
Comparative Transcriptome Profile of the Cytoplasmic Male Sterile and Fertile Floral Buds of Radish (Raphanus sativus L.)


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. The Floral Bud Size Is Associated with the Microspore Developmental Stage


2.2. Illumina Sequencing
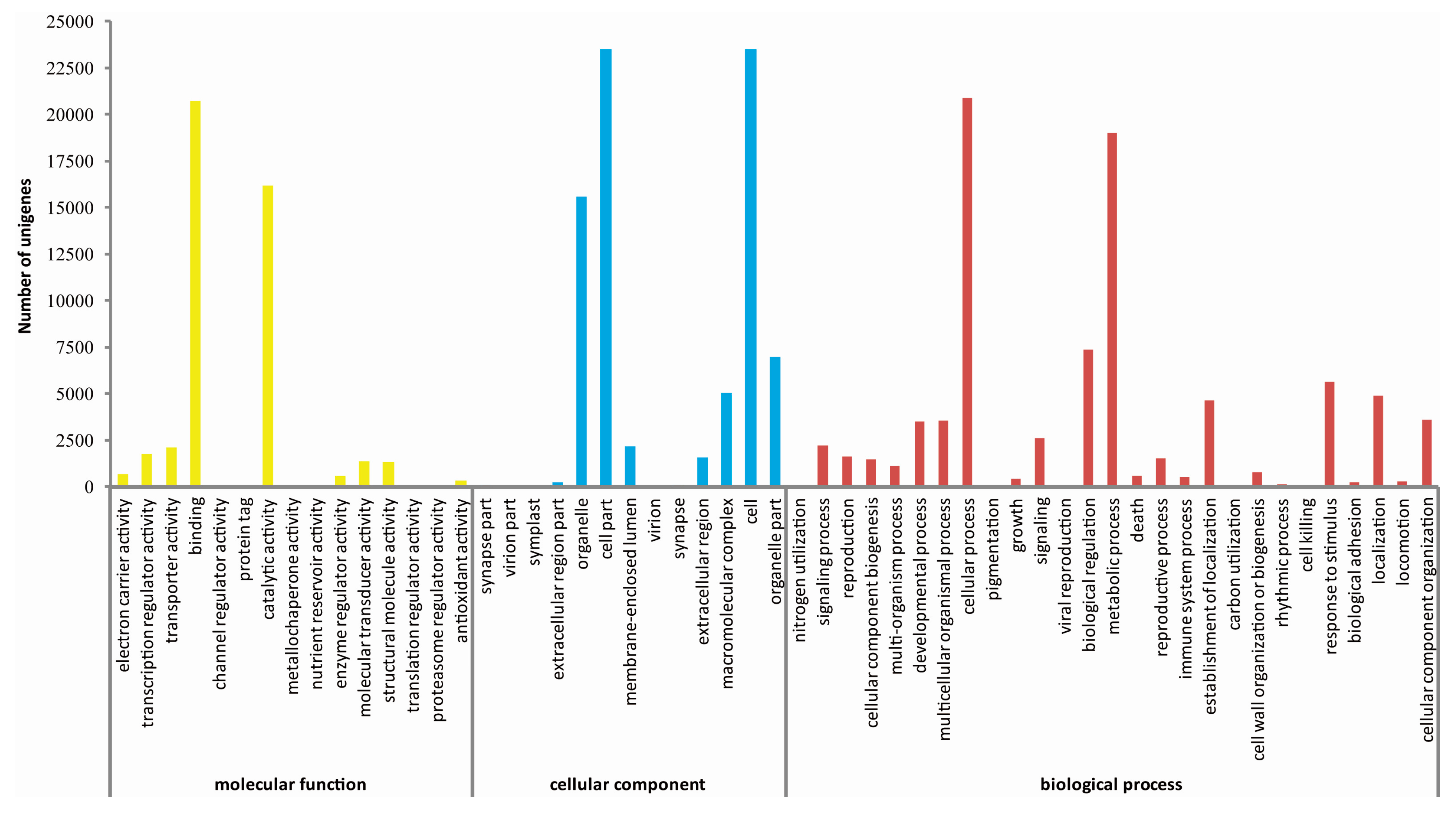
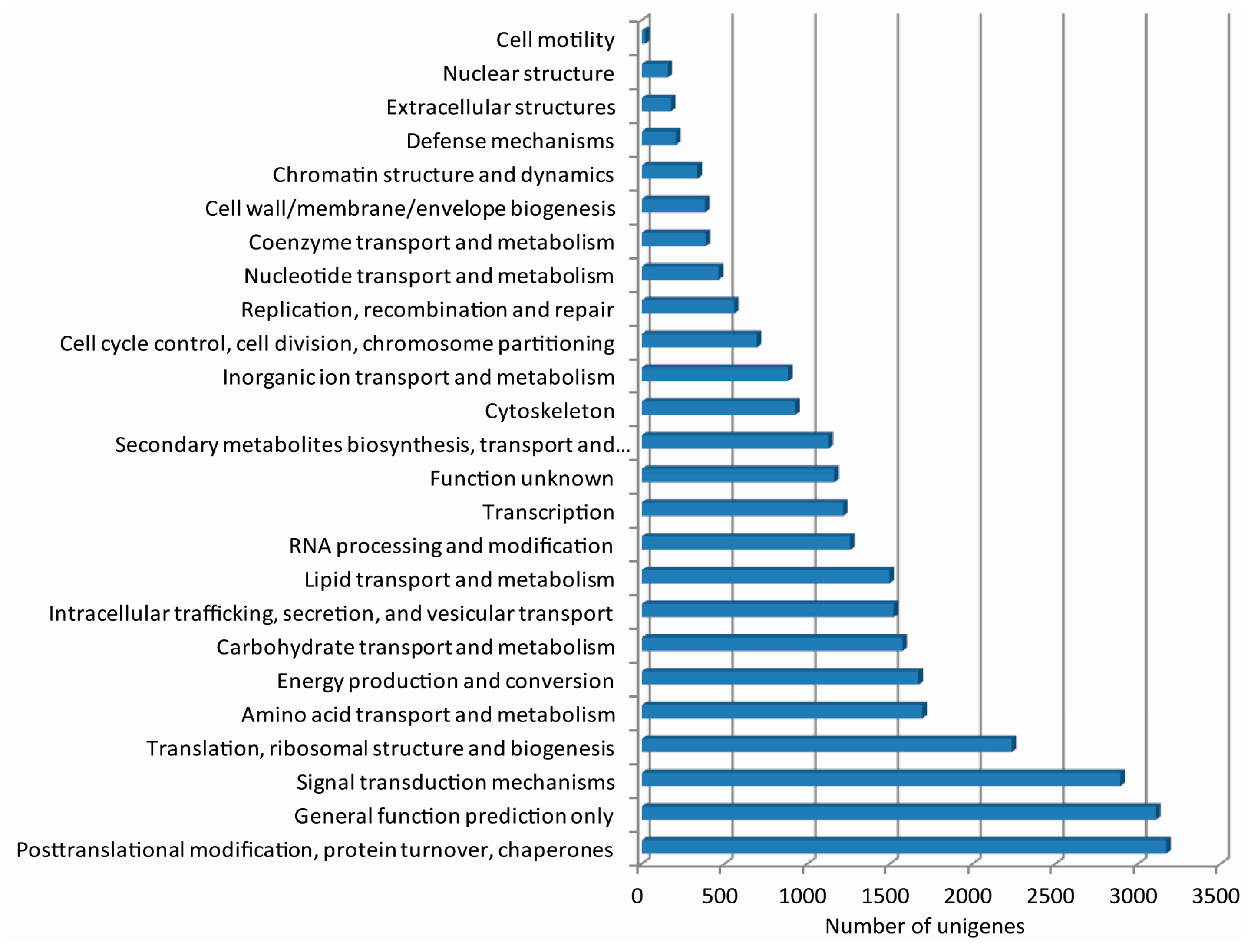
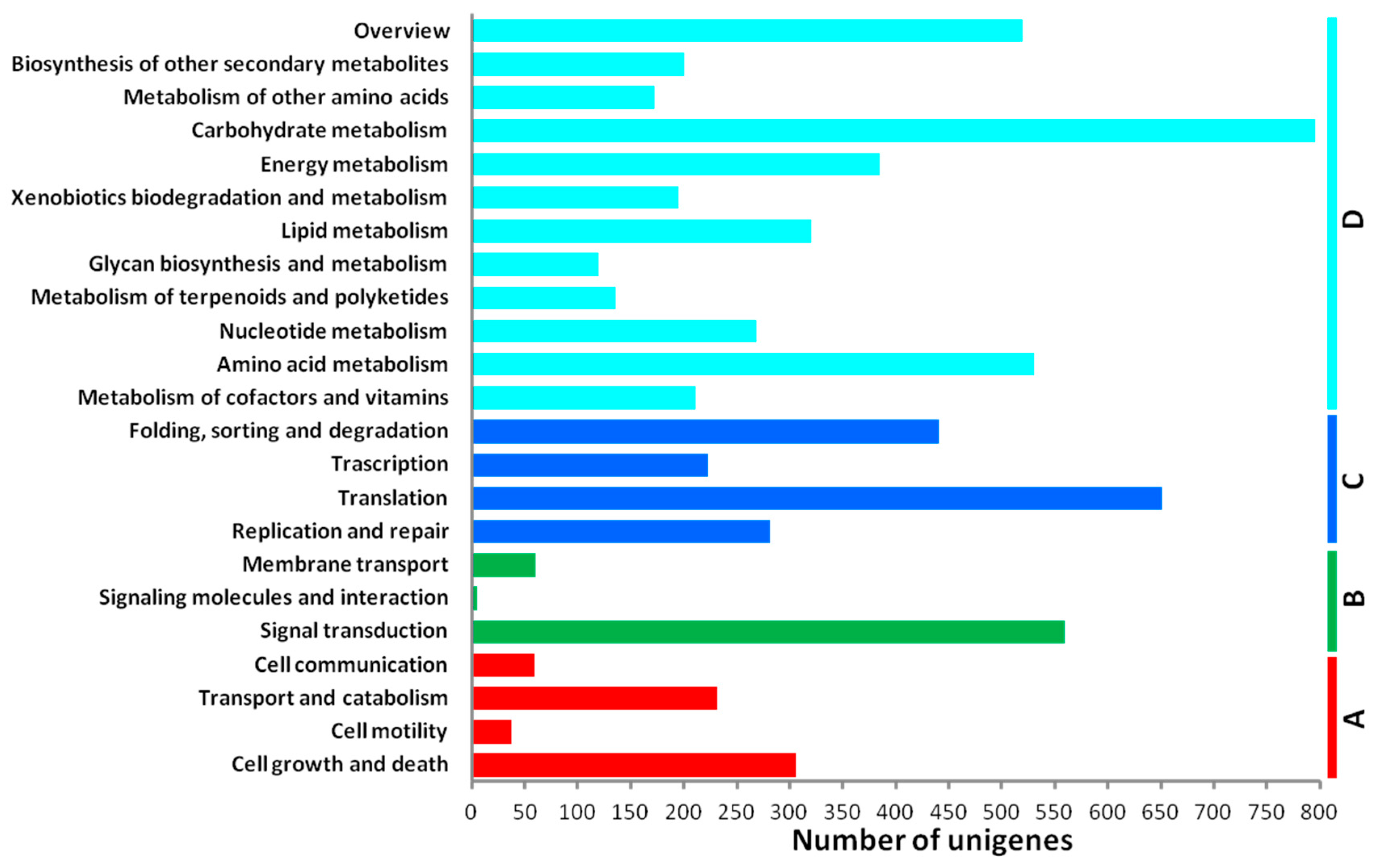
2.3. De Novo Assembly and Functional Classification
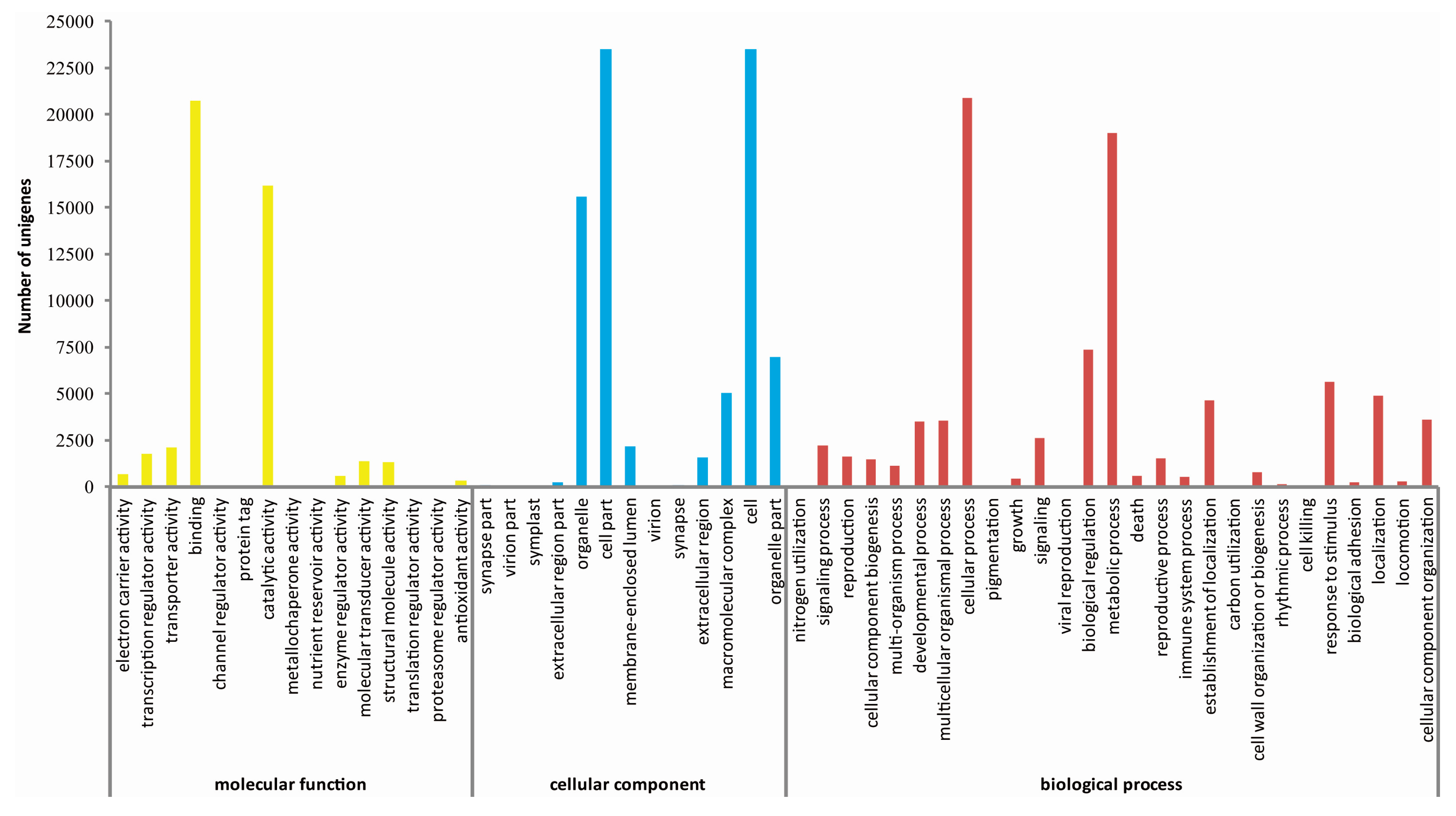
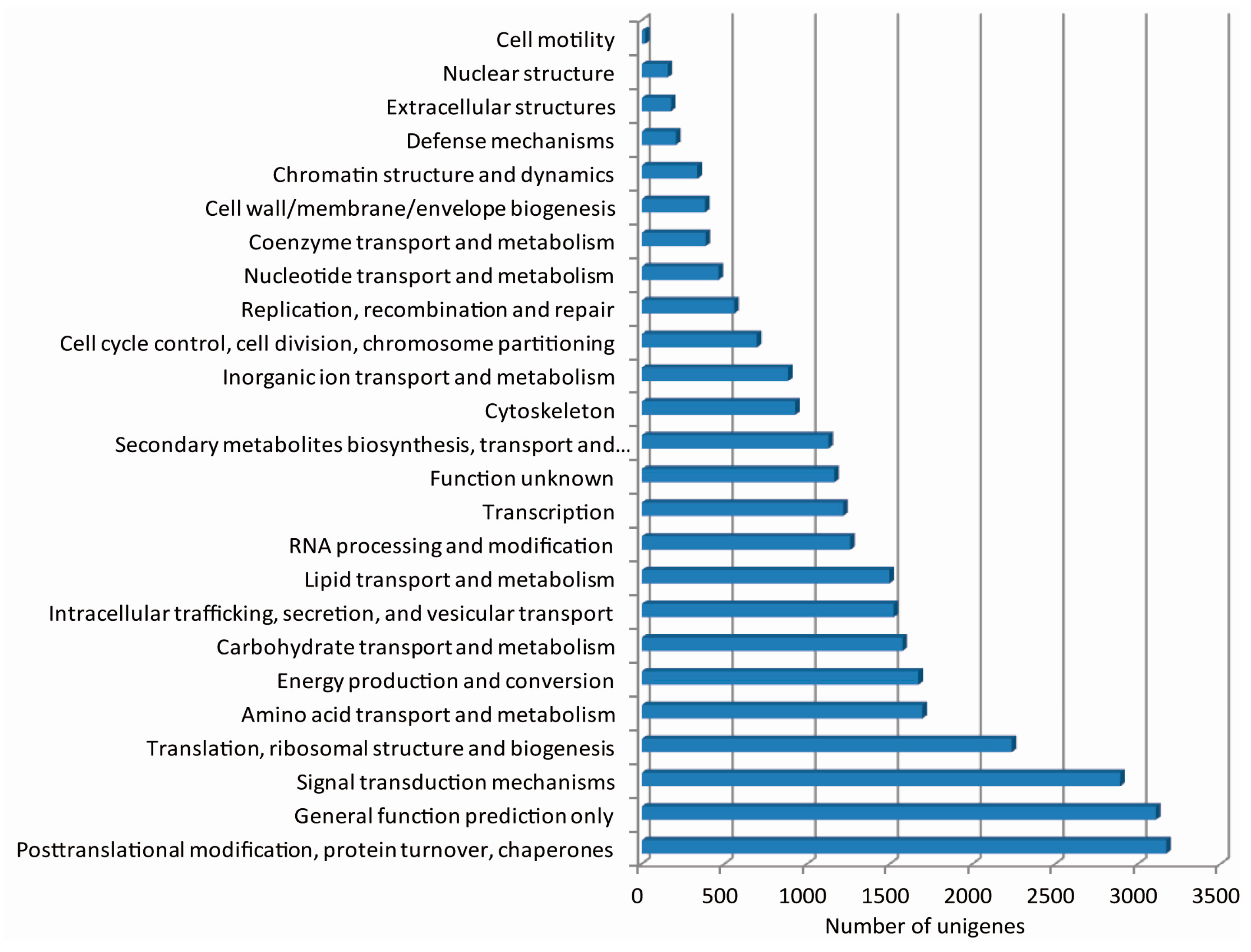
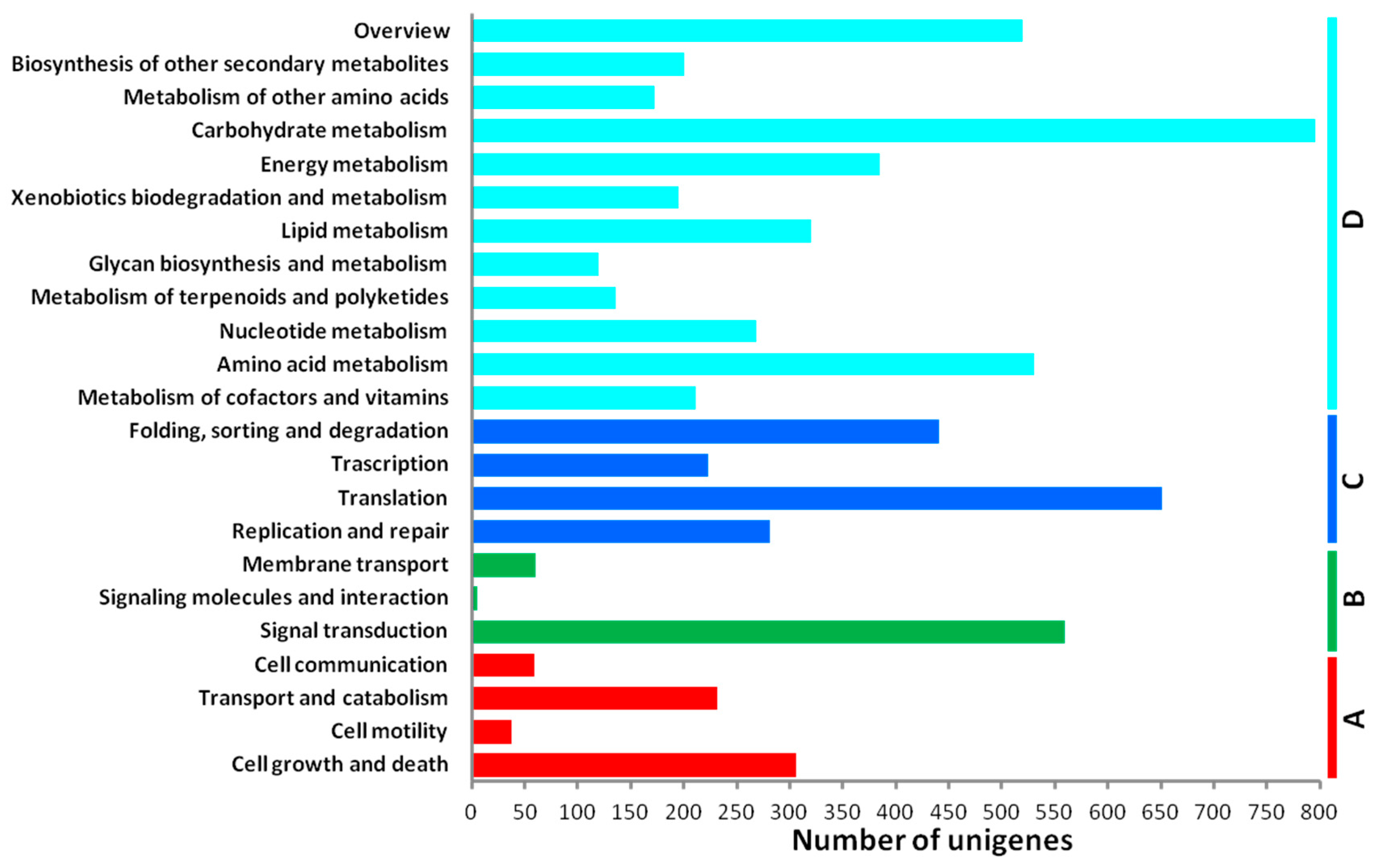
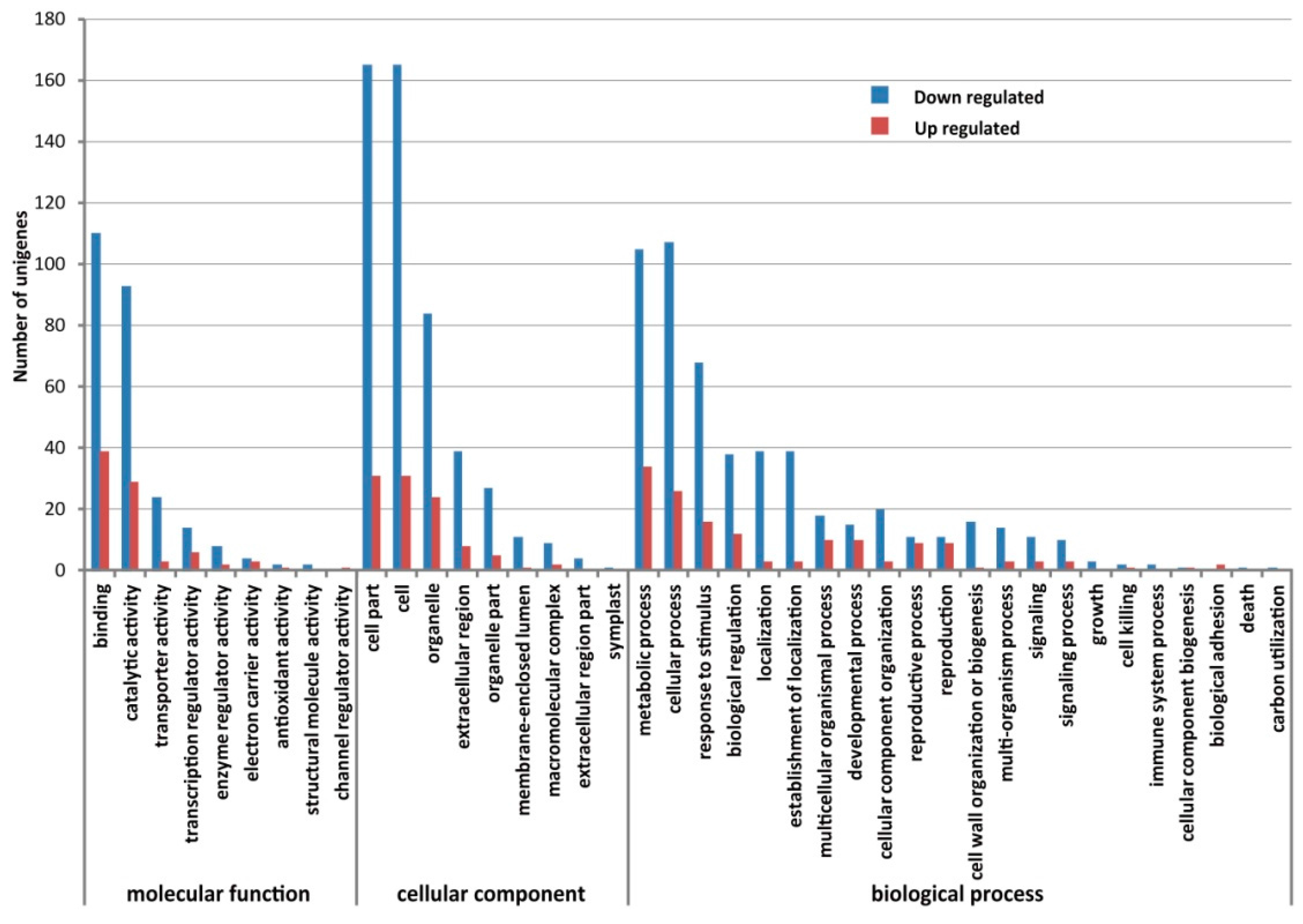
2.4. Comparison of Gene Expression Levels between CMS and Maintainer Lines
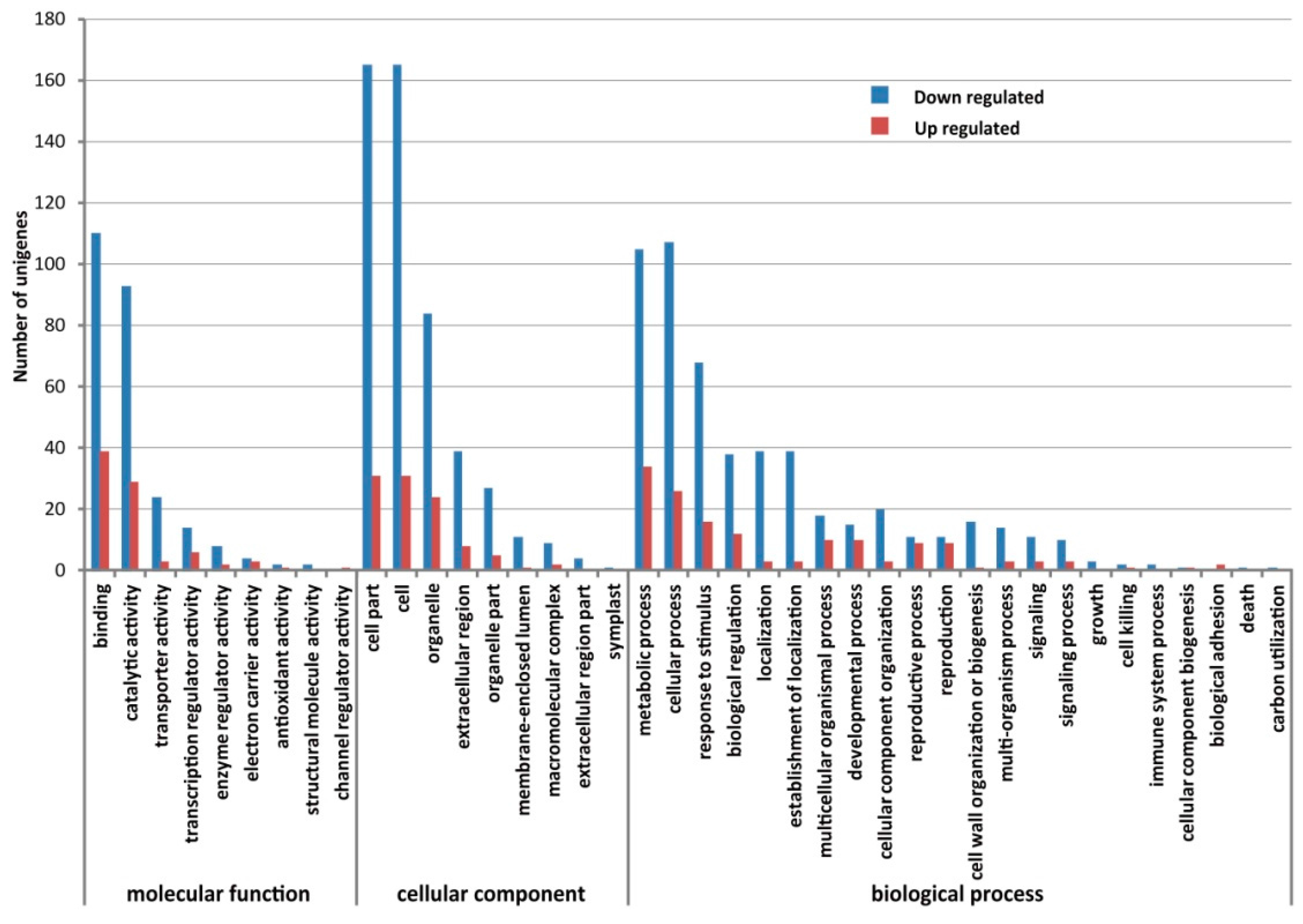
2.5. Validation of the Expression Difference of DEGs
3. Experimental Section
3.1. Plant Materials and RNA Extraction
3.2. Histological Analysis
3.3. RNA Extraction and RNA-Seq
3.4. De Novo Assembly and Database Search
3.5. Differential Gene Expression
3.6. Real-Time Quantitative PCR (qRT-PCR) Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, L.; Liu, Y.G. Male sterility and fertility restoration in crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.; Dymshits, G. Cytoplasmic male sterility and restoration of pollen fertility in higher plants. Russ. J. Genet. 2007, 43, 354–368. [Google Scholar] [CrossRef]
- Luo, D.; Xu, H.; Liu, Z.; Guo, J.; Li, H.; Chen, L.; Fang, C.; Zhang, Q.; Bai, M.; Yao, N. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat. Genet. 2013, 45, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Schnable, P.S.; Wise, R.P. The molecular basis of cytoplasmic male sterility and fertility restoration. Trends Plant Sci. 1998, 3, 175–180. [Google Scholar] [CrossRef]
- Hanson, M.R.; Bentolila, S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 2004, 16, S154–S169. [Google Scholar] [CrossRef] [PubMed]
- L’Homme, Y.; Stahl, R.J.; Li, X.Q.; Hameed, A.; Brown, G.G. Brassica nap cytoplasmic male sterility is associated with expression of a mtDNA region containing a chimeric gene similar to the pol CMS-associated orf224 gene. Curr. Genet. 1997, 31, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Nivison, H.T.; Sutton, C.A.; Wilson, R.K.; Hanson, M.R. Sequencing, processing, and localization of the petunia CMS-associated mitochondrial protein. Plant J. 1994, 5, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zou, Y.; Li, X.; Zhang, Q.; Chen, L.; Wu, H.; Su, D.; Chen, Y.; Guo, J.; Luo, D.; et al. Cytoplasmic male sterility of rice with Boro II cytoplasm is caused by a cytotoxic peptide and is restored by two related PPR motif genes via distinct modes of mRNA silencing. Plant Cell 2006, 18, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Wise, R.; Fliss, A.; Pring, D.; Gengenbach, B. urf13-T of T cytoplasm maize mitochondria encodes a 13 kD polypeptide. Plant Mol. Biol. 1987, 9, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Wolf-Litman, O.; Soferman, O.; Tabib, Y.; Izhar, S. Interaction of the mitochondrial S-Pcf locus for cytoplasmic male sterility in Petunia with multiple fertility-restoration genes in somatic hybrid plants. Theor. Appl. Genet. 1992, 84, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Gao, F.; Ji, Y.; Liu, Y.; Dan, Z.; Yang, P.; Zhu, Y.; Li, S. ORFH79 impairs mitochondrial function via interaction with a subunit of electron transport chain complex III in Honglian cytoplasmic male sterile rice. New Phytol. 2013, 198, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Wise, R.P.; Schnable, P.S. The rf2 nuclear restorer gene of male-sterile T-cytoplasm maize. Science 1996, 272, 1334–1335. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Toriyama, K. Suppressed expression of RETROGRADE-REGULATED MALE STERILITY restores pollen fertility in cytoplasmic male sterile rice plants. Proc. Natl. Acad. Sci. USA 2009, 106, 9513–9518. [Google Scholar] [CrossRef] [PubMed]
- Itabashi, E.; Iwata, N.; Fujii, S.; Kazama, T.; Toriyama, K. The fertility restorer gene, Rf2, for Lead Rice-type cytoplasmic male sterility of rice encodes a mitochondrial glycine-rich protein. Plant J. 2011, 65, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Kitazaki, K.; Arakawa, T.; Matsunaga, M.; Yui-Kurino, R.; Matsuhira, H.; Mikami, T.; Kubo, T. Post-translational mechanisms are associated with fertility restoration of cytoplasmic male sterility in sugar beet. Plant J. 2015, 83, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Matsuhira, H.; Kagami, H.; Kurata, M.; Kitazaki, K.; Matsunaga, M.; Hamaguchi, Y.; Hagihara, E.; Ueda, M.; Harada, M.; Muramatsu, A. Unusual and typical features of a novel restorer-of-fertility gene of sugar beet (Beta vulgaris L.). Genetics 2012, 192, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Bentolila, S.; Alfonso, A.A.; Hanson, M.R. A pentatricopeptide repeat-containing gene restores fertility to cytoplasmic male-sterile plants. Proc. Natl. Acad. Sci. USA 2002, 99, 10887–10892. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.G.; Formanová, N.; Jin, H.; Wargachuk, R.; Dendy, C.; Patil, P.; Laforest, M.; Zhang, J.; Cheung, W.Y.; Landry, B.S. The radish Rfo restorer gene of Ogura cytoplasmic male sterility encodes a protein with multiple pentatricopeptide repeats. Plant J. 2003, 35, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Desloire, S.; Gherbi, H.; Laloui, W.; Marhadour, S.; Clouet, V.; Cattolico, L.; Falentin, C.; Giancola, S.; Renard, M.; Budar, F. Identification of the fertility restoration locus, Rfo, in radish, as a member of the pentatricopeptide-repeat protein family. EMBO Rep. 2003, 4, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Koizuka, N.; Imai, R.; Fujimoto, H.; Hayakawa, T.; Kimura, Y.; Kohno-Murase, J.; Sakai, T.; Kawasaki, S.; Imamura, J. Genetic characterization of a pentatricopeptide repeat protein gene, orf687, that restores fertility in the cytoplasmic male-sterile Kosena radish. Plant J. 2003, 34, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, K.; Huang, W.; Liu, G.; Gao, Y.; Wang, J.; Huang, Q.; Ji, Y.; Qin, X.; Wan, L. The rice pentatricopeptide repeat protein RF5 restores fertility in Hong-Lian cytoplasmic male-sterile lines via a complex with the glycine-rich protein GRP162. Plant Cell 2012, 24, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Luo, D.; Zhou, D.; Zhang, Q.; Tian, D.; Zheng, X.; Chen, L.; Liu, Y.G. The rice restorer Rf4 for wild-abortive cytoplasmic male sterility encodes a mitochondrial-localized PPR protein that functions in reduction of WA352 transcripts. Mol. Plant Pathol. 2014, 7, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Yu, C.; Hu, J.; Wang, L.; Dan, Z.; Zhou, W.; He, C.; Zeng, Y.; Yao, G.; Qi, J. Pentatricopeptide-repeat family protein RF6 functions with hexokinase 6 to rescue rice cytoplasmic male sterility. Proc. Natl. Acad. Sci. USA 2015, 112, 14984–14989. [Google Scholar] [CrossRef] [PubMed]
- Curtis, I.S. Genetic engineering of radish: Current achievements and future goals. Plant Cell Rep. 2011, 30, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Khanum, F.; Swamy, M.S.; Krishna, K.S.; Santhanam, K.; Viswanathan, K. Dietary fiber content of commonly fresh and cooked vegetables consumed in India. Plant Foods Hum. Nutr. 2000, 55, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Vardhini, B.V.; Sujatha, E.; Rao, S.; Ram, S. Studies on the effect of brassinosteroids on the qualitative changes in the storage roots of radish. Bulg. J. Agric. Sci. 2012, 18, 63–69. [Google Scholar]
- Nahm, S.H.; Lee, H.J.; Lee, S.W.; Joo, G.Y.; Harn, C.H.; Yang, S.G.; Min, B.W. Development of a molecular marker specific to a novel CMS line in radish (Raphanus sativus L.). Theor. Appl. Genet. 2005, 111, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Lee, Y.P.; Lee, J.; Choi, B.S.; Kim, S.; Yang, T.J. Complete mitochondrial genome sequence and identification of a candidate gene responsible for cytoplasmic male sterility in radish (Raphanus sativus L.) containing DCGMS cytoplasm. Theor. Appl. Genet. 2013, 126, 1763–1774. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.W.; Wang, C.D.; Gao, L.; Mei, S.Y.; Zhou, Y.; Xiang, C.P.; Wang, T. Heterozygous alleles restore male fertility to cytoplasmic male-sterile radish (Raphanus sativus L.): A case of overdominance. J. Exp. Bot. 2013, 64, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.M.; Chung, W.H.; Mun, J.H.; Kim, N.; Yu, H.J. De novo assembly and characterization of the complete chloroplast genome of radish (Raphanus sativus L.). Gene 2014, 551, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Tsuda, M.; Yasumoto, K.; Yamagishi, H.; Terachi, T. A complete mitochondrial genome sequence of Ogura-type male-sterile cytoplasm and its comparative analysis with that of normal cytoplasm in radish (Raphanus sativus L.). BMC Genom. 2012, 13, 352. [Google Scholar] [CrossRef] [PubMed]
- Franco-Zorrilla, J.M.; López-Vidriero, I.; Carrasco, J.L.; Godoy, M.; Vera, P.; Solano, R. DNA-binding specificities of plant transcription factors and their potential to define target genes. Proc. Natl. Acad. Sci. USA 2014, 111, 2367–2372. [Google Scholar] [CrossRef] [PubMed]
- Honys, D.; Twell, D. Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol. 2004, 5, R85. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Han, M.J.; Lee, Y.S.; Kim, Y.W.; Hwang, I.; Kim, M.J.; Kim, Y.K.; Nahm, B.H.; An, G. Rice Undeveloped Tapetum1 is a major regulator of early tapetum development. Plant Cell 2005, 17, 2705–2722. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, A.M.; Kröber, S.; Unte, U.S.; Huijser, P.; Dekker, K.; Saedler, H. The Arabidopsis ABORTED MICROSPORES (AMS) gene encodes a MYC class transcription factor. Plant J. 2003, 33, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sun, Y.; Timofejeva, L.; Chen, C.; Grossniklaus, U.; Ma, H. Regulation of Arabidopsis tapetum development and function by DYSFUNCTIONAL TAPETUM1 (DYT1) encoding a putative bHLH transcription factor. Development 2006, 133, 3085–3095. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhang, D.S.; Liu, H.S.; Yin, C.S.; Li, X.X.; Liang, W.Q.; Yuan, Z.; Xu, B.; Chu, H.W.; Wang, J.; et al. The rice tapetum degeneration retardation gene is required for tapetum degradation and anther development. Plant Cell 2006, 18, 2999–3014. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.A.; Iacuone, S.; Li, S.F.; Parish, R.W. The MYB80 transcription factor is required for pollen development and the regulation of tapetal programmed cell death in Arabidopsis thaliana. Plant Cell 2011, 23, 2209–2224. [Google Scholar] [CrossRef] [PubMed]
- Borg, M.; Rutley, N.; Kagale, S.; Hamamura, Y.; Gherghinoiu, M.; Kumar, S.; Sari, U.; Esparza-Franco, M.A.; Sakamoto, W.; Rozwadowski, K. An EAR-dependent regulatory module promotes male germ cell division and sperm fertility in Arabidopsis. Plant Cell 2014, 26, 2098–2113. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.F.; Meng, X.Z.; Khanna, R.; LaMontagne, E.; Liu, Y.D.; Zhang, S.Q. Phosphorylation of a WRKY transcription factor by MAPKs is required for pollen development and function in Arabidopsis. PLoS Genet. 2014, 10, e1004384. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, M.; Groß-Hardt, R.; Schöffl, F. Heat shock factor HSFB2a involved in gametophyte development of Arabidopsis thaliana and its expression is controlled by a heat-inducible long non-coding antisense RNA. Plant Mol. Biol. 2014, 85, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Marrs, K.A.; Casey, E.S.; Capitant, S.A.; Bouchard, R.A.; Dietrich, P.S.; Mettler, I.J.; Sinibaldi, R.M. Characterization of two maize HSP90 heat shock protein genes: Expression during heat shock, embryogenesis, and pollen development. Dev. Genet. 1993, 14, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Giorno, F.; Wolters-Arts, M.; Grillo, S.; Scharf, K.D.; Vriezen, W.H.; Mariani, C. Developmental and heat stress-regulated expression of HsfA2 and small heat shock proteins in tomato anthers. J. Exp. Bot. 2010, 61, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Bonen, L.; Waters, E.R.; Nguyen, S.L.; Eskandar, R.; Behan, J.; Sanders-Reed, Z. The recent evolution of a pseudogene: Diversity and divergence of a mitochondria-localized small heat shock protein in Arabidopsis thaliana. Genome 2008, 51, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, M.; Verbitskiy, D.; Zehrmann, A.; Brennicke, A. Reverse genetic screening identifies five E-class PPR proteins involved in RNA editing in mitochondria of Arabidopsis thaliana. J. Biol. Chem. 2010, 285, 27122–27129. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.W.; Zhang, Y.J.; Xiang, C.P.; Mei, S.Y.; Zhou, Y.; Chen, G.P.; Wang, T. A new fertility restorer locus linked closely to the Rfo locus for cytoplasmic male sterility in radish. Theor. Appl. Genet. 2008, 117, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mei, S.; Liu, T.; Wang, Z. Comparative Transcriptome Profile of the Cytoplasmic Male Sterile and Fertile Floral Buds of Radish (Raphanus sativus L.). Int. J. Mol. Sci. 2016, 17, 42. https://doi.org/10.3390/ijms17010042
Mei S, Liu T, Wang Z. Comparative Transcriptome Profile of the Cytoplasmic Male Sterile and Fertile Floral Buds of Radish (Raphanus sativus L.). International Journal of Molecular Sciences. 2016; 17(1):42. https://doi.org/10.3390/ijms17010042
Chicago/Turabian StyleMei, Shiyong, Touming Liu, and Zhiwei Wang. 2016. "Comparative Transcriptome Profile of the Cytoplasmic Male Sterile and Fertile Floral Buds of Radish (Raphanus sativus L.)" International Journal of Molecular Sciences 17, no. 1: 42. https://doi.org/10.3390/ijms17010042
APA StyleMei, S., Liu, T., & Wang, Z. (2016). Comparative Transcriptome Profile of the Cytoplasmic Male Sterile and Fertile Floral Buds of Radish (Raphanus sativus L.). International Journal of Molecular Sciences, 17(1), 42. https://doi.org/10.3390/ijms17010042