
Isoliquiritigenin Attenuates Atherogenesis in Apolipoprotein E-Deficient Mice


Abstract
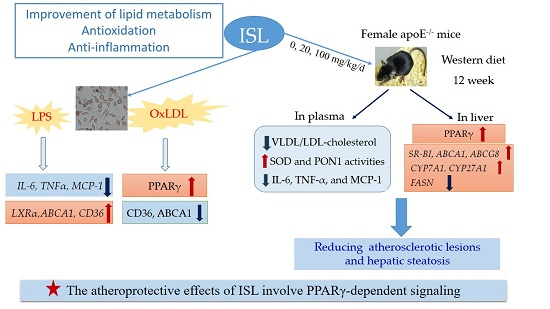
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
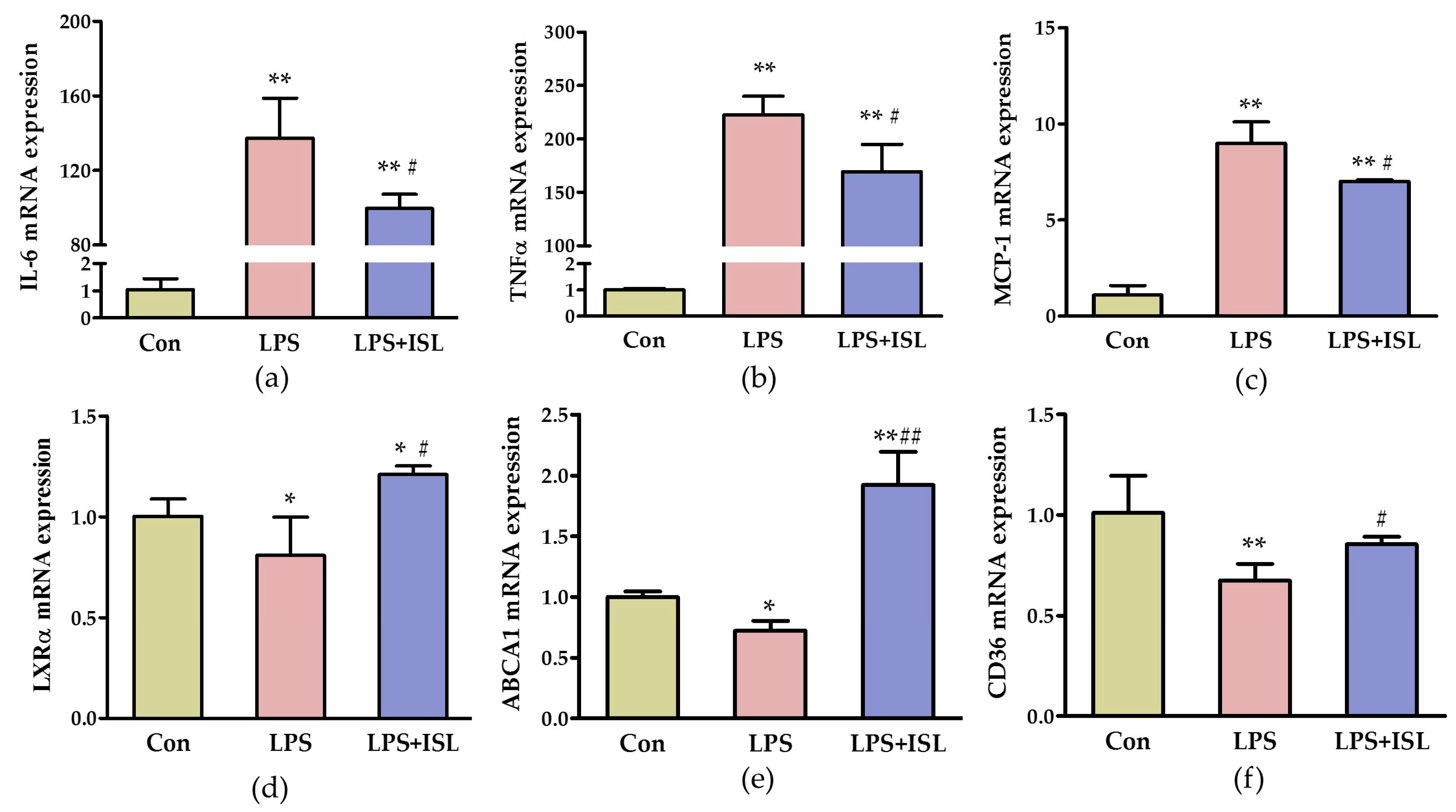
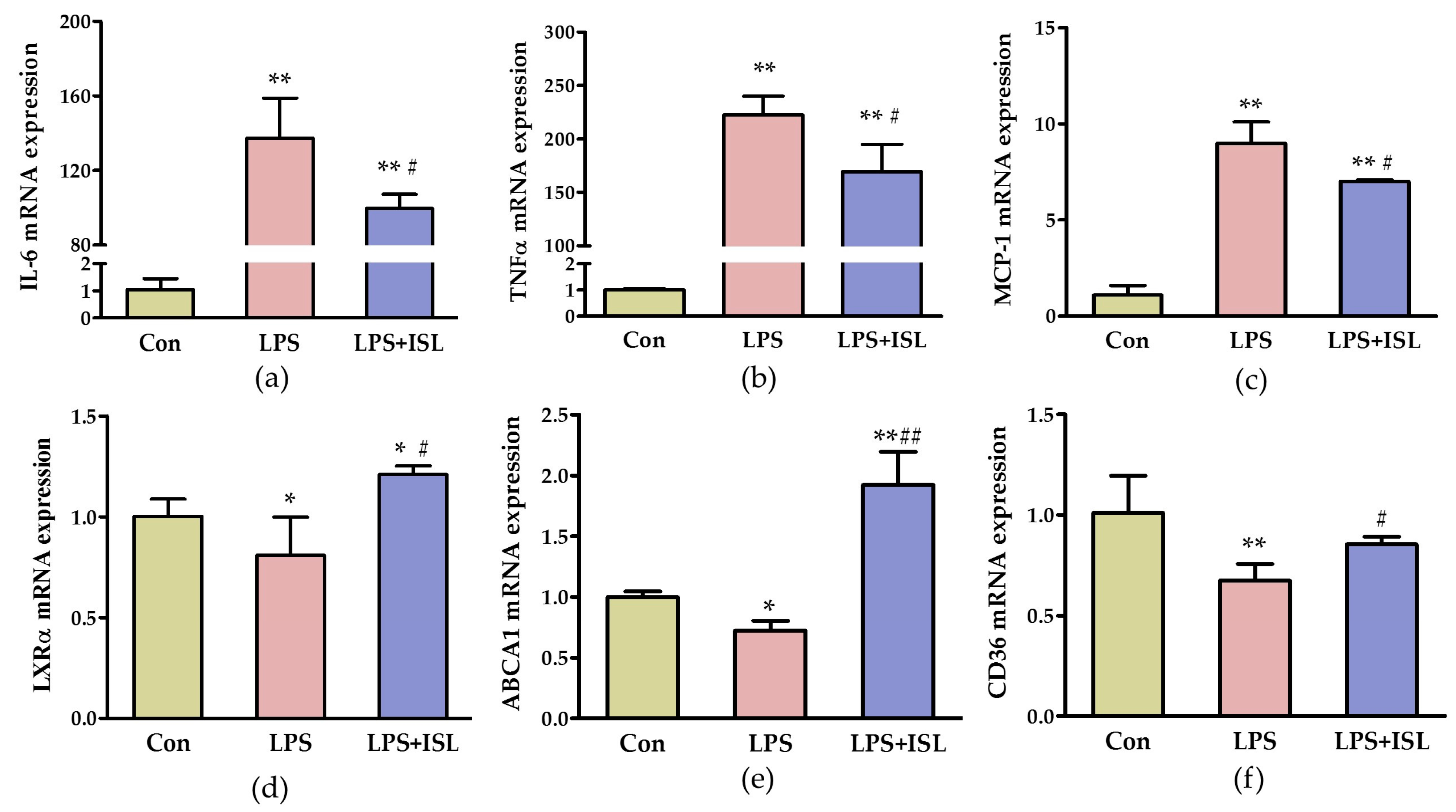
2.1. ISL Suppresses the Inflammatory Response of Macrophages to Lipopolysaccharide (LPS)
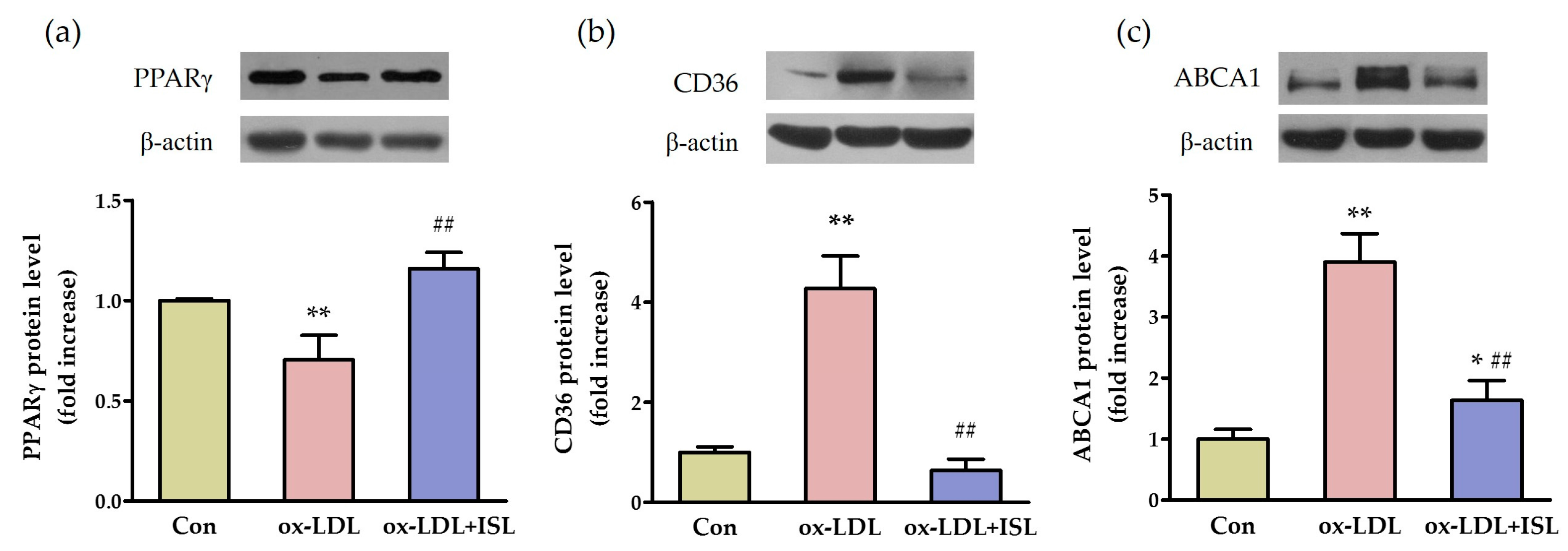
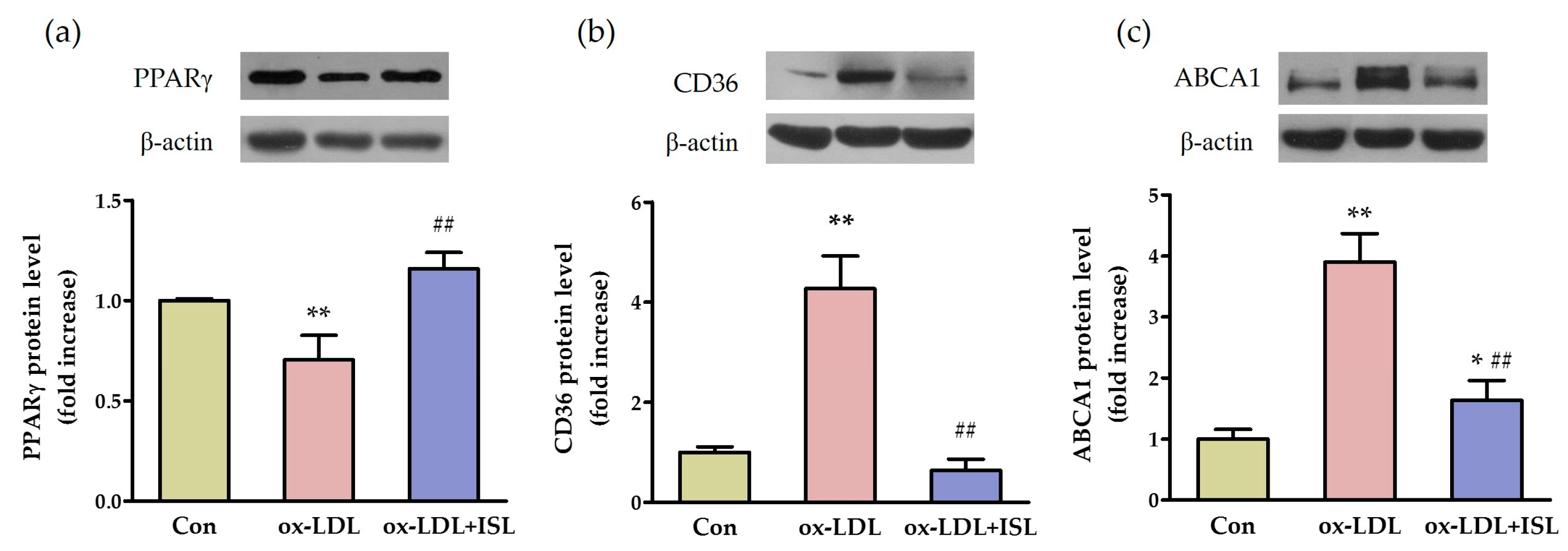
2.2. ISL Affects Cholesterol-Flux-Related Protein Expression in Macrophages Treated with Oxidative-LDL
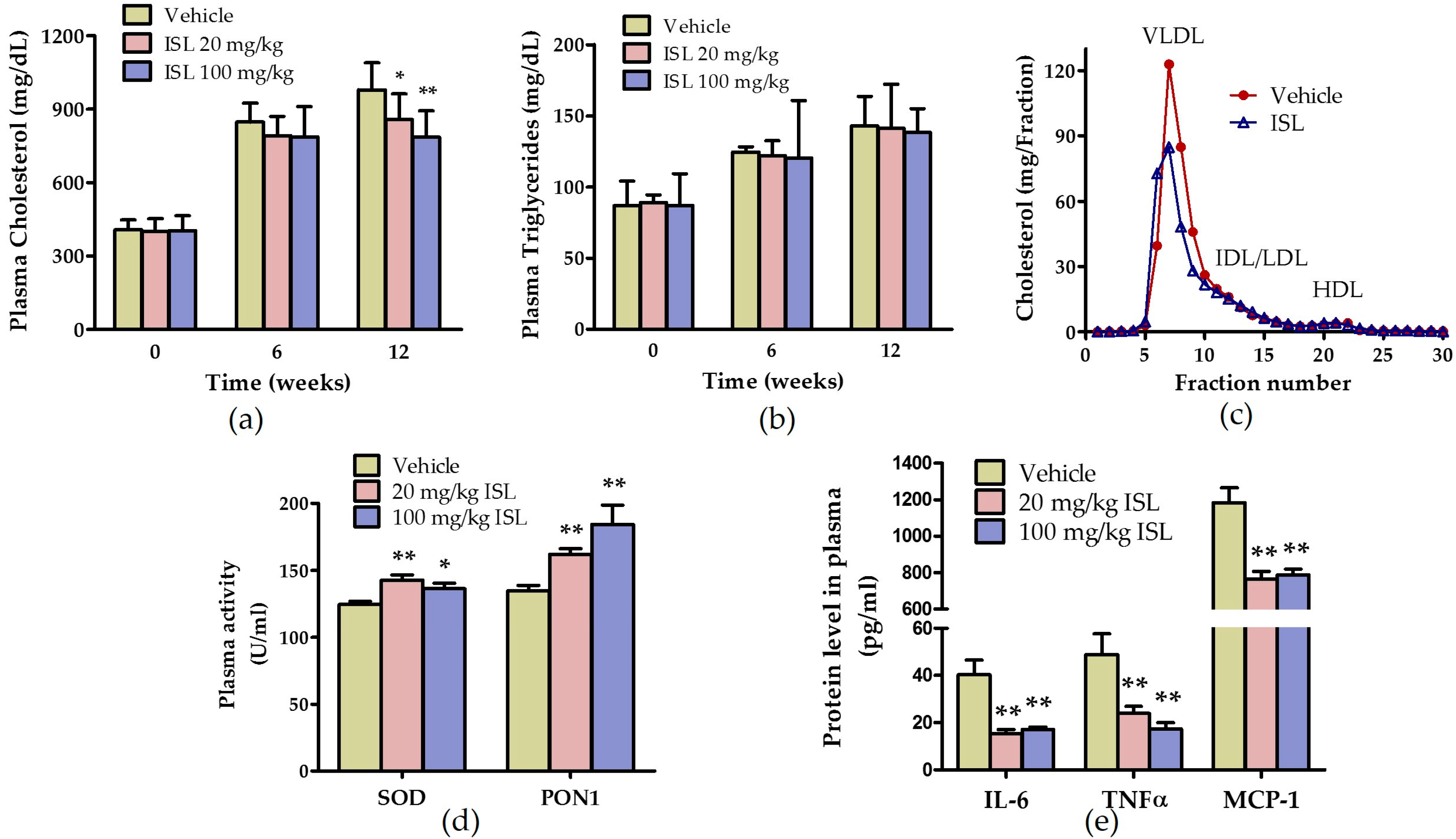
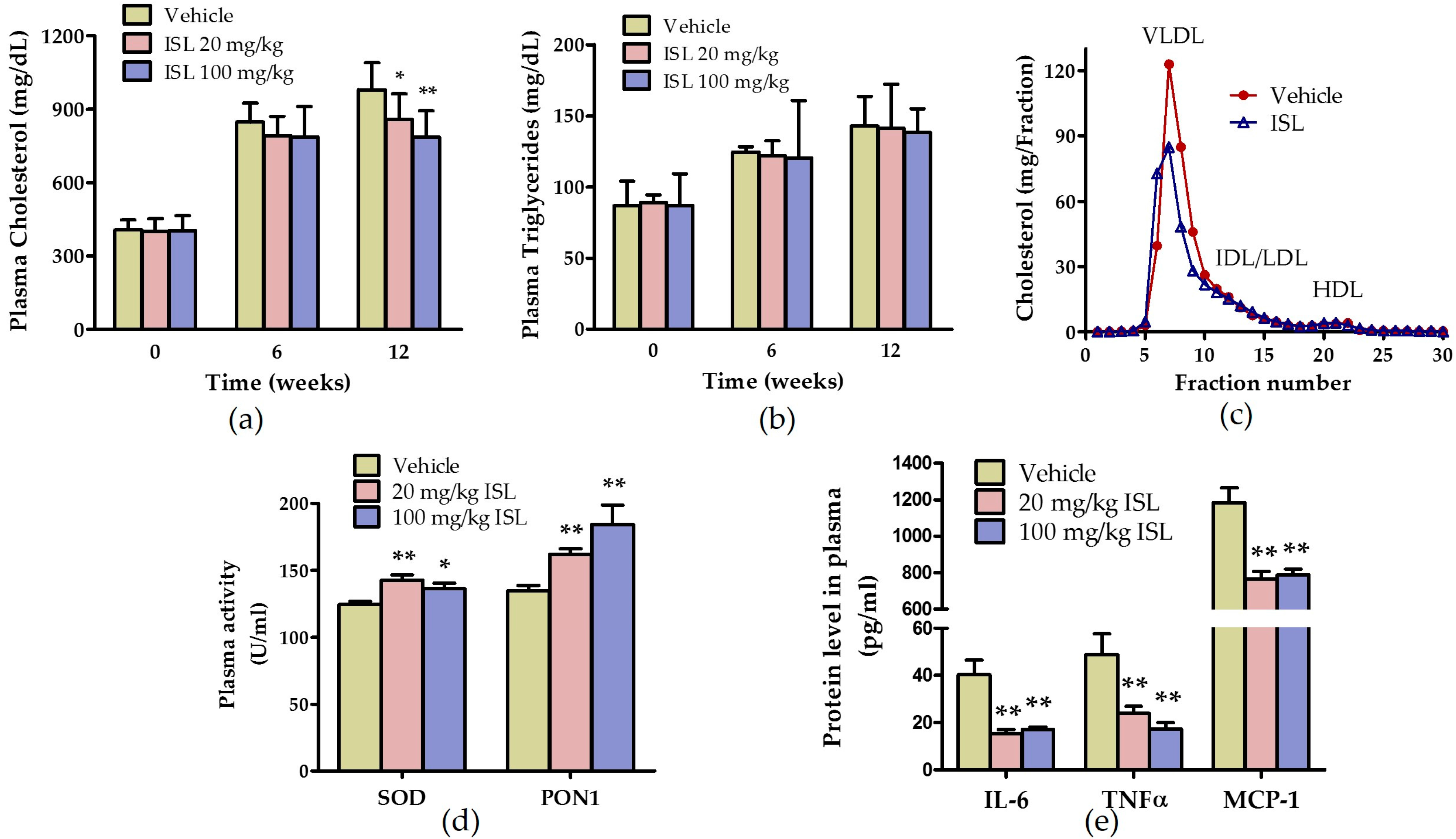
2.3. ISL Ameliorates Plasma Lipid Levels and Inhibits Oxidative Stress and Inflammation in ApoE−/− Mice Fed a Western Diet
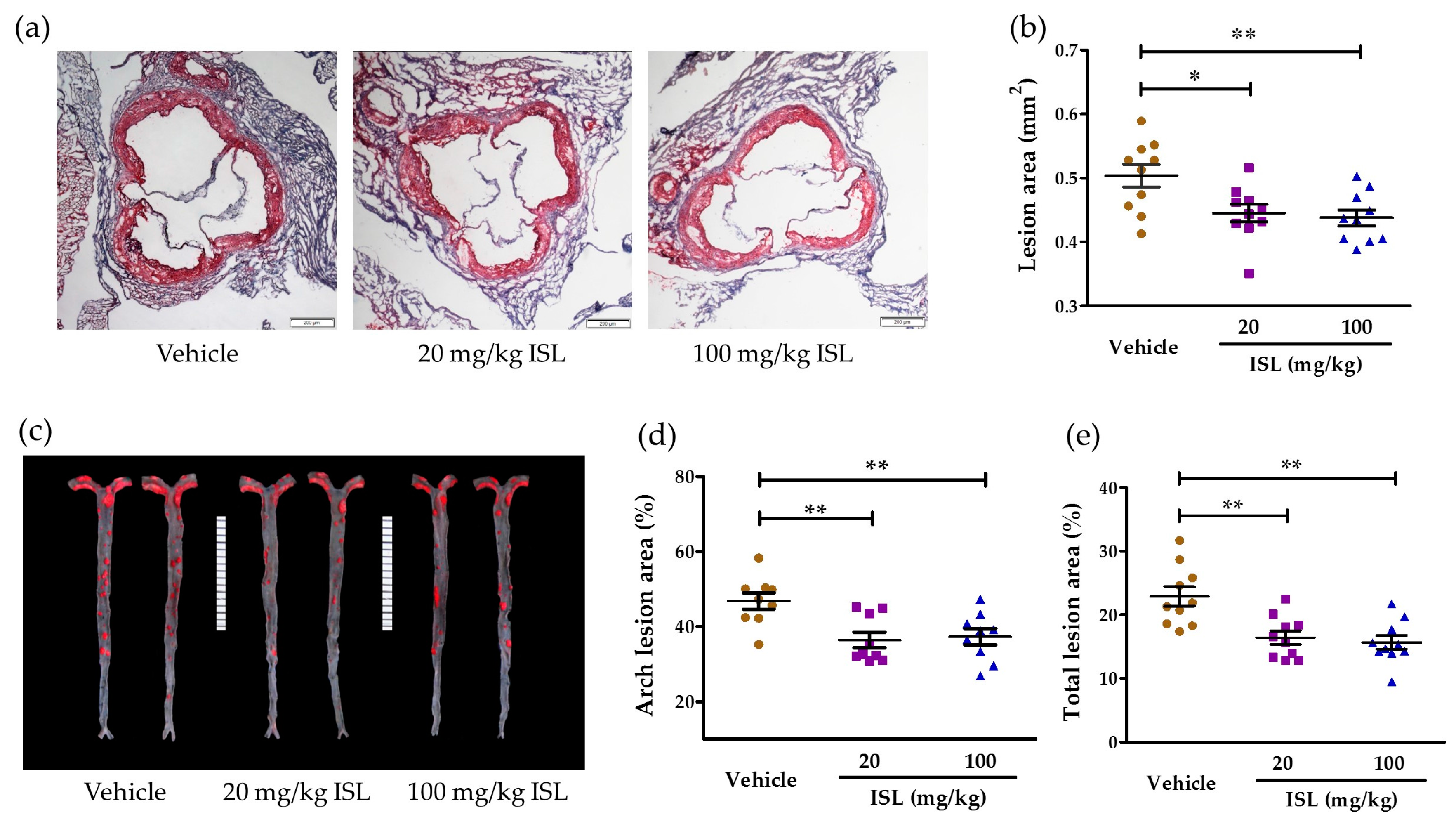
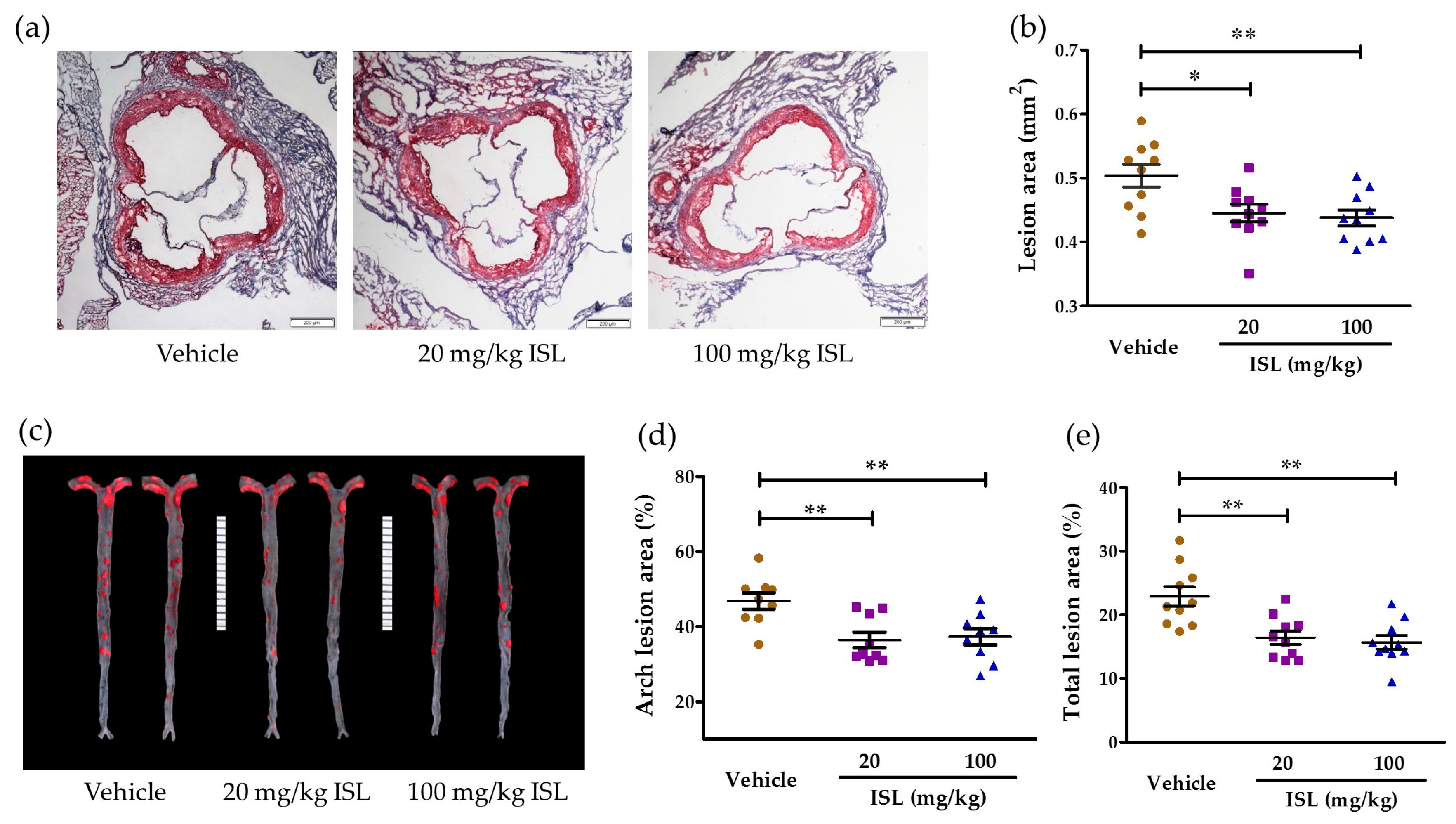
2.4. ISL Attenuates Atherosclerosis in ApoE−/− Mice
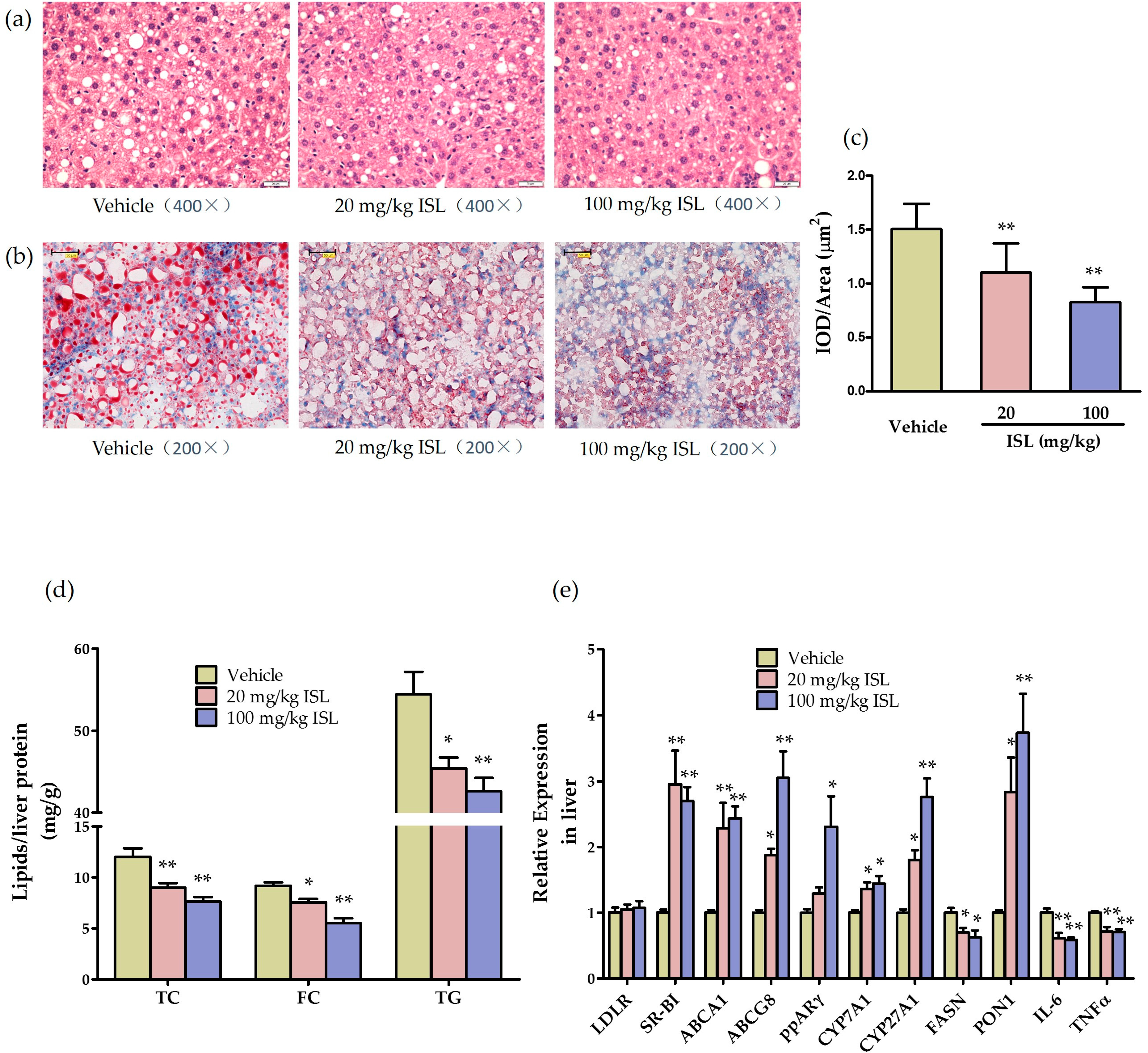
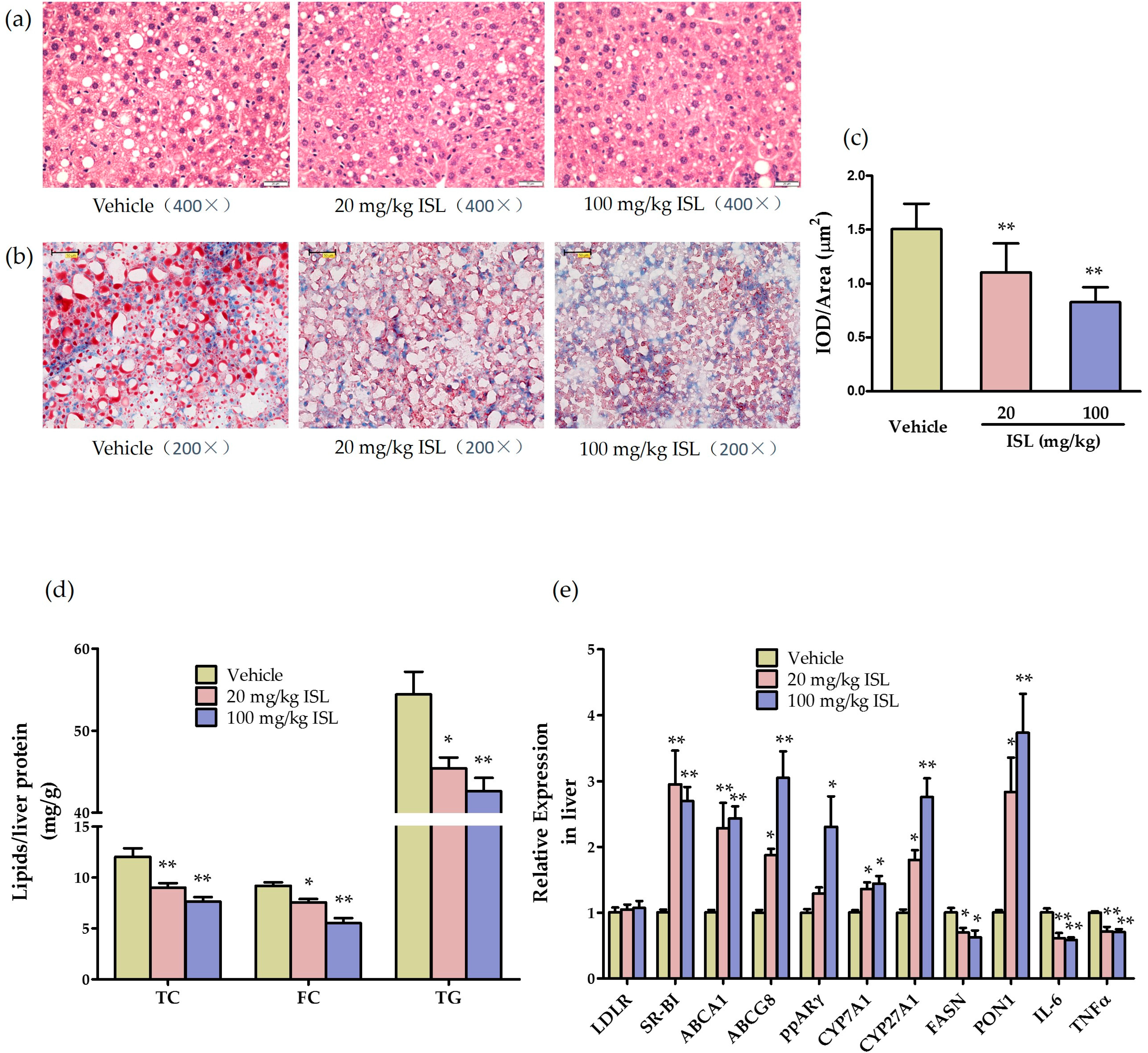
2.5. ISL Reduces Hepatic Steatosis and Alters Hepatic Gene Expression in ApoE−/− Mice
3. Discussion
3.1. ISL Inhibits the Inflammatory Responses In Vitro and In Vivo
3.2. ISL Ameliorates Oxidative Stress In Vitro and In Vivo
3.3. ISL Regulates Lipid Metabolism In Vivo
4. Materials and Methods
4.1. Materials and Reagents
4.2. Mice
4.3. Cell Culture
4.4. Determination of Cytokine Expression Levels by Real-Time PCR
4.5. Determination of Protein Expression by Western Blot
4.6. Plasma Analysis
4.7. Atherosclerosis Lesion Analysis
4.8. Gene Expression, Biochemical, and Histochemical Analyses of the Liver
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ABCA1 | ATP-binding cassette transporter A1 |
| apoE−/− | apolipoprotein E deficient |
| FPLC | fast protein liquid chromatography |
| HDL | high density lipoprotein |
| HE | hematoxylin and eosin |
| HRP | horseradish peroxidase |
| IL | interleukin |
| ISL | isoliquiritigenin |
| LDL | low density lipoproteins |
| LPS | lipopolysaccharide |
| LXRα | liver X receptor α |
| MCP-1 | monocyte chemotactic protein-1 |
| TNF | tumor necrosis factor |
| ox-LDL | oxidative low density lipoproteins |
| PPARγ | peroxisome proliferator-activated receptor gamma |
| PON1 | Paraoxonase-1 |
| qPCR | quantitative real-time PCR |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| SR-BI | scavenger receptor class B type I |
| TC | total cholesterol |
| TG | triglyceride |
References
- Madamanchi, N.R.; Vendrov, A.; Runge, M.S. Oxidative stress and vascular disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D. Atherogenesis in perspective: Hypercholesterolemia and inflammation as partners in crime. Nat. Med. 2002, 8, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Jacob, R.F.; Mason, R.P. Inflammation and the development of atherosclerosis. J. Atheroscler. Thromb. 2011, 18, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Tedgui, A.; Mallat, Z. Cytokines in atherosclerosis: Pathogenic and regulatory pathways. Physiol. Rev. 2006, 86, 515–581. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Dudek, K.; Mayr, M. Oxidative stress in atherosclerosis: The role of micrornas in arterial remodeling. Free Radic. Biol. Med. 2013, 64, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.L.; Singh, V.; Barthwal, M.K. Macrophages: An elusive yet emerging therapeutic target of atherosclerosis. Med. Res. Rev. 2008, 28, 483–544. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Feng Yeh, C.; Wang, K.C.; Chiang, L.C.; Shieh, D.E.; Yen, M.H.; San Chang, J. Water extract of licorice had anti-viral activity against human respiratory syncytial virus in human respiratory tract cell lines. J. Ethnopharmacol. 2013, 148, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Du, Q.; Peng, C.; Wang, N.; Tang, H.; Xie, X.; Shen, J.; Chen, J. A review: The pharmacology of isoliquiritigenin. Phytother. Res. 2015, 29, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Park, S.M.; Guan, L.; Wu, Y.; Lee, J.R.; Kim, S.C.; Kim, Y.W.; Zhao, R. Isoliquiritigenin attenuates oxidative hepatic damage induced by carbon tetrachloride with or without buthionine sulfoximine. Chem. Biol. Interact. 2015, 225, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Nagai, Y.; Matsunaga, T.; Okamoto, N.; Watanabe, Y.; Tsuneyama, K.; Hayashi, H.; Fujii, I.; Ikutani, M.; Hirai, Y.; et al. Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. J. Leukoc. Biol. 2014, 96, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Nagai, Y.; Honda, H.; Okamoto, N.; Yamamoto, S.; Hamashima, T.; Ishii, Y.; Tanaka, M.; Suganami, T.; Sasahara, M.; et al. Isoliquiritigenin attenuates adipose tissue inflammation in vitro and adipose tissue fibrosis through inhibition of innate immune responses in mice. Sci. Rep. 2016, 6, 23097–23113. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Xue, J.; Yang, Y.; Zhu, H.; Chen, F.; Wang, J.; Lou, G.; Liu, Y.; Shi, Y.; Yu, Y.; et al. Isoliquiritigenin inhibits interferon-γ-inducible genes expression in hepatocytes through down-regulating activation of JAK1/STAT1, IRF3/MyD88, ERK/MAPK, JNK/MAPK and PI3K/Akt signaling pathways. Cell. Physiol. Biochem. 2015, 37, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Choe, Y.G.; Kim, J.H.; Chang, K.T.; Lee, H.S.; Lee, D.S. Isoliquiritigenin impairs insulin signaling and adipocyte differentiation through the inhibition of protein-tyrosine phosphatase 1B oxidation in 3T3-L1 preadipocytes. Food Chem. Toxicol. 2016, 93, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.-J.; Li, H.-D.; Yan, M.; Li, Z.-H.; Gong, H.; Jiang, P.; Deng, Y.; Fang, P.-F.; Zhang, B.-K. The protective effects of isoliquiritigenin and glycyrrhetinic acid against triptolide-induced oxidative stress in HepG2 cells involve Nrf2 activation. Evid.-Based Complement. Altern. Med. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.J.; Min, J.S.; Ku, H.Y.; Choi, H.S.; Park, M.K.; Kim, M.K.; Song, K.S.; Lee, D.S. Isoliquiritigenin isolated from glycyrrhiza uralensis protects neuronal cells against glutamate-induced mitochondrial dysfunction. Biochem. Biophys. Res. Commun. 2012, 421, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Kim, T.H.; Kim, Y.W.; Yang, Y.M.; Ryu, D.H.; Hwang, S.J.; Lee, J.R.; Kim, S.C.; Kim, S.G. Inhibition of liver X receptor-α-dependent hepatic steatosis by isoliquiritigenin, a licorice antioxidant flavonoid, as mediated by JNK1 inhibition. Free Radic. Biol. Med. 2010, 49, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Webb, N.R.; Moore, K.J. Macrophage-derived foam cells in atherosclerosis: Lessons from murine models and implications for therapy. Curr. Drug Targets 2007, 8, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Vittone, F.; Liberman, A.; Vasic, D.; Ostertag, R.; Esser, M.; Walcher, D.; Ludwig, A.; Marx, N.; Burgmaier, M. Sitagliptin reduces plaque macrophage content and stabilises arteriosclerotic lesions in Apoe−/− mice. Diabetologia 2012, 55, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Qu, D.; Liu, J.; Lau, C.W.; Huang, Y. IL-6 in diabetes and cardiovascular complications. Br. J. Pharmacol. 2014, 171, 3595–3603. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Kakkar, V.; Lu, X. Impact of MCP-1 in atherosclerosis. Curr. Pharm. Des. 2014, 20, 4580–4588. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; de Caterina, R. Relevance of new drug discovery to reduce NF-κB activation in cardiovascular disease. Vascul. Pharmacol. 2012, 57, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Luscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014, 35, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Tietge, U.J. Hyperlipidemia and cardiovascular disease: Inflammation, dyslipidemia, and atherosclerosis. Curr. Opin. Lipidol. 2014, 25, 94–95. [Google Scholar] [CrossRef] [PubMed]
- Maitra, U.; Li, L. Molecular mechanisms responsible for the reduced expression of cholesterol transporters from macrophages by low-dose endotoxin. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Im, S.S.; Osborne, T.F. Liver X receptors in atherosclerosis and inflammation. Circ. Res. 2011, 108, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.L.; Ruan, X.Z.; Powis, S.H.; Chen, Y.; Moorhead, J.F.; Varghese, Z. Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of apolipoprotein e knockout mice. Hepatology 2008, 48, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Moser, A.H.; Shigenaga, J.K.; Grunfeld, C.; Feingold, K.R. Downregulation of liver X receptor-α in mouse kidney and HK-2 proximal tubular cells by lps and cytokines. J. Lipid Res. 2005, 46, 2377–2387. [Google Scholar] [CrossRef] [PubMed]
- Kunjathoor, V.V.; Febbraio, M.; Podrez, E.A.; Moore, K.J.; Andersson, L.; Koehn, S.; Rhee, J.S.; Silverstein, R.; Hoff, H.F.; Freeman, M.W. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 2002, 277, 49982–49988. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.; Chang, L.; Fan, Y.; Zhang, J.; Chen, Y.E. PPARs and the cardiovascular system. Antioxid. Redox Signal. 2009, 11, 1415–1452. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.; Chen, H.; Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARγ. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef]
- Tontonoz, P.; Nagy, L.; Alvarez, J.G.; Thomazy, V.A.; Evans, R.M. PPARγ promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 1998, 93, 241–252. [Google Scholar] [CrossRef]
- Chawla, A.; Boisvert, W.A.; Lee, C.H.; Laffitte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A.; Curtiss, L.K.; et al. A PPAR γ-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell 2001, 7, 161–171. [Google Scholar] [CrossRef]
- Chawla, A.; Barak, Y.; Nagy, L.; Liao, D.; Tontonoz, P.; Evans, R.M. PPARγ dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat. Med. 2001, 7, 48–52. [Google Scholar] [PubMed]
- Abreu, I.A.; Cabelli, D.E. Superoxide dismutases—A review of the metal—Associated mechanistic variations. Biochim. Biophys. Acta 2010, 1804, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Vakili, L.; Hama, S.; Kim, J.B.; Tien, D.; Safarpoor, S.; Ly, N.; Vakili, G.; Hough, G.; Navab, M. The effect of HDL mimetic peptide 4F on PON1. Adv. Exp. Med. Biol. 2010, 660, 167–172. [Google Scholar] [PubMed]
- Litvinov, D.; Mahini, H.; Garelnabi, M. Antioxidant and anti-inflammatory role of paraoxonase 1: Implication in arteriosclerosis diseases. N. Am. J. Med. Sci. 2012, 4, 523–532. [Google Scholar] [PubMed]
- Mackness, B.; Mackness, M. Anti-inflammatory properties of paraoxonase-1 in atherosclerosis. Adv. Exp. Med. Biol. 2010, 660, 143–151. [Google Scholar] [PubMed]
- Zhou, C.; Cao, J.; Shang, L.; Tong, C.; Hu, H.; Wang, H.; Fan, D.; Yu, H. Reduced paraoxonase 1 activity as a marker for severe coronary artery disease. Dis. Markers 2013, 35, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.L.; Mujawar, Z.; Tamehiro, N. ABC transporters, atherosclerosis and inflammation. Atherosclerosis 2010, 211, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Trigatti, B.L.; Krieger, M.; Rigotti, A. Influence of the HDL receptor SR-BI on lipoprotein metabolism and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1732–1738. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Shaul, P.W. Functions of scavenger receptor class B, type I in atherosclerosis. Curr. Opin. Lipidol. 2012, 23, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Oram, J.F.; Vaughan, A.M. ATP-binding cassette cholesterol transporters and cardiovascular disease. Circ. Res. 2006, 99, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Graf, G.A.; Yu, L.; Li, W.P.; Gerard, R.; Tuma, P.L.; Cohen, J.C.; Hobbs, H.H. ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J. Biol. Chem. 2003, 278, 48275–48282. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Yu, F.; Wang, Y.; Hui, Y.; Carnevale, K.; Fu, M.; Lu, H.; Fan, D. MicroRNA-155 deficiency results in decreased macrophage inflammation and attenuated atherogenesis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Y.; Du, F.; Meng, B.; Xie, G.H.; Cao, J.; Fan, D.; Yu, H. Hepatic overexpression of methionine sulfoxide reductase a reduces atherosclerosis in apolipoprotein E-deficient mice. J. Lipid Res. 2015, 56, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, W.; Yancey, P.G.; Koury, M.J.; Zhang, Y.; Fazio, S.; Linton, M.F. Macrophage apolipoprotein E reduces atherosclerosis and prevents premature death in apolipoprotein E and scavenger receptor-class BI double-knockout mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 150–156. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, F.; Gesang, Q.; Cao, J.; Qian, M.; Ma, L.; Wu, D.; Yu, H. Isoliquiritigenin Attenuates Atherogenesis in Apolipoprotein E-Deficient Mice. Int. J. Mol. Sci. 2016, 17, 1932. https://doi.org/10.3390/ijms17111932
Du F, Gesang Q, Cao J, Qian M, Ma L, Wu D, Yu H. Isoliquiritigenin Attenuates Atherogenesis in Apolipoprotein E-Deficient Mice. International Journal of Molecular Sciences. 2016; 17(11):1932. https://doi.org/10.3390/ijms17111932
Chicago/Turabian StyleDu, Fen, Quzhen Gesang, Jia Cao, Mei Qian, Li Ma, Dongfang Wu, and Hong Yu. 2016. "Isoliquiritigenin Attenuates Atherogenesis in Apolipoprotein E-Deficient Mice" International Journal of Molecular Sciences 17, no. 11: 1932. https://doi.org/10.3390/ijms17111932
APA StyleDu, F., Gesang, Q., Cao, J., Qian, M., Ma, L., Wu, D., & Yu, H. (2016). Isoliquiritigenin Attenuates Atherogenesis in Apolipoprotein E-Deficient Mice. International Journal of Molecular Sciences, 17(11), 1932. https://doi.org/10.3390/ijms17111932