
15,16-Dihydrotanshinone I from the Functional Food Salvia miltiorrhiza Exhibits Anticancer Activity in Human HL-60 Leukemia Cells: in Vitro and in Vivo Studies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
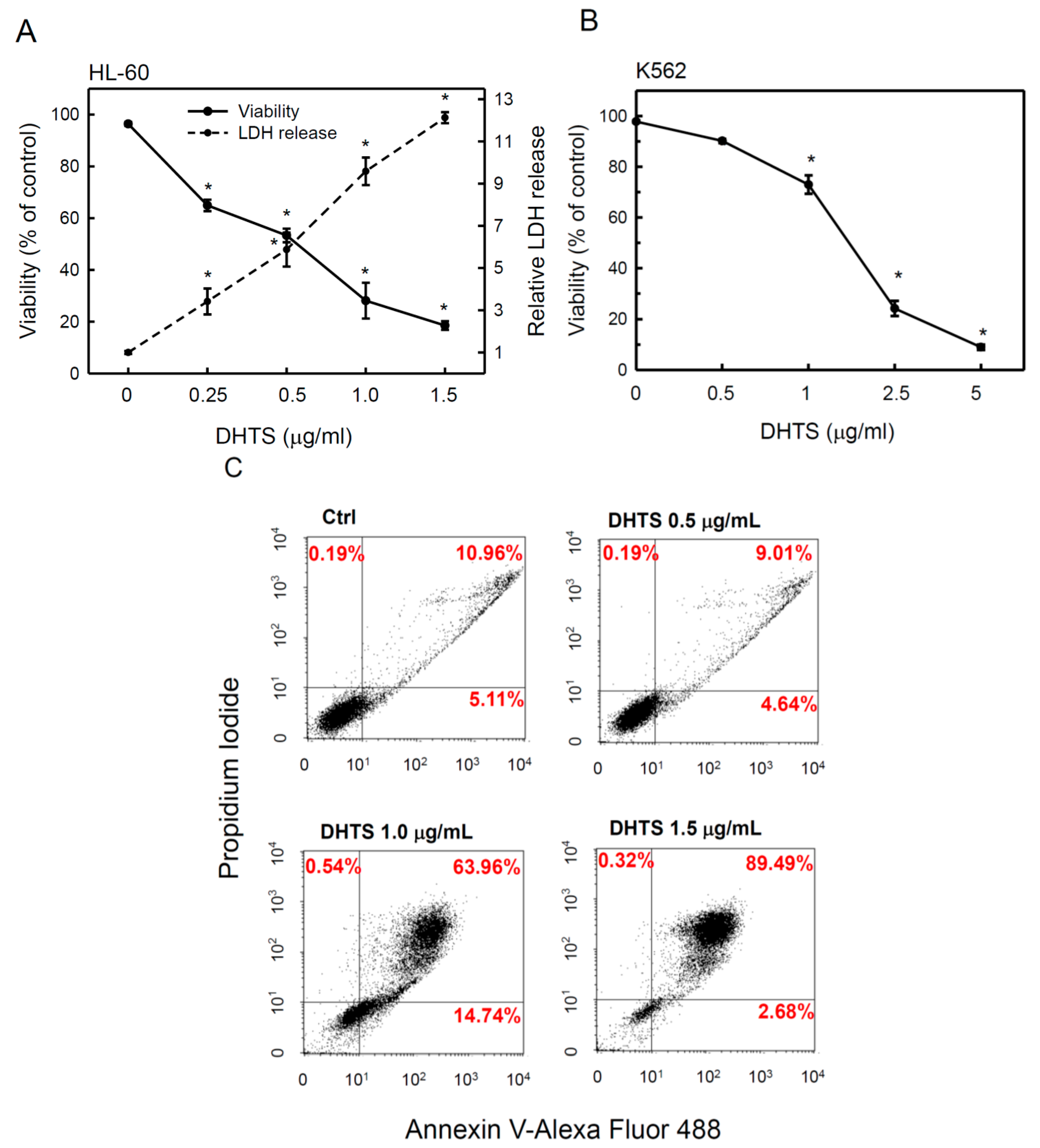
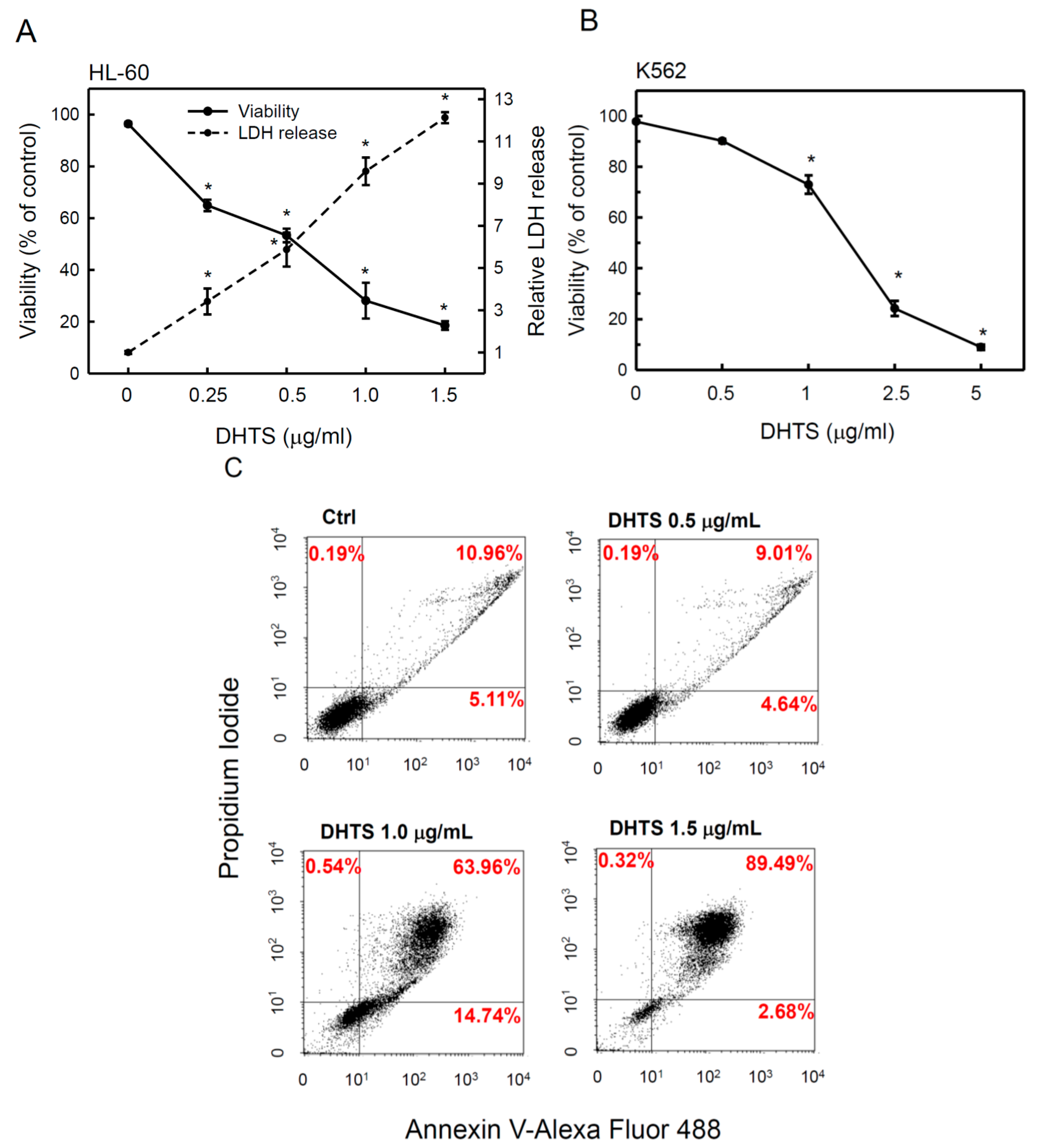
2.1. 15,16-Dihydrotanshinone I (DHTS) Inhibited Cell Proliferation and Triggered Apoptosis
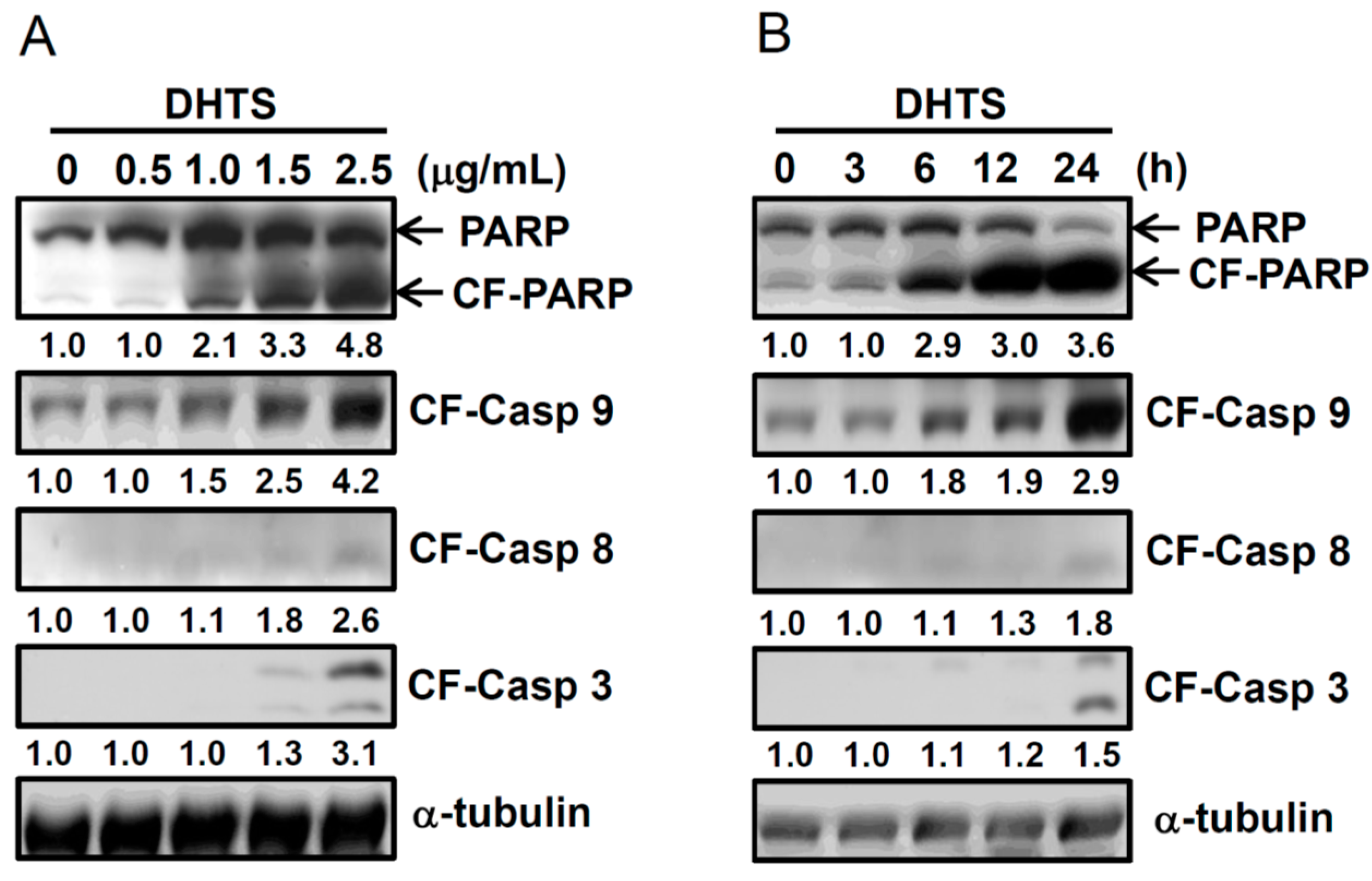
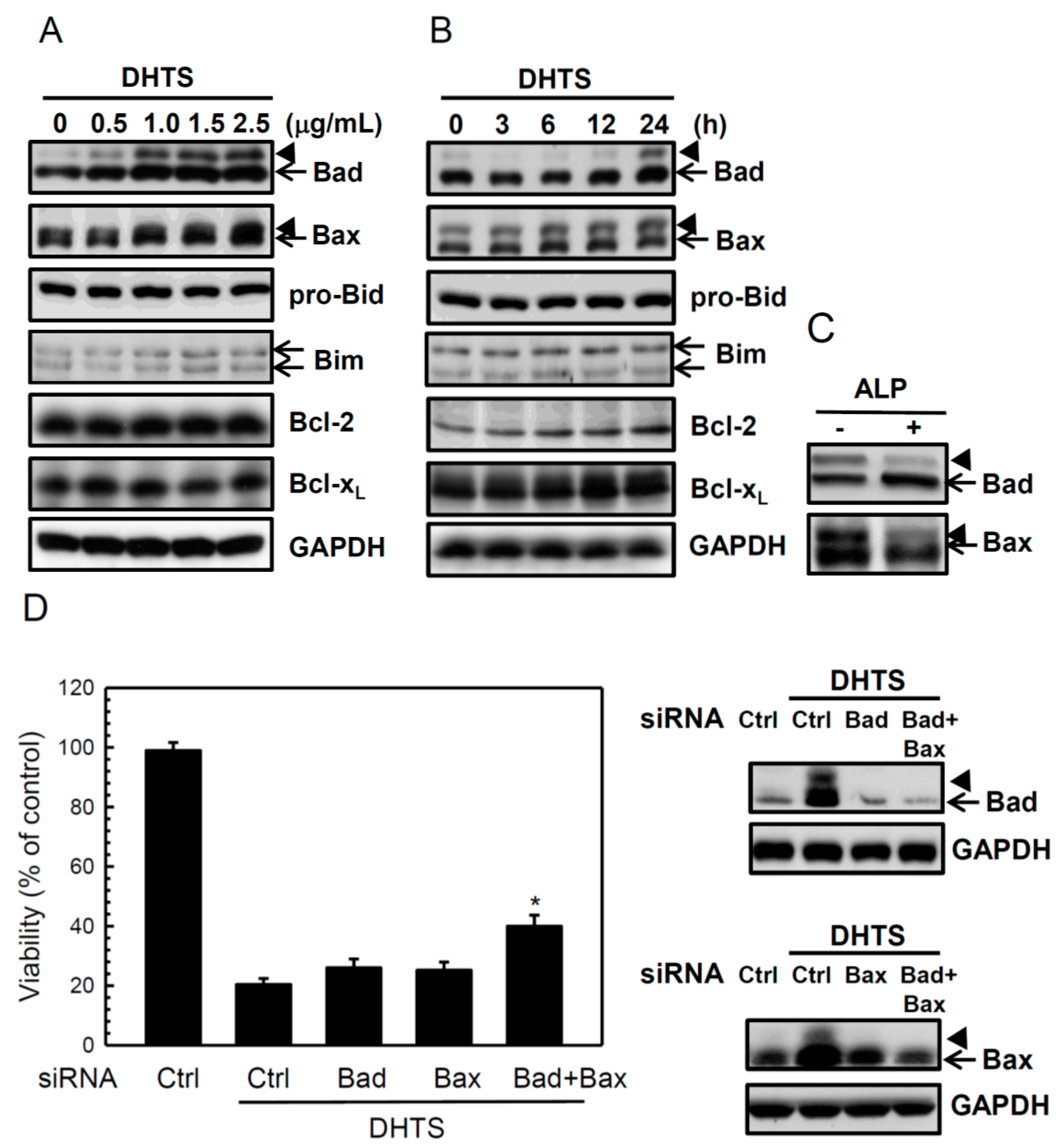
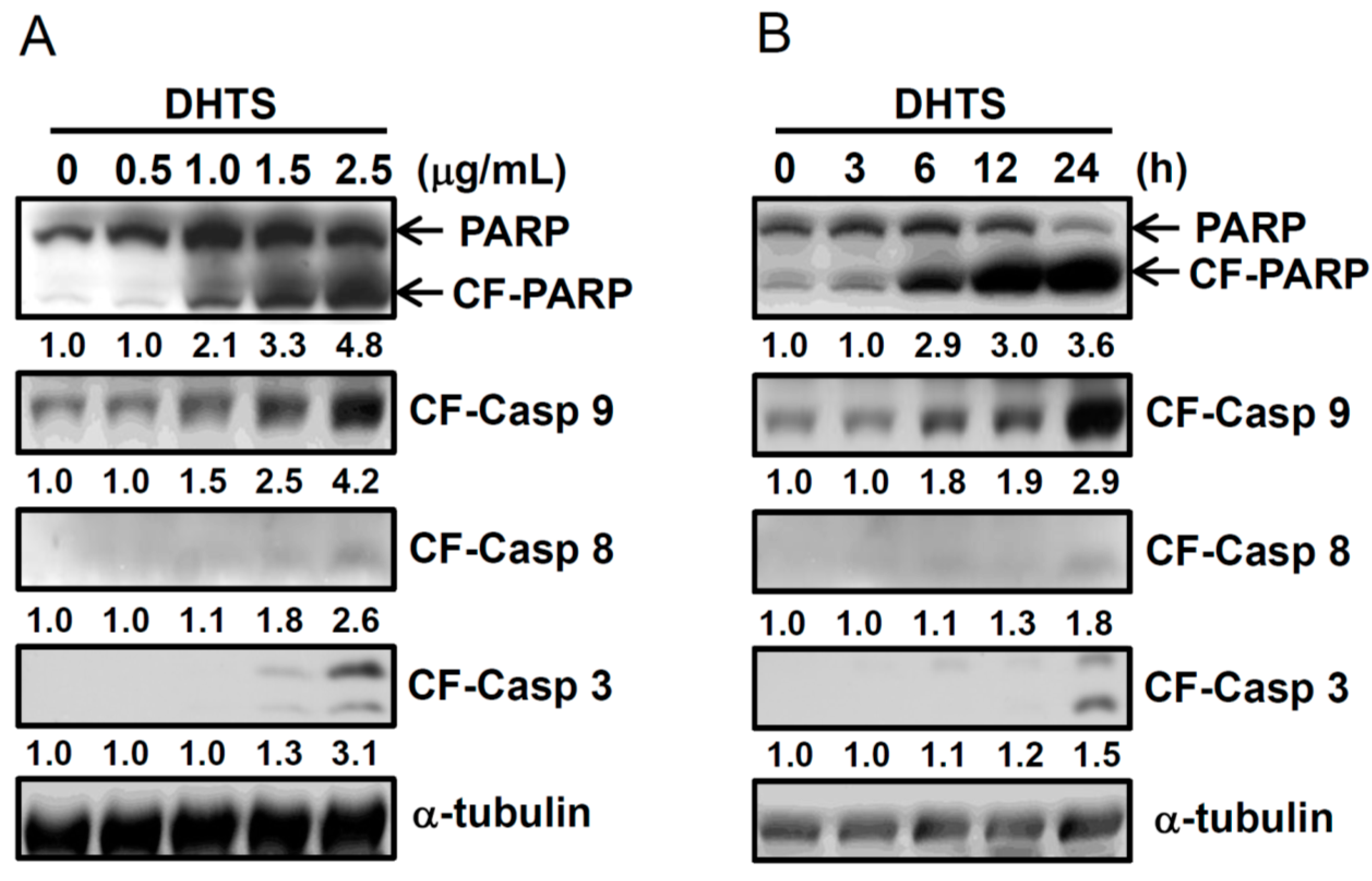
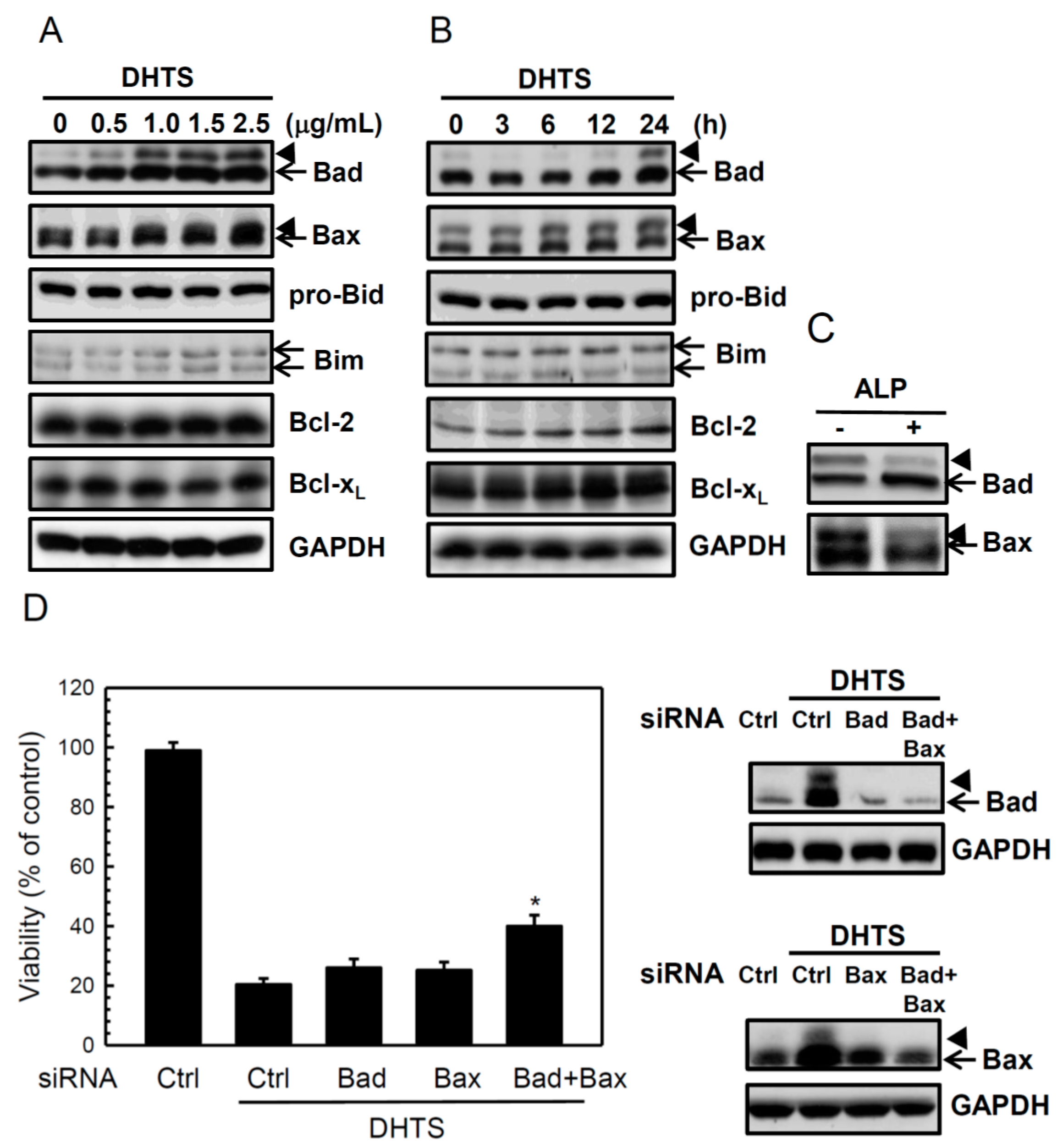
2.2. DHTS Induced Apoptosis through Increased Bad/Bax Expression and Activation of Caspases
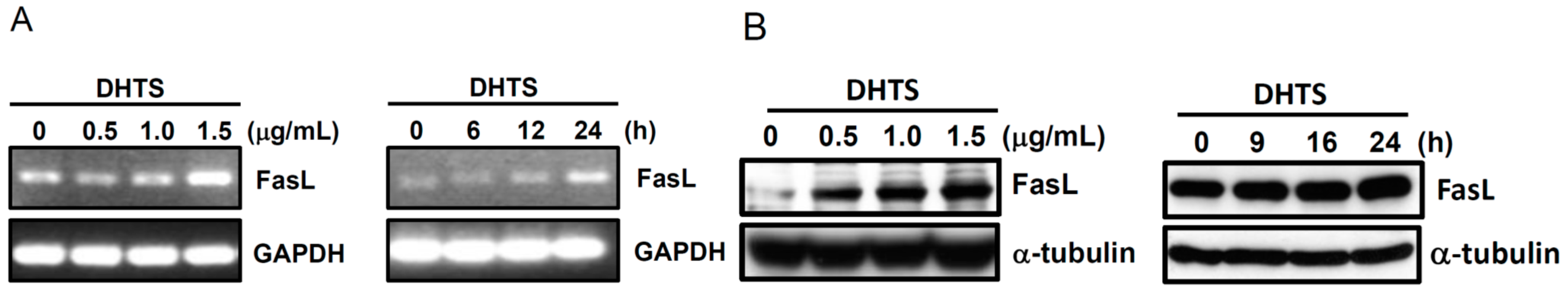
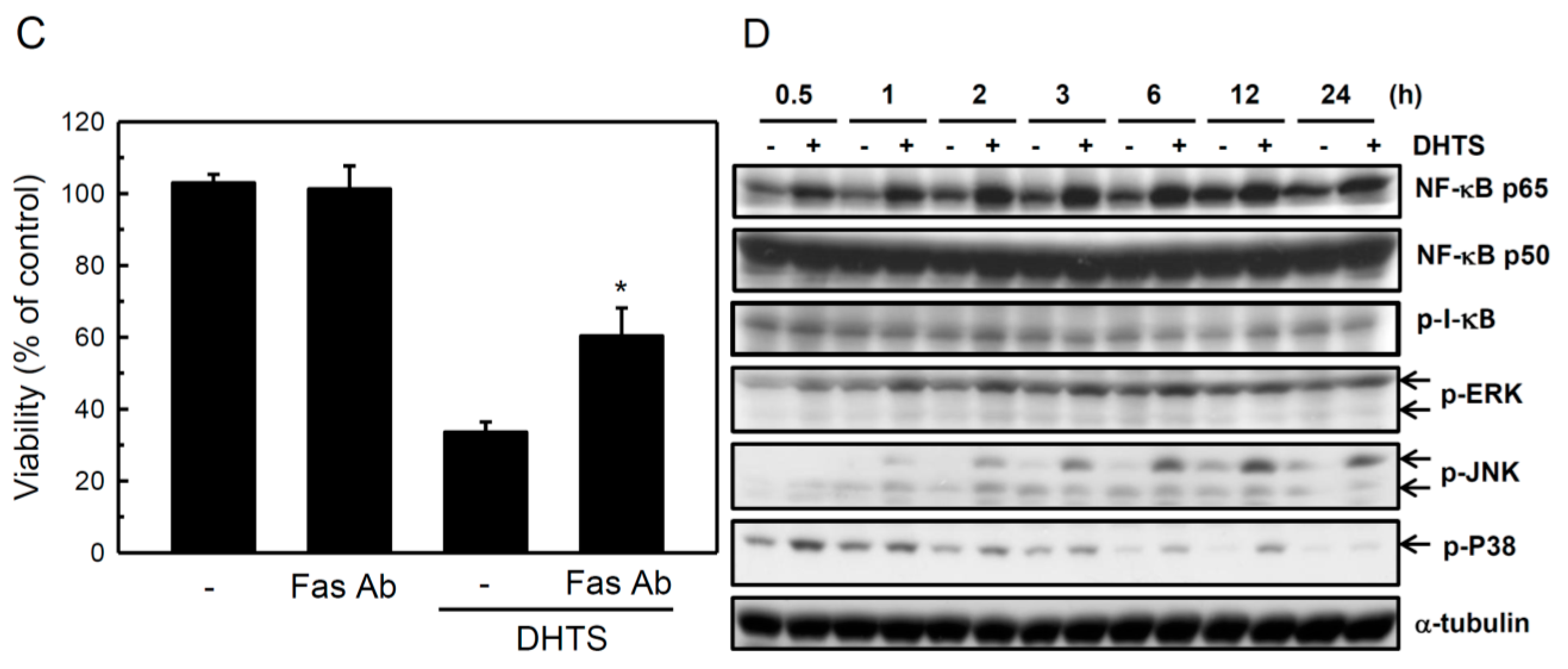
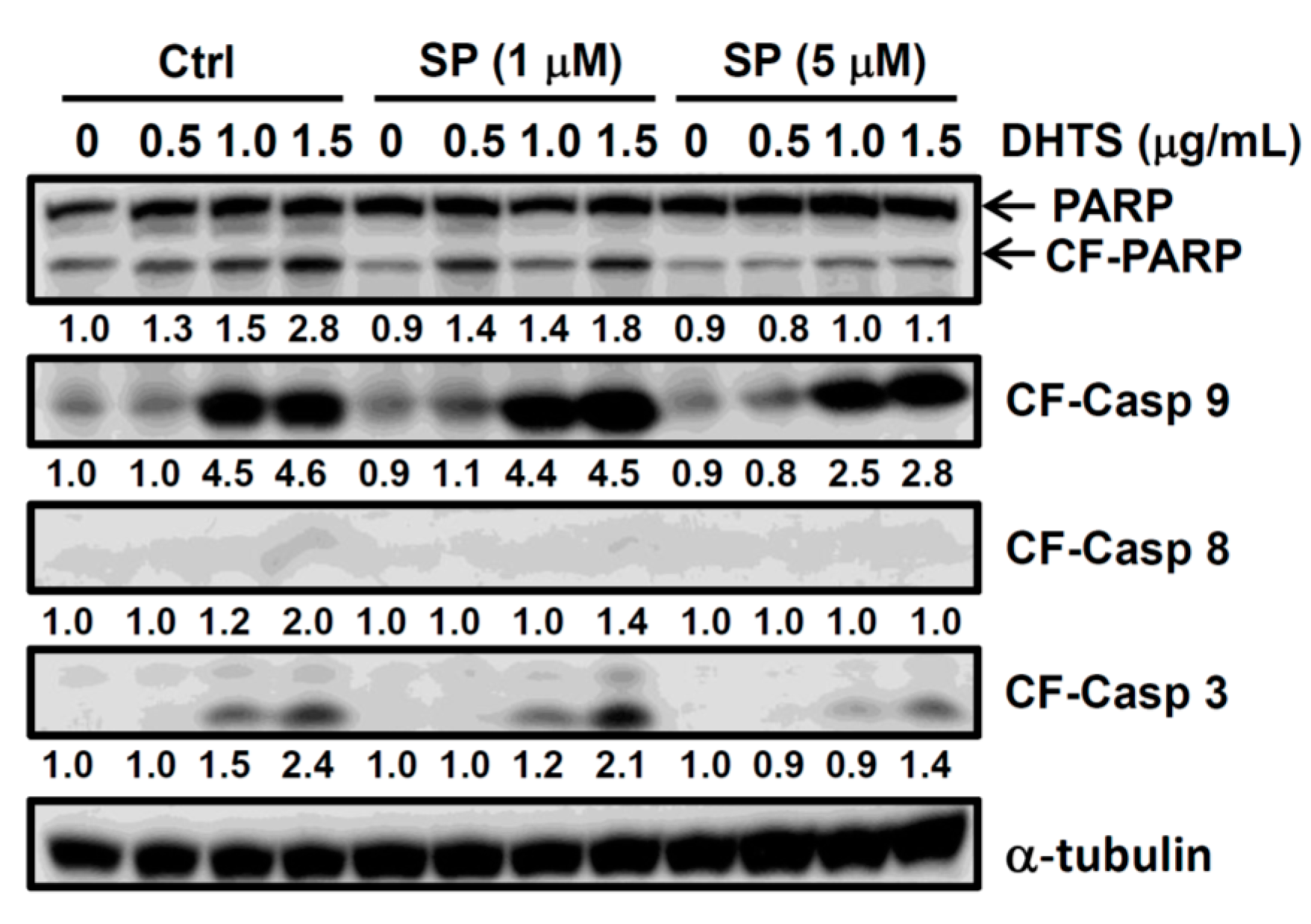
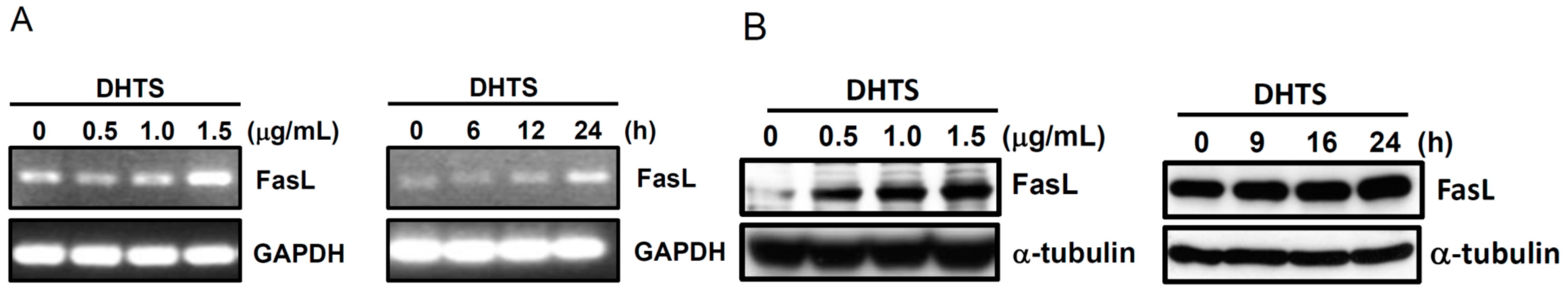
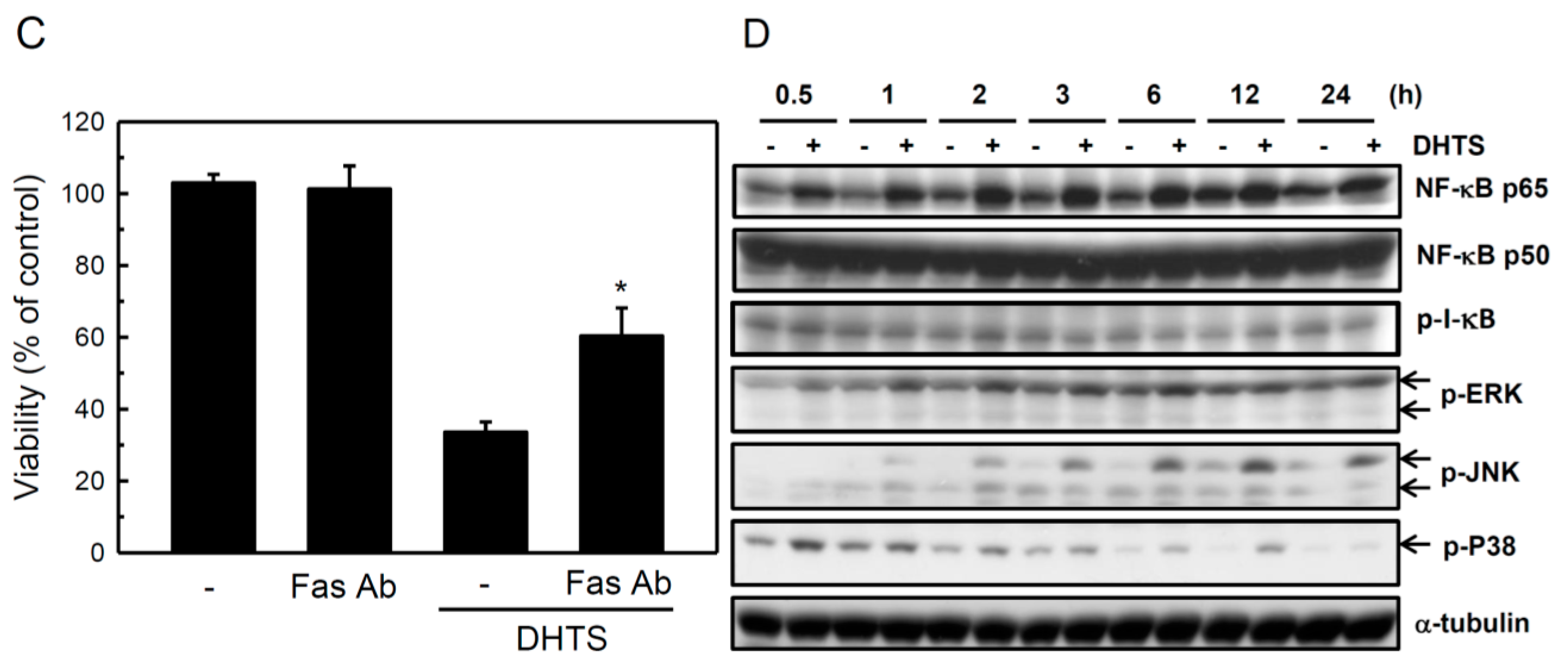
2.3. Both FasL and c-Jun N-Terminal Kinase (JNK) Contributed to DHTS-Induced Apoptosis
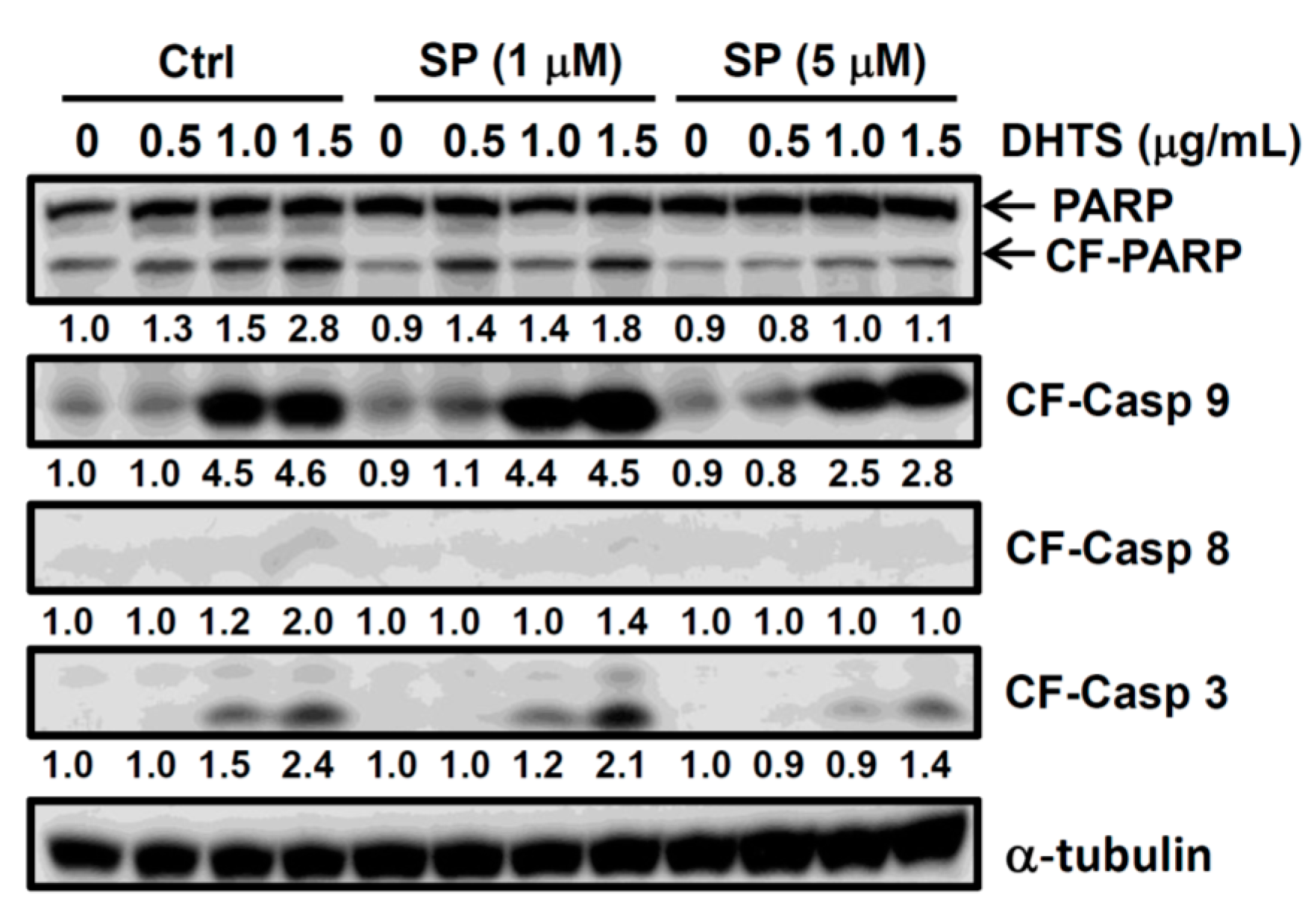
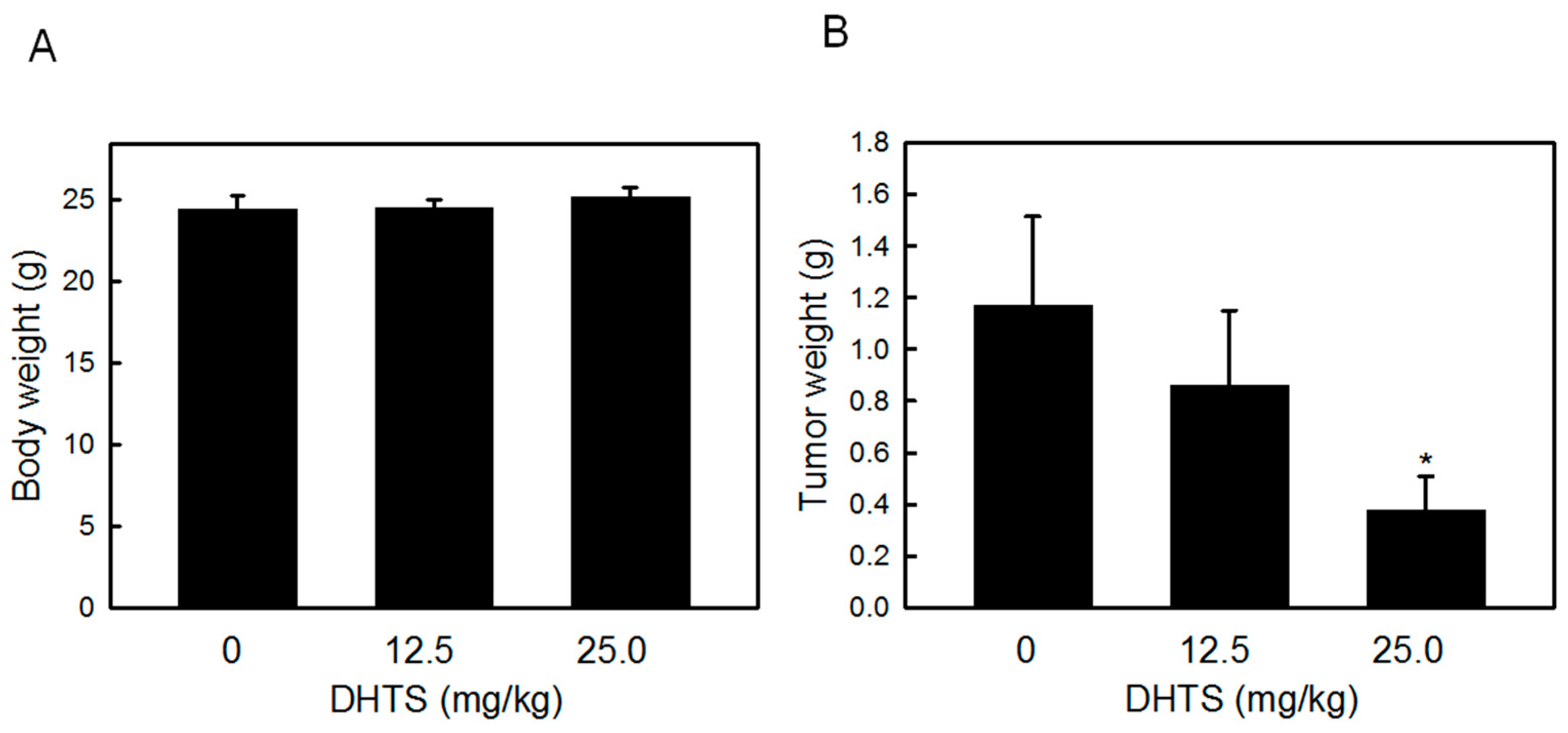
2.4. DHTS Inhibited Leukemia Tumor Growth in Nude Mice
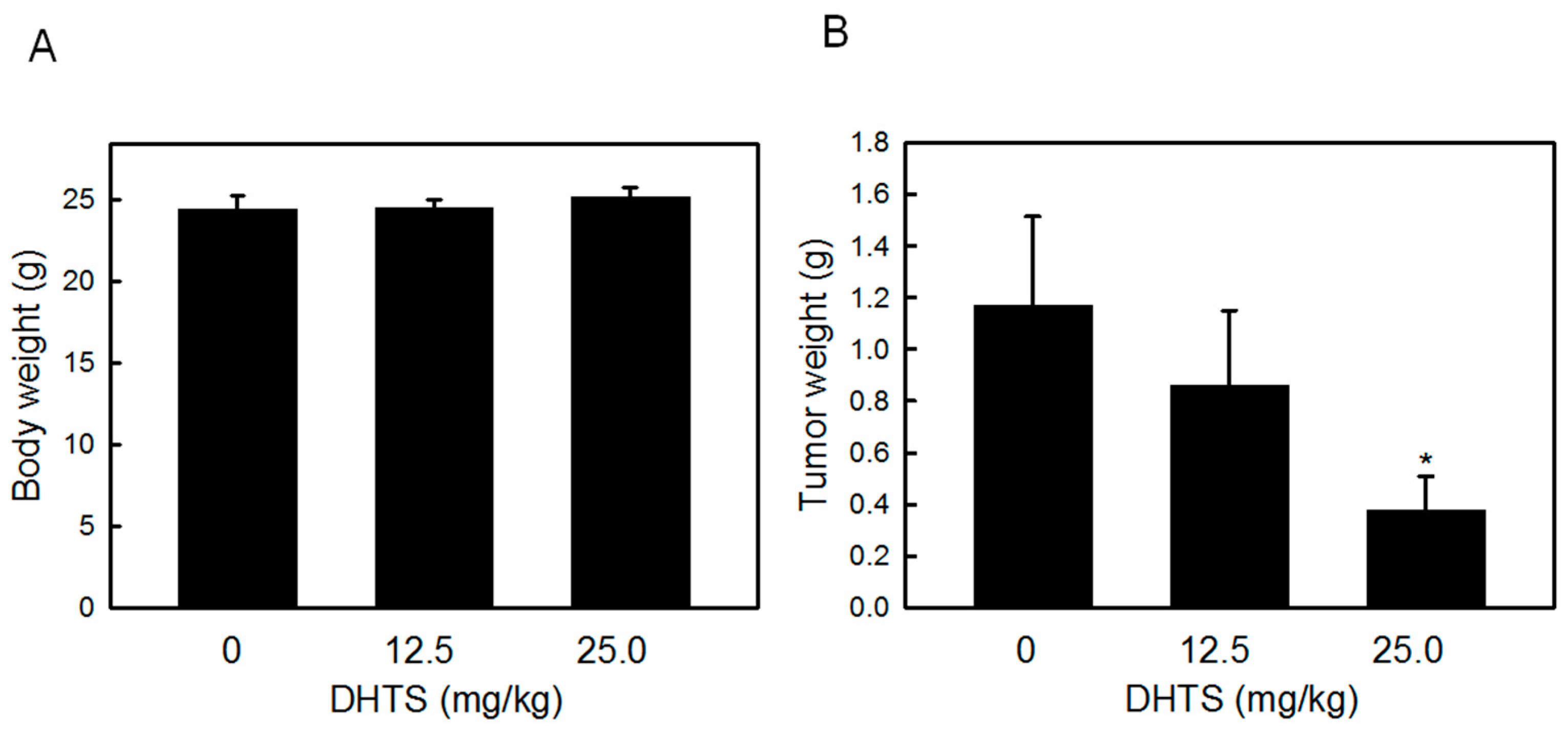
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Transient Transfection
4.3. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl Tetrazolium Bromide (MTT) Assay and Lactate Dehydrogenase (LDH) Release Assay
4.4. Western Blot Analysis
4.5. RNA Isolation and a Semiquantitative Reverse-Transcription Polymerase Chain Reaction (RT-PCR)
4.6. Flow Cytometric Analysis
4.7. Antitumor Nude Mice Experiment
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tzen, J.T.; Jinn, T.R.; Chen, Y.C.; Li, F.Y.; Cheng, F.C.; Shi, L.S.; She, H.Kh.; Chen, B.C.; Hsieh, V.; Tu, M.L. Magnesium lithospermate B possesses inhibitory activity on Na+, K+-ATPase and neuroprotective effects against ischemic stroke. Acta Pharmacol. Sin. 2007, 28, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Morris-Natschke, S.L.; Lee, K.H. New developments in the chemistry and biology of the bioactive constituents of Tanshen. Med. Res. Rev. 2007, 27, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.L.; Suk, F.M.; Wang, C.I.; Liu, D.Z.; Hou, W.C.; Lin, P.J.; Hung, L.F.; Liang, Y.C. Anti-tumor potential of 15,16-dihydrotanshinone I against breast adenocarcinoma through inducing G1 arrest and apoptosis. Biochem. Pharmacol. 2007, 74, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Suk, F.M.; Jou, W.J.; Lin, R.J.; Lin, S.Y.; Tzeng, F.Y.; Liang, Y.C. 15,16-Dihydrotanshinone I-induced apoptosis in human colorectal cancer cells: Involvement of ATF3. Anticancer Res. 2013, 33, 3225–3231. [Google Scholar] [PubMed]
- Chuang, M.T.; Ho, F.M.; Wu, C.C.; Zhuang, S.Y.; Lin, S.Y.; Suk, F.M.; Liang, Y.C. 15,16-Dihydrotanshinone I, a compound of Salvia miltiorrhiza Bunge, induces apoptosis through inducing endoplasmic reticular stress in human prostate carcinoma cells. Evid. Based Complement. Altern. Med. 2011, 2011, 865435. [Google Scholar]
- Lee, W.Y.; Liu, K.W.; Yeung, J.H. Reactive oxygen species-mediated kinase activation by dihydrotanshinone in tanshinones-induced apoptosis in HepG2 cells. Cancer Lett. 2009, 285, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Liu, W.D.; Yang, H.Z.; Zhang, Y.; Fang, Z.G.; Liu, P.Q.; Lin, D.J.; Xiao, R.Z.; Hu, Y.; Wang, C.Z.; et al. Inactivation of PI3k/Akt signaling pathway and activation of caspase-3 are involved in tanshinone I-induced apoptosis in myeloid leukemia cells in vitro. Ann. Hematol. 2010, 89, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Sher, H.F.; Chen, H.W.; Liu, C.C.; Chen, C.H.; Lin, C.S.; Yang, P.C.; Tsay, H.S.; Chen, J.J. Anticancer effects of tanshinone I in human non-small cell lung cancer. Mol. Cancer Ther. 2008, 7, 3527–3538. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.L.; Hung, Y.C.; Wng, P.W.; Chen, S.T.; Hsu, T.K.; Sintupisut, N.; Cheng, C.S.; Lyu, P.C. Functional proteomic and structural insights into molecular targets related to the growth inhibitory effect of tanshinone IIA on HeLa cells. Proteomic 2010, 10, 914–929. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Xue, H.L.; Huang, H.B.; Wang, X.G. Tanshinone IIA inhibits constitutive STAT3 activation, suppresses proliferation, and induces apoptosis in rat C6 glioma cells. Neurosci. Lett. 2010, 470, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S. Molecular signaling in death receptor and mitochondrial pathways of apoptosis (Review). Int. J. Oncol. 2003, 22, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, T.; Follis, A.V.; Kriwacki, R.W.; Green, D.R. Many players in BCL-2 family affairs. Trends Biochem. Sci. 2014, 39, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; Strasser, A. Keeping killers on a tight leash: Transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 2002, 9, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Konishi, Y.; Lehtinen, M.; Donovan, N.; Bonni, A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol. Cell 2002, 9, 1005–1016. [Google Scholar] [CrossRef]
- Renault, T.T.; Manon, S. Bax: Addressed to kill. Biochimie 2011, 93, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Deng, X. Nicotine inactivation of the proapoptotic function of Bax through phosphorylation. J. Biol. Chem. 2005, 280, 10781–10789. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Deng, X. Protein phosphatase 2A enhances the proapoptotic function of Bax through dephosphorylation. J. Biol. Chem. 2006, 281, 18859–18867. [Google Scholar] [CrossRef] [PubMed]
- Linseman, D.A.; Butts, B.D.; Precht, T.A.; Phelps, R.A.; Le, S.S.; Laessig, T.A.; Bouchard, R.J.; Florez-McClure, M.L.; Heidenreich, K.A. Glycogen synthase kinase-3β phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J. Neurosci. 2004, 24, 9993–10002. [Google Scholar] [CrossRef] [PubMed]
- Workman, L.M.; Habelhah, H. TNFR1 signaling kinetics: Spatiotemporal control of three phases of IKK activation by posttranslational modification. Cell Signal. 2013, 25, 1654–1664. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Ziros, P.G.; Tsopra, O.A.; Dimas, K.; Kokkinou, D.; Thanopoulou, E.; Karakantza, M.; Pantazis, P.; Spyridonidis, A.; Zoumbos, N.C. c-Jun N-terminal kinase activation failure is a new mechanism of anthracycline resistance in acute myeloid leukemia. Leukemia 2008, 22, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Suarez, G.; Sha, J.; Sierra, J.C.; Peterson, J.W.; Chopra, A.K. Phospholipase A2-activating protein (PLAA) enhances cisplatin-induced apoptosis in HeLa cells. Cell Signal. 2009, 21, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Park, G.B.; Lee, H.K.; Song, H.; Choi, I.H.; Lee, W.J.; Hur, D.Y. Cross-linking of B7-H1 on EBV-transformed B cells induces apoptosis through reactive oxygen species production, JNK signaling activation, and FasL expression. J. Immunol. 2008, 181, 6158–6169. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Jeong, E.Y.; Kim, Y.S.; Roh, G.S.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S. c-Jun N-terminal kinase regulates the interaction between 14-3-3 and Bad in ethanol-induced cell death. J. Neurosci. Res. 2008, 86, 3221–3229. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Zhu, B.T. Mechanism of 2-methoxyestradiol-induced apoptosis and growth arrest in human breast cancer cells. Mol. Carcinog. 2009, 48, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Takada, Y.; Boriek, A.M.; Aggarwal, B.B. Nuclear factor-kappaB: Its role in health and disease. J. Mol. Med. 2004, 82, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. NF-κB and cancer: Mechanisms and targets. Mol. Carcinog. 2006, 45, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Chu, J.S.; Chien, H.L.; Tseng, C.H.; Ko, P.C.; Mei, Y.Y.; Tang, W.C.; Kao, Y.T.; Cheng, H.Y.; Liang, Y.C.; et al. MCPIP1 suppresses hepatitis C virus replication and negatively regulates virus-induced proinflammatory cytokine responses. J. Immunol. 2014, 193, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Ahn, E.Y.; Chen, Y.; Feng, G.; Reddy, V.; Jhala, N.C.; McDonald, J.M. Reciprocal co-expression of Fas and Fas ligand in human cholangiocarcinoma. Int. J. Oncol. 2007, 31, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Y.; Huang, W.J.; Ho, F.M.; Lin, R.J.; Lin, S.Y.; Suk, F.M.; Liang, Y.C. N-Hydroxycinnamide derivatives of osthole inhibit cell migration and invasion by suppressing Smad2 and Akt pathways in human colorectal adenocarcinoma cells. Chem. Biol. Interact. 2014, 217, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Y.; Huang, W.J.; Lin, R.J.; Lin, S.Y.; Liang, Y.C. N-hydroxycinnamide derivatives of osthole presenting genotoxicity and cytotoxicity against human colon adenocarcinoma cells in vitro and in vivo. Chem. Res. Toxicol. 2013, 26, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.-J.; Wu, H.-H.; Chen, T.-H.; Leung, W.; Liang, Y.-C. 15,16-Dihydrotanshinone I from the Functional Food Salvia miltiorrhiza Exhibits Anticancer Activity in Human HL-60 Leukemia Cells: in Vitro and in Vivo Studies. Int. J. Mol. Sci. 2015, 16, 19387-19400. https://doi.org/10.3390/ijms160819387
Liu J-J, Wu H-H, Chen T-H, Leung W, Liang Y-C. 15,16-Dihydrotanshinone I from the Functional Food Salvia miltiorrhiza Exhibits Anticancer Activity in Human HL-60 Leukemia Cells: in Vitro and in Vivo Studies. International Journal of Molecular Sciences. 2015; 16(8):19387-19400. https://doi.org/10.3390/ijms160819387
Chicago/Turabian StyleLiu, Jun-Jen, Hsueh-Hsia Wu, Tzu-Ho Chen, Wan Leung, and Yu-Chih Liang. 2015. "15,16-Dihydrotanshinone I from the Functional Food Salvia miltiorrhiza Exhibits Anticancer Activity in Human HL-60 Leukemia Cells: in Vitro and in Vivo Studies" International Journal of Molecular Sciences 16, no. 8: 19387-19400. https://doi.org/10.3390/ijms160819387
APA StyleLiu, J.-J., Wu, H.-H., Chen, T.-H., Leung, W., & Liang, Y.-C. (2015). 15,16-Dihydrotanshinone I from the Functional Food Salvia miltiorrhiza Exhibits Anticancer Activity in Human HL-60 Leukemia Cells: in Vitro and in Vivo Studies. International Journal of Molecular Sciences, 16(8), 19387-19400. https://doi.org/10.3390/ijms160819387