
Potential of LC Coupled to Fluorescence Detection in Food Metabolomics: Determination of Phenolic Compounds in Virgin Olive Oil

 , ,
, , 
Abstract
:
1. Introduction
2. Results and Discussion
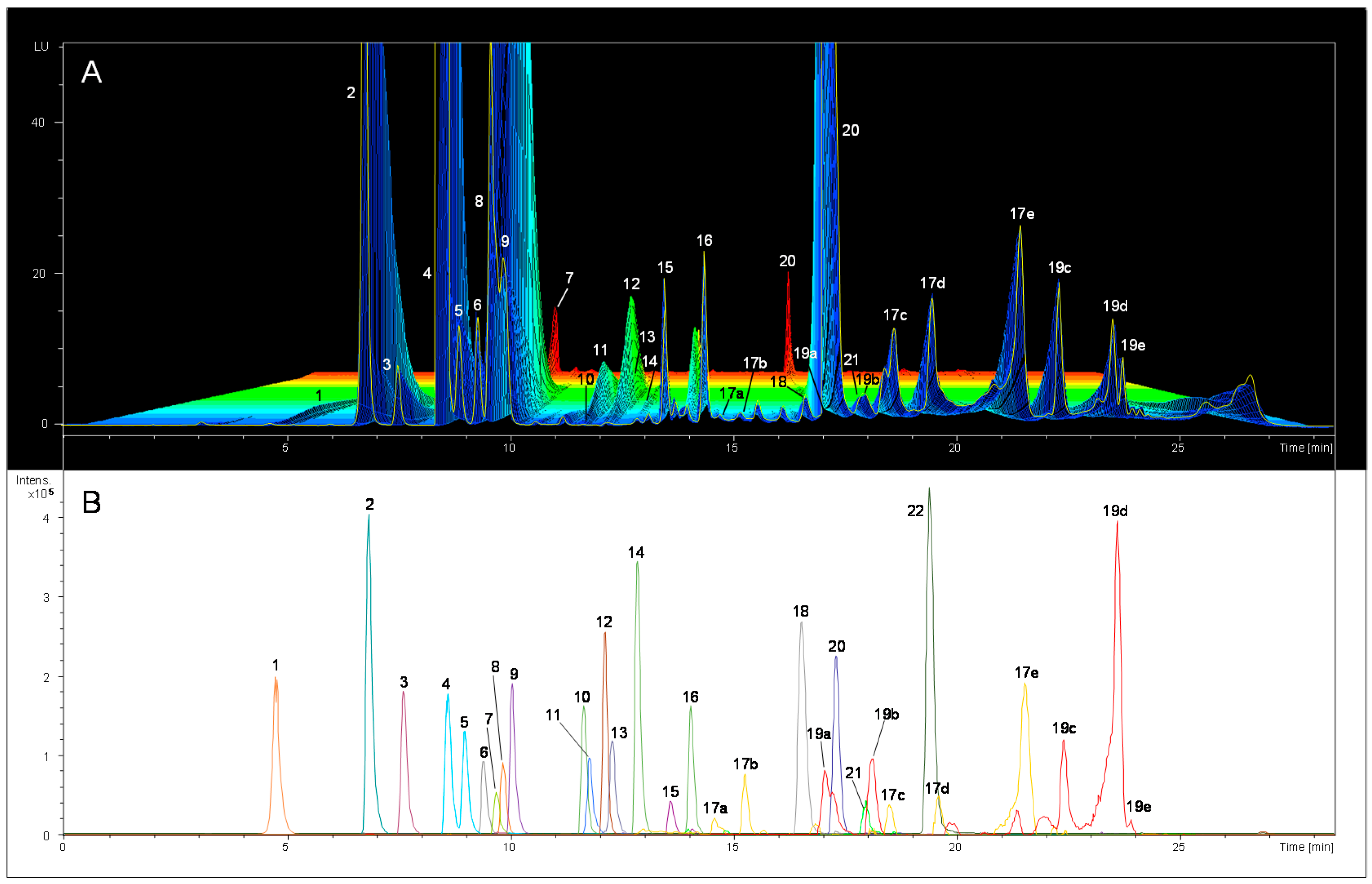
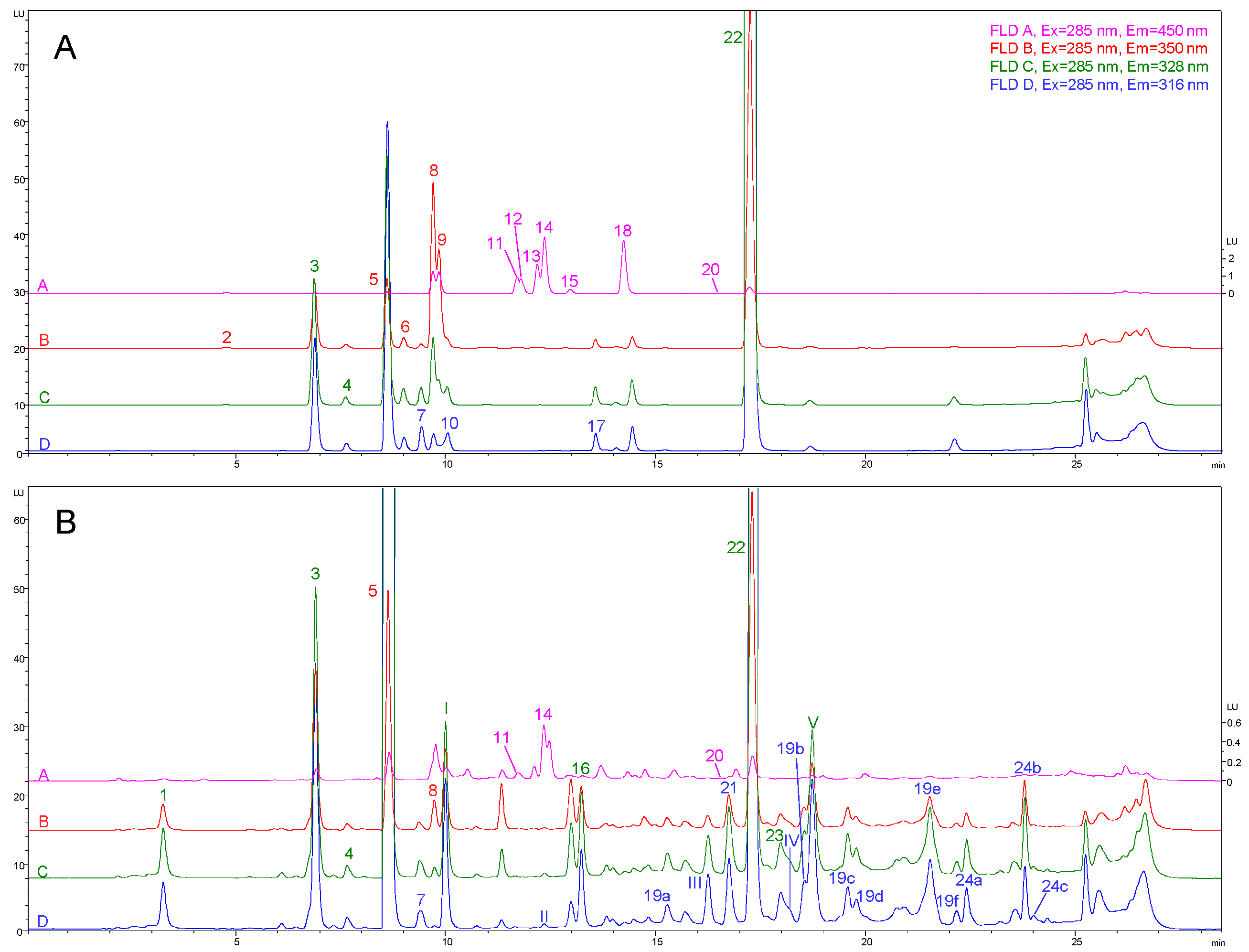
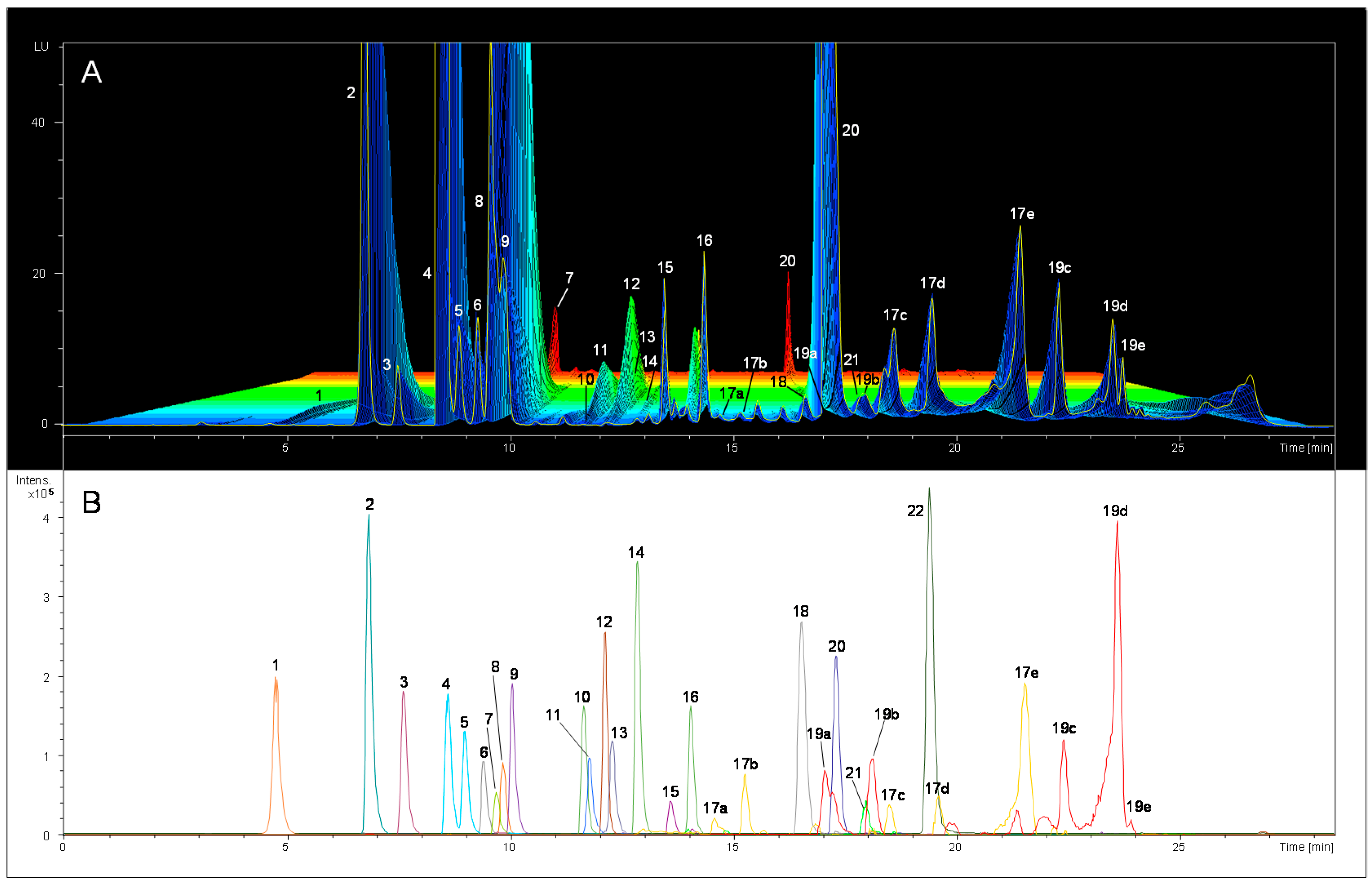
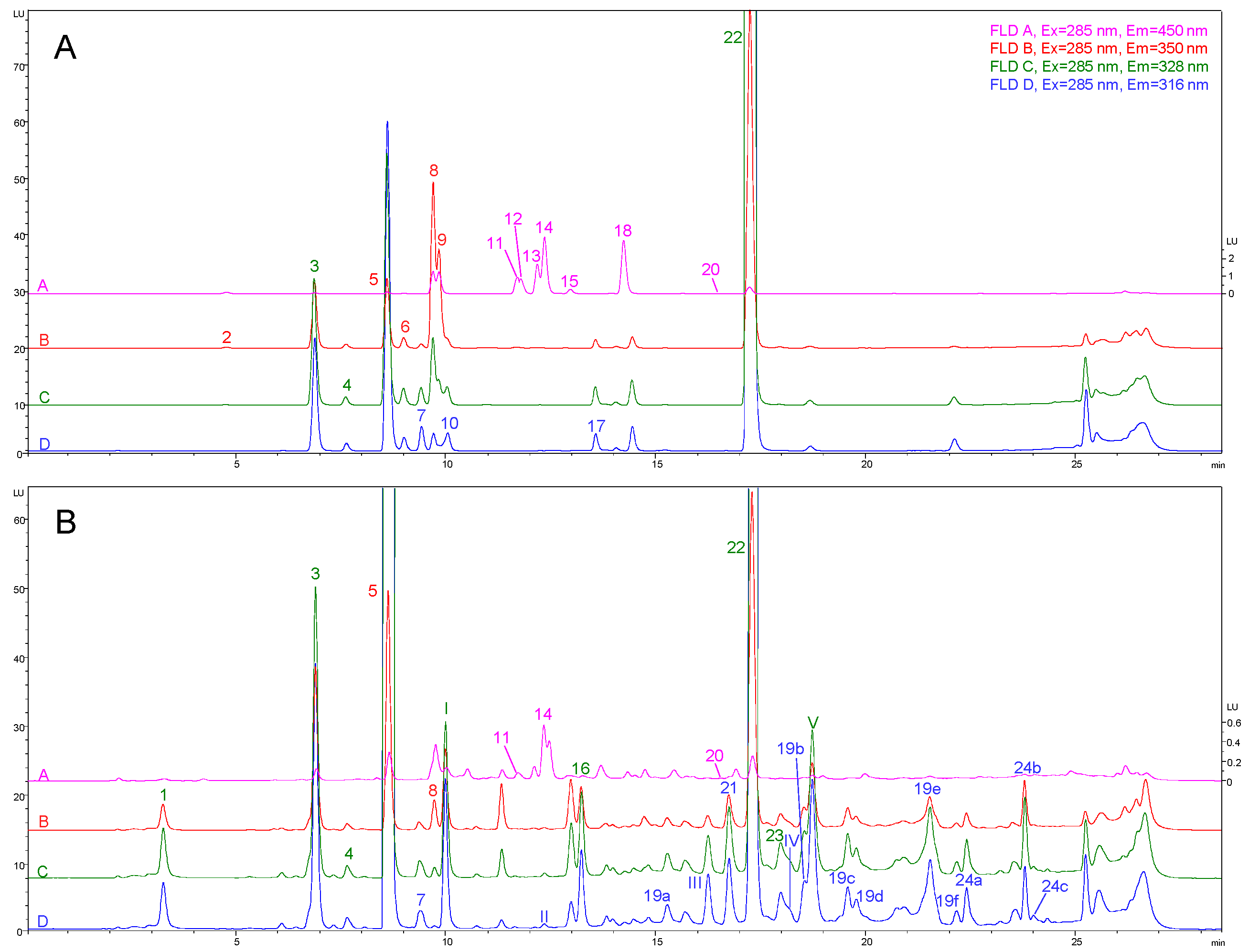
2.1. Preliminary FLD Study and Compounds Identification
2.2. Optimization of Detection Conditions
2.3. Method Validation
2.4. Analysis of Phenolic Compounds in VOO Samples
3. Materials and Methods
3.1. Reagents and Materials
3.2. LC-FLD/MS Analysis
3.3. Samples and Sample Preparation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bendini, A.; Cerretani, L.; Carrasco-Pancorbo, A.; Gómez-Caravaca, A.M.; Segura-Carretero, A.; Fernández-Gutiérrez, A.; Lercker, G. Phenolic molecules in virgin olive oils: A survey of their sensory properties, health effects, antioxidant activity and analytical methods. An overview of the last decade. Molecules 2007, 12, 1679–1719. [Google Scholar] [CrossRef] [PubMed]
- Martín-Peláez, S.; Covas, M.I.; Fitó, M.; Kusar, A.; Pravst, I. Health effects of olive oil polyphenols: Recent advances and possibilities for the use of health claims. Mol. Nutr. Food Res. 2013, 57, 760–771. [Google Scholar] [CrossRef] [PubMed]
- Servili, M.; Sordini, B.; Esposto, S.; Urbani, S.; Veneziani, G.; Di Maio, I.; Selvaggini, R.; Taticchi, A. Biological activities of phenolic compounds of extra virgin olive oil. Antioxidants 2013, 3, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Cerretani, L.; Salvador, M.D.; Bendini, A.; Fregapane, G. Relationship between sensory evaluation performed by italian and spanish official panels and volatile and phenolic profiles of virgin olive oils. Chemosens. Percept. 2008, 1, 258–267. [Google Scholar] [CrossRef]
- Bendini, A.; Valli, E. Sensory analysis of virgin olive oil. In Olive Oil—Constituents, Quality, Health Properties and Bioconversions; InTech: Rijeka, Croatia, 2012; pp. 109–130. [Google Scholar]
- Bajoub, A.; Hurtado-Fernández, E.; Ajal, E.A.; Fernández-Gutiérrez, A.; Carrasco-Pancorbo, A.; Ouazzani, N. Quality and chemical profiles of monovarietal north moroccan olive oils from “picholine marocaine” cultivar: Registration database development and geographical discrimination. Food Chem. 2015, 179, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Bajoub, A.; Carrasco-Pancorbo, A.; Ajal, E.A.; Ouazzani, N.; Fernández-Gutiérrez, A. Potential of LC-MS phenolic profiling combined with multivariate analysis as an approach for the determination of the geographical origin of north moroccan virgin olive oils. Food Chem. 2015, 166, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Oliveras-López, M.J.; Innocenti, M.; Giaccherini, C.; Ieri, F.; Romani, A.; Mulinacci, N. Study of the phenolic composition of spanish and italian monocultivar extra virgin olive oils: Distribution of lignans, secoiridoidic, simple phenols and flavonoids. Talanta 2007, 73, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Sánchez De Medina, V.; Priego-Capote, F.; De Castro, M.D.L. Characterization of monovarietal virgin olive oils by phenols profiling. Talanta 2015, 132, 424–432. [Google Scholar] [CrossRef] [PubMed]
- EFSA. Nda panel, scientific opinion on the substantiation of health claims related to polyphenols in olive and protection of LDL particles from oxidative damage. EFSA J. 2011, 9, 1–25. [Google Scholar]
- Commission Regulation (EU) No 432/2012 of 16 May 2012 Establishing a List of Permitted Health Claims Made on Foods, Other Than those Referring to the Reduction of Disease Risk and to Child. Available online: https://www.fsai.ie/uploadedfiles/consol_reg432_2012.pdf (accessed on 8 July 2016).
- Tsimidou, M.Z.; Boskou, D. The health claim on “olive oil polyphenols” and the need for meaningful terminology and effective analytical protocols. Eur. J. Lipid Sci. Technol. 2015, 117, 1091–1094. [Google Scholar] [CrossRef]
- Motilva, M.-J.; Serra, A.; Maciá, A. Analysis of food polyphenols by ultra high-performance liquid chromatography coupled to mass spectrometry: An overview. J. Chromatogr. A 2013, 1292, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Pancorbo, A.; Cerretani, L.; Bendini, A.; Segura-Carretero, A.; Gallina-Toschi, T.; Fernández-Gutiérrez, A. Analytical determination of polyphenols in olive oil. J. Sep. Sci. 2005, 28, 837–858. [Google Scholar] [CrossRef] [PubMed]
- Brenes, M.; García, A.; Rios, J.; García, P.; Garrido, A. Use of 1-acetoxypinoresinol to authenticate picual olive oils. Int. J. Food Sci. Technol. 2002, 37, 615–625. [Google Scholar] [CrossRef]
- García, A.; Brenes, M.; García, P.; Romero, C.; Garrido, A. Phenolic content of commercial olive oils. Eur. Food Res. Technol. 2003, 216, 520–525. [Google Scholar]
- Ryan, D.; Robards, K.; Lavee, S. Determination of phenolic compounds in olives by reversed-phase chromatography and mass spectrometry. J. Chromatogr. A 1999, 832, 87–96. [Google Scholar] [CrossRef]
- Godoy-Caballero, M.P.; Acedo-Valenzuela, M.I.; Durán-Merás, I.; Galeano-Díaz, T. Development of a non-aqueous capillary electrophoresis method with UV-Visible and fluorescence detection for phenolics compounds in olive oil. Anal. Bioanal. Chem. 2012, 403, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Caballero, M.P.; Acedo-Valenzuela, M.I.; Galeano-Díaz, T. Simple quantification of phenolic compounds present in the minor fraction of virgin olive oil by LC-DAD-FLD. Talanta 2012, 101, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Caballero, M.P.; Galeano-Díaz, T.; Acedo-Valenzuela, M.I. Simple and fast determination of phenolic compounds from different varieties of olive oil by nonaqueous capillary electrophoresis with UV-visible and fluorescence detection. J. Sep. Sci. 2012, 35, 3529–3539. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.W.; Tuck, K.L.; Stupans, I.; Hayball, P.J. Simultaneous determination of oleuropein and hydroxytyrosol in rat plasma using liquid chromatography with fluorescence detection. J. Chromatogr. B 2003, 785, 187–191. [Google Scholar] [CrossRef]
- Selvaggini, R.; Servili, M.; Urbani, S.; Esposto, S.; Taticchi, A.; Montedoro, G. Evaluation of phenolic compounds in virgin olive oil by direct injection in high-performance liquid chromatography with fluorometric detection. J. Agric. Food Chem. 2006, 54, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- El Riachy, M.; Priego-Capote, F.; Rallo, L.; Luque-de Castro, M.D.; León, L. Phenolic profile of virgin olive oil from advanced breeding selections. Span. J. Agric. Res. 2012, 10, 443–453. [Google Scholar] [CrossRef]
- El Riachy, M.; Priego-Capote, F.; Rallo, L.; Luque-de Castro, M.D.; León, L. Phenolic composition of virgin olive oils in cultivars for narrow hedgerow olive orchards. Eur. J. Lipid Sci. Technol. 2013, 115, 800–810. [Google Scholar] [CrossRef]
- Zandomeneghi, M.; Carbonaro, L.; Caffarata, C. Fluorescence of vegetable oils: Olive oils. J. Agric. Food Chem. 2005, 53, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, E.; Khmelinskii, I.; Sikorski, M. Analysis of olive oils by fluorescence spectroscopy: Methods and applications. In Olive Oil—Constituents, Quality, Health Properties and Bioconversions; InTech: Rijeka, Croatia, 2012; pp. 63–88. [Google Scholar]
- Christodouleas, D.; Fotakis, C.; Papadopoulos, K.; Dimotikali, D.; Calokerinos, A.C. Luminescent methods in the analysis of untreated edible oils: A review. Anal. Lett. 2012, 45, 625–641. [Google Scholar] [CrossRef]
- Lipka, E.; Vaccher, C. Quantitative analysis of drugs in biological matrices by HPLC hyphenated to fluorescence detection. Bioanalysis 2015, 7, 743–762. [Google Scholar] [CrossRef] [PubMed]
- Kmellár, B.; Fodor, P.; Pareja, L.; Ferrer, C.; Martínez-Uroz, M.A.; Valverde, A.; Fernandez-Alba, A.R. Validation and uncertainty study of a comprehensive list of 160 pesticide residues in multi-class vegetables by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2008, 1215, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Bajoub, A.; Ajal, E.A.; Fernández-Gutiérrez, A.; Carrasco-Pancorbo, A. Evaluating the potential of phenolic profiles as discriminant features among extra virgin olive oils from moroccan controlled designations of origin. Food Res. Int. 2016, 84, 41–51. [Google Scholar] [CrossRef]
- García-Villalba, R.; Pacchiarotta, T.; Carrasco-Pancorbo, A.; Segura-Carretero, A.; Fernández-Gutiérrez, A.; Deelder, A.M.; Mayboroda, O.A. Gas chromatography-atmospheric pressure chemical ionization-time of flight mass spectrometry for profiling of phenolic compounds in extra virgin olive oil. J. Chromatogr. A 2011, 1218, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Pancorbo, A.; Arráez-Román, D.; Segura-Carretero, A.; Fernández-Gutiérrez, A. Capillary electrophoresis-electrospray ionization-mass spectrometry method to determine the phenolic fraction of extra-virgin olive oil. Electrophoresis 2006, 27, 2182–2196. [Google Scholar] [CrossRef] [PubMed]
- Bajoub, A.; Hurtado-Fernández, E.; Ajal, E.A.; Ouazzani, N.; Fernández-Gutiérrez, A.; Carrasco-Pancorbo, A. Comprehensive 3-year study of the phenolic profile of moroccan monovarietal virgin olive oils from the Meknès region. J. Agric. Food Chem. 2015, 63, 4376–4385. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Compound | Accuracy | Matrix Effect | ||||||
|---|---|---|---|---|---|---|---|---|
| Intra-Day Repeatability (% RSD) a | Inter-Day Repeatability (% RSD) b | Trueness (%) c | Solvent Calibration Slope | Matrix Calibration Slope | Matrix Slope/Solvent Slope | |||
| Area | tr | Area | tr | |||||
| Gal | 2.72 | 0.39 | 5.70 | 0.41 | 92.83 | 0.127 | 0.137 | 1.08 |
| HTY | 6.80 | 0.34 | 6.91 | 0.40 | 103.66 | 10.226 | 9.041 | 0.88 |
| TY | 2.17 | 0.26 | 3.40 | 0.47 | 90.08 | 6.239 | 5.257 | 0.86 |
| 4-HBA | 3.34 | 0.17 | 4.27 | 0.46 | 101.98 | 0.880 | 0.983 | 1.12 |
| 4-HPA | 3.68 | 0.19 | 4.66 | 0.50 | 85.49 | 2.012 | 2.104 | 1.05 |
| Van | 3.68 | 0.21 | 3.74 | 0.51 | 101.01 | 14.276 | 13.384 | 0.94 |
| Syr | 3.49 | 0.25 | 4.80 | 0.54 | 104.71 | 6.770 | 6.748 | 1.00 |
| Hmvan | 9.05 | 0.24 | 11.98 | 0.56 | 87.53 | 1.244 | 1.273 | 1.02 |
| p-Cou | 2.94 | 0.18 | 3.51 | 0.58 | 86.03 | 0.272 | 0.285 | 1.05 |
| Val | 2.94 | 0.23 | 4.42 | 0.53 | 85.28 | 0.299 | 0.323 | 1.08 |
| Sin | 3.75 | 0.26 | 4.38 | 0.63 | 80.55 | 0.618 | 0.678 | 1.10 |
| Fer | 2.35 | 0.24 | 2.79 | 0.61 | 88.06 | 1.287 | 1.410 | 1.10 |
| m-Cou | 3.33 | 0.21 | 5.93 | 0.63 | 111.81 | 0.083 | 0.087 | 1.05 |
| Ole | 4.32 | 0.34 | 6.83 | 0.81 | 112.28 | 1.246 | 1.411 | 1.13 |
| o-Cou | 2.52 | 0.24 | 2.92 | 0.70 | 84.27 | 1.170 | 1.288 | 1.10 |
| Lut | * | * | * | * | 100.74 | 0.009 | 0.011 | 1.16 |
| Pin | 2.35 | 0.36 | 2.13 | 0.99 | 100.46 | 40.452 | 44.258 | 1.09 |
| Analyte | Detector | External Calibration Curve | R2 | LOD (μg·mL−1) | LOQ (μg·mL−1) | Linear Range (μg·mL−1) a |
|---|---|---|---|---|---|---|
| Gal | FLD | y = 0.127x + 0.1263 | 0.9987 | 0.625 | 2.083 | 250 |
| MS | y = 40519x + 37647 | 0.9965 | 0.051 | 0.171 | 100 | |
| HTY | FLD | y = 10.226x + 2.1124 | 0.9989 | 0.035 | 0.115 | 20 |
| MS | y = 52263x + 55720 | 0.9945 | 0.017 | 0.057 | 50 | |
| TY | FLD | y = 6.239x + 3.3395 | 0.9993 | 0.009 | 0.029 | 100 |
| MS | y = 20379x + 17714 | 0.9924 | 0.058 | 0.195 | 50 | |
| 4-HBA | FLD | y = 0.880x + 2.804 | 0.9954 | 0.036 | 0.121 | 250 |
| MS | y = 16457x + 49330 | 0.9911 | 0.061 | 0.204 | 150 | |
| 4-HPA | FLD | y = 2.012x + 2.6778 | 0.9982 | 0.357 | 1.190 | 250 |
| MS | y = 13242x − 2729.7 | 0.9938 | 0.122 | 0.407 | 150 | |
| Van | FLD | y = 14.276x − 0.4684 | 0.9989 | 0.004 | 0.013 | 20 |
| MS | y = 16919x + 11618 | 0.9956 | 0.025 | 0.084 | 10 | |
| Syr | FLD | y = 6.770x + 8.7496 | 0.9861 | 0.007 | 0.023 | 50 |
| MS | y = 27088x + 14618 | 0.9928 | 0.025 | 0.083 | 10 | |
| Hmvan | FLD | y = 1.244x + 0.4454 | 0.9981 | 0.556 | 1.852 | 250 |
| MS | y = 31576x − 42195 | 0.9983 | 0.155 | 0.515 | 50 | |
| p-Cou | FLD | y = 0.272x + 1.036 | 0.9932 | 0.031 | 0.103 | 250 |
| MS | y = 40281x + 21180 | 0.9941 | 0.018 | 0.059 | 20 | |
| Val | FLD | y = 0.299x + 0.631 | 0.9974 | 0.090 | 0.301 | 250 |
| MS | y = 12440x + 7246.2 | 0.9927 | 0.055 | 0.182 | 10 | |
| Sin | FLD | y = 0.618x + 0.8065 | 0.9986 | 0.039 | 0.132 | 250 |
| MS | y = 58259x + 28312 | 0.9939 | 0.021 | 0.069 | 10 | |
| Fer | FLD | y = 1.287x + 3.1028 | 0.9965 | 0.021 | 0.069 | 250 |
| MS | y = 39342x + 13674 | 0.9949 | 0.022 | 0.073 | 10 | |
| m-Cou | FLD | y = 0.083x + 0.122 | 0.9995 | 0.306 | 1.020 | 250 |
| MS | y = 58990x + 130287 | 0.9934 | 0.013 | 0.043 | 100 | |
| Ole | FLD | y = 1.246x + 0.1575 | 0.9998 | 0.273 | 0.909 | 250 |
| MS | y = 6709x + 86 | 0.9990 | 0.050 | 0.165 | 20 | |
| o-Cou | FLD | y = 1.170x + 3.713 | 0.9954 | 0.022 | 0.072 | 250 |
| MS | y = 33291x + 12614 | 0.9946 | 0.077 | 0.255 | 100 | |
| Lut | FLD | y = 0.009x + 0.0064 | 0.9984 | 7.143 | 23.810 | 250 |
| MS | y = 201100x + 44947 | 0.9994 | 0.005 | 0.016 | 10 | |
| Pin | FLD | y = 40.452x + 7.795 | 0.9991 | 0.005 | 0.016 | 10 |
| MS | y = 34634x + 63107 | 0.9912 | 0.032 | 0.108 | 50 |
| Compound | Detector | Arbequina 1 | Arbequina 2 | Arauco 1 | Arauco 2 | Changlot | Hojiblanca | Picual | Blend 1 | Blend 2 | Blend 3 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HTY | FLD | 0.46 ± 0.03 | 5.0 ± 0.3 | 5.8 ± 0.4 | 4.3 ± 0.3 | 5.0 ± 0.3 | 0.66 ± 0.04 | 11.2 ± 0.8 | 3.0 ± 0.2 | 19.2 ± 1.2 | 1.6 ± 0.1 |
| MS | 0.48 ± 0.03 | 4.8 ± 0.3 | 6.3 ± 0.4 | 4.2 ± 0.3 | 5.0 ± 0.3 | 0.74 ± 0.05 | 11.4 ± 0.5 | 2.8 ± 0.2 | 17 ± 1 | 1.5 ± 0.1 | |
| TY | FLD | 2.8 ± 0.2 | 4.9 ± 0.3 | 13.9 ± 0.8 | 11.7 ± 0.8 | 6.8 ± 0.3 | 2.0 ± 0.1 | 14.5 ± 0.7 | 15.3 ± 0.9 | 24 ± 1 | 1.2 ± 0.1 |
| MS | 3.1 ± 0.2 | 4.7 ± 0.2 | 15 ± 1 | 10.2 ± 0.7 | 6.2 ± 0.3 | 2.3 ± 0.2 | 15.8 ± 0.7 | 14.8 ± 0.6 | 24 ± 1 | 1.2 ± 0.1 | |
| 4-HBA | FLD | n.d. | n.d. | n.d. | n.d. | n.d. | 0.29 ± 0.01 | n.d. | n.d. | n.d. | n.d. |
| MS | n.d. | n.d. | n.d. | n.d. | n.d. | 0.31 ± 0.01 | n.d. | n.d. | n.d. | n.d. | |
| 4-HPA | FLD | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 1.68 ± 0.07 | n.d. | n.d. | n.d. |
| MS | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 1.72 ± 0.07 | n.d. | n.d. | n.d. | |
| Van | FLD | 0.29 ± 0.02 | 0.45 ± 0.03 | 0.91 ± 0.06 | 0.39 ± 0.02 | n.q. | 0.43 ± 0.02 | 0.76 ± 0.04 | 2.0 ± 0.1 | 0.61 ± 0.02 | n.d. |
| MS | 0.26 ± 0.02 | 0.41 ± 0.02 | 0.82 ± 0.04 | 0.43 ± 0.02 | n.d. | 0.40 ± 0.02 | 0.68 ± 0.04 | 1.8 ± 0.1 | 0.58 ± 0.03 | n.d. | |
| p-Cou | FLD | 0.11 ± 0.01 | 0.21 ± 0.01 | 0.61 ± 0.04 | 0.32 ± 0.02 | 0.25 ± 0.01 | 0.28 ± 0.02 | 0.38 ± 0.02 | n.q. | 0.15 ± 0.01 | n.q. |
| MS | 0.13 ± 0.01 | 0.21 ± 0.01 | 0.57 ± 0.03 | 0.35 ± 0.02 | 0.25 ± 0.01 | 0.25 ± 0.02 | 0.36 ± 0.02 | 0.061 ± 0.003 | 0.17 ± 0.01 | 0.093 ± 0.005 | |
| Val | FLD | n.q. | 0.51 ± 0.02 | n.q. | n.q. | n.q. | n.q. | n.d. | n.q. | n.q. | 0.66 ± 0.03 |
| MS | 0.134 ± 0.007 | 0.52 ± 0.03 | 0.102 ± 0.004 | 0.113 ± 0.006 | 0.135 ± 0.007 | n.q. | n.q. | n.q. | 0.143 ± 0.008 | 0.70 ± 0.03 | |
| Fer | FLD | 0.058 ± 0.002 | 0.081 ± 0.004 | 0.166 ± 0.005 | 0.076 ± 0.002 | 0.126 ± 0.005 | n.q. | 0.071 ± 0.002 | n.q. | n.q. | n.d. |
| MS | 0.055 ± 0.002 | 0.080 ± 0.004 | 0.176 ± 0.006 | 0.074 ± 0.002 | 0.122 ± 0.005 | n.q. | 0.069 ± 0.002 | n.q. | n.q. | n.d. | |
| Lut | FLD | n.q. | n.q. | n.q. | n.q. | n.d. | n.q. | n.d. | n.d. | n.d. | n.d. |
| MS | 5.7 ± 0.3 | 4.4 ± 0.1 | 6.2 ± 0.3 | 5.3 ± 0.3 | 2.6 ± 0.1 | 7.0 ± 0.4 | 3.4 ± 0.2 | 1.22 ± 0.07 | 1.20 ± 0.06 | 3.0 ± 0.1 | |
| Pin | FLD | 2.09 ± 0.09 * | 3.0 ± 0.1 | 0.95 ± 0.04 | 0.90 ± 0.04 * | 2.7 ± 0.1 * | 0.81 ± 0.05 | 4.0 ± 0.2 * | 3.1 ± 0.1 * | 0.77 ± 0.05 | 3.3 ± 0.2 |
| MS | 0.91 ± 0.05 * | 3.0 ± 0.1 | 0.87 ± 0.05 | 0.39 ± 0.02 * | 1.6 ± 0.1 * | 0.71 ± 0.05 | 2.4 ± 0.1 * | 1.3 ± 0.1 * | 0.68 ± 0.05 | 2.9 ± 0.2 |
| Compound | Arbequina 1 | Arbequina 2 | Arauco 1 | Arauco 2 | Changlot | Hojiblanca | Picual | Blend 1 | Blend 2 | Blend 3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| OxHTY | 0.163 ± 0.008 | 1.87 ± 0.09 | 1.91 ± 0.09 | 0.19 ± 0.01 | 0.34 ± 0.02 | 0.122 ± 0.006 | 2.3 ± 0.1 | n.q. | 1.9 ± 0.1 | 0.51 ± 0.02 | |
| AcHTY | 1.34 ± 0.07 | 0.47 ± 0.02 | 1.10 ± 0.06 | 0.41 ± 0.02 | n.d. | 0.27 ± 0.01 | 2.9 ± 0.2 | n.d. | 9.2 ± 0.5 | 0.20 ± 0.01 | |
| OleAgly | Isomer 1 | 0.74 ± 0.04 | 1.04 ± 0.05 | 2.7 ± 0.1 | 5.4 ± 0.3 | n.d. | n.d. | n.q. | 10.8 ± 0.5 | 1.10 ± 0.06 | 6.1 ± 0.3 |
| Isomer 2 | n.d. | n.d. | n.d. | 25 ± 1 | 19.2 ± 0.9 | n.d. | 15.8 ± 0.6 | 115 ± 6 | 48 ± 2 | 43 ± 2 | |
| Isomer 3 | 1.50 ± 0.07 | 2.6 ± 0.1 | 11.7 ± 0.6 | 85 ± 4 | 58 ± 3 | 2.1 ± 0.1 | n.q. | 7.8 ± 0.4 | 13.3 ± 0.6 | 3.4 ± 0.2 | |
| Isomer 4 | 2.0 ± 0.1 | 2.4 ± 0.2 | 5.5 ± 0.3 | 8.9 ± 0.5 | 3.3 ± 0.2 | 1.46 ± 0.07 | 12.7 ± 0.8 | 10.1 ± 0.5 | n.d. | 3.9 ± 0.2 | |
| Main isomer | 12.7 ± 0.6 | 7.5 ± 0.4 | 32 ± 2 | 147 ± 7 | 87 ± 4 | 6.5 ± 0.3 | 36 ± 2 | 24 ± 1 | 28 ± 1 | 11.1 ± 0.6 | |
| Isomer 5 | 4.1 ± 0.2 | 3.1 ± 0.1 | 1.16 ± 0.04 | 3.1 ± 0.1 | 2.5 ± 0.1 | 2.6 ± 0.1 | 4.3 ± 0.2 | 19.1 ± 0.8 | 3.7 ± 0.2 | 3.8 ± 0.1 | |
| Total | 21.1 ± 0.6 | 16.6 ± 0.4 | 53 ± 2 | 274 ± 8 | 170 ± 5 | 12.8 ± 0.3 | 69 ± 2 | 187 ± 6 | 94 ± 1 | 72 ± 2 | |
| DOA | 12.1 ± 0.6 | 23 ± 1 | 11.5 ± 0.6 | 11.8 ± 0.6 | 16.6 ± 0.9 | 5.4 ± 0.2 | 25 ± 1 | 18.4 ± 0.9 | 10.0 ± 0.4 | 11.6 ± 0.6 | |
| AcPin | 11.1 ± 0.6 | 2.5 ± 0.1 | 1.21 ± 0.06 | 1.02 ± 0.05 | 3.2 ± 0.1 | 0.82 ± 0.05 | 1.08 ± 0.05 | 3.2 ± 0.1 | 1.52 ± 0.08 | 2.14 ± 0.09 | |
| LigAgly | Isomer 1 | 10.6 ± 0.6 | 2.8 ± 0.2 | 10.8 ± 0.6 | 103 ± 6 | 38 ± 2 | 5.9 ± 0.4 | 17 ± 1 | 19 ± 1 | 7.1 ± 0.4 | 4.7 ± 0.2 |
| Main isomer | 7.6 ± 0.4 | 18.7 ± 0.9 | 9.6 ± 0.5 | 11.3 ± 0.6 | 10.3 ± 0.5 | 4.4 ± 0.2 | 18.8 ± 0.9 | 36 ± 2 | 6.4 ± 0.3 | 16.2 ± 0.8 | |
| Isomer 2 | 6.7 ± 0.3 | n.d. | 5.9 ± 0.3 | 5.1 ± 0.3 | 6.1 ± 0.3 | 5.8 ± 0.3 | 8.4 ± 0.4 | 11.4 ± 0.5 | 16.1 ± 0.8 | n.d. | |
| Total | 24.8 ± 0.8 | 21.4 ± 0.9 | 26.2 ± 0.8 | 119 ± 6 | 54 ± 2 | 16.1 ± 0.5 | 44 ± 1 | 67 ± 2 | 30 ± 1 | 20.9 ± 0.8 | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monasterio, R.P.; Olmo-García, L.; Bajoub, A.; Fernández-Gutiérrez, A.; Carrasco-Pancorbo, A. Potential of LC Coupled to Fluorescence Detection in Food Metabolomics: Determination of Phenolic Compounds in Virgin Olive Oil. Int. J. Mol. Sci. 2016, 17, 1627. https://doi.org/10.3390/ijms17101627
Monasterio RP, Olmo-García L, Bajoub A, Fernández-Gutiérrez A, Carrasco-Pancorbo A. Potential of LC Coupled to Fluorescence Detection in Food Metabolomics: Determination of Phenolic Compounds in Virgin Olive Oil. International Journal of Molecular Sciences. 2016; 17(10):1627. https://doi.org/10.3390/ijms17101627
Chicago/Turabian StyleMonasterio, Romina P., Lucía Olmo-García, Aadil Bajoub, Alberto Fernández-Gutiérrez, and Alegría Carrasco-Pancorbo. 2016. "Potential of LC Coupled to Fluorescence Detection in Food Metabolomics: Determination of Phenolic Compounds in Virgin Olive Oil" International Journal of Molecular Sciences 17, no. 10: 1627. https://doi.org/10.3390/ijms17101627
APA StyleMonasterio, R. P., Olmo-García, L., Bajoub, A., Fernández-Gutiérrez, A., & Carrasco-Pancorbo, A. (2016). Potential of LC Coupled to Fluorescence Detection in Food Metabolomics: Determination of Phenolic Compounds in Virgin Olive Oil. International Journal of Molecular Sciences, 17(10), 1627. https://doi.org/10.3390/ijms17101627