
Tension in Cancer


Abstract
:1. Introduction
2. Integrins
2.1. Structure and Ligand Affinity of Integrins
2.2. Integrin-Mediated Adhesion and Physical Forces
3. Interfering Integrin-ECM Interactions as a Potential Therapeutic Strategy
4. Changing the Tension in Cancer
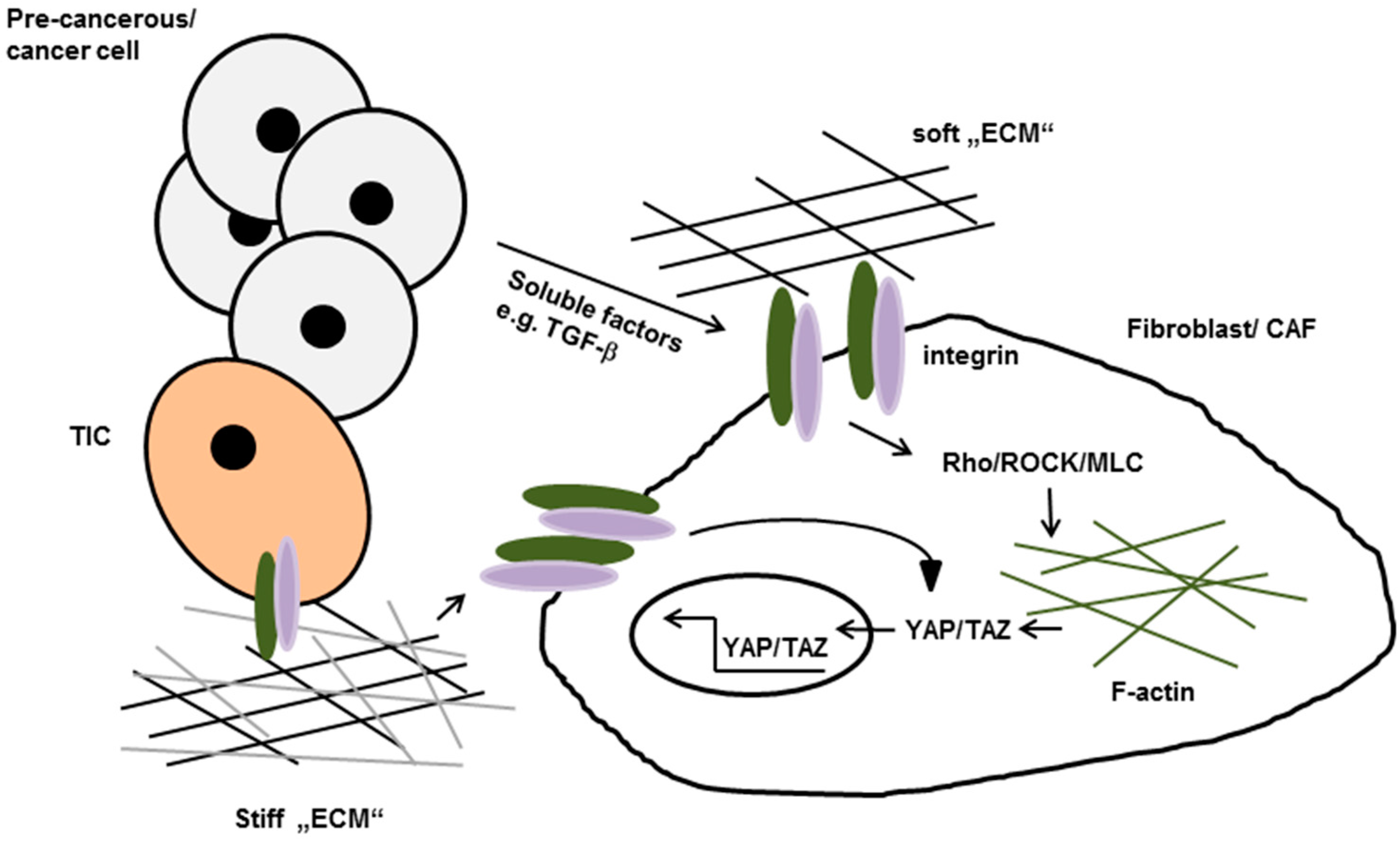
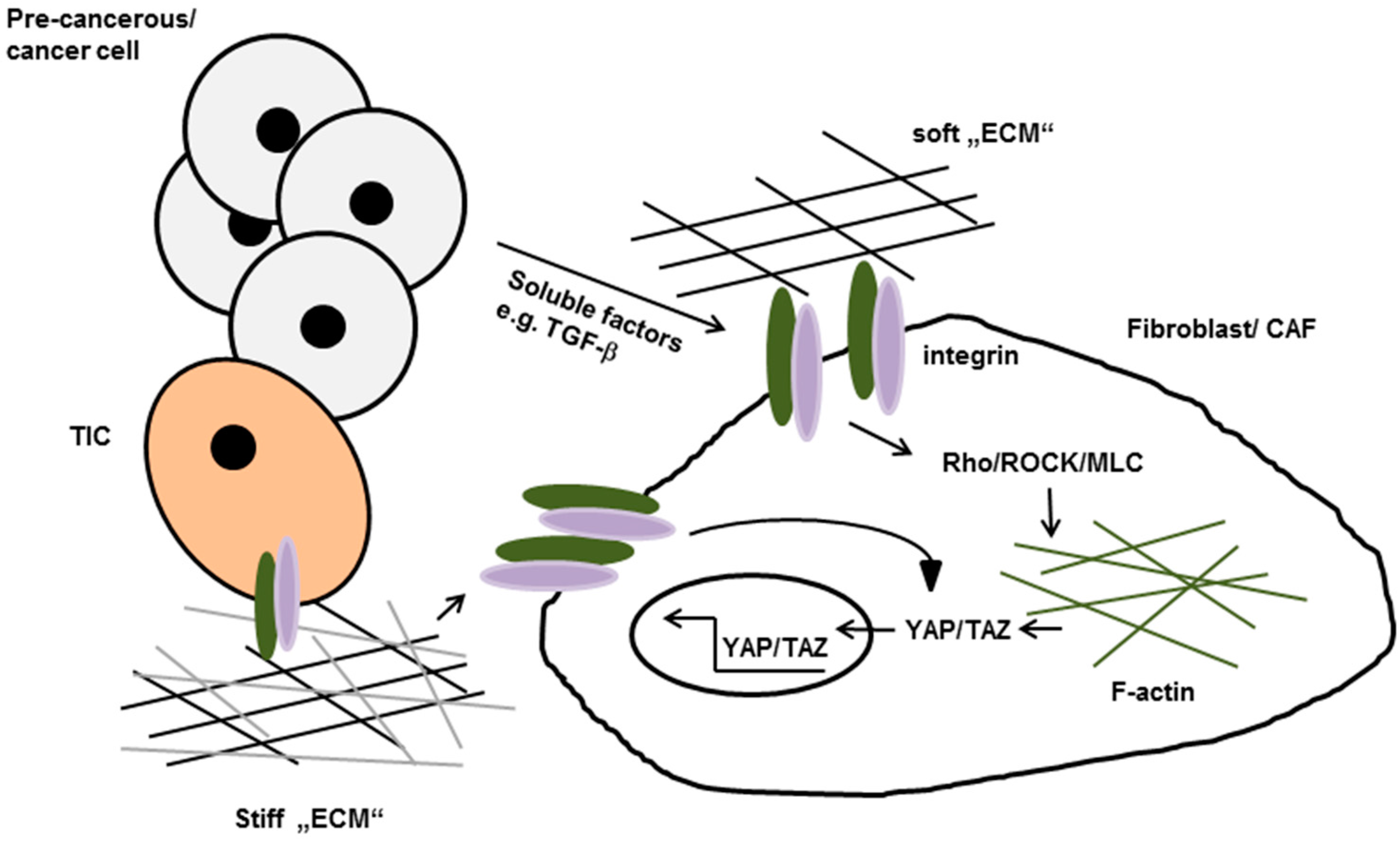
4.1. Stroma-Driven, Integrin-Mediated Neoplastic Transformation
4.2. Matrix-Stiffening Regulates Malignancy by Enhancing Integrin-Dependent Mechano-Transduction
4.3. Matrix-Stiffening Induced Integrin-Signalling Drives Tumor Therapy Resistance
5. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tamkun, J.W.; DeSimone, D.W.; Fonda, D.; Patel, R.S.; Buck, C.; Horwitz, A.F.; Hynes, R.O. Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell 1986, 46, 271–282. [Google Scholar] [CrossRef]
- Brooks, P.C.; Clark, R.A.; Cheresh, D.A. Requirement of vascular integrin αvβ3 for angiogenesis. Science 1994, 264, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.J.; Cheresh, D.A.; Little, C.D. An antagonist of integrin αvβ3 prevents maturation of blood vessels during embryonic neovascularization. J. Cell Sci. 1995, 108, 2655–2661. [Google Scholar] [PubMed]
- Leavesley, D.I.; Schwartz, M.A.; Rosenfeld, M.; Cheresh, D.A. Integrin β1- and β3-mediated endothelial cell migration is triggered through distinct signaling mechanisms. J. Cell Biol. 1993, 121, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Bu, X.Y.; Khankaldyyan, V.; Gonzales-Gomez, I.; McComb, J.G.; Laug, W.E. Effect of the angiogenesis inhibitor Cilengitide (EMD 121974) on glioblastoma growth in nude mice. Neurosurgery 2006, 59, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Arnaout, M.A.; Goodman, S.L.; Xiong, J.P. Coming to grips with integrin binding to ligands. Curr. Opin. Cell Biol. 2002, 14, 641–651. [Google Scholar] [CrossRef]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Carman, C.V.; Springer, T.A. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 2003, 301, 1720–1725. [Google Scholar] [CrossRef] [PubMed]
- Mould, A.P.; Humphries, M.J. Regulation of integrin function through conformational complexity: Not simply a knee-jerk reaction? Curr. Opin. Cell Biol. 2004, 16, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Xie, C.; Shimaoka, M.; Cheng, Y.; Walz, T.; Springer, T.A. Activation of leukocyte β2 integrins by conversion from bent to extended conformations. Immunity 2006, 25, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Shimaoka, M.; Lu, C.; Salas, A.; Xiao, T.; Takagi, J.; Springer, T.A. Stabilizing the integrin α M inserted domain in alternative conformations with a range of engineered disulfide bonds. Proc. Natl. Acad. Sci. USA 2002, 99, 16737–16741. [Google Scholar] [CrossRef] [PubMed]
- Takagi, J.; Petre, B.M.; Walz, T.; Springer, T.A. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 2002, 110, 599–611. [Google Scholar] [CrossRef]
- Zhu, J.; Boylan, B.; Luo, B.H.; Newman, P.J.; Springer, T.A. Tests of the extension and deadbolt models of integrin activation. J. Biol. Chem. 2007, 282, 11914–11920. [Google Scholar] [CrossRef] [PubMed]
- Humphries, M.J.; McEwan, P.A.; Barton, S.J.; Buckley, P.A.; Bella, J.; Mould, A.P. Integrin structure: Heady advances in ligand binding, but activation still makes the knees wobble. Trends Biochem. Sci. 2003, 28, 313–320. [Google Scholar] [CrossRef]
- Luo, B.H.; Springer, T.A. Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 2006, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Walko, G.; Castanon, M.J.; Wiche, G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2015, 360, 529–544. [Google Scholar] [CrossRef] [PubMed]
- Nishie, W.; Sawamura, D.; Goto, M.; Ito, K.; Shibaki, A.; McMillan, J.R.; Sakai, K.; Nakamura, H.; Olasz, E.; Yancey, K.B.; et al. Humanization of autoantigen. Nat. Med. 2007, 13, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Pulkkinen, L.; Uitto, J. Hemidesmosomal variants of epidermolysis bullosa. Mutations in the α6β4 integrin and the 180-kD bullous pemphigoid antigen/type XVII collagen genes. Exp. Dermatol. 1998, 7, 46–64. [Google Scholar] [CrossRef] [PubMed]
- Raymond, K.; Kreft, M.; Janssen, H.; Calafat, J.; Sonnenberg, A. Keratinocytes display normal proliferation, survival and differentiation in conditional β4-integrin knockout mice. J. Cell Sci. 2005, 118, 1045–1060. [Google Scholar] [CrossRef] [PubMed]
- Uitto, J. Dohi memorial lecture. Clinical implications of basic research on heritable skin diseases. J. Dermatol. 1997, 24, 690–700. [Google Scholar] [CrossRef] [PubMed]
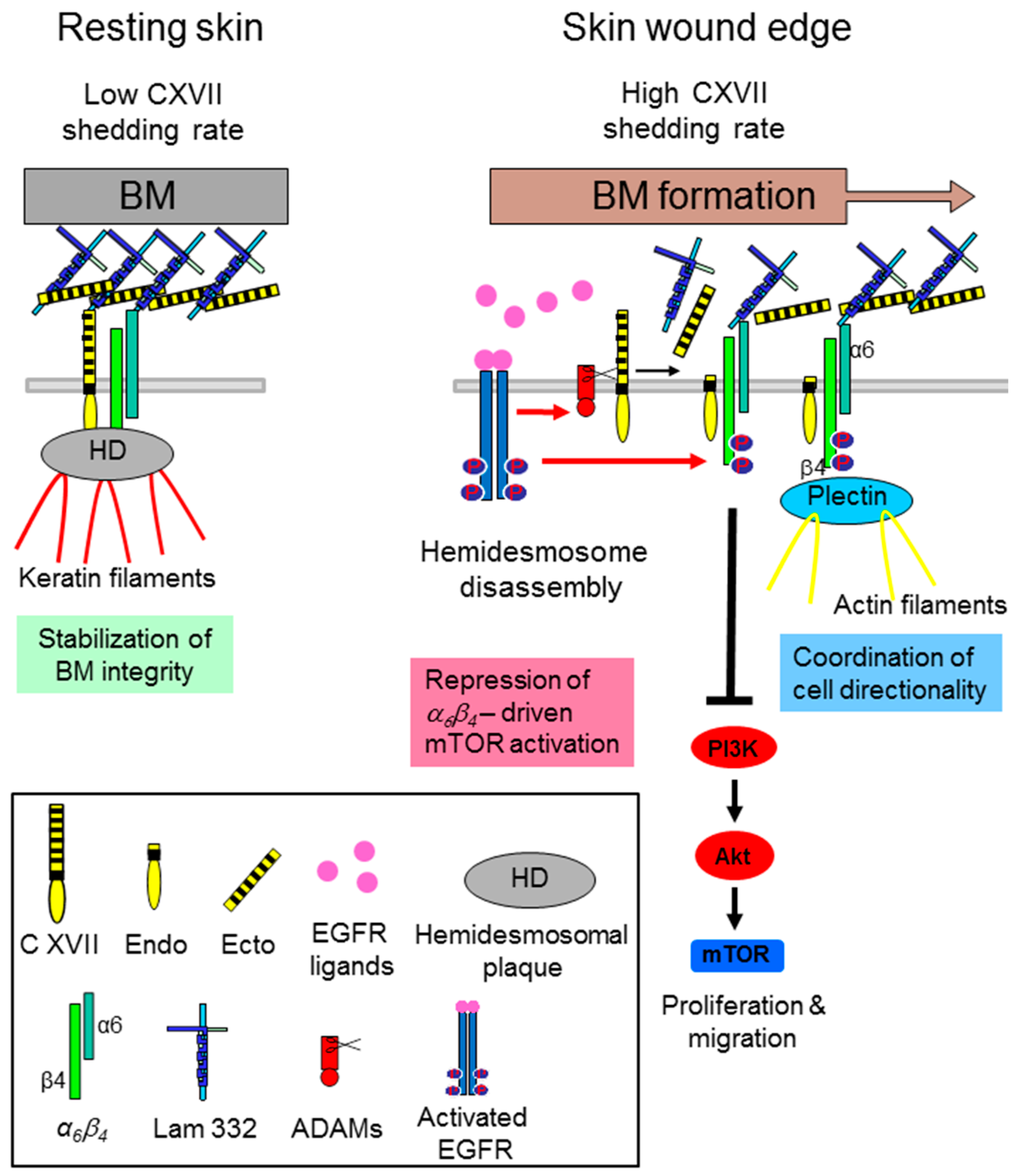
- Jackow, J.; Löffek, S.; Nystrom, A.; Bruckner-Tuderman, L.; Franzke, C.W. Collagen XVII Shedding suppresses re-epithelialization by directing keratinocyte migration and dampening mTOR signaling. J. Investig. Dermatol. 2016, 136, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Jackow, J.; Schlosser, A.; Sormunen, R.; Nystrom, A.; Sitaru, C.; Tasanen, K.; Bruckner-Tuderman, L.; Franzke, C.W. Generation of a functional non-shedding collagen XVII mouse model: Relevance of collagen XVII shedding in wound healing. J. Investig. Dermatol. 2016, 136, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Underwood, R.A.; Carter, W.G.; Usui, M.L.; Olerud, J.E. Ultrastructural localization of integrin subunits β 4 and α3 within the migrating epithelial tongue of in vivo human wounds. J. Histochem. Cytochem. 2009, 57, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Löffek, S.; Hurskainen, T.; Jackow, J.; Sigloch, F.C.; Schilling, O.; Tasanen, K.; Bruckner-Tuderman, L.; Franzke, C.W. Transmembrane collagen XVII modulates integrin dependent keratinocyte migration via PI3K/Rac1 signaling. PLoS ONE 2014, 9, e87263. [Google Scholar] [CrossRef] [PubMed]
- Margadant, C.; Frijns, E.; Wilhelmsen, K.; Sonnenberg, A. Regulation of hemidesmosome disassembly by growth factor receptors. Curr. Opin. Cell Biol. 2008, 20, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Parikka, M.; Nissinen, L.; Kainulainen, T.; Bruckner-Tuderman, L.; Salo, T.; Heino, J.; Tasanen, K. Collagen XVII promotes integrin-mediated squamous cell carcinoma transmigration—A novel role for αIIb integrin and tirofiban. Exp. Cell Res. 2006, 312, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Rousselle, P.; Nokelainen, P.; Tallapragada, S.; Nguyen, N.T.; Fincher, E.F.; Marinkovich, M.P. Targeting a tumor-specific laminin domain critical for human carcinogenesis. Cancer Res. 2008, 68, 2885–2894. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.E.; Carter, W.G. Laminin 5 deposition regulates keratinocyte polarization and persistent migration. J. Cell Sci. 2004, 117, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Pullar, C.E.; Baier, B.S.; Kariya, Y.; Russell, A.J.; Horst, B.A.; Marinkovich, M.P.; Isseroff, R.R. β4 integrin and epidermal growth factor coordinately regulate electric field-mediated directional migration via Rac1. Mol. Biol. Cell 2006, 17, 4925–4935. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, B.U.; DeBiase, P.J.; Matzno, S.; Chew, T.L.; Claiborne, J.N.; Hopkinson, S.B.; Russell, A.; Marinkovich, M.P.; Jones, J.C. Integrin β4 regulates migratory behavior of keratinocytes by determining laminin-332 organization. J. Biol. Chem. 2006, 281, 35487–35498. [Google Scholar] [CrossRef] [PubMed]
- Tasanen, K.; Tunggal, L.; Chometon, G.; Bruckner-Tuderman, L.; Aumailley, M. Keratinocytes from patients lacking collagen XVII display a migratory phenotype. Am. J. Pathol. 2004, 164, 2027–2038. [Google Scholar] [CrossRef]
- Theodosiou, M.; Widmaier, M.; Bottcher, R.T.; Rognoni, E.; Veelders, M.; Bharadwaj, M.; Lambacher, A.; Austen, K.; Muller, D.J.; Zent, R.; et al. Kindlin-2 cooperates with talin to activate integrins and induces cell spreading by directly binding paxillin. eLife 2016, 5, e10130. [Google Scholar] [CrossRef] [PubMed]
- Ciobanasu, C.; Faivre, B.; Le, C.C. Integrating actin dynamics, mechanotransduction and integrin activation: The multiple functions of actin binding proteins in focal adhesions. Eur. J. Cell Biol. 2013, 92, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Pellegrin, S.; Mellor, H. Actin stress fibers. J. Cell Sci. 2007, 120, 3491–3499. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le, D.J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Elbediwy, A.; Vincent-Mistiaen, Z.I.; Spencer-Dene, B.; Stone, R.K.; Boeing, S.; Wculek, S.K.; Cordero, J.; Tan, E.H.; Ridgway, R.; Brunton, V.G.; et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 2016, 143, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Frijns, E.; Sachs, N.; Kreft, M.; Wilhelmsen, K.; Sonnenberg, A. EGF-induced MAPK signaling inhibits hemidesmosome formation through phosphorylation of the integrin β4. J. Biol. Chem. 2010, 285, 37650–37662. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitz, I.; Tsomo, L.; Mercurio, A.M. Protein kinase C-α phosphorylation of specific serines in the connecting segment of the β4 integrin regulates the dynamics of type II hemidesmosomes. Mol. Cell. Biol. 2004, 24, 4351–4360. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsen, K.; Litjens, S.H.; Kuikman, I.; Margadant, C.; van Rheenen, J.; Sonnenberg, A. Serine phosphorylation of the integrin β4 subunit is necessary for epidermal growth factor receptor induced hemidesmosome disruption. Mol. Biol. Cell 2007, 18, 3512–3522. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.M.; Rabinovitz, I.; Wang, H.H.; Toker, A.; Mercurio, A.M. Activation of phosphoinositide 3-OH kinase by the α6β4 integrin promotes carcinoma invasion. Cell 1997, 91, 949–960. [Google Scholar] [CrossRef]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An integrin β3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, A.M.; Verhoef, E.I.; Roobol, M.J.; Schroder, F.H.; Wildhagen, M.F.; van der Kwast, T.H.; Jenster, G.; van Leenders, G.J. Validation of stem cell markers in clinical prostate cancer: α6-integrin is predictive for non-aggressive disease. Prostate 2014, 74, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; MacSwords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin α6 regulates glioblastoma stem cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Jiang, W.G. Evaluation of the expression of stem cell markers in human breast cancer reveals a correlation with clinical progression and metastatic disease in ductal carcinoma. Oncol. Rep. 2014, 31, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Reigstad, I.; Smeland, H.Y.; Skogstrand, T.; Sortland, K.; Schmid, M.C.; Reed, R.K.; Stuhr, L. Stromal Integrin α11β1 Affects RM11 Prostate and 4T1 Breast Xenograft Tumors Differently. PLoS ONE 2016, 11, e0151663. [Google Scholar] [CrossRef] [PubMed]
- Schober, M.; Fuchs, E. Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-β and integrin/focal adhesion kinase (FAK) signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 10544–10549. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, N.; Ishii, H.; Mimori, K.; Ohta, K.; Uemura, M.; Nishimura, J.; Hata, T.; Takemasa, I.; Mizushima, T.; Yamamoto, H.; et al. CD49f-positive cell population efficiently enriches colon cancer-initiating cells. Int. J. Oncol. 2013, 43, 425–430. [Google Scholar] [PubMed]
- Zheng, Y.; de la Cruz, C.C.; Sayles, L.C.; Alleyne-Chin, C.; Vaka, D.; Knaak, T.D.; Bigos, M.; Xu, Y.; Hoang, C.D.; Shrager, J.B.; et al. A rare population of CD24+ITGB4+Notchhi cells drives tumor propagation in NSCLC and requires Notch3 for self-renewal. Cancer Cell 2013, 24, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.Y.; Shih, D.T.; Meier, F.E.; Van, B.P.; Hsu, J.Y.; Elder, D.E.; Buck, C.A.; Herlyn, M. Adenoviral gene transfer of β3 integrin subunit induces conversion from radial to vertical growth phase in primary human melanoma. Am. J. Pathol. 1998, 153, 1435–1442. [Google Scholar] [CrossRef]
- Seftor, R.E.; Seftor, E.A.; Gehlsen, K.R.; Stetler-Stevenson, W.G.; Brown, P.D.; Ruoslahti, E.; Hendrix, M.J. Role of the αvβ3 integrin in human melanoma cell invasion. Proc. Natl. Acad. Sci. USA 1992, 89, 1557–1561. [Google Scholar] [CrossRef] [PubMed]
- Seftor, R.E.; Seftor, E.A.; Hendrix, M.J. Molecular role(s) for integrins in human melanoma invasion. Cancer Metastasis Rev. 1999, 18, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Enomoto, A.; Watanabe, T.; Haga, H.; Ishida, S.; Kondo, Y.; Furukawa, K.; Urano, T.; Mii, S.; Weng, L.; et al. TRIM27/MRTF-B-dependent integrin β1 expression defines leading cells in cancer cell collectives. Cell Rep. 2014, 7, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Aguirre Ghiso, J.A.; Kovalski, K.; Ossowski, L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J. Cell Biol. 1999, 147, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Chu, I.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J.; et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 2010, 70, 5706–5716. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. Integrin β1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 10290–10295. [Google Scholar] [CrossRef] [PubMed]
- Park, C.C.; Zhang, H.; Pallavicini, M.; Gray, J.W.; Baehner, F.; Park, C.J.; Bissell, M.J. β1 integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimensional cultures and in vivo. Cancer Res. 2006, 66, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 1996, 12, 697–715. [Google Scholar] [CrossRef] [PubMed]
- Humphries, M.J.; Olden, K.; Yamada, K.M. A synthetic peptide from fibronectin inhibits experimental metastasis of murine melanoma cells. Science 1986, 233, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Saiki, I.; Murata, J.; Iida, J.; Sakurai, T.; Nishi, N.; Matsuno, K.; Azuma, I. Antimetastatic effects of synthetic polypeptides containing repeated structures of the cell adhesive Arg-Gly-Asp (RGD) and Tyr-Ile-Gly-Ser-Arg (YIGSR) sequences. Br. J. Cancer 1989, 60, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Reinmuth, N.; Liu, W.; Ahmad, S.A.; Fan, F.; Stoeltzing, O.; Parikh, A.A.; Bucana, C.D.; Gallick, G.E.; Nickols, M.A.; Westlin, W.F.; et al. αvβ3 integrin antagonist S247 decreases colon cancer metastasis and angiogenesis and improves survival in mice. Cancer Res. 2003, 63, 2079–2087. [Google Scholar] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071–22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Wong, P.P.; Demircioglu, F.; Ghazaly, E.; Alrawashdeh, W.; Stratford, M.R.; Scudamore, C.L.; Cereser, B.; Crnogorac-Jurcevic, T.; McDonald, S.; Elia, G.; et al. Dual-action combination therapy enhances angiogenesis while reducing tumor growth and spread. Cancer Cell 2015, 27, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Berlin, J.; de Bono, J.S.; Cohen, R.B.; Keedy, V.; Mugundu, G.; Zhang, L.; Abbattista, A.; Davis, C.; Gallo, S.C.; et al. A first-in-human study of the anti-α5β1 integrin monoclonal antibody PF-04605412 administered intravenously to patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.C.; Schoumacher, R.A.; Tiller, R.E. Pancreatic adenocarcinoma in a patient with cystic fibrosis. Am. J. Med. 1988, 85, 592. [Google Scholar] [CrossRef]
- Ricart, A.D.; Tolcher, A.W.; Liu, G.; Holen, K.; Schwartz, G.; Albertini, M.; Weiss, G.; Yazji, S.; Ng, C.; Wilding, G. Volociximab, a chimeric monoclonal antibody that specifically binds α5β1 integrin: A phase I, pharmacokinetic, and biological correlative study. Clin. Cancer Res. 2008, 14, 7924–7929. [Google Scholar] [CrossRef] [PubMed]
- Bell-McGuinn, K.M.; Matthews, C.M.; Ho, S.N.; Barve, M.; Gilbert, L.; Penson, R.T.; Lengyel, E.; Palaparthy, R.; Gilder, K.; Vassos, A.; et al. A phase II, single-arm study of the anti-α5β1 integrin antibody volociximab as monotherapy in patients with platinum-resistant advanced epithelial ovarian or primary peritoneal cancer. Gynecol. Oncol. 2011, 121, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.L.; Picard, M. Integrins as therapeutic targets. Trends Pharmacol. Sci. 2012, 33, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Millard, M.; Odde, S.; Neamati, N. Integrin targeted therapeutics. Theranostics 2011, 1, 154–188. [Google Scholar] [CrossRef] [PubMed]
- Demircioglu, F.; Hodivala-Dilke, K. αvβ3 Integrin and tumour blood vessels-learning from the past to shape the future. Curr. Opin. Cell Biol. 2016, 42, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Cianfrocca, M.E.; Kimmel, K.A.; Gallo, J.; Cardoso, T.; Brown, M.M.; Hudes, G.; Lewis, N.; Weiner, L.; Lam, G.N.; Brown, S.C.; et al. Phase 1 trial of the antiangiogenic peptide ATN-161 (Ac-PHSCN-NH(2)), aβ integrin antagonist, in patients with solid tumours. Br. J. Cancer 2006, 94, 1621–1626. [Google Scholar] [PubMed]
- McNeel, D.G.; Eickhoff, J.; Lee, F.T.; King, D.M.; Alberti, D.; Thomas, J.P.; Friedl, A.; Kolesar, J.; Marnocha, R.; Volkman, J.; et al. Phase I trial of a monoclonal antibody specific for αvβ3 integrin (MEDI-522) in patients with advanced malignancies, including an assessment of effect on tumor perfusion. Clin. Cancer Res. 2005, 11, 7851–7860. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, K.S.; Yuen, E.H.; Cho, C.C.; Tong, C.S.; Lee, Y.Y.; Ahuja, A.T. A pilot study evaluating real-time shear wave ultrasound elastography of miscellaneous non-nodal neck masses in a routine head and neck ultrasound clinic. Ultrasound Med. Biol. 2012, 38, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Levental, I.; Levental, K.R.; Klein, E.A.; Assoian, R.; Miller, R.T.; Wells, R.G.; Janmey, P.A. A simple indentation device for measuring micrometer-scale tissue stiffness. J. Phys. Condens. Matter 2010, 22, 194120. [Google Scholar] [CrossRef] [PubMed]
- Pohlers, D.; Brenmoehl, J.; Loffler, I.; Muller, C.K.; Leipner, C.; Schultze-Mosgau, S.; Stallmach, A.; Kinne, R.W.; Wolf, G. TGF-β and fibrosis in different organs—Molecular pathway imprints. Biochim. Biophys. Acta 2009, 1792, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Siraganian, P.A.; Miller, R.W.; Swender, P.T. Cystic fibrosis and ileal carcinoma. Lancet 1987, 2, 1158. [Google Scholar] [CrossRef]
- Fine, J.D.; Johnson, L.B.; Weiner, M.; Li, K.P.; Suchindran, C. Epidermolysis bullosa and the risk of life-threatening cancers: The National EB Registry experience, 1986–2006. J. Am. Acad. Dermatol. 2009, 60, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Mittapalli, V.R.; Madl, J.; Löffek, S.; Kiritsi, D.; Kern, J.S.; Romer, W.; Nystrom, A.; Bruckner-Tuderman, L. Injury-driven stiffening of the dermis expedites skin carcinoma progression. Cancer Res. 2016, 76, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Onishi, A.; Sugiyama, D.; Kumagai, S.; Morinobu, A. Cancer incidence in systemic sclerosis: Meta-analysis of population-based cohort studies. Arthritis Rheumatol. 2013, 65, 1913–1921. [Google Scholar] [CrossRef] [PubMed]
- Stern, E.P.; Denton, C.P. The Pathogenesis of Systemic Sclerosis. Rheum. Dis. Clin. N. Am. 2015, 41, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Boyd, N.F. Mammographic density. Potential mechanisms of breast cancer risk associated with mammographic density: Hypotheses based on epidemiological evidence. Breast Cancer Res. 2008, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Weder, G.; Hendriks-Balk, M.C.; Smajda, R.; Rimoldi, D.; Liley, M.; Heinzelmann, H.; Meister, A.; Mariotti, A. Increased plasticity of the stiffness of melanoma cells correlates with their acquisition of metastatic properties. Nanomedicine 2014, 10, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Zheng, Q.; Dong, Y.; Wang, Y.; Zhang, L.; Xue, T.; Xie, X.; Hu, C.; Wang, Z.; Chen, R.; et al. Higher matrix stiffness upregulates osteopontin expression in hepatocellular carcinoma cells mediated by integrin β1/GSK3β/β-catenin signaling pathway. PLoS ONE 2015, 10, e0134243. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, O.; Koshy, S.T.; Branco da, C.C.; Shin, J.W.; Verbeke, C.S.; Allison, K.H.; Mooney, D.J. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat. Mater. 2014, 13, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Navab, R.; Strumpf, D.; To, C.; Pasko, E.; Kim, K.S.; Park, C.J.; Hai, J.; Liu, J.; Jonkman, J.; Barczyk, M.; et al. Integrin α11β1 regulates cancer stromal stiffness and promotes tumorigenicity and metastasis in non-small cell lung cancer. Oncogene 2016, 35, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Fraczek, N.; Bronisz, I.; Pietryka, M.; Kepinska, D.; Strzala, P.; Mielnicka, K.; Korga, A.; Dudka, J. An outline of main factors of drug resistance influencing cancer therapy. J. Chemother. 2016, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Paschen, A.; Landsberg, J.; Helfrich, I.; Becker, J.C.; Schadendorf, D. Phenotypic tumour cell plasticity as a resistance mechanism and therapeutic target in melanoma. Eur. J. Cancer 2016, 59, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.V.; Sleiman, M.; Moriarty, T.; Herrick, W.G.; Peyton, S.R. Sorafenib resistance and JNK signaling in carcinoma during extracellular matrix stiffening. Biomaterials 2014, 35, 5749–5759. [Google Scholar] [CrossRef] [PubMed]
- Kanda, R.; Kawahara, A.; Watari, K.; Murakami, Y.; Sonoda, K.; Maeda, M.; Fujita, H.; Kage, M.; Uramoto, H.; Costa, C.; et al. Erlotinib resistance in lung cancer cells mediated by integrin β1/Src/Akt-driven bypass signaling. Cancer Res. 2013, 73, 6243–6253. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Park, C.C.; Hilsenbeck, S.G.; Ward, R.; Rimawi, M.F.; Wang, Y.C.; Shou, J.; Bissell, M.J.; Osborne, C.K.; Schiff, R. β1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast Cancer Res. 2011, 13, R84. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Integrin | Prototypic Ligands |
|---|---|
| α1β1 (CD49a, VLA1) | Collagen IV, I and IX |
| α2β1 (CD49b, VLA2) | Collagen I, IV and IX |
| α3β1 (CD49c, VLA3) | Laminin-511, -332, -211 |
| α4β1 (CD49d, VLA4) | Fibronectin, VCAM-1 |
| α5β1 (CD49e, VLA5) | Fibronectin |
| α6β1 (CD49f, VLA6) | Laminin-511, -332, -111, -411 |
| α7β1 | Laminin-511, -211, -411, -111 |
| α8β1 | Fibronectin, vitronectin, |
| α9β1 | Tenascin-C, VEGF-C, VEGF-D |
| α10β1 | Collagen I, IV, II and IX |
| α11β1 | Collagen I, IV and IX |
| α6β4 | Laminin-332, -511 |
| αvβ1 (CD51) | Fibronectin, vitronectin |
| αvβ3 | Vitronectin, fibronectin, fibrinogen |
| αvβ5 | Vitronectin |
| αvβ6 | Fibronectin, TGF-β-LAP |
| αvβ8 | Vitronectin, TGF-β-LAP |
| αEβ7 (CD103, HML-1) | E-cadherin |
| α4β7 | MadCAM-1, fibronectin, VCAM-1 |
| αLβ2 (CD11a) | ICAM-1, -2, -3, -5 |
| αMβ2 (CD11b) | Fibrinogen |
| αXβ2 (CD11c) | Fibrinogen |
| αDβ2 (CD11d) | ICAM-3, VCAM-1 |
| αIIBβ3 (CD41) | Fibrinogen, fibronectin |
| Target | Inhibitor | Clinical Trial | Ref. |
|---|---|---|---|
| α5β1 | ATN-161 (small peptide antagonist) | Phase II: patients with advanced solid malignancies | [73] |
| α5β1 | Volociximab (mAb) | Phase I: patients with advanced solid malignancies | [68] |
| α5β1 | Volociximab (mAb) | Phase II: patients with therapy-resistant epithelial ovarian cancer and primary peritoneal cancer | [69] |
| αvβ3, αvβ5 | Cilengitide (EMD 121974) | Phase III: glioblastoma patients | [64] |
| α5β1 | PF-04605412 (mAb) | Safety study: advanced solid tumors | [66] |
| αvβ3 | Etaracizumab (MEDI-522) | Phase I: metastatic solid tumors | [74] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Löffek, S.; Franzke, C.-W.; Helfrich, I. Tension in Cancer. Int. J. Mol. Sci. 2016, 17, 1910. https://doi.org/10.3390/ijms17111910
Löffek S, Franzke C-W, Helfrich I. Tension in Cancer. International Journal of Molecular Sciences. 2016; 17(11):1910. https://doi.org/10.3390/ijms17111910
Chicago/Turabian StyleLöffek, Stefanie, Claus-Werner Franzke, and Iris Helfrich. 2016. "Tension in Cancer" International Journal of Molecular Sciences 17, no. 11: 1910. https://doi.org/10.3390/ijms17111910
APA StyleLöffek, S., Franzke, C.-W., & Helfrich, I. (2016). Tension in Cancer. International Journal of Molecular Sciences, 17(11), 1910. https://doi.org/10.3390/ijms17111910