
Pterostilbene Inhibits Human Multiple Myeloma Cells via ERK1/2 and JNK Pathway In Vitro and In Vivo


Abstract
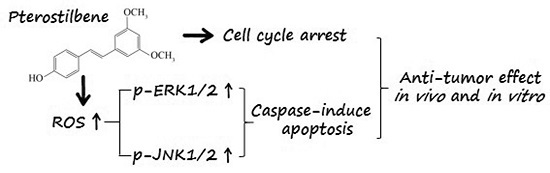
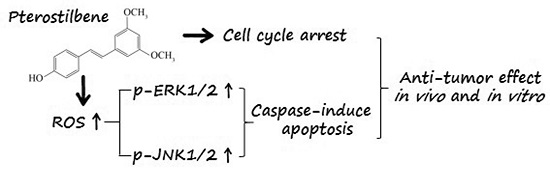
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
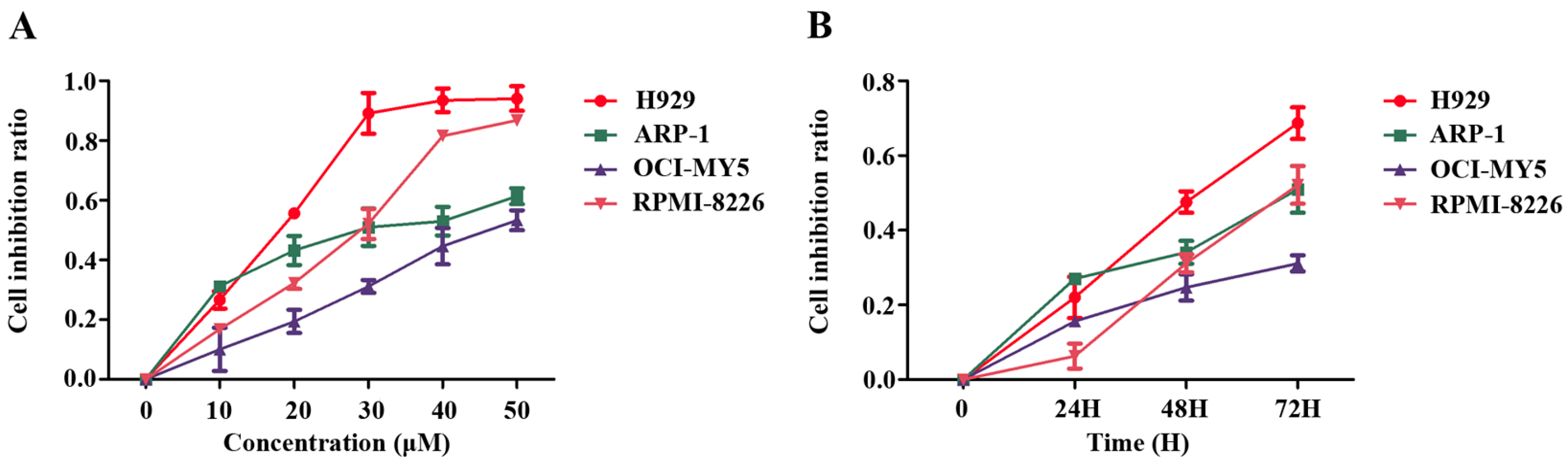
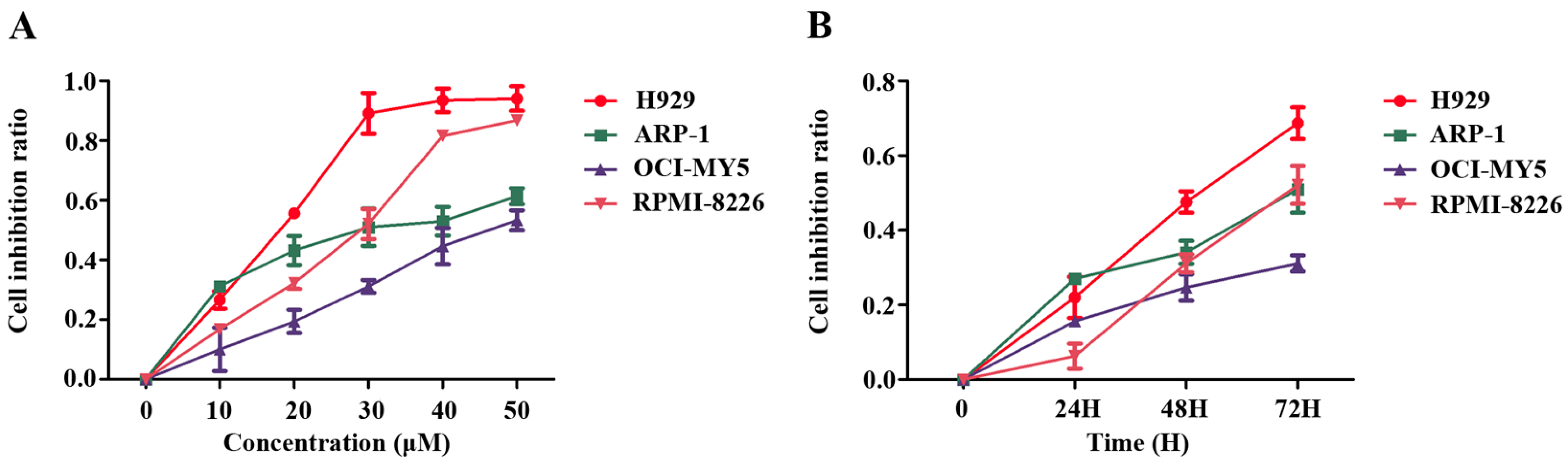
2.1. Pterostilbene (PTE) Inhibits the Proliferation of Multiple Myeloma (MM) Cell Lines
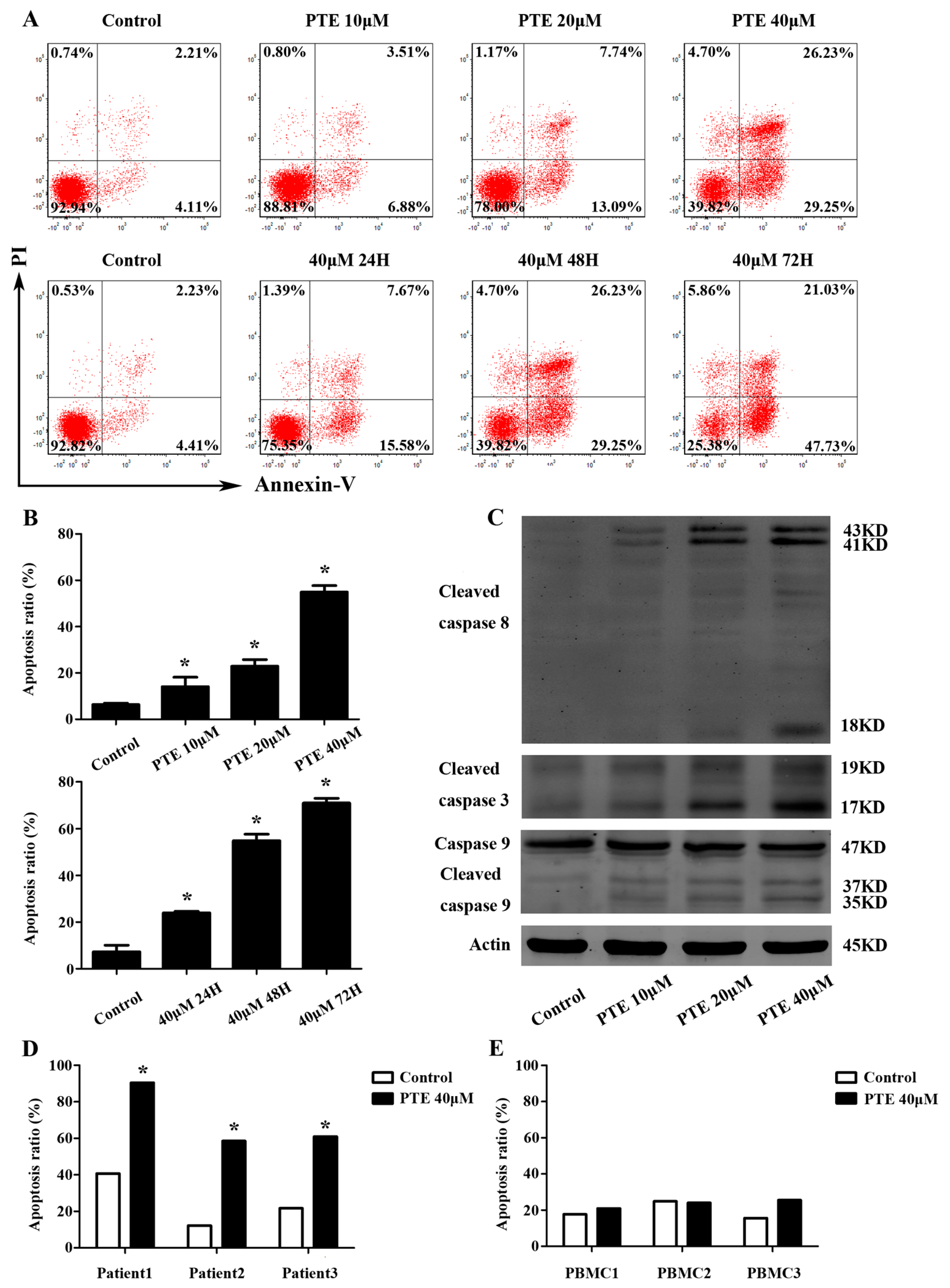
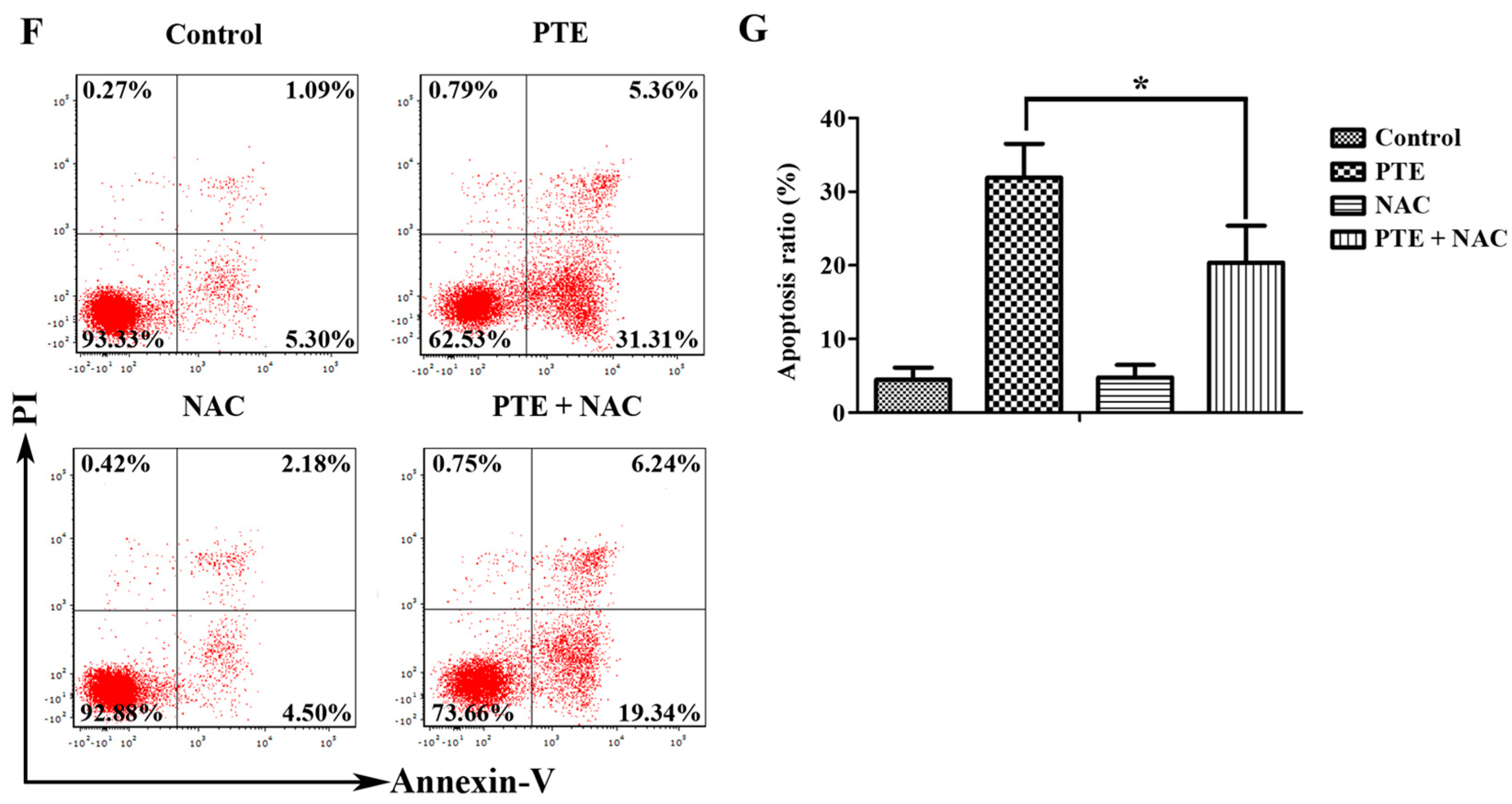
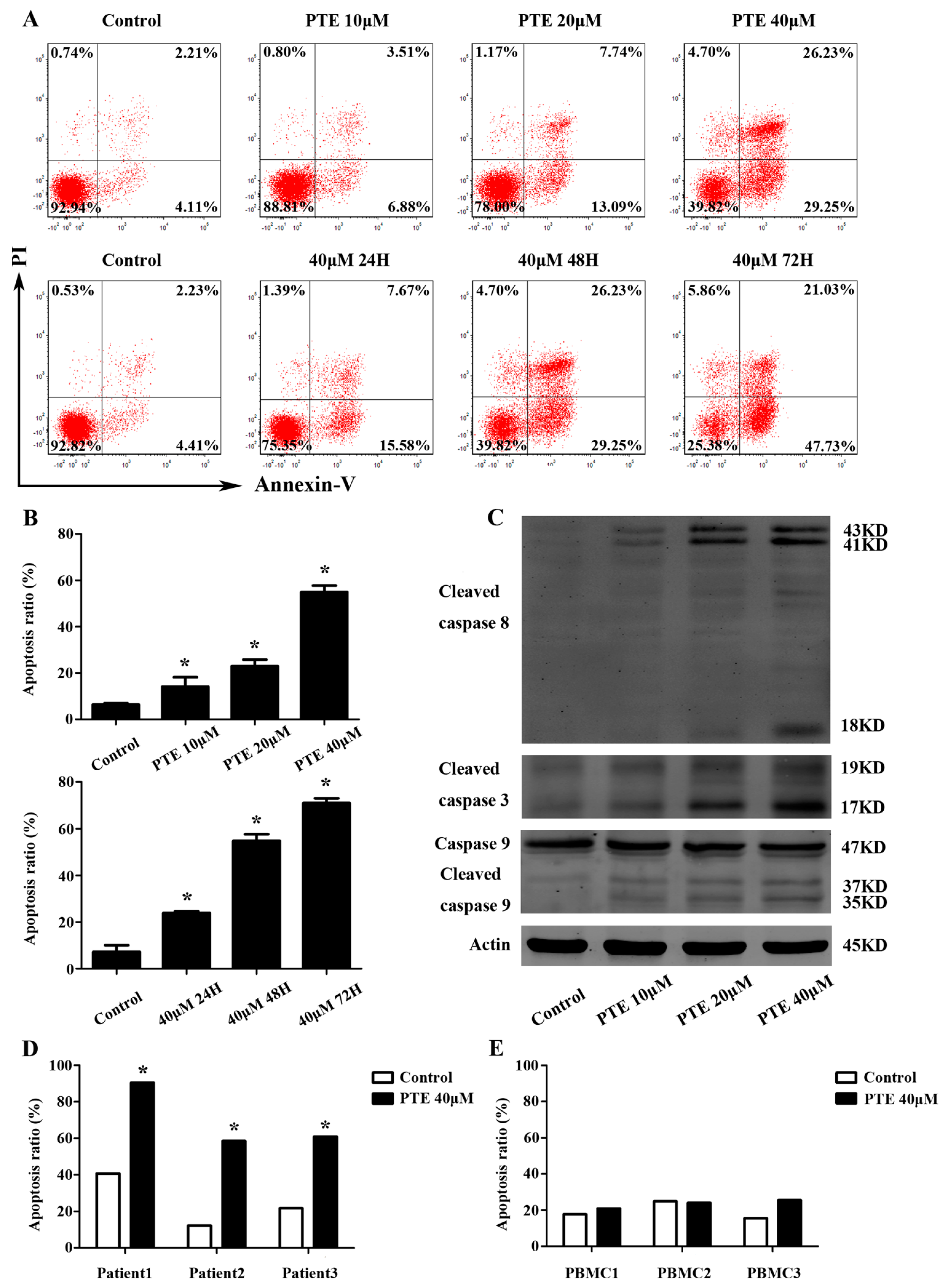
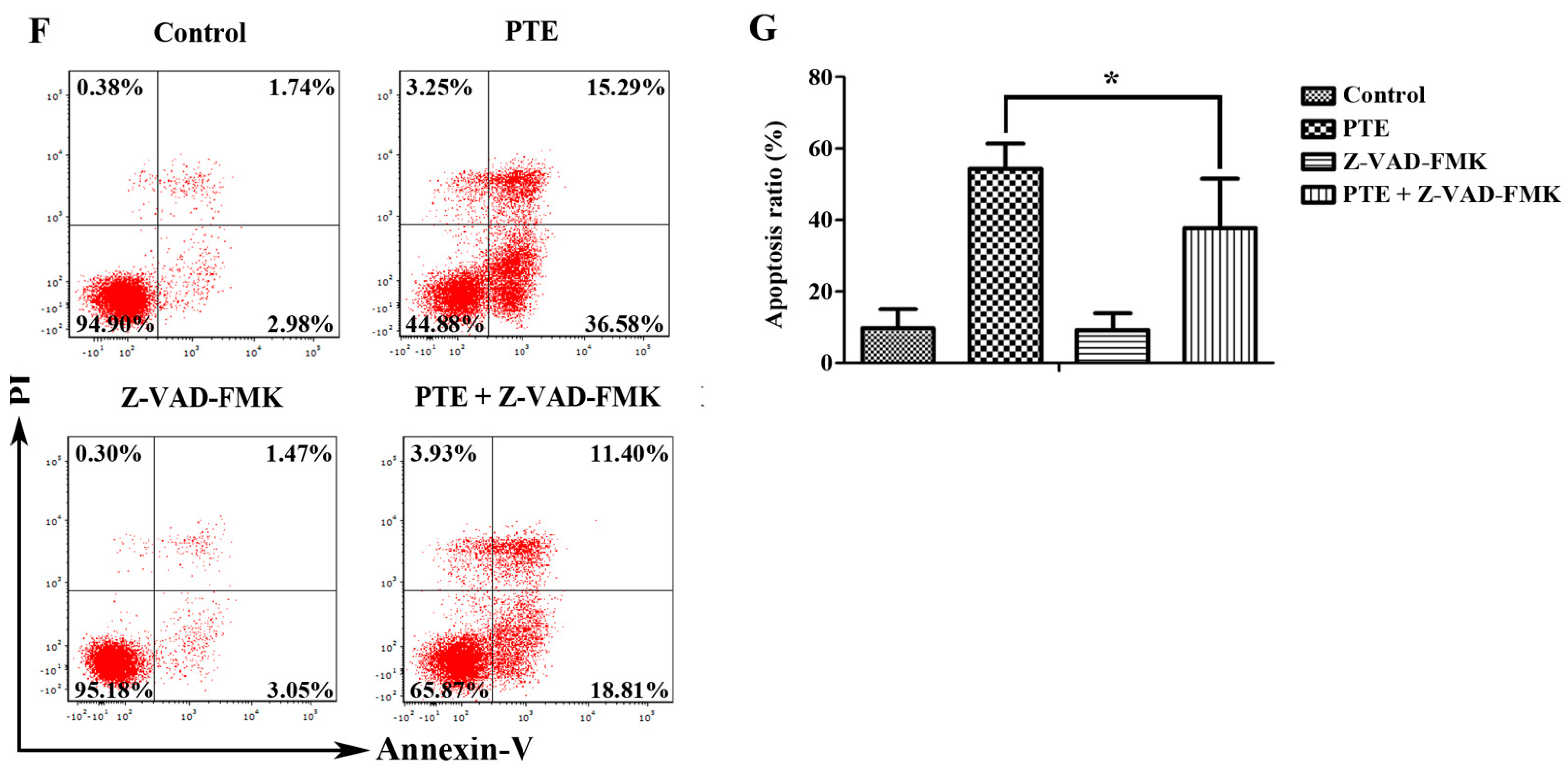
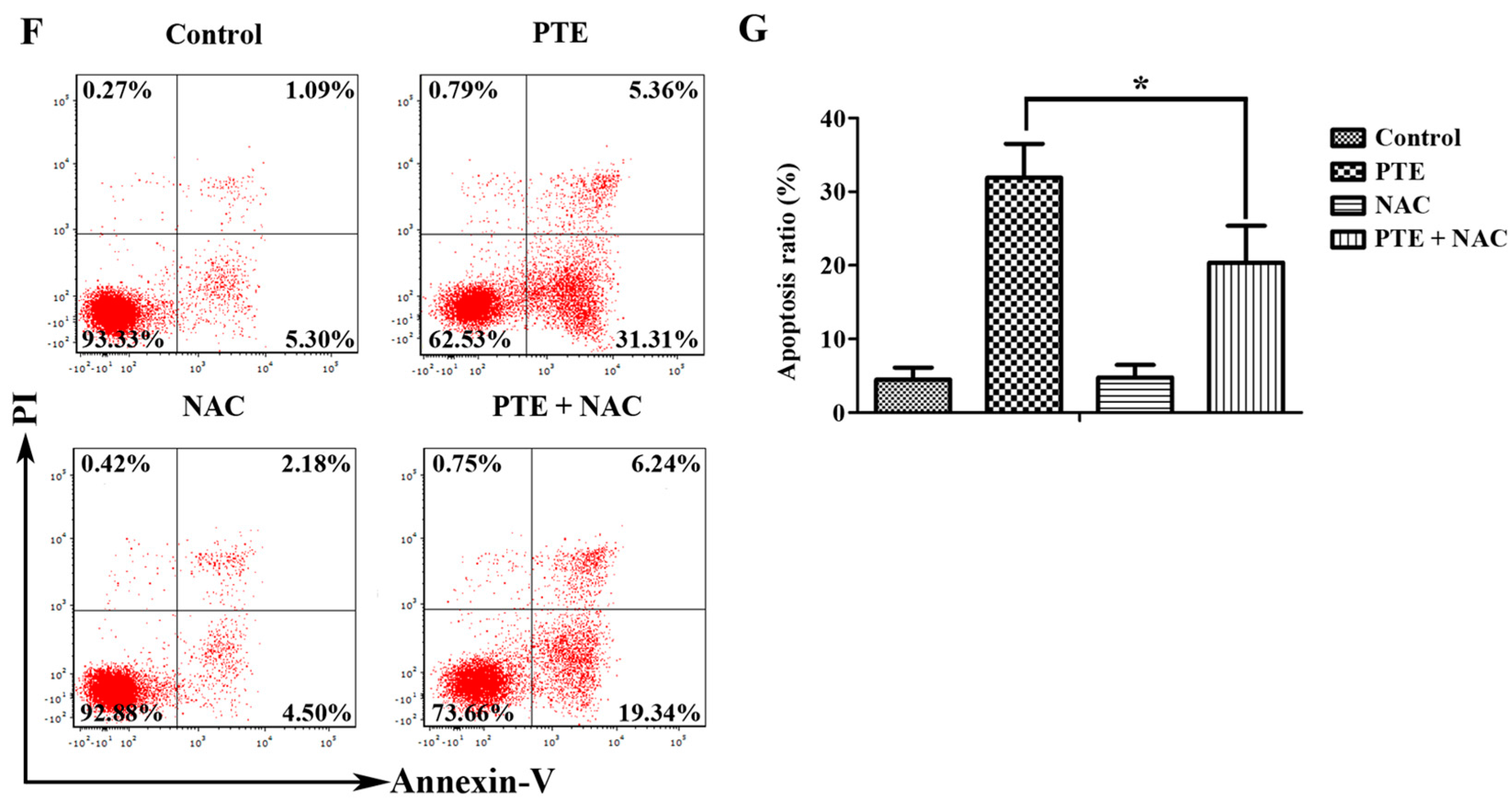
2.2. PTE Enhances Caspase Activation and Induces Apoptosis in H929 Cells
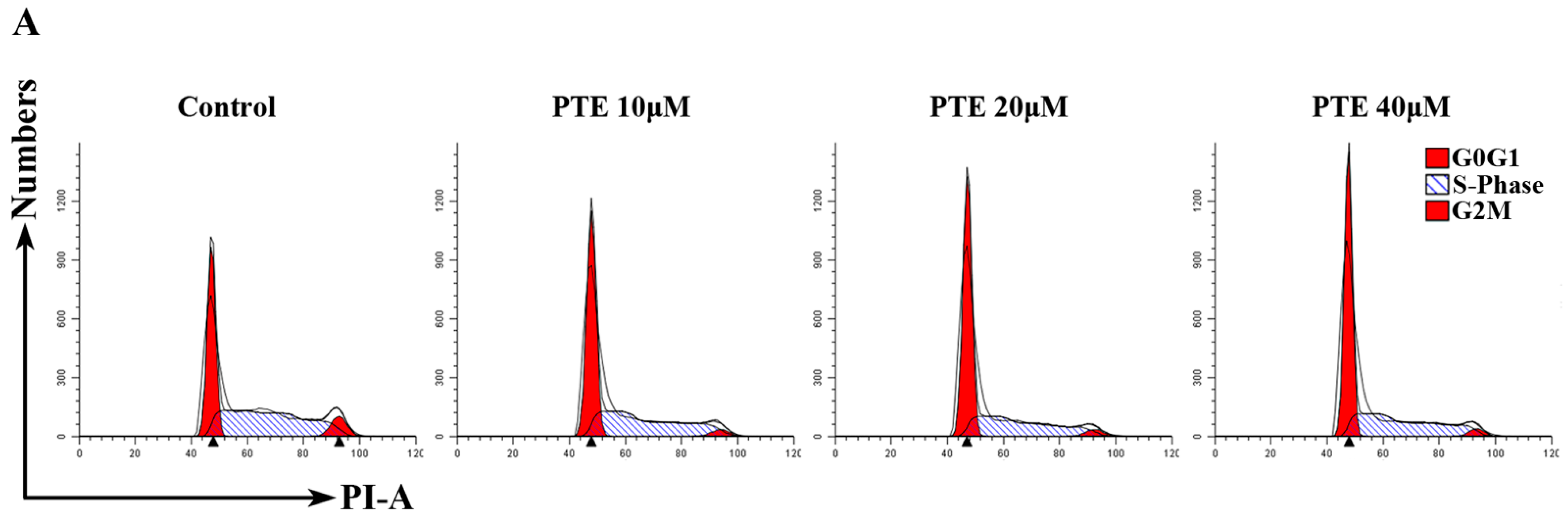
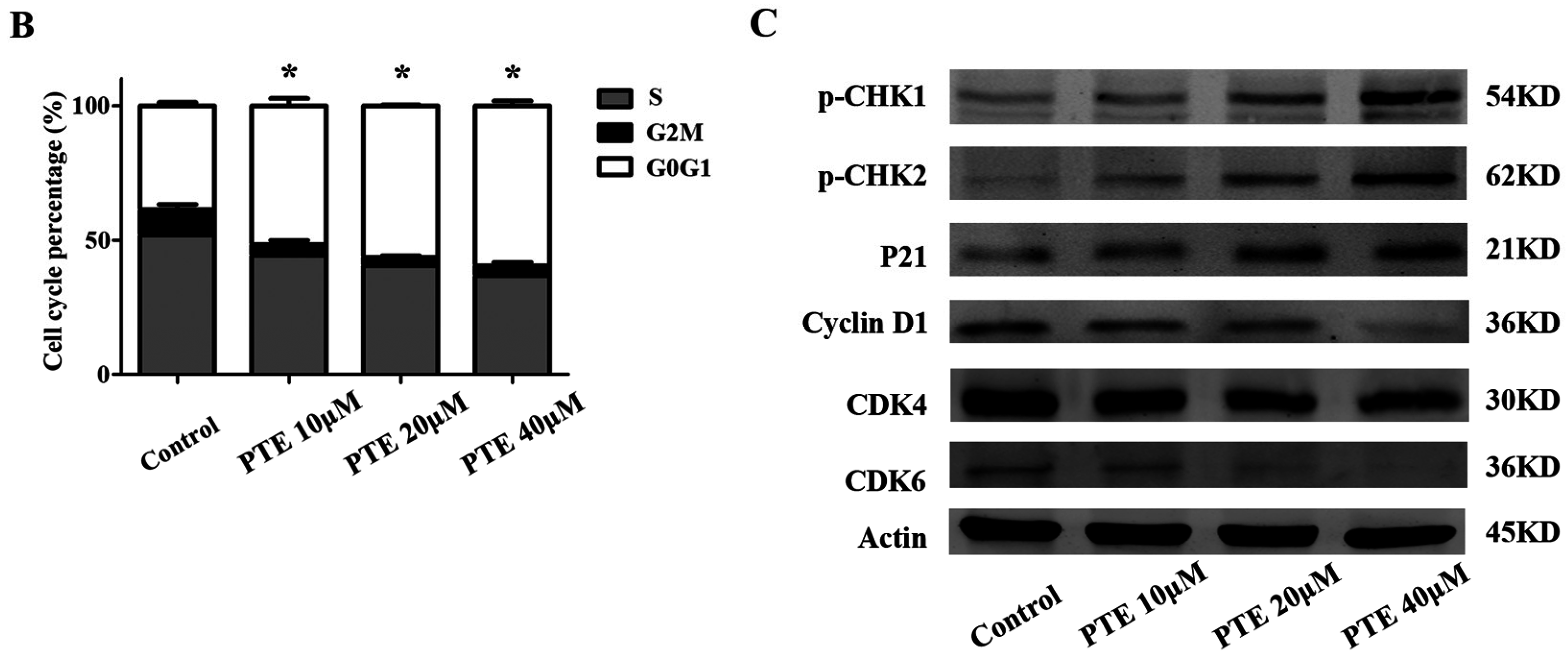
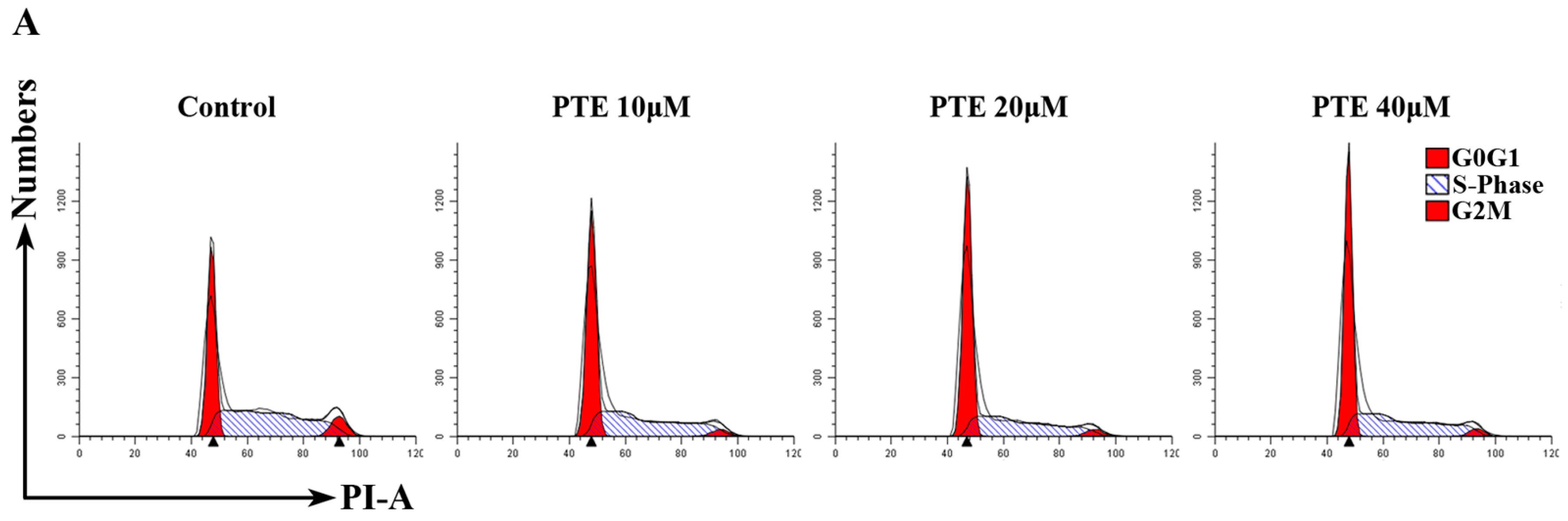
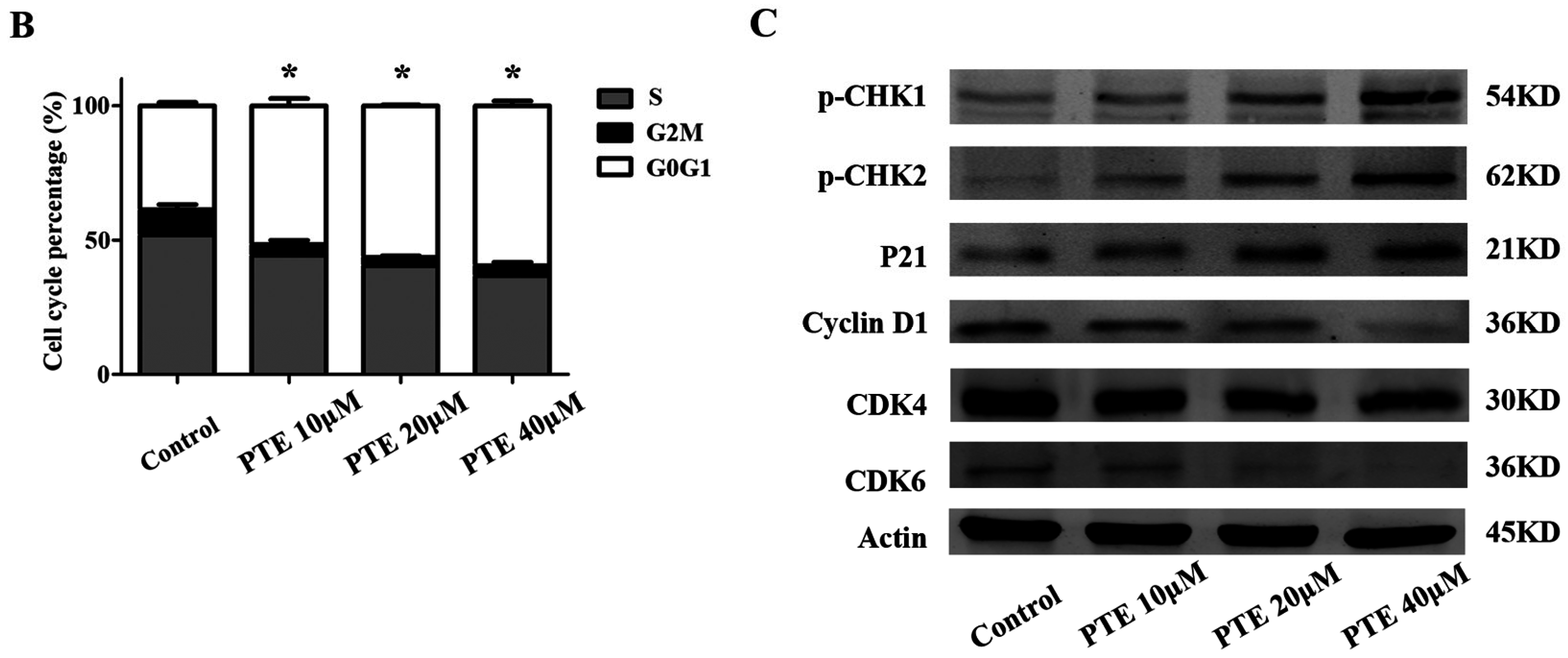
2.3. PTE Arrests the Cell Cycle at G0/G1 Phase
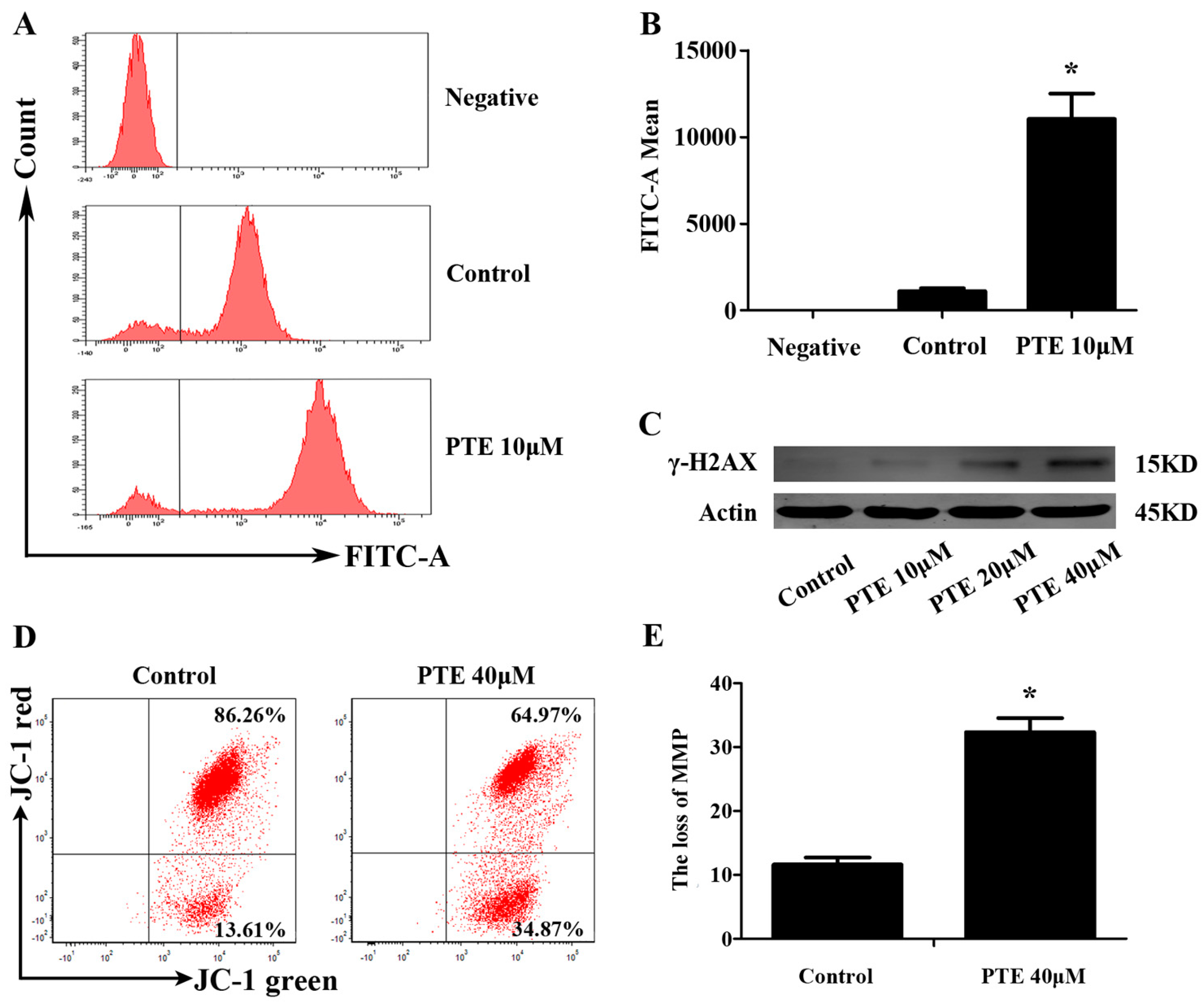
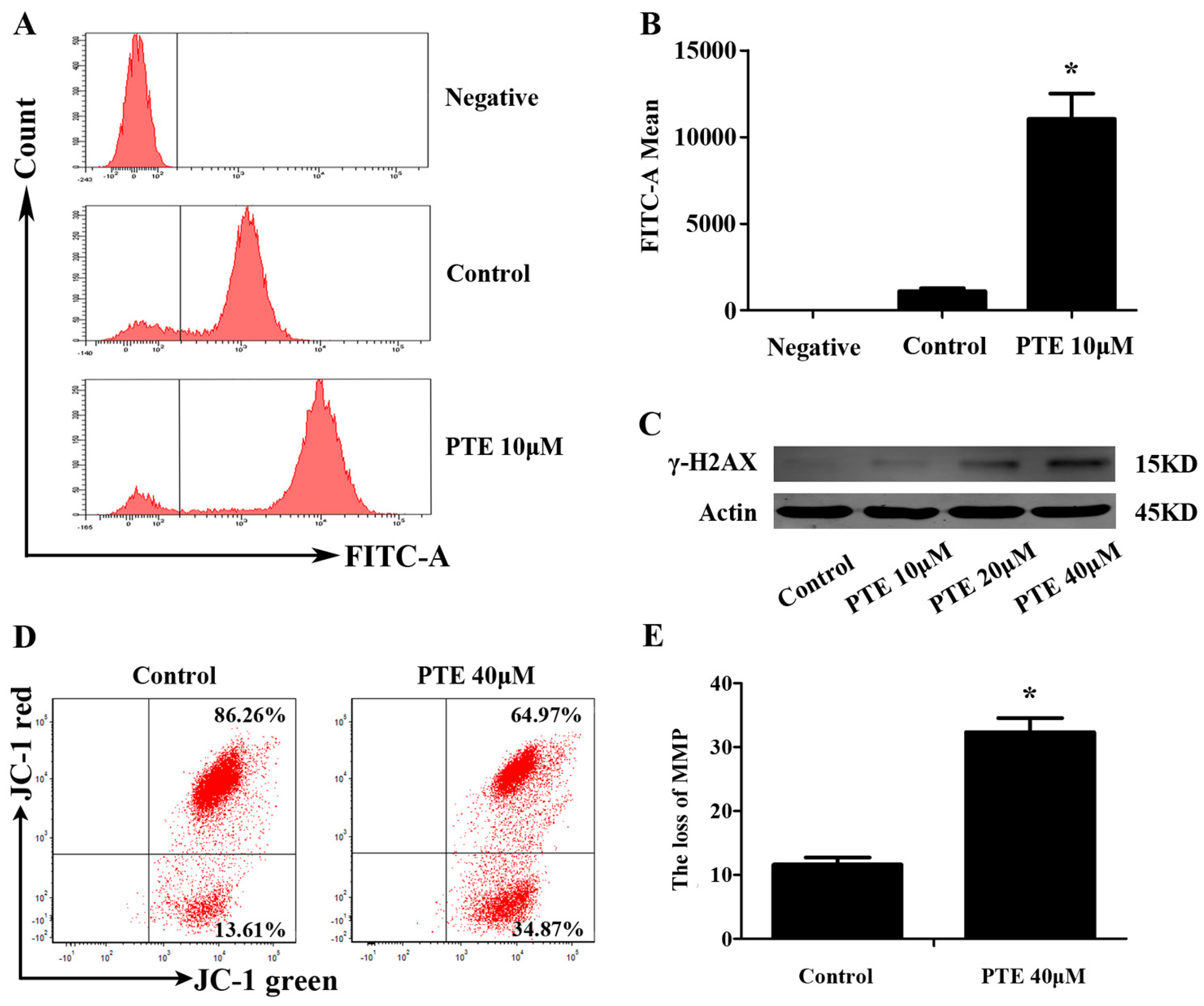
2.4. PTE Causes DNA Damage and Enhances Reactive Oxygen Species (ROS) Generation
2.5. PTE Induces the Loss of Mitochondrial Membrane Potential
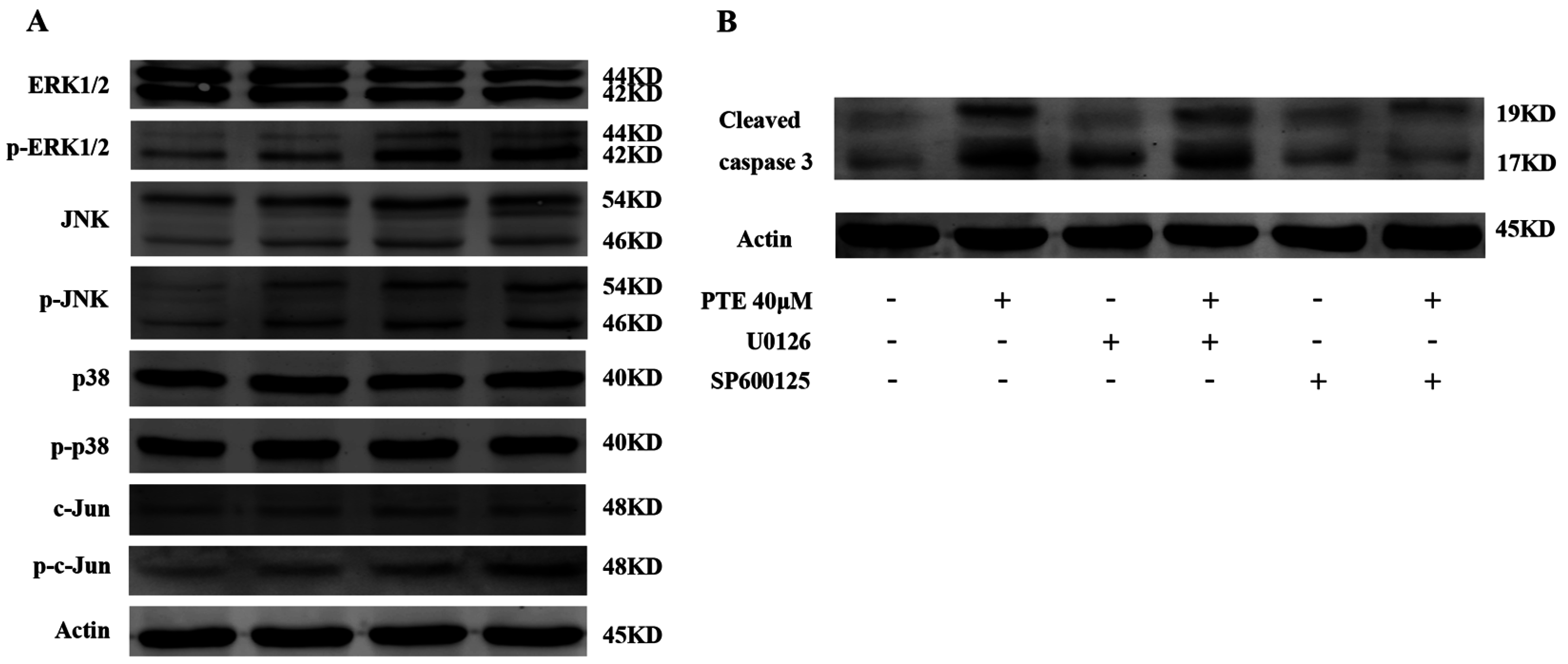
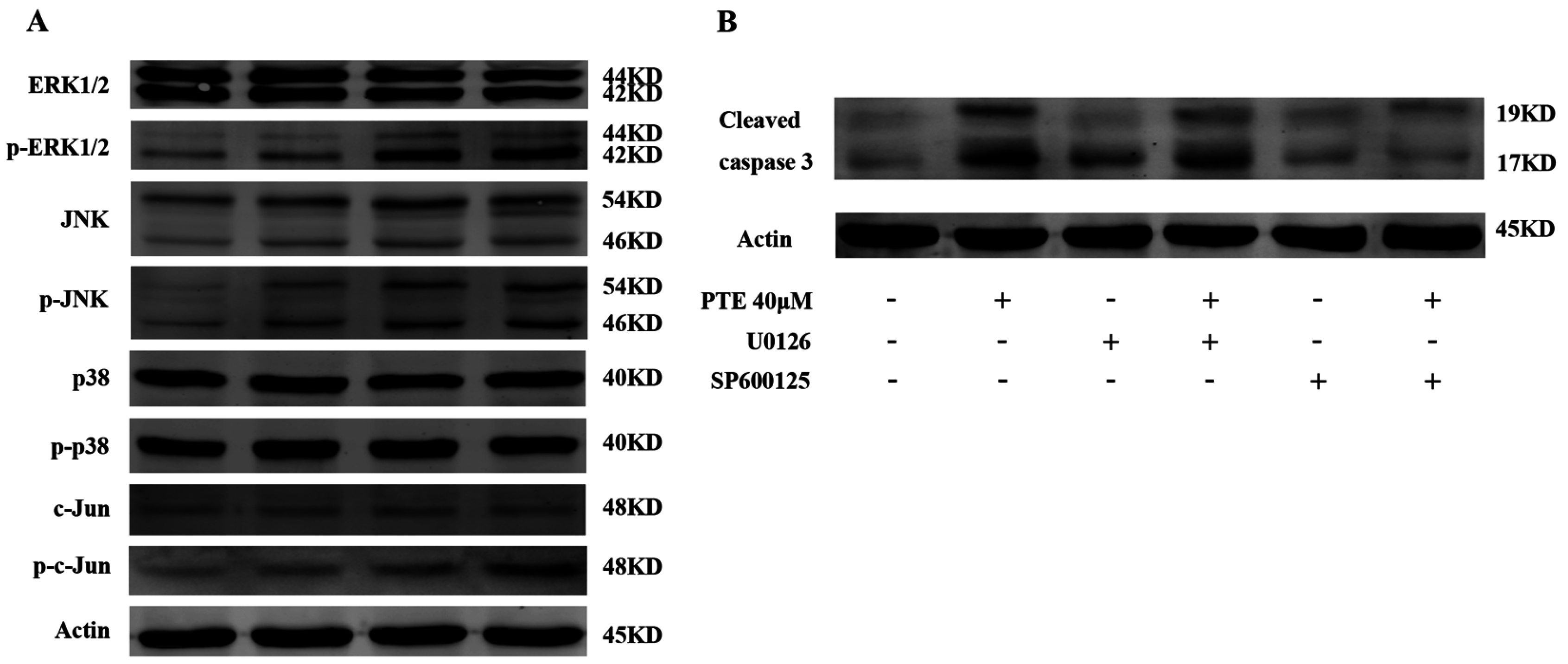
2.6. PTE Enhances the Activation of Extracellular Regulated Protein Kinases (ERK) 1/2 and c-Jun N-Terminal Kinase (JNK) Pathway
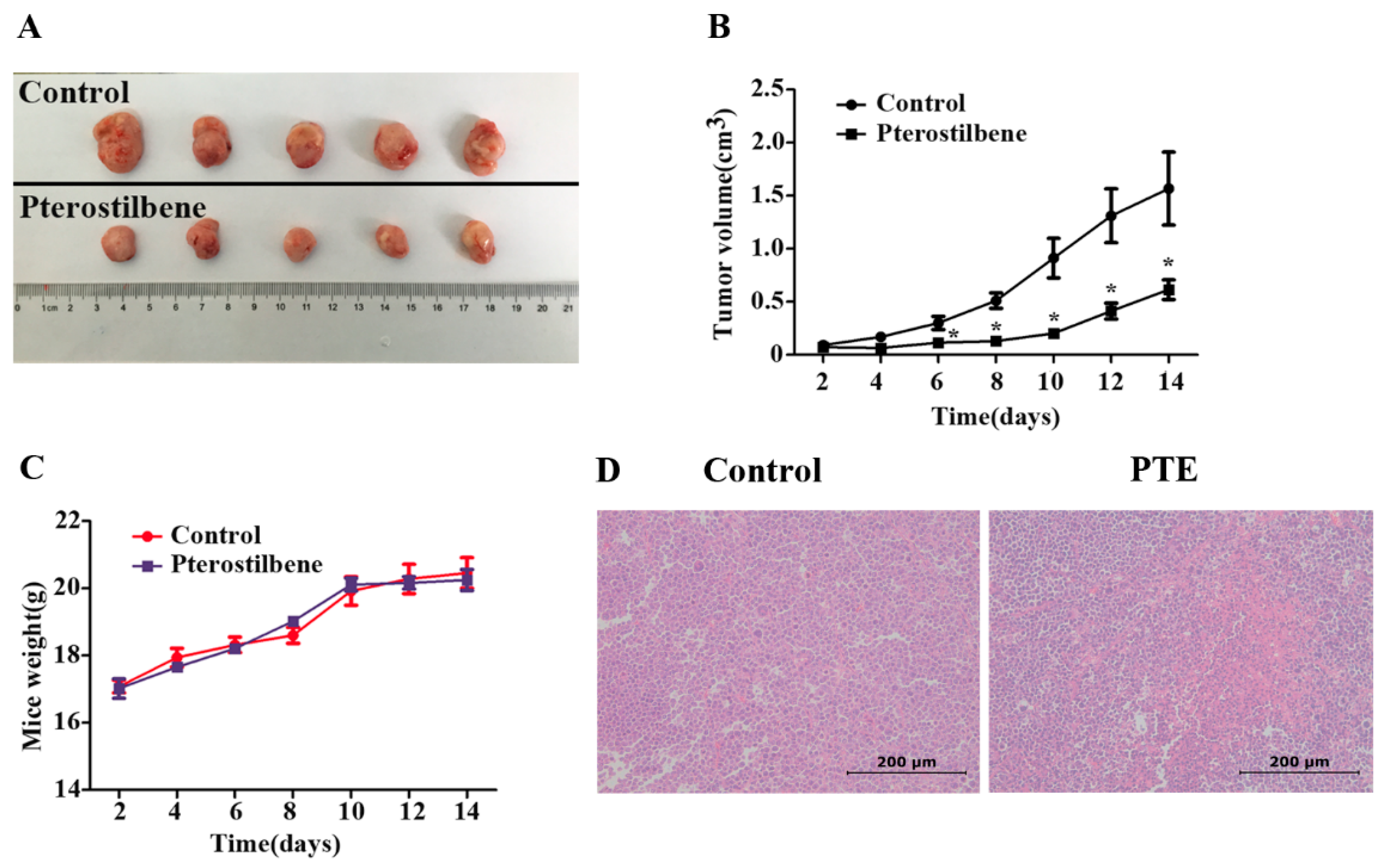
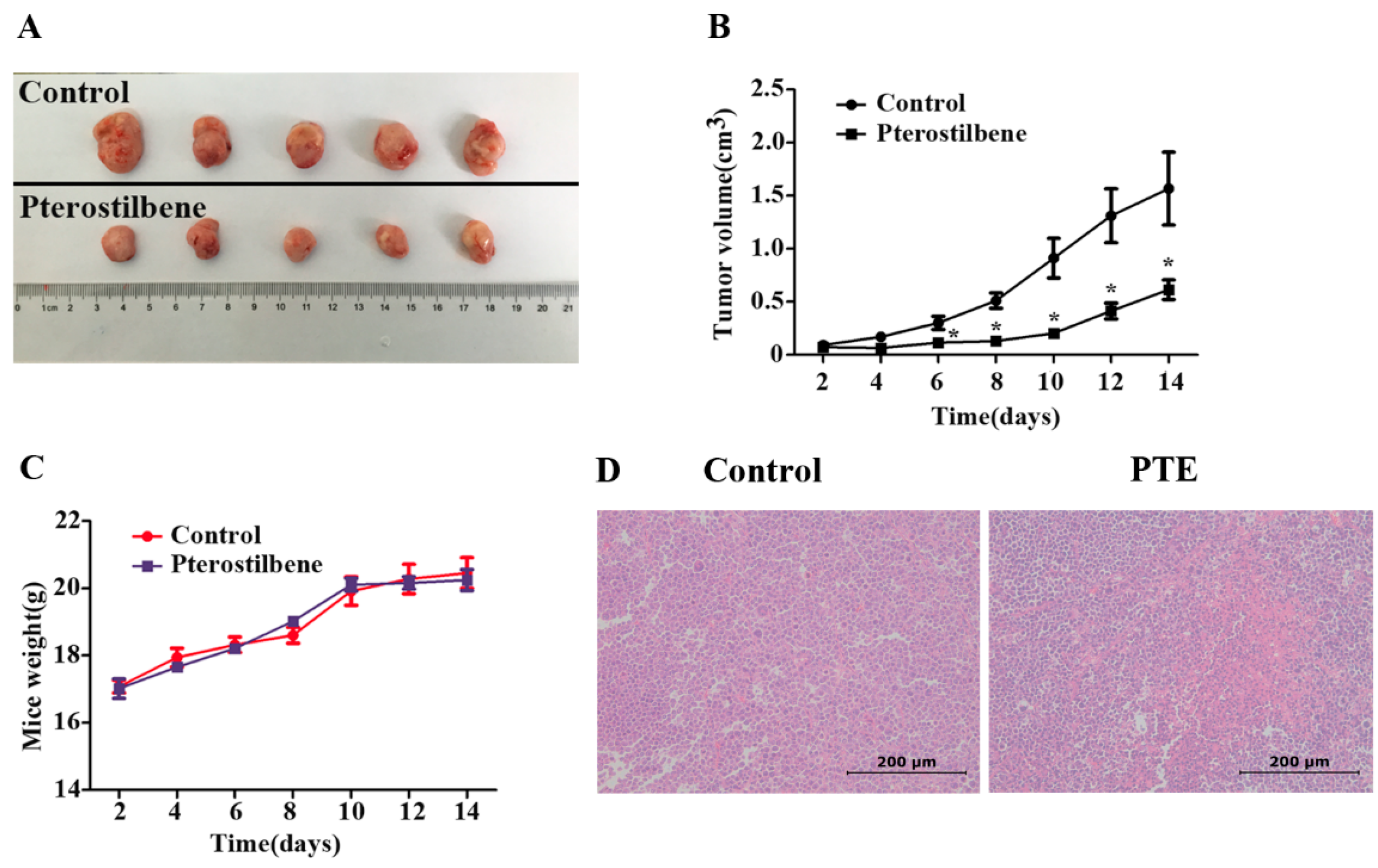
2.7. The Anti-Tumor Effect of PTE In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Proliferation Assay
4.3. Cell Apoptosis Assay
4.4. Cell Cycle Assay
4.5. Measurement of ROS Generation
4.6. JC-1 Assay
4.7. Western Blot Analysis
4.8. Tumor Xenograft Models
4.9. Pterostilbene
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, J.; Shao, L.; Wu, C.; Lu, H.; Xu, R. Hypericin-mediated photodynamic therapy induces apoptosis of myoloma SP2/0 cells depended on caspase activity in vitro. Cancer Cell Int. 2015, 15, 58. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Sethi, G.; Vadhan-Raj, S.; Bueso-Ramos, C.; Takada, Y.; Gaur, U.; Nair, A.S.; Shishodia, S.; Aggarwal, B.B. Resveratrol inhibits proliferation, induces apoptosis, and overcomes chemoresistance through down-regulation of STAT3 and nuclear factor-κB-regulated antiapoptotic and cell survival gene products in human multiple myeloma cells. Blood 2007, 109, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, W.M.; Bergsagel, P.L. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J. Clin. Investig. 2012, 122, 3456–3463. [Google Scholar] [CrossRef] [PubMed]
- Meinel, F.G.; Mandl-Weber, S.; Baumann, P.; Leban, J.; Schmidmaier, R. The Novel, Proteasome-Independent NF-κB Inhibitor V1810 Induces Apoptosis and Cell Cycle Arrest in Multiple Myeloma and Overcomes NF-κB-Mediated Drug Resistance. Mol. Cancer Ther. 2010, 9, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.Z.; Jandova, J.; Janda, J.; Vleugels, F.R.; Elliott, D.A.; Sligh, J.E. Resveratrol-sensitized UVA induced apoptosis in human keratinocytes through mitochondrial oxidative stress and pore opening. J. Photochem. Photobiol. B Biol. 2012, 113, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Barjot, C.; Tournaire, M.; Castagnino, C.; Vigor, C.; Vercauteren, J.; Rossi, J. Evaluation of antitumor effects of two vine stalk oligomers of resveratrol on a panel of lymphoid and myeloid cell lines: Comparison with resveratrol. Life Sci. 2007, 81, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.; Lin, C.; Chen, M.; Yang, S.; Chiou, H.; Hsieh, M. Pterostilbene induce autophagy on human oral cancer cells through modulation of Akt and mitogen-activated protein kinase pathway. Oral Oncol. 2015, 51, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Benlloch, M.; Obrador, E.; Valles, S.L.; Rodriguez, M.L.; Sirerol, J.A.; Alcácer, J.; Pellicer, J.A.; Salvador, R.; Cerdá, C.; Sáez, G.T.; et al. Pterostilbene Decreases the Antioxidant Defenses of Aggressive Cancer CellsIn Vivo: A Physiological Glucocorticoids- and Nrf2-Dependent Mechanism. Antioxid. Redox Signal. 2016, 24, 974–990. [Google Scholar] [CrossRef] [PubMed]
- Nikhil, K.; Sharan, S.; Roy, P. A pterostilbene derivative suppresses osteoclastogenesis by regulating RANKL-mediated NFκB and MAPK signaling in RAW264.7 cells. Pharmacol. Rep. 2015, 67, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Chan, M.L.; Chen, M.J.; Tsai, T.H.; Chen, Y.J. Modulation of macrophage polarization and lung cancer cell stemness by MUC1 and development of a related small-molecule inhibitor pterostilbene. Oncotarget 2016, 7, 39363–39375. [Google Scholar] [CrossRef] [PubMed]
- Kala, R.; Shah, H.N.; Martin, S.L.; Tollefsbol, T.O. Epigenetic-based combinatorial resveratrol and pterostilbene alters DNA damage response by affecting SIRT1 and DNMT enzyme expression, including SIRT1-dependent γ-H2AX and telomerase regulation in triple-negative breast cancer. BMC Cancer 2015, 15, 672. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Kumar, A.; Rimando, A.M.; Zhang, X.; Levenson, A.S. Resveratrol and pterostilbene epigenetically restore PTEN expression by targeting oncomiRs of the miR-17 family in prostate cancer. Oncotarget 2015, 6, 27214–27226. [Google Scholar] [CrossRef] [PubMed]
- Siedlecka-Kroplewska, K.; Jozwik, A.; Boguslawski, W.; Wozniak, M.; Zauszkiewicz-Pawlak, A.; Spodnik, J.H.; Rychlowski, M.; Kmiec, Z. Pterostilbene induces accumulation of autophagic vacuoles followed by cell death in HL60 human leukemia cells. J. Physiol. Pharmacol. 2013, 64, 545–556. [Google Scholar] [PubMed]
- Hsiao, P.C.; Chou, Y.E.; Tan, P.; Lee, W.J.; Yang, S.F.; Chow, J.M.; Chen, H.Y.; Lin, C.H.; Lee, L.M.; Chien, M.H. Pterostilbene simultaneously induced G0/G1-phase arrest and MAPK-mediated mitochondrial-derived apoptosis in human acute myeloid leukemia cell lines. PLoS ONE 2014, 9, e105342. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Yang, Y.; Fan, C.; Di, S.; Hu, W.; Jiang, S.; Li, T.; Ma, Z.; Chao, D.; Feng, X.; et al. Pterostilbene Inhibits the Growth of Human Esophageal Cancer Cells by Regulating Endoplasmic Reticulum Stress. Cell. Physiol. Biochem. 2016, 38, 1226–1244. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Lee, W.; Wu, A.T.H.; Lin, Y.; Wang, L.; Wu, C.; Yeh, C. Pterostilbene inhibits triple-negative breast cancer metastasis via inducing microRNA-205 expression and negatively modulates epithelial-to-mesenchymal transition. J. Nutr. Biochem. 2015, 26, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B.B.; Schuler, M.; Echeverri, F.; Green, D.R. Caspase-3 is the primary activator of apoptotic DNA fragmentation via DNA fragmentation factor-45/inhibitor of caspase-activated DNase inactivation. J. Biol. Chem. 1999, 274, 30651–30656. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Jin, S.; Li, Y.; Zhang, L.; Jiang, W.; Yang, C.; Du, J. Magnesium inhibits the calcification of the extracellular matrix in tendon-derived stem cells via the ATP-P2R and mitochondrial pathways. Biochem. Biophys. Res. Commun. 2016, 478, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.J.; Neuzil, J. Mitochondria as targets for cancer therapy. Mol. Nutr. Food Res. 2009, 53, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Evens, A.M.; Prachand, S.; Shi, B.; Paniaqua, M.; Gordon, L.I.; Gartenhaus, R.B. Imexon-induced apoptosis in multiple myeloma tumor cells is caspase-8 dependent. Clin. Cancer Res. 2004, 10, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.S.; Hong, S.W.; Kim, S.M.; Jin, D.H.; Shin, J.S.; Yoon, D.H.; Kim, K.P.; Lee, J.L.; Heo, D.S.; Lee, J.S.; et al. Bortezomib induces G2-M arrest in human colon cancer cells through ROS-inducible phosphorylation of ATM-CHK1. Int. J. Oncol. 2012, 41, 76–82. [Google Scholar] [PubMed]
- Dewi, N.I.; Yagasaki, K.; Miura, Y. Anti-proliferative effect of pterostilbene on rat hepatoma cells in culture. Cytotechnology 2015, 67, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Bag, A.K.; Tripathi, R.; Samanta, S.K.; Pal, B.C.; Shaha, C.; Mandal, C. Mahanine, a novel mitochondrial complex-III inhibitor induces G0/G1 arrest through redox alteration-mediated DNA damage response and regresses glioblastoma multiforme. Am. J. Cancer Res. 2014, 4, 629–647. [Google Scholar] [PubMed]
- Di Rora, A.G.; Iacobucci, I.; Imbrogno, E.; Papayannidis, C.; Derenzini, E.; Ferrari, A.; Guadagnuolo, V.; Robustelli, V.; Parisi, S.; Sartor, C.; et al. Prexasertib, a Chk1/Chk2 inhibitor, increases the effectiveness of conventional therapy in B-/T- cell progenitor acute lymphoblastic leukemia. Oncotarget 2016, 7, 53377–53391. [Google Scholar]
- Prince, E.W.; Balakrishnan, I.; Shah, M.; Mulcahy, L.J.; Griesinger, A.M.; Alimova, I.; Harris, P.S.; Birks, D.K.; Donson, A.M.; Davidson, N.; et al. Checkpoint kinase 1 expression is an adverse prognostic marker and therapeutic target in MYC-driven medulloblastoma. Oncotarget 2016, 7, 53881–53894. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Seah, S.; Loh, X.; Chan, C.W.; Hartman, M.; Goh, B.C.; Lee, S.C. Simvastatin-induced breast cancer cell death and deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by metabolic products of the mevalonate pathway. Oncotarget 2016, 7, 2532–2544. [Google Scholar] [PubMed]
- Ahronian, L.G.; Sennott, E.M.; van Allen, E.M.; Wagle, N.; Kwak, E.L.; Faris, J.E.; Godfrey, J.T.; Nishimura, K.; Lynch, K.D.; Mermel, C.H.; et al. Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer Discov. 2015, 5, 358–367. [Google Scholar] [CrossRef] [PubMed]
- McCormack, D.E.; Mannal, P.; McDonald, D.; Tighe, S.; Hanson, J.; McFadden, D. Genomic analysis of pterostilbene predicts its antiproliferative effects against pancreatic cancer in vitro and in vivo. J. Gastrointest. Surg. 2012, 16, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Dias, S.J.; Rimando, A.M.; Dhar, S.; Mizuno, C.S.; Penman, A.D.; Lewin, J.R.; Levenson, A.S. Pterostilbene acts through metastasis-associated protein 1 to inhibit tumor growth, progression and metastasis in prostate cancer. PLoS ONE 2013, 8, e57542. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yan, X.; Duan, W.; Yan, J.; Yi, W.; Liang, Z.; Wang, N.; Li, Y.; Chen, W.; Yu, S.; et al. Pterostilbene exerts antitumor activity via the Notch1 signaling pathway in human lung adenocarcinoma cells. PLoS ONE 2013, 8, e62652. [Google Scholar] [CrossRef] [PubMed]
- Ernst, T.J.; Gazdar, A.; Ritz, J.; Shipp, M.A. Identification of a second transforming gene, rasn, in a human multiple myeloma line with a rearranged C-MYC allele. Blood 1988, 72, 1163–1167. [Google Scholar] [PubMed]
- Bellamy, W.T.; Dalton, W.S.; Gleason, M.C.; Grogan, T.M.; Trent, J.M. Development and characterization of a melphalan-resistant human multiple myeloma cell line. Cancer Res. 1991, 51, 995–1002. [Google Scholar] [PubMed]
- Wang, W. PC Cell-Derived Growth Factor Confers Resistance to Dexamethasone and Promotes Tumorigenesis in Human Multiple Myeloma. Clin. Cancer Res. 2006, 12, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Wandl, U.; Hoang, T.; Minden, M.; Messner, H.A. Segregation of precursors with high proliferative potential from their differentiated progeny in a myeloma cell line. J. Cell. Physiol. 1988, 136, 384–388. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, B.; Xu, Z.; Hu, L.; Chen, G.; Wei, R.; Yang, G.; Li, B.; Chang, G.; Sun, X.; Wu, H.; et al. Pterostilbene Inhibits Human Multiple Myeloma Cells via ERK1/2 and JNK Pathway In Vitro and In Vivo. Int. J. Mol. Sci. 2016, 17, 1927. https://doi.org/10.3390/ijms17111927
Xie B, Xu Z, Hu L, Chen G, Wei R, Yang G, Li B, Chang G, Sun X, Wu H, et al. Pterostilbene Inhibits Human Multiple Myeloma Cells via ERK1/2 and JNK Pathway In Vitro and In Vivo. International Journal of Molecular Sciences. 2016; 17(11):1927. https://doi.org/10.3390/ijms17111927
Chicago/Turabian StyleXie, Bingqian, Zhijian Xu, Liangning Hu, Gege Chen, Rong Wei, Guang Yang, Bo Li, Gaomei Chang, Xi Sun, Huiqun Wu, and et al. 2016. "Pterostilbene Inhibits Human Multiple Myeloma Cells via ERK1/2 and JNK Pathway In Vitro and In Vivo" International Journal of Molecular Sciences 17, no. 11: 1927. https://doi.org/10.3390/ijms17111927
APA StyleXie, B., Xu, Z., Hu, L., Chen, G., Wei, R., Yang, G., Li, B., Chang, G., Sun, X., Wu, H., Zhang, Y., Dai, B., Tao, Y., Shi, J., & Zhu, W. (2016). Pterostilbene Inhibits Human Multiple Myeloma Cells via ERK1/2 and JNK Pathway In Vitro and In Vivo. International Journal of Molecular Sciences, 17(11), 1927. https://doi.org/10.3390/ijms17111927