
iTRAQ-Based Quantitative Proteomic Analysis of the Potentiated and Dormant Antler Stem Cells



Abstract
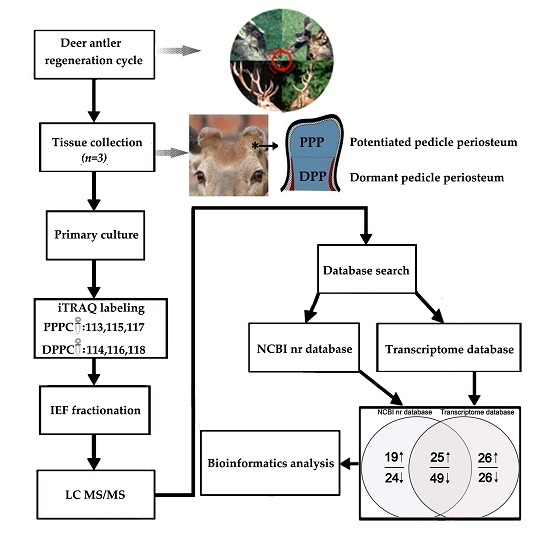
:
1. Introduction
2. Results
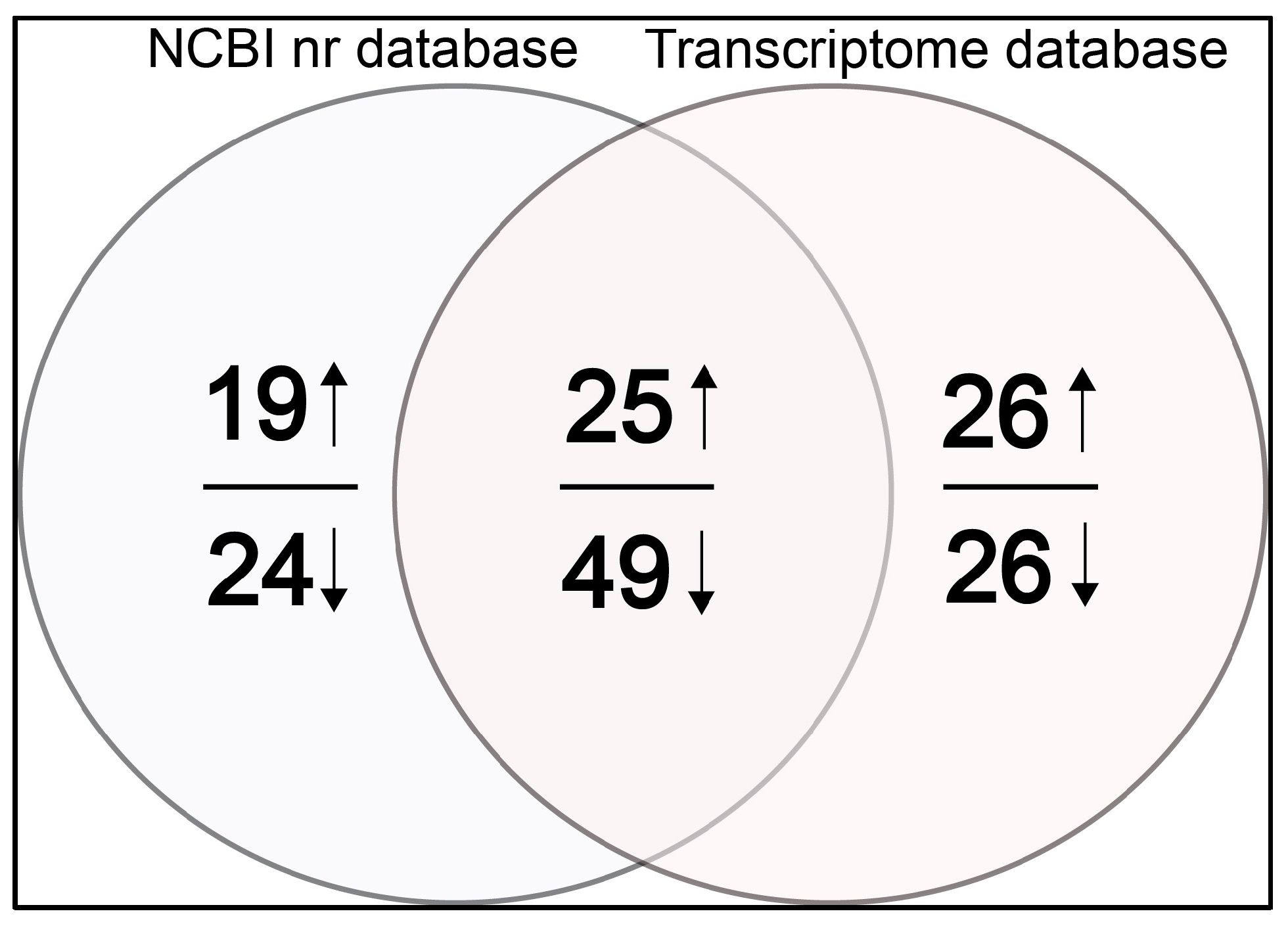
2.1. Identified Differentially Expressed Proteins
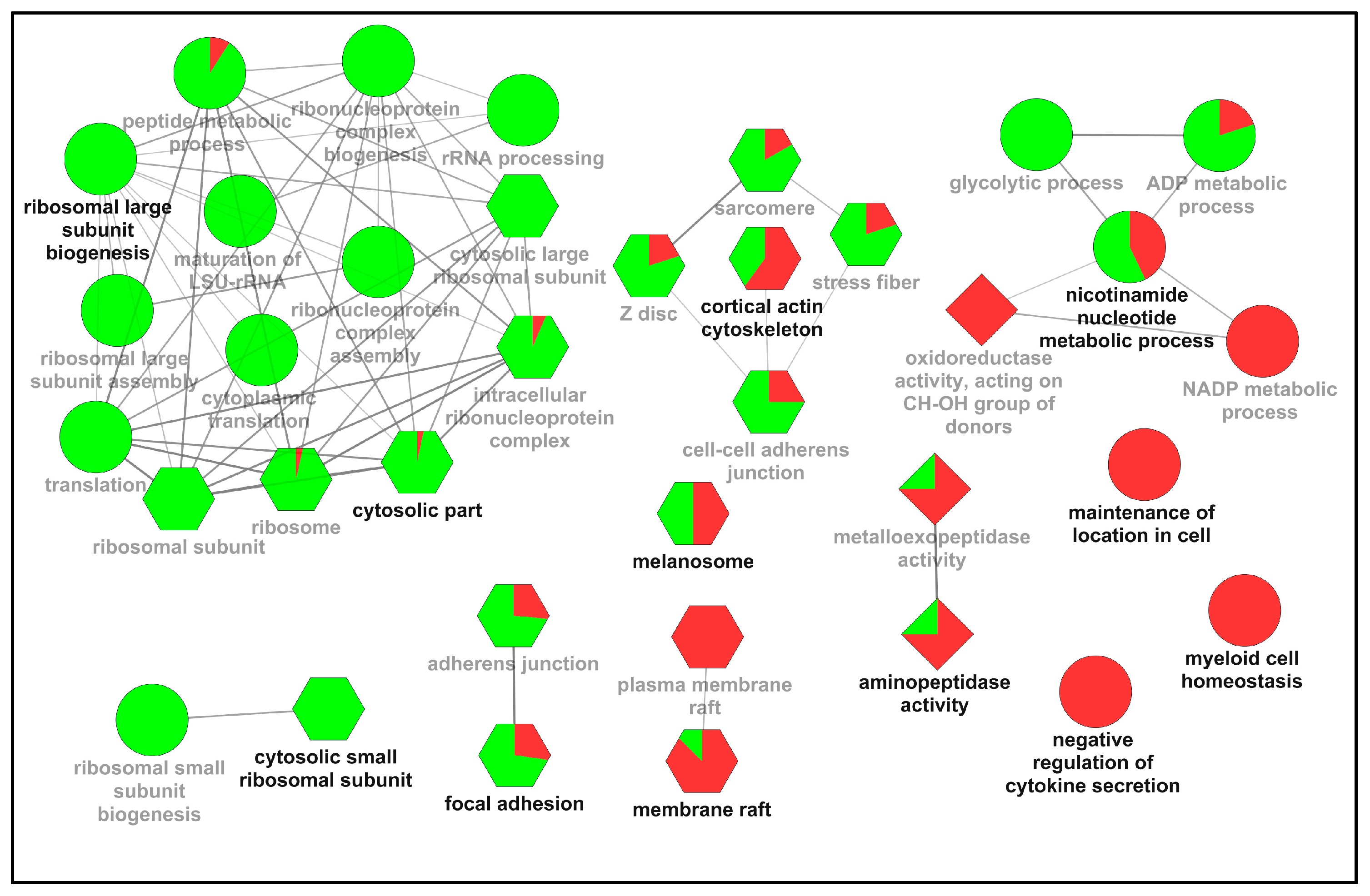
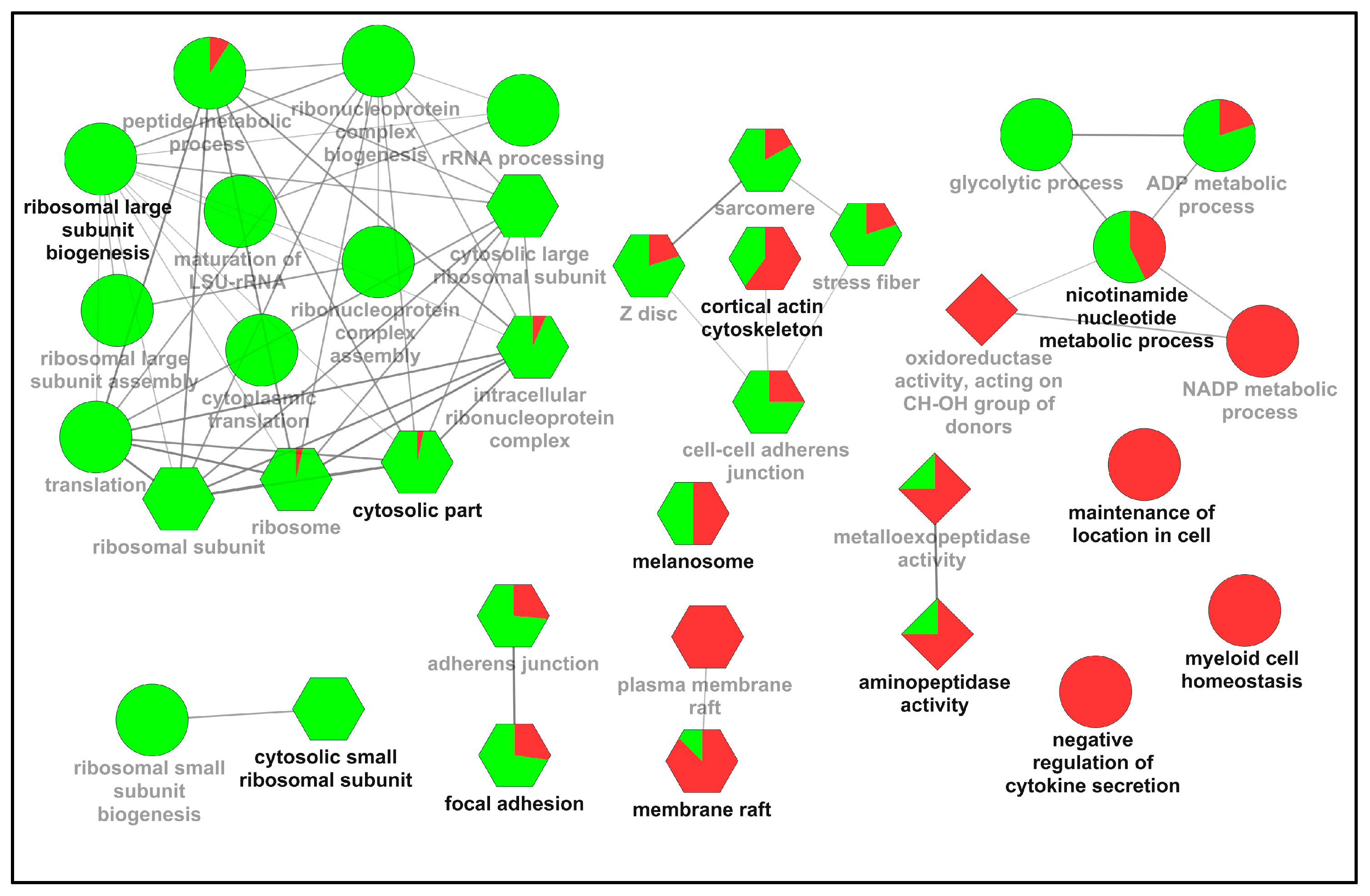
2.2. Functional Classification of the Differentially Expressed Proteins
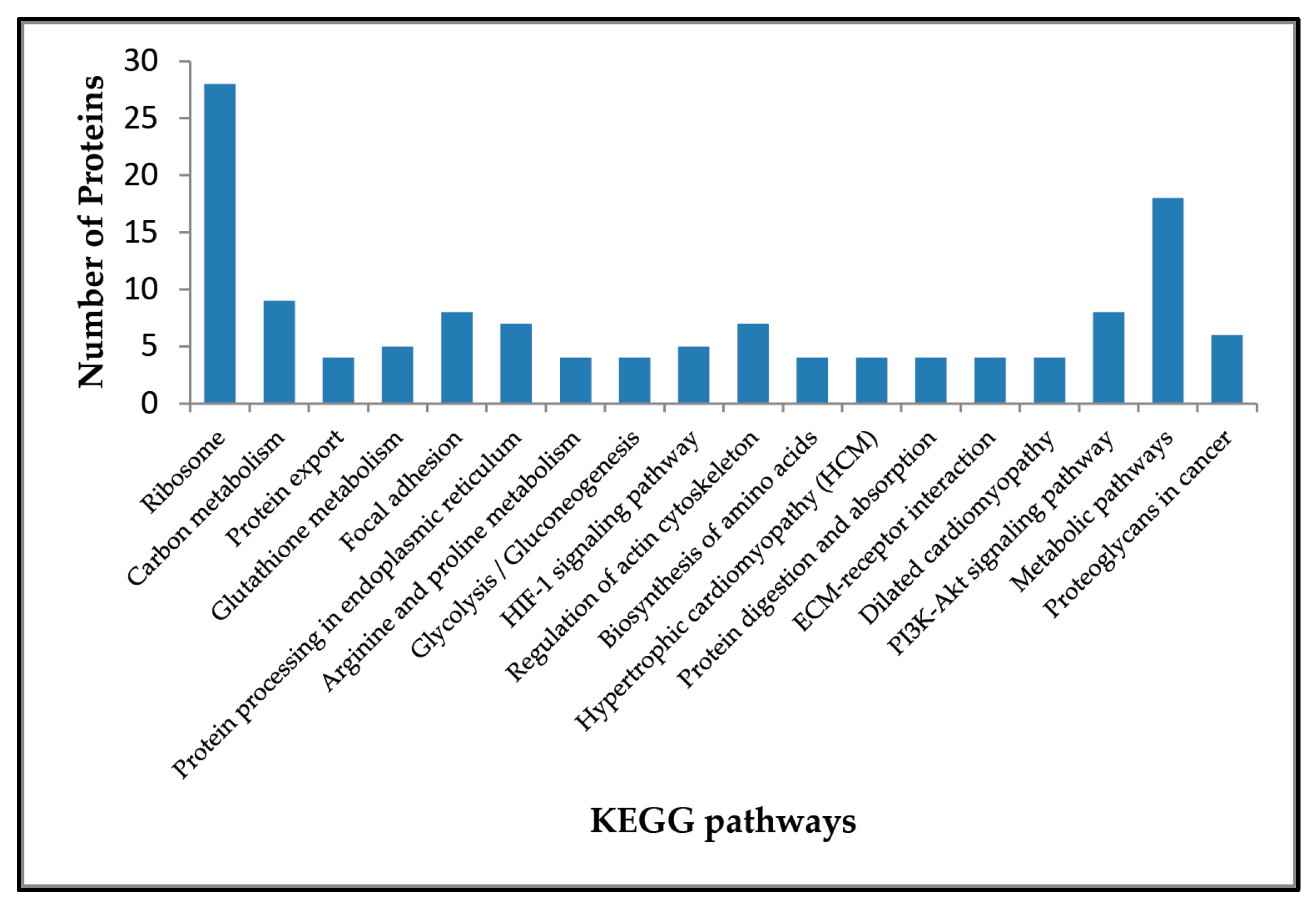
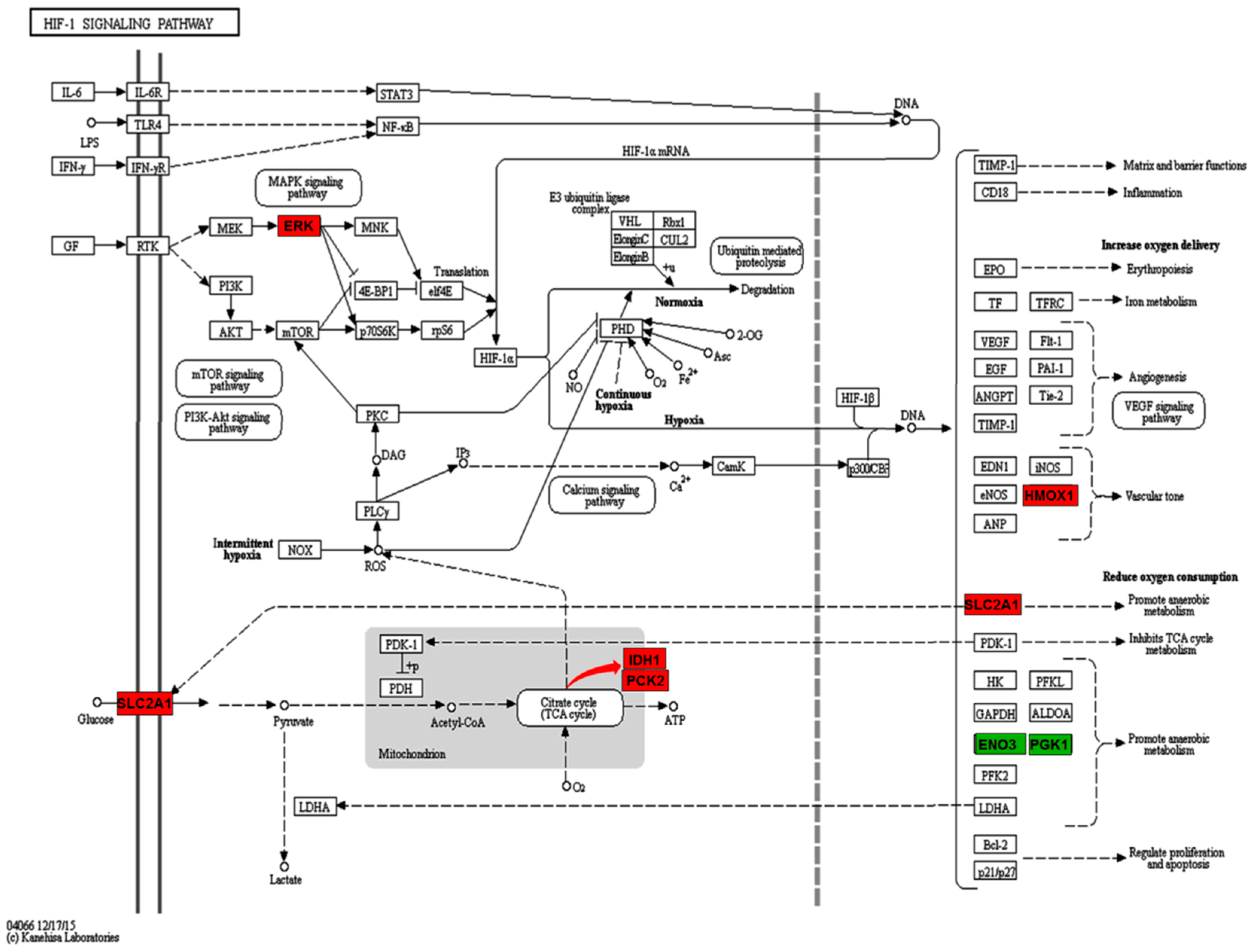
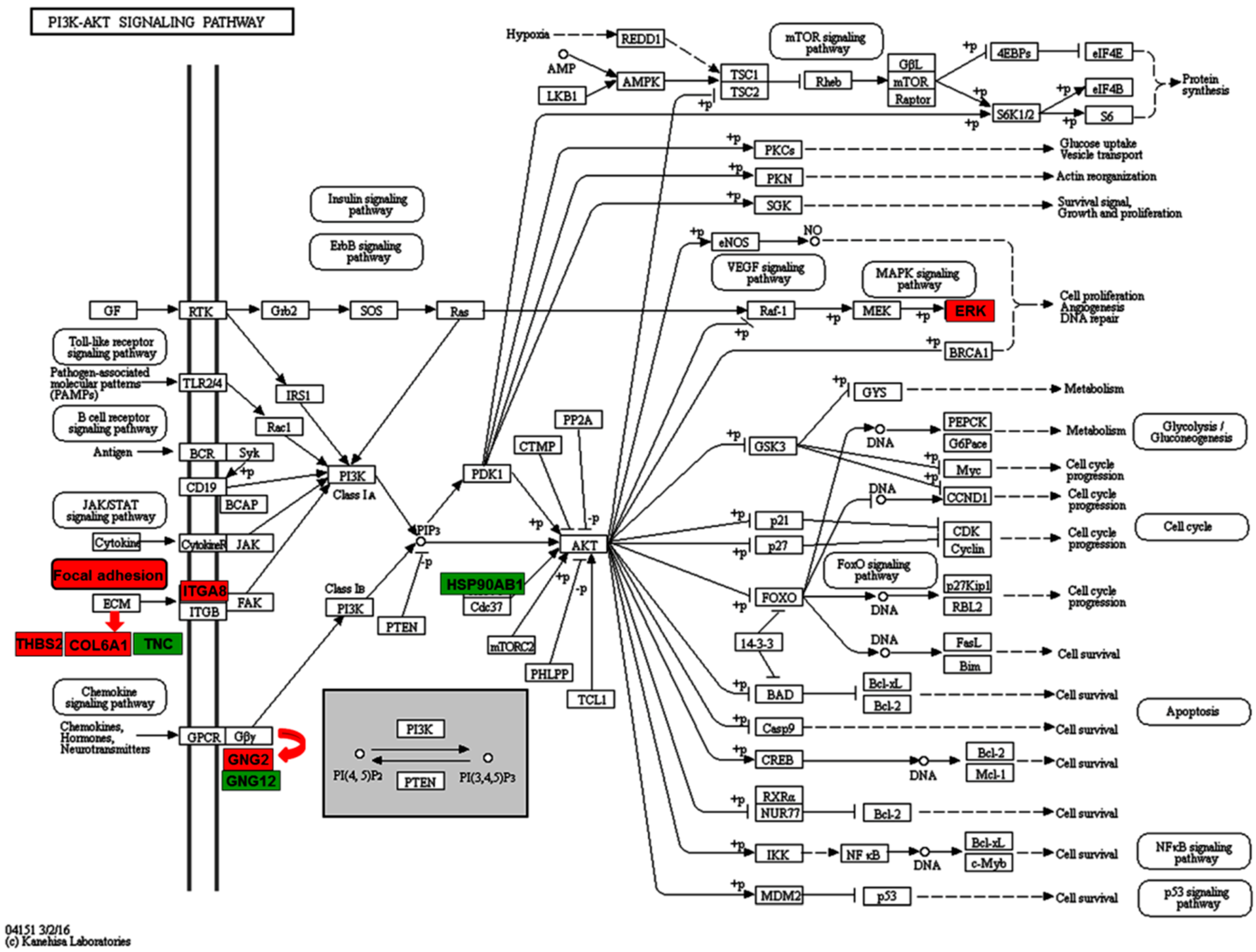
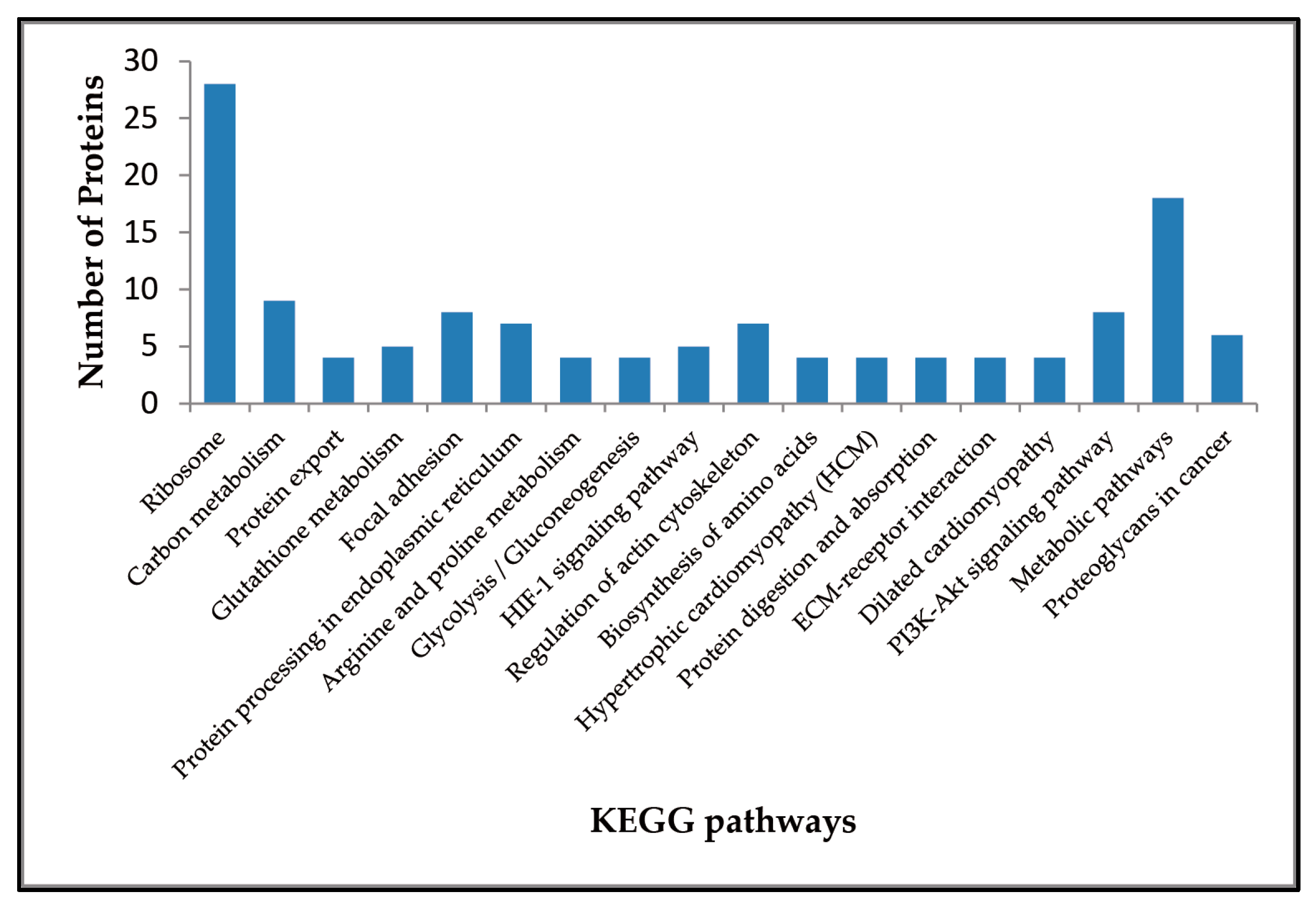
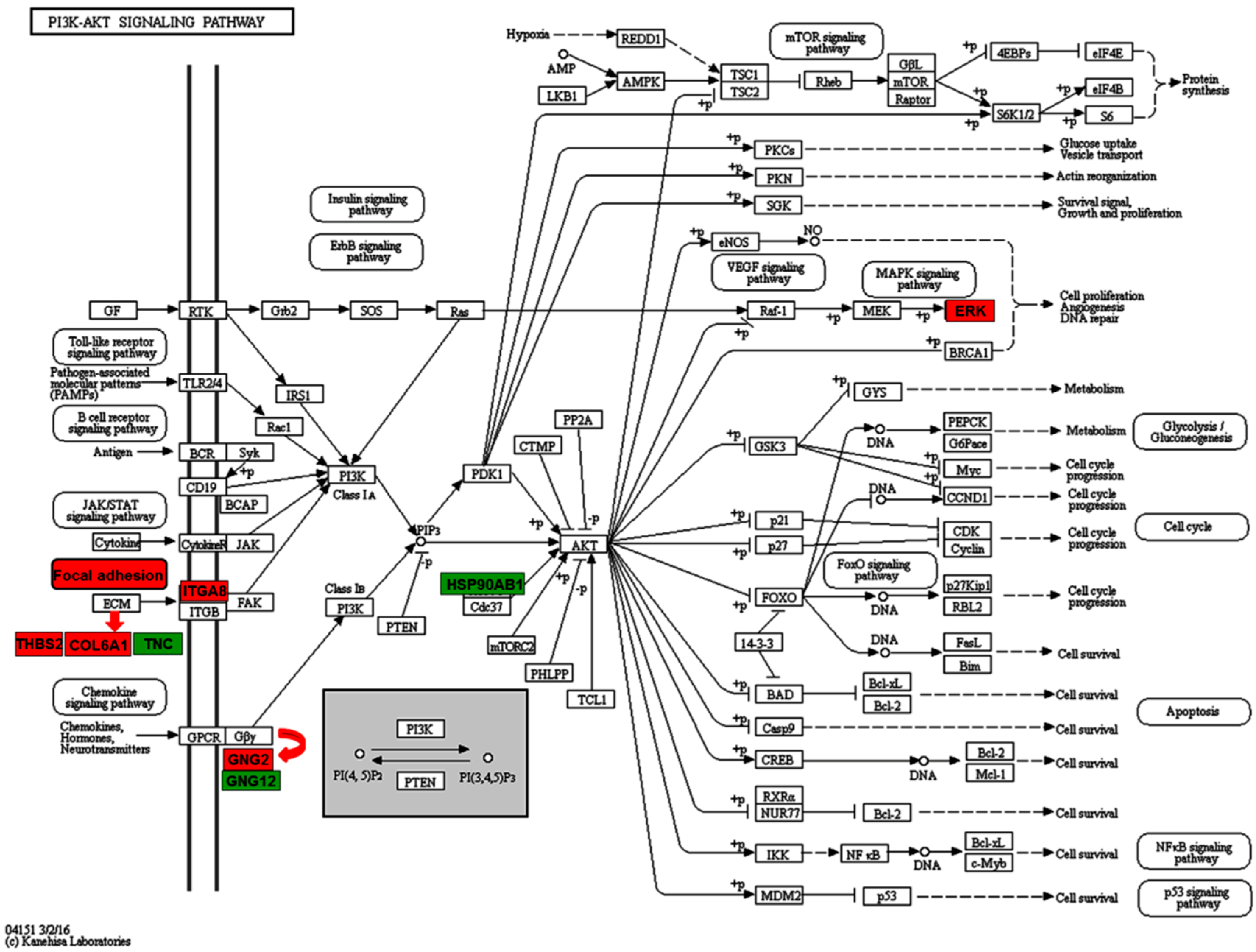
2.3. Enriched Pathways Participated by the Differentially Expressed Proteins
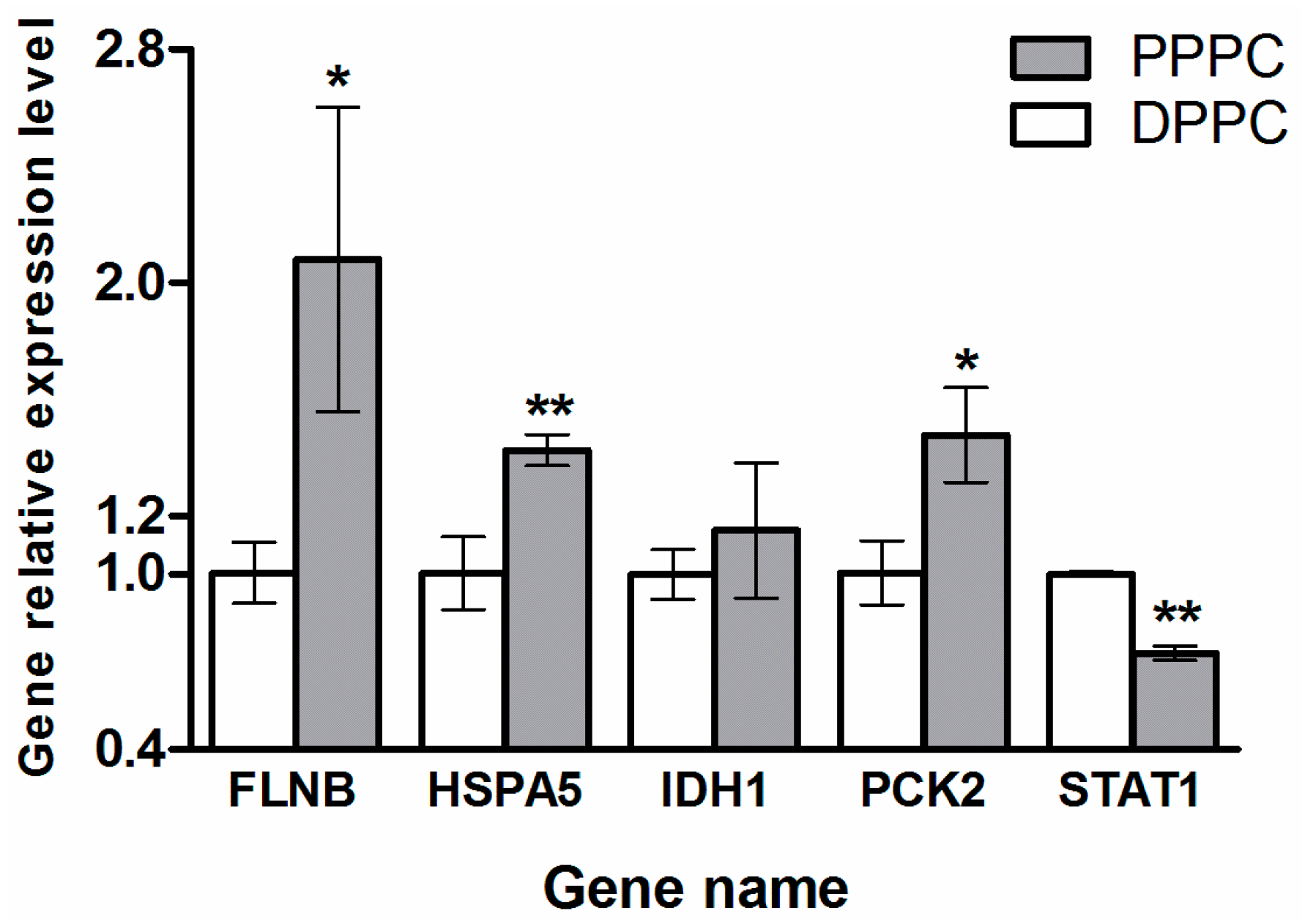
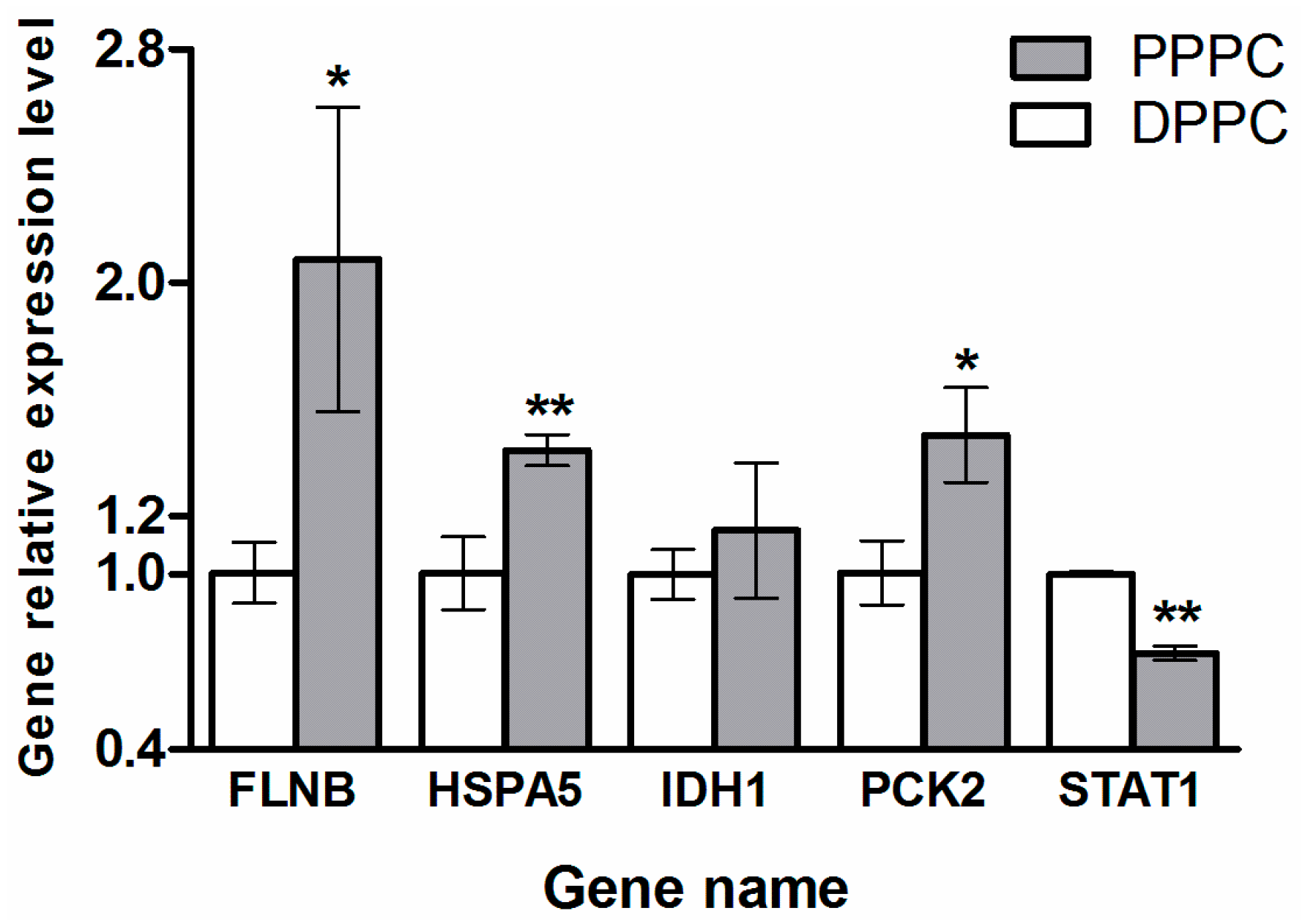
2.4. Validation of the Five Selected Differentially Expressed Proteins
3. Discussion
4. Materials and Methods
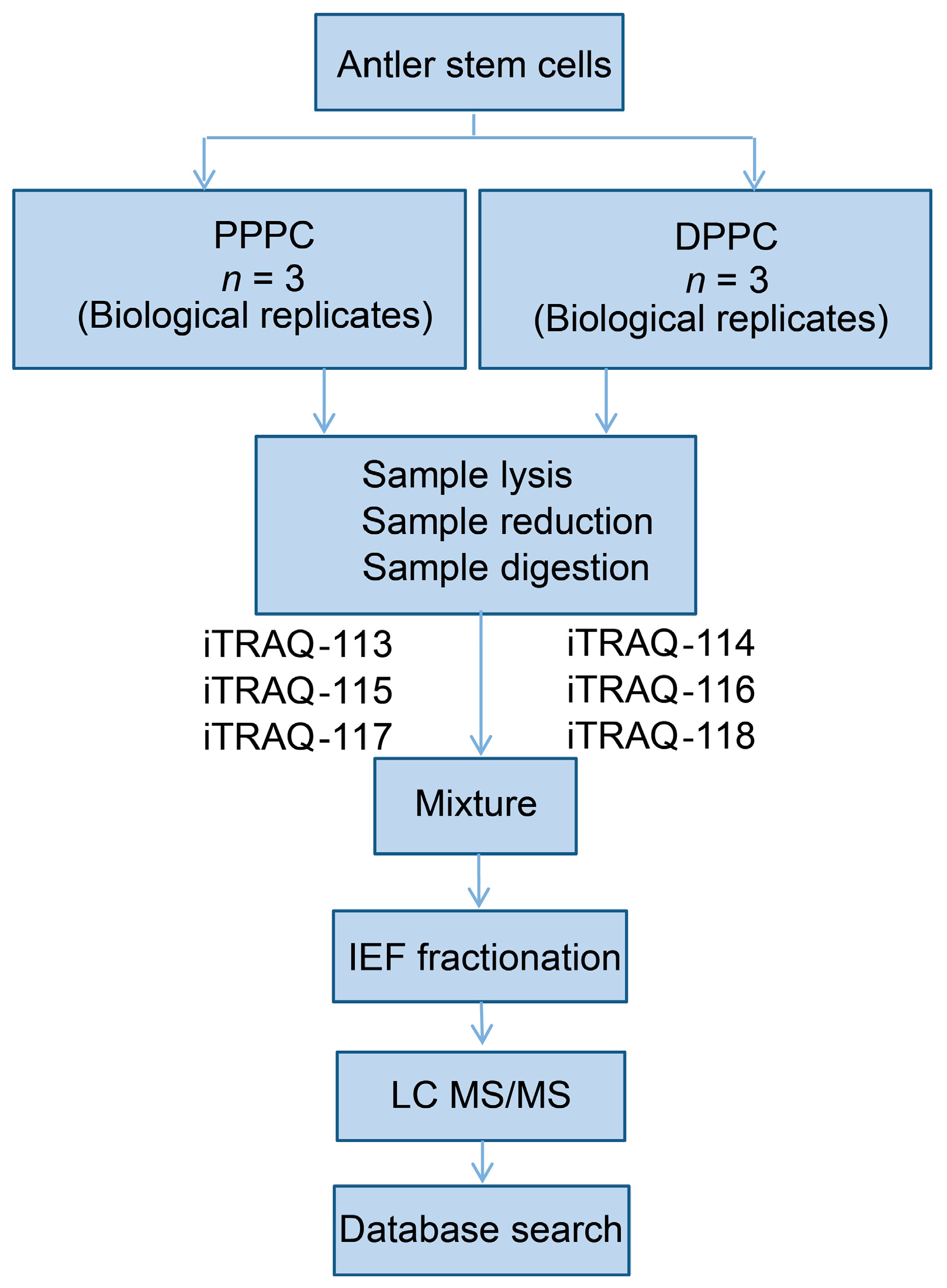
4.1. Tissue Sampling and Primary Culture of the PPCs
4.2. Protein Extraction and Labeling
4.3. Analysis of Proteins Using a LC-ESI-MS/MS
4.4. Database Search
4.5. Bioinformatics Analysis
4.6. Validation by qRT-PCR
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stocum, D.L. Stem cells in CNS and cardiac regeneration. Adv. Biochem. Eng. Biotechnol. 2005, 93, 135–159. [Google Scholar] [PubMed]
- Goss, R.J. Future-directions in antler research. Anat. Rec. 1995, 241, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Goss, R.J. Deer Antlers: Regeneration, Function and Evolution; Academic Press: New York, NY, USA, 1983. [Google Scholar]
- Li, C.; Suttie, J.M.; Clark, D.E. Histological examination of antler regeneration in red deer (cervus elaphus). Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2005, 282, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Mackintosh, C.G.; Martin, S.K.; Clark, D.E. Identification of key tissue type for antler regeneration through pedicle periosteum deletion. Cell Tissue Res. 2007, 328, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yang, F.; Sheppard, A. Adult stem cells and mammalian epimorphic regeneration-insights from studying annual renewal of deer antlers. Curr. Stem Cell Res. Ther. 2009, 4, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Suttie, J.M. Tissue collection methods for antler research. Eur. J. Morphol. 2003, 41, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yang, F.; Li, G.; Gao, X.; Xing, X.; Wei, H.; Deng, X.; Clark, D.E. Antler regeneration: A dependent process of stem tissue primed via interaction with its enveloping skin. J. Exp. Zool. A Ecol. Genet. Physiol. 2007, 307, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Brewis, I.A.; Brennan, P. Proteomics technologies for the global identification and quantification of proteins. Adv. Protein Chem. Struct. Biol. 2010, 80, 1–44. [Google Scholar] [PubMed]
- Treumann, A.; Thiede, B. Isobaric protein and peptide quantification: Perspectives and issues. Expert Rev. Proteom. 2010, 7, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.; Unwin, R.D.; Evans, C.A.; Griffiths, S.; Carney, L.; Zhang, L.; Jaworska, E.; Lee, C.F.; Blinco, D.; Okoniewski, M.J.; et al. Eight-channel iTRAQ enables comparison of the activity of six leukemogenic tyrosine kinases. Mol. Cell. Proteom. 2008, 7, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.S.; Liu, X.H.; Liu, L.X.; Lou, W.H.; Jin, D.Y.; Yang, P.Y.; Wang, X.L. iTRAQ-based quantitative proteomics reveals myoferlin as a novel prognostic predictor in pancreatic adenocarcinoma. J. Proteom. 2013, 91, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Battle, T.E.; Lynch, R.A.; Frank, D.A. Signal transducer and activator of transcription 1 activation in endothelial cells is a negative regulator of angiogenesis. Cancer Res. 2006, 66, 3649–3657. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Park, O.; Horiguchi, N.; Kulkarni, S.; Jeong, W.I.; Sun, H.Y.; Radaeva, S.; Gao, B. STAT1 contributes to dsRNA inhibition of liver regeneration after partial hepatectomy in mice. Hepatology 2006, 44, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.G.; Ma, K.W.; Wang, H.X.; Xiao, F.; Gao, Y.; Zhang, W.; Wang, K.P.; Gao, X.; Ip, N.; Wu, Z.G. JAK1-STAT1-STAT3, a key pathway promoting proliferation and preventing premature differentiation of myoblasts. J. Cell Biol. 2007, 179, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, Y.F.; Guo, X.; Wu, Z.G.; Zhang, W. Loss of STAT1 in bone marrow-derived cells accelerates skeletal muscle regeneration. PLoS ONE 2012, 7, e37656. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Koga, T.; Isobe, M.; Kern, B.E.; Yokochi, T.; Chin, Y.E.; Karsenty, G.; Taniguchi, T.; Takayanagi, H. STAT1 functions as a attenuator of Runx2 in the transcriptional program of osteoblast cytoplasmic differentiation. Gene Dev. 2003, 17, 1979–1991. [Google Scholar] [CrossRef] [PubMed]
- Fujie, A.; Funayama, A.; Miyauchi, Y.; Sato, Y.; Kobayashi, T.; Kanagawa, H.; Katsuyama, E.; Hao, W.; Tando, T.; Watanabe, R.; et al. BCL6 promotes osteoblastogenesis through STAT1 inhibition. Biochem. Biophys. Res. Commun. 2015, 457, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef]
- Ducy, P.; Schinke, T.; Karsenty, G. The osteoblast: A sophisticated fibroblast under central surveillance. Science 2000, 289, 1501–1504. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Yang, F.; Chu, W.; Zhao, H.; McMahon, C.; Li, C. Lentiviral-mediated rnai knockdown of Cbfa1 gene inhibits endochondral ossification of antler stem cells in micromass culture. PLoS ONE 2012, 7, e47367. [Google Scholar] [CrossRef] [PubMed]
- Brusselmans, K.; Bono, F.; Collen, D.; Herbert, J.M.; Carmeliet, P.; Dewerchin, M. A novel role for vascular endothelial growth factor as an autocrine survival factor for embryonic stem cells during hypoxia. J. Biol. Chem. 2005, 280, 3493–3499. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.L.; Wu, L.Y.; Yew, D.T.; Fan, M. Effects of hypoxia on the proliferation and differentiation of NSCs. Mol. Neurobiol. 2005, 31, 231–242. [Google Scholar] [CrossRef]
- Grayson, W.L.; Zhao, F.; Izadpanah, R.; Bunnell, B.; Ma, T. Effects of hypoxia on human mesenchymal stem cell expansion and plasticity in 3D constructs. J. Cell. Physiol. 2006, 207, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.W.; Fermor, B.; Gimble, J.M.; Awad, H.A.; Guilak, F. Influence of oxygen on the proliferation and metabolism of adipose derived adult stem cells. J. Cell. Physiol. 2005, 204, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, H.; Krishnamachary, B.; Zhou, Y.F.; Nagasawa, H.; Bosch-Marce, M.; Semenza, G.L. Expression of vascular endothelial growth factor receptor 1 in bone marrow-derived mesenchymal cells is dependent on hypoxia-inducible factor 1. J. Biol. Chem. 2006, 281, 15554–15563. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Haynie, J.; Yang, X.M.; Han, B.G.; Kiatchoosakun, S.; Restivo, J.; Yuan, S.Y.; Prabhakar, N.R.; Herrup, K.; Conlon, R.A.; et al. The essential role of Cited2, a negative regulator for HIF-1α, in heart development and neurulation. Proc. Natl. Acad. Sci. USA 2002, 99, 10488–10493. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.L.; Midwinter, R.G.; Ni, J.; Hamid, H.A.; Parish, C.R.; Stocker, R. New insights into intracellular locations and functions of heme oxygenase-1. Antioxid. Redox Signal. 2014, 20, 1723–1742. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E.; Kemp, R.B. Anaerobic metabolism in aerobic mammalian cells: Information from the ratio of calorimetric heat flux and respirometric oxygen flux. Biochim. Biophys. Acta 1990, 1016, 328–332. [Google Scholar] [CrossRef]
- Teslaa, T.; Teitell, M.A. Pluripotent stem cell energy metabolism: An update. EMBO J. 2015, 34, 138–153. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Mentel, M.; van Hellemond, J.J.; Henze, K.; Woehle, C.; Gould, S.B.; Yu, R.Y.; van der Giezen, M.; Tielens, A.G.; Martin, W.F. Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. Microbiol. Mol. Biol. Rev. 2012, 76, 444–495. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; Grimaldi, C.; Cappellini, A.; Ognibene, A.; McCubrey, J.A. The emerging role of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in normal myelopoiesis and leukemogenesis. Biochim. Biophys. Acta 2010, 1803, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.F.; Qi, Y.Y.; Li, Y.; Xu, K. PI3 kinase regulation of neural regeneration and muscle hypertrophy after spinal cord injury. Mol. Biol. Rep. 2012, 39, 3541–3547. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. Multiple signal pathways in obesity-associated cancer. Obes. Rev. 2011, 12, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.P. Filamin A: Phenotypic diversity. Curr. Opin. Genet. Dev. 2005, 15, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Stossel, T.P.; Hartwig, J.H. The filamins organizers of cell structure and function. Cell Adhes. Migr. 2011, 5, 160–169. [Google Scholar] [CrossRef]
- Van der Flier, A.; Sonnenberg, A. Structural and functional aspects of filamins. Biochim. Biophys. Acta 2001, 1538, 99–117. [Google Scholar] [CrossRef]
- Del Valle-Perez, B.; Martinez, V.G.; Lacasa-Salavert, C.; Figueras, A.; Shapiro, S.S.; Takafuta, T.; Casanovas, O.; Capella, G.; Ventura, F.; Vinals, F. Filamin B plays a key role in vascular endothelial growth factor-induced endothelial cell motility through its interaction with Rac-1 and Vav-2. J. Biol. Chem. 2010, 285, 10748–10760. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Harper, A.; Puddick, J.; Wang, W.; McMahon, C. Proteomes and signalling pathways of antler stem cells. PLoS ONE 2012, 7, e30026. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. Cluego: A cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID gene functional classification tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. String v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, 447–452. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KEGG Pathway | Up-Regulated Proteins | Down-Regulated Proteins |
|---|---|---|
| Ribosome | – | RPL35, RPL7A, RPL29, RPL7, RPL3L, RPL19, RPL10, RPL13A, RPL8, RPL13, RPL4, RPS18, RPL34, RPS2, RPL21, RPL28, RPL35A, RPL24, RPL26L1, RPS8, RPL18A, RPL6, RPL23A, RPS13, RPL14, RPS5, RPS16, RPS27L |
| Protein export | HSPA5 | SPCS2, SRPRB, SEC61B |
| Protein processing in endoplasmic reticulum | RPN2, HSPA5 | SAR1A, SEC61B, SEC23B, RRBP1, CRYAB, HSP90AB1 |
| Focal adhesion | THBS2, FLNB, COL6A1, ITGA8, MAPK3 | ACTN1, TNC, ZYX |
| HIF-1 signaling pathway | SLC2A1, MAPK3, HMOX1 | ENO3, PGK1 |
| ECM-receptor interaction | THBS2, ITGA8, COL6A1 | TNC |
| PI3K-Akt signaling pathway | THBS2, GNG2, COL6A1, ITGA8, MAPK3 | TNC, HSP90AB1, GNG12 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, Z.; Ba, H.; Zhang, W.; Coates, D.; Li, C. iTRAQ-Based Quantitative Proteomic Analysis of the Potentiated and Dormant Antler Stem Cells. Int. J. Mol. Sci. 2016, 17, 1778. https://doi.org/10.3390/ijms17111778
Dong Z, Ba H, Zhang W, Coates D, Li C. iTRAQ-Based Quantitative Proteomic Analysis of the Potentiated and Dormant Antler Stem Cells. International Journal of Molecular Sciences. 2016; 17(11):1778. https://doi.org/10.3390/ijms17111778
Chicago/Turabian StyleDong, Zhen, Hengxing Ba, Wei Zhang, Dawn Coates, and Chunyi Li. 2016. "iTRAQ-Based Quantitative Proteomic Analysis of the Potentiated and Dormant Antler Stem Cells" International Journal of Molecular Sciences 17, no. 11: 1778. https://doi.org/10.3390/ijms17111778
APA StyleDong, Z., Ba, H., Zhang, W., Coates, D., & Li, C. (2016). iTRAQ-Based Quantitative Proteomic Analysis of the Potentiated and Dormant Antler Stem Cells. International Journal of Molecular Sciences, 17(11), 1778. https://doi.org/10.3390/ijms17111778