spectrum and other psychotic disorders in the DSM-5. Tijdschr. Psychiatr. 2013, 56, 167–172. [Google Scholar]
- First, M.B. Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; DSM IV; American Psychological Association: Washington, DC, USA, 1994. [Google Scholar]
- Brewerton, T.D. Hyperreligiosity in psychotic disorders. J. Nerv. Ment. Dis. 1994, 182, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Kay, S.R.; Flszbein, A.; Opfer, L.A. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr. Bull. 1987, 13, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Hoch, P.H. Experimentally produced psychoses. Am. J. Psychiatry 1951, 107, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Gouzoulis-Mayfrank, E.; Habermeyer, E.; Hermle, L.; Steinmeyer, A.; Kunert, H.; Sass, H. Hallucinogenic drug induced states resemble acute endogenous psychoses: Results of an empirical study. Eur. Psychiatry 1998, 13, 399–406. [Google Scholar] [CrossRef]
- Weil-Malherbe, H.; Szara, S.I. The biochemistry of functional and experimental psychoses. J. Pharm. Sci. 1971, 61, 819. [Google Scholar]
- Unger, S. The Current Scientific Status of Psychedelic Drug Research; Unpublished Paper Read to the Conference on Method in Philosophy and the Sciences in New York City on 3 May 1964; The University of Edinburgh: Edinburgh, UK, 1964; Volume 3. [Google Scholar]
- Klee, G.; Weintraub, W. Paranoid response following lysergic acid diethylamide (LSD-25). In Neuro-Psychopharmacology; Elsevier-Van Nostrand: Princeton, NJ, USA, 1959; pp. 457–460. [Google Scholar]
- Langs, R.J.; Barr, H.L. lysergic acid diethylamide (LSD-25) and schizophrenic reactions. J. Nerv. Ment. Dis. 1968, 147, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Anastasopoulos, G.; Photiades, H. Effects of LSD-25 on relatives of schizophrenic patients. Br. J. Psychiatry 1962, 108, 95–98. [Google Scholar] [CrossRef]
- Power, R.A.; Verweij, K.J.; Zuhair, M.; Montgomery, G.W.; Henders, A.K.; Heath, A.C.; Madden, P.A.; Medland, S.E.; Wray, N.R.; Martin, N.G. Genetic predisposition to schizophrenia associated with increased use of cannabis. Mol. Psychiatry 2014, 19, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Henquet, C.; di Forti, M.; Morrison, P.; Kuepper, R.; Murray, R.M. Gene-environment interplay between cannabis and psychosis. Schizophr. Bull. 2008, 34, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Vardy, M.M.; Kay, S.R. LSD Psychosis or LSD-Induced Schizophrenia?: A Multimethod Inquiry. Arch. Gen. Psychiatry 1983, 40, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Ungerleider, J.T.; Fisher, D.D.; Fuller, M.; Caldwell, A. The “bad trip”-the etiology of the adverse LSD reaction. Am. J. Psychiatry 1968, 124, 1483–1490. [Google Scholar] [CrossRef] [PubMed]
- Smart, R.G.; Jones, D. Illicit LSD users: Their personality characteristics and psychopathology. J. Abnorm. Psychol. 1970, 75, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Tucker, G.J.; Hanover, N.; Quinlan, D.; Harrow, M. Chronic hallucinogenic drug use and thought disturbance. Arch. Gen. Psychiatry 1972, 27, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Stone, M. Drug-related schizophrenic syndromes. Int. J. Psychiatry 1973, 11, 391–437. [Google Scholar] [PubMed]
- Cohen, S. A classification of LSD complications. Psychosomatics 1966, 7, 182–186. [Google Scholar] [CrossRef]
- Johansen, P.-Ø.; Krebs, T.S. Psychedelics not linked to mental health problems or suicidal behavior: A population study. J. Psychopharmacol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Blacker, K.; Jones, R.T.; Stone, G.C.; Pfefferbaum, D. Chronic users of LSD: The “acidheads”. Am. J. Psychiatry 1968, 125, 341–351. [Google Scholar] [CrossRef]
- McGlothlin, W.H.; Arnold, D.O.; Freedman, D.X. Organicity measures following repeated LSD ingestion. Arch. Gen. Psychiatry 1969, 21, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Abraham, H.D. A chronic impairment of colour vision in users of LSD. Br. J. Psychiatry 1982, 140, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Halpern, J.H.; Pope, H.G. Hallucinogen persisting perception disorder: What do we know after 50 years? Drug Alcohol Depend. 2003, 69, 109–119. [Google Scholar] [CrossRef]
- Behan, W.; Bakheit, A.; Behan, P.; More, I. The muscle findings in the neuroleptic malignant syndrome associated with lysergic acid diethylamide. J. Neurol. Neurosurg. Psychiatry 1991, 54, 741–743. [Google Scholar] [CrossRef] [PubMed]
- Carhart-Harris, R.; Kaelen, M.; Bolstridge, M.; Williams, T.; Williams, L.; Underwood, R.; Feilding, A.; Nutt, D. The paradoxical psychological effects of lysergic acid diethylamide (LSD). Psychol. Med. 2016, 46, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Giannini, A.J.; Eighan, M.S.; Loiselle, R.H.; Giannini, M.C. Comparison of haloperidol and chlorpromazine in the treatment of phencyclidine psychosis. J. Clin. Pharmacol. 1984, 24, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Marona-Lewicka, D.; Nichols, C.D.; Nichols, D.E. An animal model of schizophrenia based on chronic LSD administration: Old idea, new results. Neuropharmacology 2011, 61, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Carhart-Harris, R.L.; Muthukumaraswamy, S.; Roseman, L.; Kaelen, M.; Droog, W.; Murphy, K.; Tagliazucchi, E.; Schenberg, E.E.; Nest, T.; Orban, C. Neural correlates of the LSD experience revealed by multimodal neuroimaging. Proc. Natl. Acad. Sci. USA 2016, 113, 4853–4858. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K. Clinical Psychopathology; Grune & Stratton: New York, NY, USA, 1959; p. 95. [Google Scholar]
- Steeds, H.; Carhart-Harris, R.L.; Stone, J.M. Drug models of schizophrenia. Ther. Adv. Psychopharmacol. 2015, 5, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Woolley, D.; Shaw, E. Some neurophysiological aspects of serotonin. Br. Med. J. 1954, 2, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Shaw, E.; Woolley, D. Some serotoninlike activities of lysergic acid diethylamide. Science 1956, 124, 121–122. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.D.; Sanders-Bush, E. Molecular genetic responses to lysergic acid diethylamide include transcriptional activation of MAP kinase phosphatase-1, C/EBP-β and ILAD-1, a novel gene with homology to arrestins. J. Neurochem. 2004, 90, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Halberstadt, A.L.; Geyer, M.A. Serotonergic hallucinogens as translational models relevant to schizophrenia. Int. J. Neuropsychopharmacol. 2013, 16, 2165–2180. [Google Scholar] [CrossRef] [PubMed]
- Sanders-Bush, E.; Burris, K.D.; Knoth, K. Lysergic acid diethylamide and 2, 5-dimethoxy-4-methylamphetamine are partial agonists at serotonin receptors linked to phosphoinositide hydrolysis. J. Pharmacol. Exp. Ther. 1988, 246, 924–928. [Google Scholar] [PubMed]
- Rickli, A.; Luethi, D.; Reinisch, J.; Buchy, D.; Hoener, M.C.; Liechti, M.E. Receptor interaction profiles of novel N-2-methoxybenzyl (NBOMe) derivatives of 2, 5-dimethoxy-substituted phenethylamines (2C drugs). Neuropharmacology 2015, 99, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Rickli, A.; Moning, O.D.; Hoener, M.C.; Liechti, M.E. Receptor interaction profiles of novel psychoactive tryptamines compared with classic hallucinogens. Eur. Neuropsychopharmacol. 2016, 26, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.; Battaglia, G.; Creese, I. [3H] WB4101 labels the 5-HT1A serotonin receptor subtype in rat brain. Guanine nucleotide and divalent cation sensitivity. Mol. Pharmacol. 1985, 28, 487–494. [Google Scholar] [PubMed]
- Reissig, C.; Eckler, J.; Rabin, R.; Winter, J. The 5-HT1A receptor and the stimulus effects of LSD in the rat. Psychopharmacology 2005, 182, 197–204. [Google Scholar] [CrossRef] [PubMed]
- BURT, D.R.; Creese, I.; Snyder, S.H. Binding interactions of lysergic acid diethylamide and related agents with dopamine receptors in the brain. Mol. Pharmacol. 1976, 12, 631–638. [Google Scholar] [PubMed]
- Aghajanian, G.K.; Marek, G.J. Serotonin model of schizophrenia: Emerging role of glutamate mechanisms. Brain Res. Rev. 2000, 31, 302–312. [Google Scholar] [CrossRef]
- Seeman, P.; Ko, F.; Tallerico, T. Dopamine receptor contribution to the action of PCP, LSD and ketamine psychotomimetics. Mol. Psychiatry 2005, 10, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Minuzzi, L.; Nomikos, G.G.; Wade, M.R.; Jensen, S.B.; Olsen, A.K.; Cumming, P. Interaction between LSD and dopamine D2/3 binding sites in pig brain. Synapse 2005, 56, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P.; Guan, H.C.; Hirbec, H. Dopamine D2High receptors stimulated by phencyclidines, lysergic acid diethylamide, salvinorin A, and modafinil. Synapse 2009, 63, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Bunzow, J.R.; Sonders, M.S.; Arttamangkul, S.; Harrison, L.M.; Zhang, G.; Quigley, D.I.; Darland, T.; Suchland, K.L.; Pasumamula, S.; Kennedy, J.L. Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor. Mol. Pharmacol. 2001, 60, 1181–1188. [Google Scholar] [PubMed]
- Lindemann, L.; Meyer, C.A.; Jeanneau, K.; Bradaia, A.; Ozmen, L.; Bluethmann, H.; Bettler, B.; Wettstein, J.G.; Borroni, E.; Moreau, J.-L. Trace amine-associated receptor 1 modulates dopaminergic activity. J. Pharmacol. Exp. Ther. 2008, 324, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Miller, G.M. Trace amine-associated receptor 1 is a modulator of the dopamine transporter. J. Pharmacol. Exp. Ther. 2007, 321, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Revel, F.; Moreau, J.; Pouzet, B.; Mory, R.; Bradaia, A.; Buchy, D.; Metzler, V.; Chaboz, S.; Zbinden, K.G.; Galley, G. A new perspective for schizophrenia: TAAR1 agonists reveal antipsychotic-and antidepressant-like activity, improve cognition and control body weight. Mol. Psychiatry 2013, 18, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.L.; Kahn, R.S.; Ko, G.; Davidson, M. Dopamine in schizophrenia: A review and reconceptualization. Am. J. Psychiatry 1991, 148, 1474–1486. [Google Scholar] [PubMed]
- Seeman, P.; Weinshenker, D.; Quirion, R.; Srivastava, L.K.; Bhardwaj, S.K.; Grandy, D.K.; Premont, R.T.; Sotnikova, T.D.; Boksa, P.; El-Ghundi, M. Dopamine supersensitivity correlates with D2High states, implying many paths to psychosis. Proc. Natl. Acad. Sci. USA 2005, 102, 3513–3518. [Google Scholar] [CrossRef] [PubMed]
- Sax, K.W.; Strakowski, S.M.; Keck, P.; Upadhyaya, V.H.; West, S.A.; McELROY, S.L. Relationships among negative, positive, and depressive symptoms in schizophrenia and psychotic depression. Br. J. Psychiatry 1996, 168, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Butler, R.W.; Mueser, K.T.; Sprock, J.; Braff, D.L. Positive symptoms of psychosis in posttraumatic stress disorder. Biol. Psychiatry 1996, 39, 839–844. [Google Scholar] [CrossRef]
- Zimmermann, G.; Favrod, J.; Trieu, V.; Pomini, V. The effect of cognitive behavioral treatment on the positive symptoms of schizophrenia spectrum disorders: A meta-analysis. Schizophr. Res. 2005, 77, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kringelbach, M.L.; Berridge, K.C. Towards a functional neuroanatomy of pleasure and happiness. Trends Cogn. Sci. 2009, 13, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Farde, L.; Nordström, A.-L.; Wiesel, F.-A.; Pauli, S.; Halldin, C.; Sedvall, G. Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine: Relation to extrapyramidal side effects. Arch. Gen. Psychiatry 1992, 49, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, A.-L.; Farde, L. Plasma prolactin and central D2 receptor occupancy in antipsychotic drug-treated patients. J. Clin. Psychopharmacol. 1998, 18, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Kapur, S.; Remington, G. Serotonin-dopamine interaction and its relevance to schizophrenia. Am. J. Psychiatry 1996, 153, 466–476. [Google Scholar] [PubMed]
- Meltzer, H.; Massey, B. The role of serotonin receptors in the action of atypical antipsychotic drugs. Curr. Opin. Pharmacol. 2011, 11, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Daskalakis, Z.; Christensen, B.; Zipursky, R.; Zhang-Wong, J.; Beiser, M. 376. Relationship between D2 occupancy and prolactin levels in first episode psychosis. Biol. Psychiatry 1998, 43, S113. [Google Scholar] [CrossRef]
- Davis, J.M.; Chen, N.; Glick, I.D. A meta-analysis of the efficacy of second-generation antipsychotics. Arch. Gen. Psychiatry 2003, 60, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Leucht, S.; Wahlbeck, K.; Hamann, J.; Kissling, W. New generation antipsychotics versus low-potency conventional antipsychotics: A systematic review and meta-analysis. Lancet 2003, 361, 1581–1589. [Google Scholar] [CrossRef]
- Artigas, F. The prefrontal cortex: A target for antipsychotic drugs. Acta Psychiatr. Scand. 2010, 121, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Borsetti, P.; Cortés, R.; Artigas, F. Pyramidal neurons in rat prefrontal cortex projecting to ventral tegmental area and dorsal raphe nucleus express 5-HT2A receptors. Cereb. Cortex 2009, 19, 1678–1686. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Mataix, L.; Scorza, M.C.; Bortolozzi, A.; Toth, M.; Celada, P.; Artigas, F. Involvement of 5-HT1A receptors in prefrontal cortex in the modulation of dopaminergic activity: Role in atypical antipsychotic action. J. Neurosci. 2005, 25, 10831–10843. [Google Scholar] [CrossRef] [PubMed]
- Rollema, H.; Lu, Y.; Schmidt, A.W.; Zorn, S.H. Clozapine increases dopamine release in prefrontal cortex by 5-HT 1A receptor activation. Eur. J. Pharmacol. 1997, 338, R3–R5. [Google Scholar] [CrossRef]
- Millan, M.J. Improving the treatment of schizophrenia: Focus on serotonin (5-HT) 1A receptors. J. Pharmacol. Exp. Ther. 2000, 295, 853–861. [Google Scholar] [PubMed]
- Bantick, R.; Deakin, J.; Grasby, P. The 5-HT1A receptor in schizophrenia: A promising target for novel atypical neuroleptics? J. Psychopharmacol. 2001, 15, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Sumiyoshi, T.; Park, S.; Jayathilake, K.; Roy, A.; Ertugrul, A.; Meltzer, H.Y. Effect of buspirone, a serotonin 1A partial agonist, on cognitive function in schizophrenia: A randomized, double-blind, placebo-controlled study. Schizophr. Res. 2007, 95, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Wadenberg, M.-L. Antagonism by 8-OH-DPAT, but not ritanserin, of catalepsy induced by SCH 23390 in the rat. J. Neural Transm. Gen. Sect. 1992, 89, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Hicks, P.B. The effect of serotonergic agents on haloperidol-induced catalepsy. Life Sci. 1990, 47, 1609–1615. [Google Scholar] [CrossRef]
- Maj, J.; Sarnek, J.; Klimek, V.; Rawlow, A. On the anticataleptic action of cyproheptadine. Pharmacol. Biochem. Behav. 1976, 5, 201–205. [Google Scholar] [CrossRef]
- Fuenmayor, L.D.; Vogt, M. the influence of cerebral 5-hydroxytryptamine on catalepsy induced by brain-amine depleting neuroleptics or by cholinomimetics. Br. J. Pharmacol. 1979, 67, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Nabeshima, T.; Kameyama, T. Potentiation of phencyclidine-induced dopamine-dependent behaviors in rats after pretreatments with serotonin-depletors. J. Pharmacobiodyn. 1986, 9, 479–489. [Google Scholar] [PubMed]
- Baldessarini, R.J.; Amatruda, T.T.; Griffith, F.F.; Gerson, S. Differential effects of serotonin on turning and stereotypy induced by apomorphine. Brain Res. 1975, 93, 158–163. [Google Scholar] [CrossRef]
- Kapur, S.; Seeman, P. Does fast dissociation from the dopamine D2 receptor explain the action of atypical antipsychotics?: A new hypothesis. Am. J. Psychiatry 2001, 158, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Cox, S.M.; Benkelfat, C.; Dagher, A.; Delaney, J.S.; Durand, F.; McKenzie, S.A.; Kolivakis, T.; Casey, K.F.; Leyton, M. Striatal dopamine responses to intranasal cocaine self-administration in humans. Biol. Psychiatry 2009, 65, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Stockton, M.E.; Rasmussen, K. Electrophysiological effects of olanzapine, a novel atypical antipsychotic, on A9 and A10 dopamine neurons. Neuropsychopharmacology 1996, 14, 97–104. [Google Scholar] [CrossRef]
- van Domburg, P.H.M.F.; ten Donkelaar, H.J. The Human Substantia Nigra and Ventral Tegmental Area. In The Human Substantia Nigra and Ventral Tegmental Area; Springer: Berlin/Heidelberg, Germany, 1991; pp. 32–69. [Google Scholar]
- Borowsky, B.; Adham, N.; Jones, K.A.; Raddatz, R.; Artymyshyn, R.; Ogozalek, K.L.; Durkin, M.M.; Lakhlani, P.P.; Bonini, J.A.; Pathirana, S. Trace amines: Identification of a family of mammalian G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 8966–8971. [Google Scholar] [CrossRef] [PubMed]
- Revel, F.G.; Moreau, J.-L.; Gainetdinov, R.R.; Bradaia, A.; Sotnikova, T.D.; Mory, R.; Durkin, S.; Zbinden, K.G.; Norcross, R.; Meyer, C.A. TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc. Natl. Acad. Sci. USA 2011, 108, 8485–8490. [Google Scholar] [CrossRef] [PubMed]
- Grandy, D.K. Trace amine-associated receptor 1—Family archetype or iconoclast? Pharmacol. Ther. 2007, 116, 355–390. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, L.; Hoener, M.C. A renaissance in trace amines inspired by a novel GPCR family. Trends Pharmacol. Sci. 2005, 26, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.M. The emerging role of trace amine-associated receptor 1 in the functional regulation of monoamine transporters and dopaminergic activity. J. Neurochem. 2011, 116, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Narang, D.; Tomlinson, S.; Holt, A.; Mousseau, D.D.; Baker, G.B. Trace amines and their relevance to psychiatry and neurology: A brief overview. Bull. Clin. Psychopharmacol. 2011, 21, 73–79. [Google Scholar] [CrossRef]
- Sotnikova, T.D.; Caron, M.G.; Gainetdinov, R.R. Trace amine-associated receptors as emerging therapeutic targets. Mol. Pharmacol. 2009, 76, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Berry, M. The potential of trace amines and their receptors for treating neurological and psychiatric diseases. Rev. Recent Clin. Trials 2007, 2, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Burchett, S.A.; Hicks, T.P. The mysterious trace amines: Protean neuromodulators of synaptic transmission in mammalian brain. Prog. Neurobiol. 2006, 79, 223–246. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, T.; Swanson, C.; Smith, K.; Zhong, H.; Borowsky, B.; Seeman, P.; Branchek, T.; Gerald, C. The Trace Amine 1 receptor knockout mouse: An animal model with relevance to schizophrenia. Genes Brain Behav. 2007, 6, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Bradaia, A.; Trube, G.; Stalder, H.; Norcross, R.D.; Ozmen, L.; Wettstein, J.G.; Pinard, A.; Buchy, D.; Gassmann, M.; Hoener, M.C. The selective antagonist EPPTB reveals TAAR1-mediated regulatory mechanisms in dopaminergic neurons of the mesolimbic system. Proc. Natl. Acad. Sci. USA 2009, 106, 20081–20086. [Google Scholar] [CrossRef] [PubMed]
- Stalder, H.; Hoener, M.C.; Norcross, R.D. Selective antagonists of mouse trace amine-associated receptor 1 (mTAAR1): Discovery of EPPTB (RO5212773). Bioorg. Med. Chem. Lett. 2011, 21, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, S.; Salahpour, A.; Masri, B.; Sotnikova, T.D.; Messa, M.; Barak, L.S.; Caron, M.G.; Gainetdinov, R.R. Functional interaction between trace amine-associated receptor 1 and dopamine D2 receptor. Mol. Pharmacol. 2011, 80, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Glennon, R.A.; Titeler, M.; McKenney, J. Evidence for 5-HT2 involvement in the mechanism of action of hallucinogenic agents. Life Sci. 1984, 35, 2505–2511. [Google Scholar] [CrossRef]
- Teitler, M.; Leonhardt, S.; Appel, N.M.; Souza, E.B.; Glennon, R.A. Receptor pharmacology of MDMA and related hallucinogensa. Ann. N. Y. Acad. Sci. 1990, 600, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.E.; Frescas, S.; Marona-Lewicka, D.; Huang, X.; Roth, B.L.; Gudelsky, G.A.; Nash, J.F. 1-(2, 5-Dimethoxy-4-(trifluoromethyl) phenyl)-2-aminopropane: A potent serotonin 5-HT2A/2C agonist. J. Med. Chem. 1994, 37, 4346–4351. [Google Scholar] [CrossRef] [PubMed]
- Kometer, M.; Schmidt, A.; Jäncke, L.; Vollenweider, F.X. Activation of serotonin 2A receptors underlies the psilocybin-induced effects on α oscillations, N170 visual-evoked potentials, and visual hallucinations. J. Neurosci. 2013, 33, 10544–10551. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.; Aghajanian, G.K. Effect of hallucinogens on spontaneous and sensory-evoked locus coeruleus unit activity in the rat: Reversal by selective 5-HT2antagonists. Brain Res. 1986, 385, 395–400. [Google Scholar] [CrossRef]
- Geyer, M.A.; Krebs, K.M. Serotonin receptor involvement in an animal model of the acute effects of hallucinogens. NIDA Res. Monogr. 1994, 146, 124–156. [Google Scholar] [PubMed]
- Marek, G.J.; Aghajanian, G.K. LSD and the phenethylamine hallucinogen DOI are potent partial agonists at 5-HT2A receptors on interneurons in rat piriform cortex. J. Pharmacol. Exp. Ther. 1996, 278, 1373–1382. [Google Scholar] [PubMed]
- Sanders-Bush, E.; Breeding, M. Choroid plexus epithelial cells in primary culture: A model of 5HT1C receptor activation by hallucinoginic drugs. Psychopharmacology 1991, 105, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Egan, C.T.; Herrick-Davis, K.; Miller, K.; Glennon, R.A.; Teitler, M. Agonist activity of LSD and lisuride at cloned 5HT2A and 5HT2C receptors. Psychopharmacology 1998, 136, 409–414. [Google Scholar] [CrossRef] [PubMed]
- De Gregorio, D.; Posa, L.; Ochoa-Sanchez, R.; McLaughlin, R.; Maione, S.; Comai, S.; Gobbi, G. The hallucinogen d-lysergic diethylamide (LSD) decreases dopamine firing activity through 5-HT1A, D2 and TAAR1 receptors. Pharmacol. Res. 2016, 113, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Fiorella, D.; Helsley, S.; Lorrain, D.; Rabin, R.A.; Winter, J. The role of the 5-HT2A and 5-HT2C receptors in the stimulus effects of hallucinogenic drugs III: The mechanistic basis for supersensitivity to the LSD stimulus following serotonin depletion. Psychopharmacology 1995, 121, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Academic: San Diego, CA, USA, 1986. [Google Scholar]
- Krall, C.; Richards, J.; Rabin, R.; Winter, J. Marked decrease of LSD-induced stimulus control in serotonin transporter knockout mice. Pharmacol. Biochem. Behav. 2008, 88, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Kyzar, E.J.; Stewart, A.M.; Kalueff, A.V. Effects of LSD on grooming behavior in serotonin transporter heterozygous (Sert+/−) mice. Behav. Brain Res. 2016, 296, 47–52. [Google Scholar] [PubMed]
- Watts, V.J.; Mailman, R.; Lawler, C.; Neve, K.A.; Nichols, D.E. LSD and structural analogs: Pharmacological evaluation at D1 dopamine receptors. Psychopharmacology 1995, 118, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Appel, J.; White, F.; Holohean, A. Analyzing mechanism(s) of hallucinogenic drug action with drug discrimination procedures. Neurosci. Biobehav. Rev. 1983, 6, 529–536. [Google Scholar] [CrossRef]
- Meert, T.; de Haes, P.; Janssen, P. Risperidone (R 64 766), a potent and complete LSD antagonist in drug discrimination by rats. Psychopharmacology 1989, 97, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Marona-Lewicka, D.; Thisted, R.A.; Nichols, D.E. Distinct temporal phases in the behavioral pharmacology of LSD: Dopamine D2 receptor-mediated effects in the rat and implications for psychosis. Psychopharmacology 2005, 180, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Marona-Lewicka, D.; Chemel, B.R.; Nichols, D.E. Dopamine D4 receptor involvement in the discriminative stimulus effects in rats of LSD, but not the phenethylamine hallucinogen DOI. Psychopharmacology 2009, 203, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Lambe, E.K.; Aghajanian, G.K. Hallucinogen-induced UP states in the brain slice of rat prefrontal cortex: Role of glutamate spillover and NR2B-NMDA receptors. Neuropsychopharmacology 2006, 31, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.L.; Holloway, T.; Rayannavar, V.; Sealfon, S.C.; González-Maeso, J. Chronic treatment with LY341495 decreases 5-HT 2A receptor binding and hallucinogenic effects of LSD in mice. Neurosci. Lett. 2013, 536, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, S.; Palmery, M.; Romanelli, L.; Cheng, C.Y.; Silvestrini, B. Lysergic acid diethylamide (LSD) is a partial agonist of D2 dopaminergic receptors and it potentiates dopamine-mediated prolactin secretion in lactotrophs in vitro. Life Sci. 1998, 63, 215–222. [Google Scholar] [CrossRef]
- Meltzer, H.Y.; Fessler, R.G.; Simonovic, M.; Doherty, J.; Fang, V.S. Lysergic acid diethylamide: Evidence for stimulation of pituitary dopamine receptors. Psychopharmacology 1977, 54, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Meert, T.; Awouters, F. Central 5-HT2 antagonists: A preclinical evaluation of a therapeutic potential. Acta Neuropsychiatr. 1990, 2, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Koek, W.; Jackson, A.; Colpaert, F.C. Behavioral pharmacology of antagonists at 5-HT2/5-HT1C receptors. Neurosci. Biobehav. Rev. 1992, 16, 95–105. [Google Scholar] [CrossRef]
- Meert, T. Serotonin Mechanisms in Antipsychotic Treatment: Evidence from Drug Discrimination Studies. In Serotonin in Antipsychotic Treatment. Mechanisms and Clinical Practice; Marcel Dekker, Inc.: New York, NY, USA, 1996; pp. 109–130. [Google Scholar]
- Marona-Lewicka, D.; Nichols, D.E. Further evidence that the delayed temporal dopaminergic effects of LSD are mediated by a mechanism different than the first temporal phase of action. Pharmacol. Biochem. Behav. 2007, 87, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Vollenweider, F.X.; Vollenweider-Scherpenhuyzen, M.F.; Bäbler, A.; Vogel, H.; Hell, D. Psilocybin induces schizophrenia-like psychosis in humans via a serotonin-2 agonist action. Neuroreport 1998, 9, 3897–3902. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.A.; Marona-Lewicka, D.; Nichols, D.E.; Nichols, C.D. Chronic LSD alters gene expression profiles in the mPFC relevant to schizophrenia. Neuropharmacology 2014, 83, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pehek, E.A.; Nocjar, C.; Roth, B.L.; Byrd, T.A.; Mabrouk, O.S. Evidence for the preferential involvement of 5-HT2A serotonin receptors in stress-and drug-induced dopamine release in the rat medial prefrontal cortex. Neuropsychopharmacology 2006, 31, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Vollenweider, F.; Leenders, K.; Scharfetter, C.; Maguire, P.; Stadelmann, O.; Angst, J. Positron emission tomography and fluorodeoxyglucose studies of metabolic hyperfrontality and psychopathology in the psilocybin model of psychosis. Neuropsychopharmacology 1997, 16, 357–372. [Google Scholar] [CrossRef]
- Simmler, L.D.; Buchy, D.; Chaboz, S.; Hoener, M.C.; Liechti, M.E. In vitro characterization of psychoactive substances at rat, mouse, and human trace amine-associated receptor 1. J. Pharmacol. Exp. Ther. 2016, 357, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Chandler, D.J.; Lamperski, C.S.; Waterhouse, B.D. Identification and distribution of projections from monoaminergic and cholinergic nuclei to functionally differentiated subregions of prefrontal cortex. Brain Res. 2013, 1522, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Steketee, J.D. Neurotransmitter systems of the medial prefrontal cortex: Potential role in sensitization to psychostimulants. Brain Res. Rev. 2003, 41, 203–228. [Google Scholar] [CrossRef]
- Arvanov, V.L.; Liang, X.; Russo, A.; Wang, R.Y. LSD and DOB: Interaction with 5-HT2A receptors to inhibit NMDA receptor-mediated transmission in the rat prefrontal cortex. Eur. J. Neurosci. 1999, 11, 3064–3072. [Google Scholar] [CrossRef] [PubMed]
- Solinas, M.; Panlilio, L.V.; Justinova, Z.; Yasar, S.; Goldberg, S.R. Using drug-discrimination techniques to study the abuse-related effects of psychoactive drugs in rats. Nat. Protoc. 2006, 1, 1194–1206. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.A.; Swerdlow, N.R.; Mansbach, R.S.; Braff, D.L. Startle response models of sensorimotor gating and habituation deficits in schizophrenia. Brain Res. Bull. 1990, 25, 485–498. [Google Scholar] [CrossRef]
- Geyer, M.A. Behavioral studies of hallucinogenic drugs in animals: Implications for schizophrenia research. Pharmacopsychiatry 1998, 31, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y.; Matsubara, S.; Lee, J. Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin2 pKi values. J. Pharmacol. Exp. Ther. 1989, 251, 238–246. [Google Scholar] [PubMed]
- Geyer, M.A.; Krebs-Thomson, K.; Braff, D.L.; Swerdlow, N.R. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: A decade in review. Psychopharmacology 2001, 156, 117–154. [Google Scholar] [CrossRef] [PubMed]
- Braff, D.L.; Swerdlow, N.R.; Geyer, M.A. Symptom correlates of prepulse inhibition deficits in male schizophrenic patients. Am. J. Psychiatry 1999, 156, 596–602. [Google Scholar] [PubMed]
- Parwani, A.; Duncan, E.J.; Bartlett, E.; Madonick, S.H.; Efferen, T.R.; Rajan, R.; Sanfilipo, M.; Chappell, P.B.; Chakravorty, S.; Gonzenbach, S. Impaired prepulse inhibition of acoustic startle in schizophrenia. Biol. Psychiatry 2000, 47, 662–669. [Google Scholar] [CrossRef]
- Quednow, B.B.; Frommann, I.; Berning, J.; Kühn, K.-U.; Maier, W.; Wagner, M. Impaired sensorimotor gating of the acoustic startle response in the prodrome of schizophrenia. Biol. Psychiatry 2008, 64, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Ouagazzal, A.; Grottick, A.; Moreau, J.; Higgins, G. Effect of LSD on prepulse inhibition and spontaneous behavior in the rat: A pharmacological analysis and comparison between two rat strains. Neuropsychopharmacology 2001, 25, 565–575. [Google Scholar] [CrossRef]
- Halberstadt, A.L.; Geyer, M.A. LSD but not lisuride disrupts prepulse inhibition in rats by activating the 5-HT2A receptor. Psychopharmacology 2010, 208, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Canal, C.E.; Morgan, D. Head-twitch response in rodents induced by the hallucinogen 2, 5-dimethoxy-4-iodoamphetamine: A comprehensive history, a re-evaluation of mechanisms, and its utility as a model. Drug Test. Anal. 2012, 4, 556–576. [Google Scholar] [CrossRef] [PubMed]
- González-Maeso, J.; Yuen, T.; Ebersole, B.J.; Wurmbach, E.; Lira, A.; Zhou, M.; Weisstaub, N.; Hen, R.; Gingrich, J.A.; Sealfon, S.C. Transcriptome fingerprints distinguish hallucinogenic and nonhallucinogenic 5-hydroxytryptamine 2A receptor agonist effects in mouse somatosensory cortex. J. Neurosci. 2003, 23, 8836–8843. [Google Scholar] [PubMed]
- Benneyworth, M.A.; Smith, R.L.; Sanders-Bush, E. Chronic phenethylamine hallucinogen treatment alters behavioral sensitivity to a metabotropic glutamate 2/3 receptor agonist. Neuropsychopharmacology 2008, 33, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Halberstadt, A.L.; Geyer, M.A. Characterization of the head-twitch response induced by hallucinogens in mice. Psychopharmacology 2013, 227, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Fantegrossi, W.E.; Harrington, A.W.; Eckler, J.R.; Arshad, S.; Rabin, R.A.; Winter, J.C.; Coop, A.; Rice, K.C.; Woods, J.H. Hallucinogen-like actions of 2, 5-dimethoxy-4-(n)-propylthiophenethylamine (2C-T-7) in mice and rats. Psychopharmacology 2005, 181, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Fribourg, M.; Moreno, J.L.; Holloway, T.; Provasi, D.; Baki, L.; Mahajan, R.; Park, G.; Adney, S.K.; Hatcher, C.; Eltit, J.M. Decoding the signaling of a GPCR heteromeric complex reveals a unifying mechanism of action of antipsychotic drugs. Cell 2011, 147, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.L.; Holloway, T.; Umali, A.; Rayannavar, V.; Sealfon, S.C.; González-Maeso, J. Persistent effects of chronic clozapine on the cellular and behavioral responses to LSD in mice. Psychopharmacology 2013, 225, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Tófoli, L.; de Araujo, D. Chapter Seven-Treating Addiction: Perspectives from EEG and Imaging Studies on Psychedelics. Int. Rev. Neurobiol. 2016, 129, 157–185. [Google Scholar] [PubMed]
- Dos Santos, R.G.; Osório, F.L.; Crippa, J.A.S.; Riba, J.; Zuardi, A.W.; Hallak, J.E. Antidepressive, anxiolytic, and antiaddictive effects of ayahuasca, psilocybin and lysergic acid diethylamide (LSD): A systematic review of clinical trials published in the last 25 years. Ther. Adv. Psychopharmacol. 2016, 6, 193–213. [Google Scholar] [CrossRef] [PubMed]
- Gasser, P.; Holstein, D.; Michel, Y.; Doblin, R.; Yazar-Klosinski, B.; Passie, T.; Brenneisen, R. Safety and efficacy of lysergic acid diethylamide-assisted psychotherapy for anxiety associated with life-threatening diseases. J. Nerv. Ment. Dis. 2014, 202, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Dolder, P.C.; Schmid, Y.; Müller, F.; Borgwardt, S.; Liechti, M.E. LSD acutely impairs fear recognition and enhances emotional empathy and sociality. Neuropsychopharmacology 2016, 41, 2638–2646. [Google Scholar] [CrossRef] [PubMed]
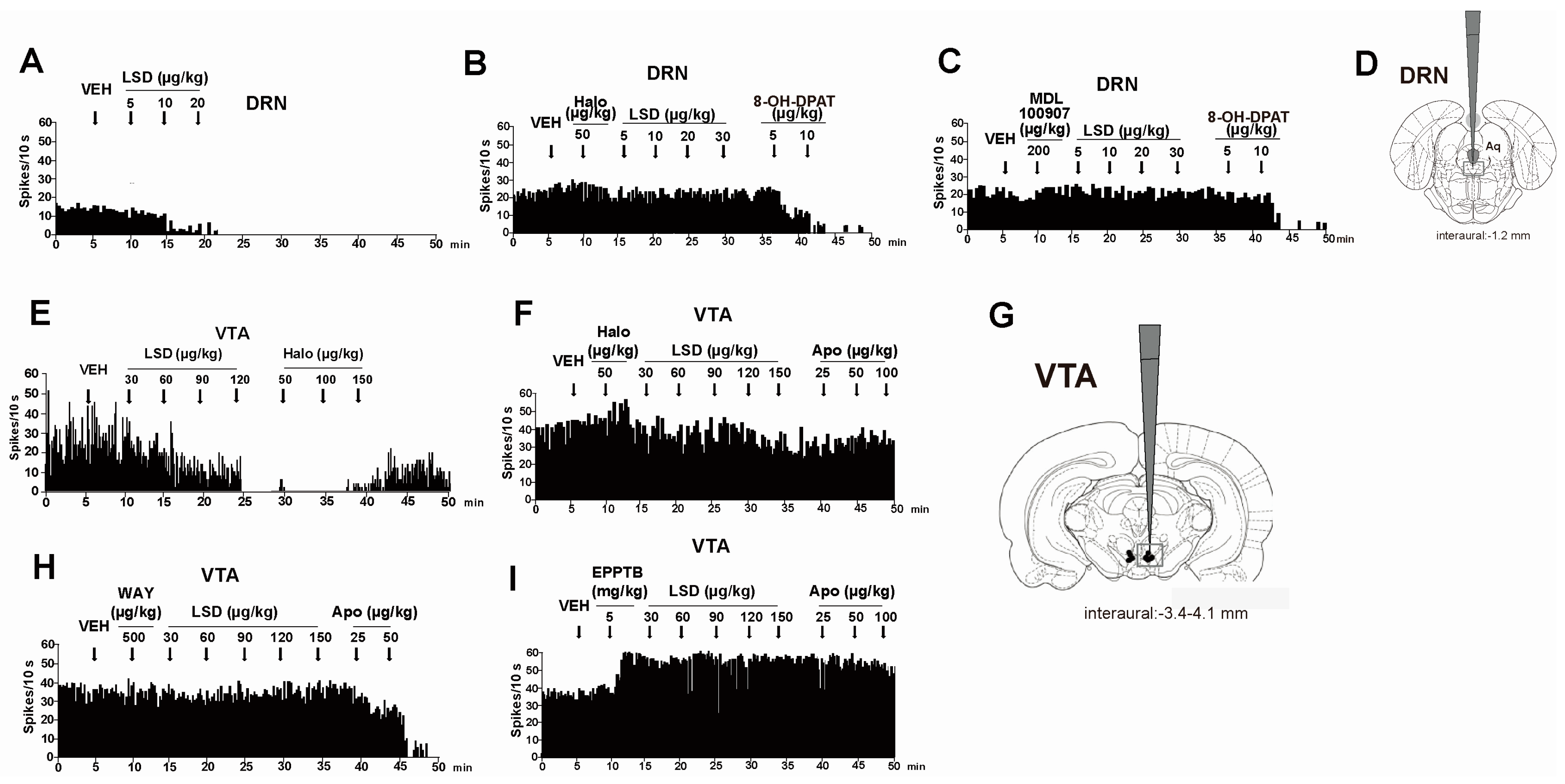

{kind=link}
{kind=link}
{kind=link}
| Parallelism between Effects Induced by LSD (100–200 µg) and Symptoms of Psychosis | |
|---|---|
| LSD-Induced Symptoms | Psychosis or Schizophrenia |
| Metamorphic alterations, unusual inner perception of bodily processes and changes in body image | Body Distortion |
| Metamorphosis-like change in objects and faces and intense (kaleidoscopic or scenic) visual imagery with transforming content | Delusions |
| Changes in perception and sensory alteration: Visual, auditory, taste, olfactory, kinaesthetic (pseudo-hallucinations) | Hallucination (Visual, auditory, taste, olfactory, kinaesthetic) |
| Depersonalization, derealisation | Depersonalization, derealisation, Cotard’s syndrome |
| Alteration of affectivity: euphoria, mood swing, anxiety | Euphoria, Dysphoria, Depression, Blunted affect |
| Mystical experience | Religious delusion, hypereligiosity |
| Suicide attempts | Depression and Suicide |
| Introjection | Interoception |
| Broader and unusual association | Clang association |
| Hyporeactivity | Psychomotor retardation, Catatonia |
| Disruption of sensorimotor gating | Deficit in sensorimotor gating |
| Disruption of pre-pulse inhibition (PPI) | Impairment in prepulse inhibition (PPI) of the acoustic startle response |
| Attention span shortened, alteration of Thinking, memory changes and decreased non-verbal abstract reasoning | Severe cognitive and memory impairments, Working memory impairment |
| Flash-back phenomena | “Déjà-vu” experiences |
| Results of LSD’s Effect on Serotonin, Dopamine, Glutamate and TAAR Systems | ||
|---|---|---|
| In Vivo Studies | In Vitro Studies | |
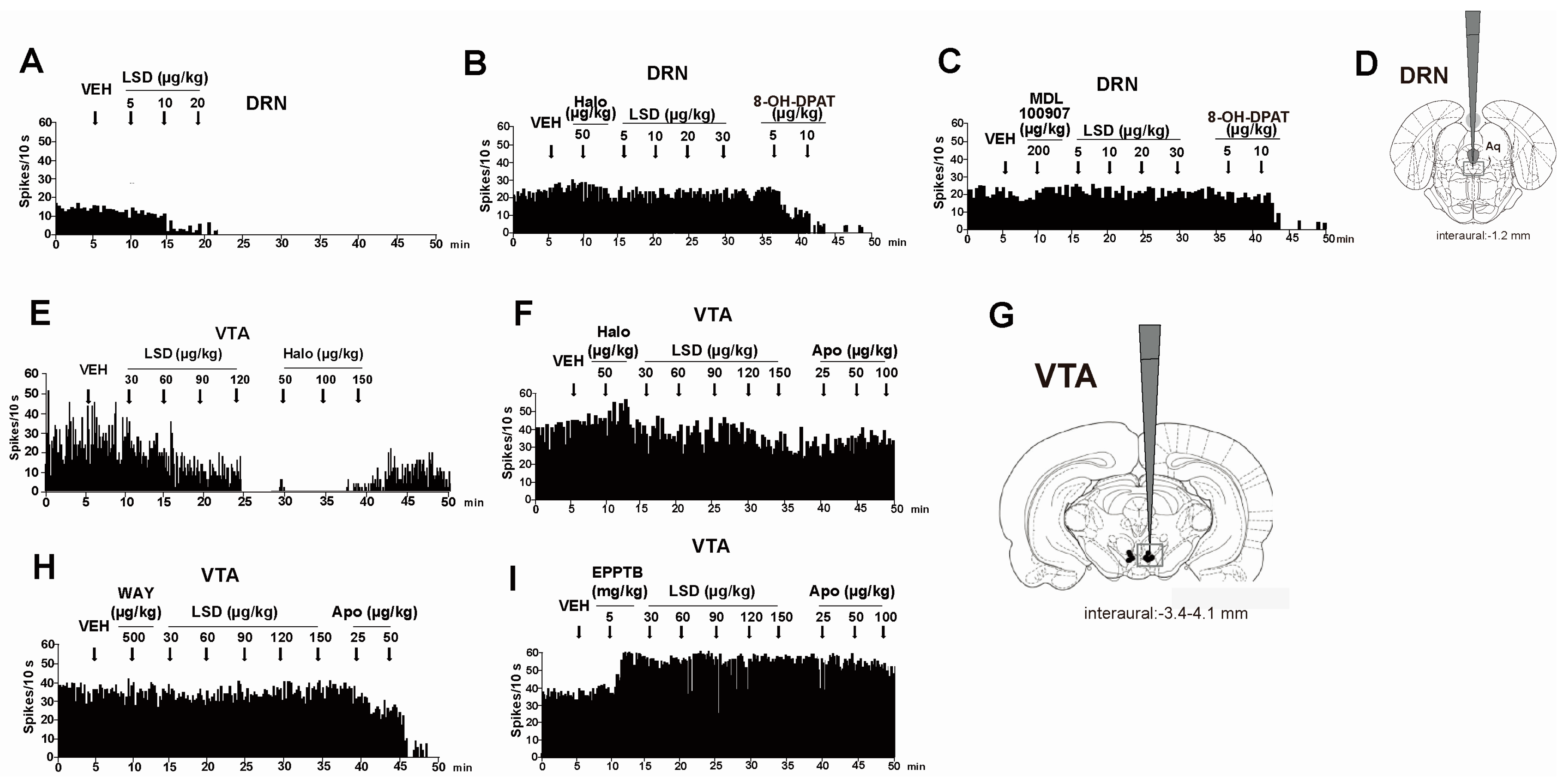
| 5-HT1A | 5-HT1A receptor agonists increase the effect of LSD (0.1 mg/kg) in stimulus control test in rats. The effect is reverted by the 5-HT1A antagonist WAY 100 635 [53] | Radio-labelled [3H]-LSD binds 5-HT1A receptors (ki: 1.1 nM) in homogenates of rat cerebral cortex [52] |
| 5-HT1A receptor antagonist WAY 100 635 (500 µg/kg) prevents the inhibitory effect of LSD (30–150 µg/kg) on VTA DA firing activity [115] | LSD binds human cloned 5-HT1A (ki: 0.0030 ± 0.0005 µM) receptor in HEK 293 cells [50] | |
| 5-HT2A | LSD (3–100 nM) excites GABAergic interneurons in the layer III of rat pyriform cortex; the effect is blocked by the 5-HT2A antagonist MDL 100 907 [112]; | Binding assay reveals affinity of radio labelled [3H]-LSD for 5-H2A receptor (ki: 2.5 nM) in parietofrontal cortex of male rats [5] |
| 5HT2 antagonist LY 53857 reverts the inhibitory effect of systemic administration of LSD (5–10 µg/kg) on the spontaneous activity of Locus Coeruleus (LC) neurons in rats [110] | ||
| 5-HT2A antagonist MDL 100 907 (200 µg/kg) prevents the inhibitory effect of LSD (5–20 µg/kg) on DRN 5-HT firing activity [115] | LSD binds Human 5-HT2A receptor-expressing NIH-3T3 cells (ki: 0.0042 ± 0.0013 µM) [50] | |
| 5-HT2c | serotonin depletion with PCPA induces supersensitivity of LSD(0.1 mg/kg)-trained rats to the stimulus effects of LSD and upregulation of the maximal level of 5-HT2C receptor [116] | Labelled LSD binds 5-HT2C receptor (ki: 10 nM) in NIH-3T3 cells transfected with rat 5-HT2C receptor [5]; |
| LSD binds human cloned 5-HT2c (ki: 0.015 ± 0.003 µM) receptor in HEK 293 cells [50] | ||
| Serotonin Transporter (SERT) | The efficacy of the stimulus control induced in SERT KO mice reveals a decreased efficacy of LSD (0.17–0.30 mg/kg)-stimulus control [118]; | LSD did not interact with SERT in HEK 293 cells [49] |
| SERT+/− mice compared to controls display a longer duration of self-grooming behaviour. The treatment with LSD (0.32 mg/kg) increases serotonin-sensitive behaviours such as head twitching, tremors and backwards gait in both SERT+/+ controls and SERT+/− mice [119] | ||
| D1 | No available studies | Labelled LSD binds D1 receptor (ki: 27.1 nM) in C-6-mD1A cells of rat striatum [120]; |
| LSD binds human cloned D1 (ki: 0.31 ± 0.1 µM) receptor in HEK 293 cells [50] | ||
| D2 | Rats trained with the DA D1/D2 agonist apomorphine (0.25 mg/kg) respond to LSD with partial generalization but the apomorphine cue is antagonized by the D2 antagonist haloperidol [121] | LSD stimulates the incorporation of [35S]GTP-γ-S into Gi coupled to D2 receptors in homogenates of rat brain striatum [56] |
| The potency of ritanserin to antagonize LSD stimulus control is markedly potentiated when administered in combination with low doses of the D2 antagonist haloperidol [122] | LSD displaces the selective D2 antagonist [3H]raclopride from pig brain cryostat sections with an IC50 of 275 Nm [57] | |
| LSD (372 nmol/kg, 0.16 mg/kg) injected 90 min before training produces a cue that is not fully blocked by 5-HT2A antagonists, but instead is significantly inhibited by the D2 antagonist haloperidol [123] | LSD stimulates the incorporation of [35S]GTP-γ-S into Gi coupled to human cloned D2 receptors [58] | |
| Haloperidol (50 µg/kg) prevents the inhibitory effect of LSD (30–150 µg/kg) on VTA DA firing activity [115] | LSD binds human cloned D2 (ki: 0.025 ± 0.0004 µM) receptor in HEK 293 cells [50,51] | |
| D4 | The D4 antagonist A-381393 attenuates the stimulus effect of LSD (0.08–0.016 mg/kg) in discriminative test [124] | 38 L Labelled LSD shows affinity for NIH3T3 fibroblast cells expressing the rat D4 receptor (ki: 56 nM) [5] |
| NMDA | NMDA receptor subunit NR2B-selective antagonists, ifenprodil and Ro25-6981 suppress the prolonged glutamate release induced by LSD onto layer V pyramidal neurons of the prefrontal cortex [125] | No available studies |
| mGlu2/mGlu3 | mGlu2/3 receptor antagonist LY341495 (1.5 mg/kg) reduces head-twitch behaviour and expression of c-fos, egr-1 and egr-2 that are both augmented by LSD (0.24 mg/kg) [126] | No available studies |
| TAAR1 | The selective TAAR1 antagonist EPPTB (5 mg/kg) prevents the inhibitory effect of LSD (30–150 µg/kg) on VTA DA firing activity [115] | LSD binds the expressed rat TAAR1 receptor on HEK-293 cells (ki: 0.8 µM) [59]; |
| LSD binds rat (ki: 0.45 ± 0.05 µM) and mouse (ki: 10 ± 2.9 µM) TAAR1 receptor in HEK 293 cells [51] | ||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Gregorio, D.; Comai, S.; Posa, L.; Gobbi, G. d-Lysergic Acid Diethylamide (LSD) as a Model of Psychosis: Mechanism of Action and Pharmacology. Int. J. Mol. Sci. 2016, 17, 1953. https://doi.org/10.3390/ijms17111953
De Gregorio D, Comai S, Posa L, Gobbi G. d-Lysergic Acid Diethylamide (LSD) as a Model of Psychosis: Mechanism of Action and Pharmacology. International Journal of Molecular Sciences. 2016; 17(11):1953. https://doi.org/10.3390/ijms17111953
Chicago/Turabian StyleDe Gregorio, Danilo, Stefano Comai, Luca Posa, and Gabriella Gobbi. 2016. "d-Lysergic Acid Diethylamide (LSD) as a Model of Psychosis: Mechanism of Action and Pharmacology" International Journal of Molecular Sciences 17, no. 11: 1953. https://doi.org/10.3390/ijms17111953
APA StyleDe Gregorio, D., Comai, S., Posa, L., & Gobbi, G. (2016). d-Lysergic Acid Diethylamide (LSD) as a Model of Psychosis: Mechanism of Action and Pharmacology. International Journal of Molecular Sciences, 17(11), 1953. https://doi.org/10.3390/ijms17111953