
From Genome to Structure and Back Again: A Family Portrait of the Transcarbamylases


Abstract
:
1. Introduction
2. Structures Deposited in the Protein Data Bank (PDB)
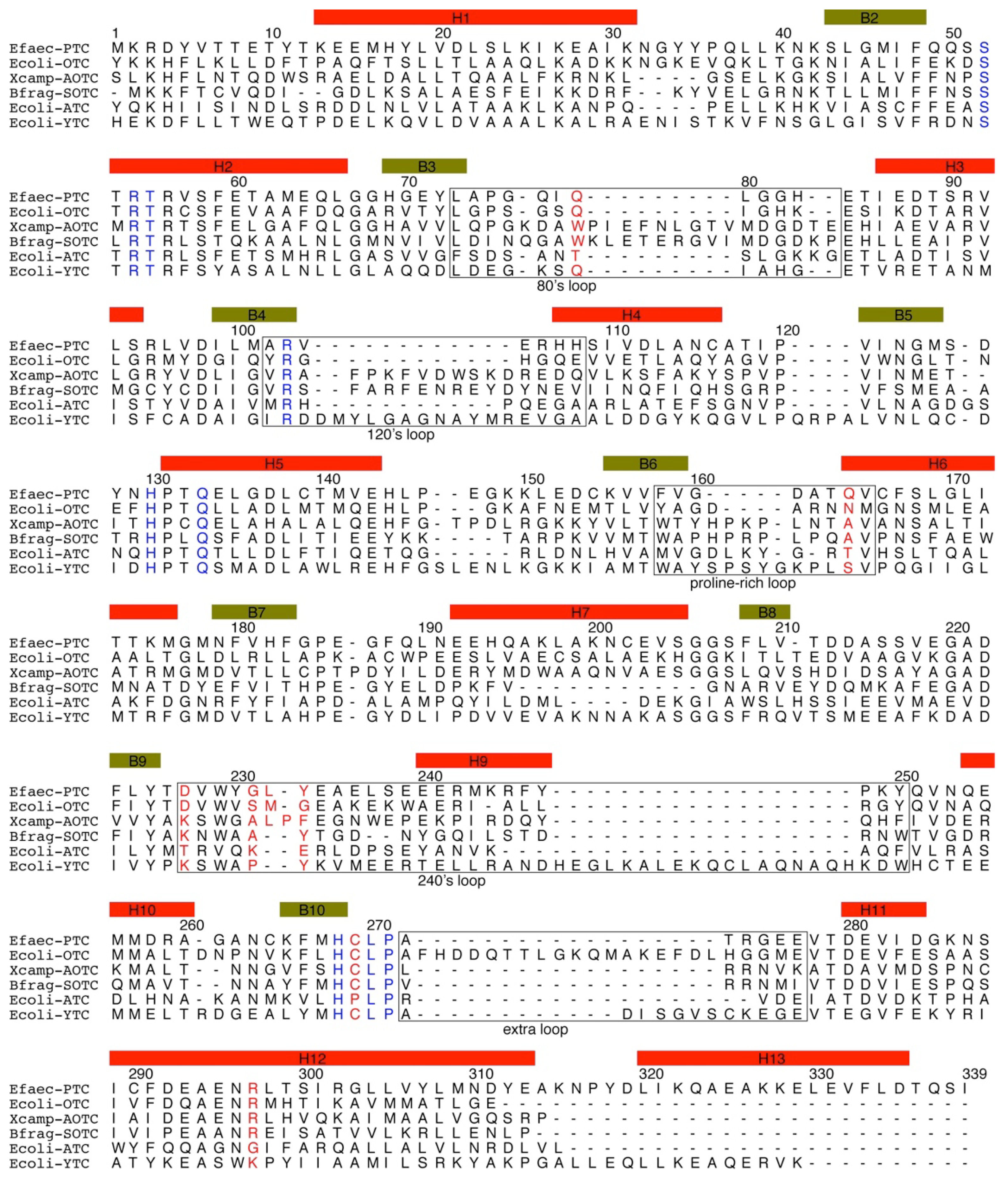
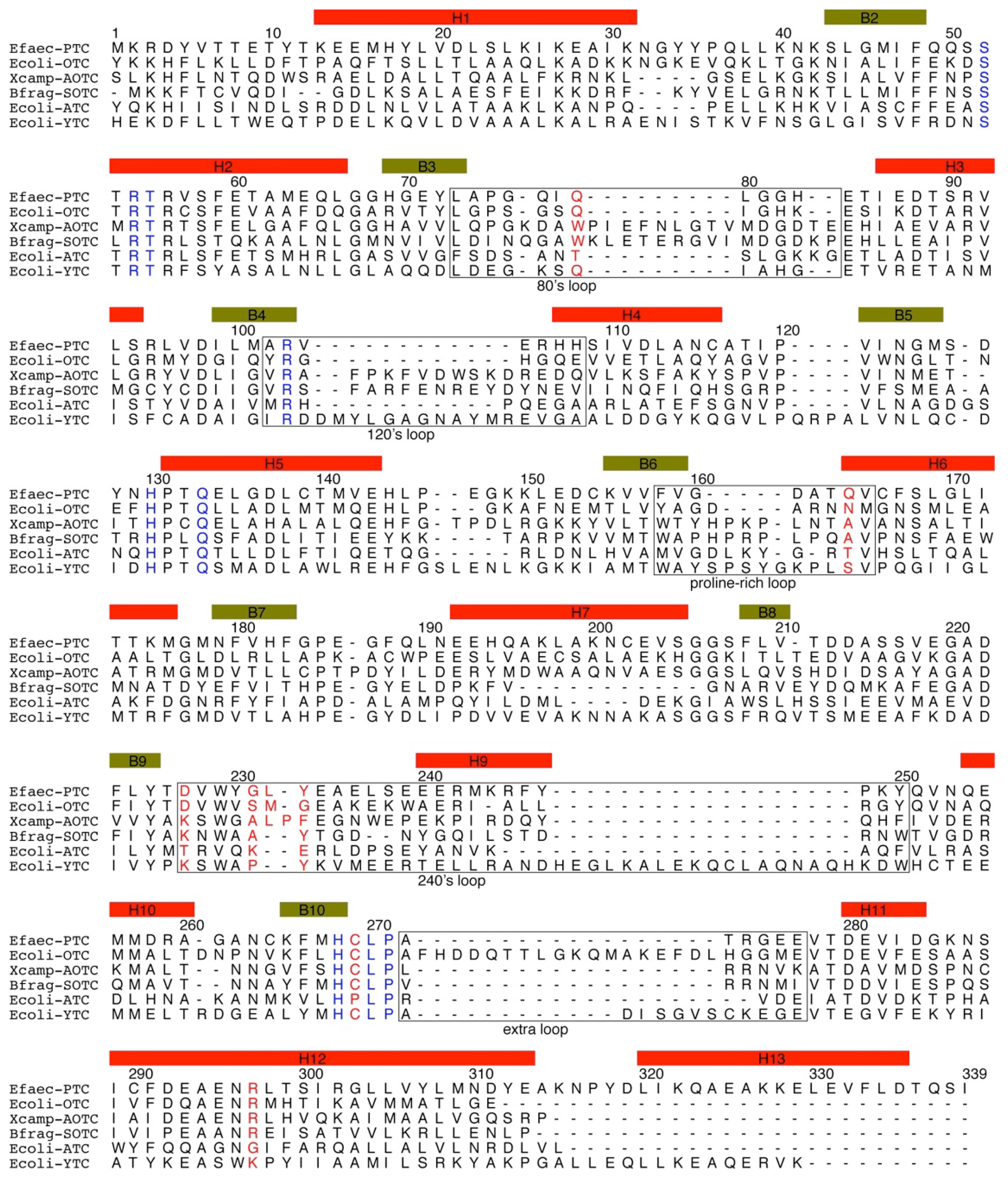
3. Sequences of Transcarbamylases
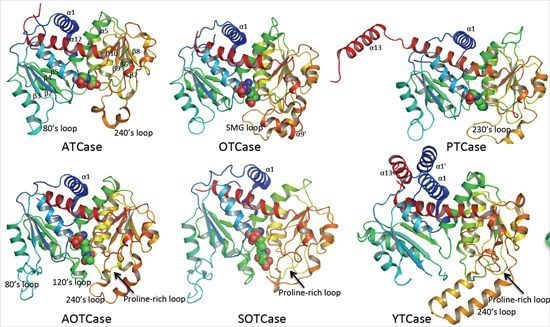
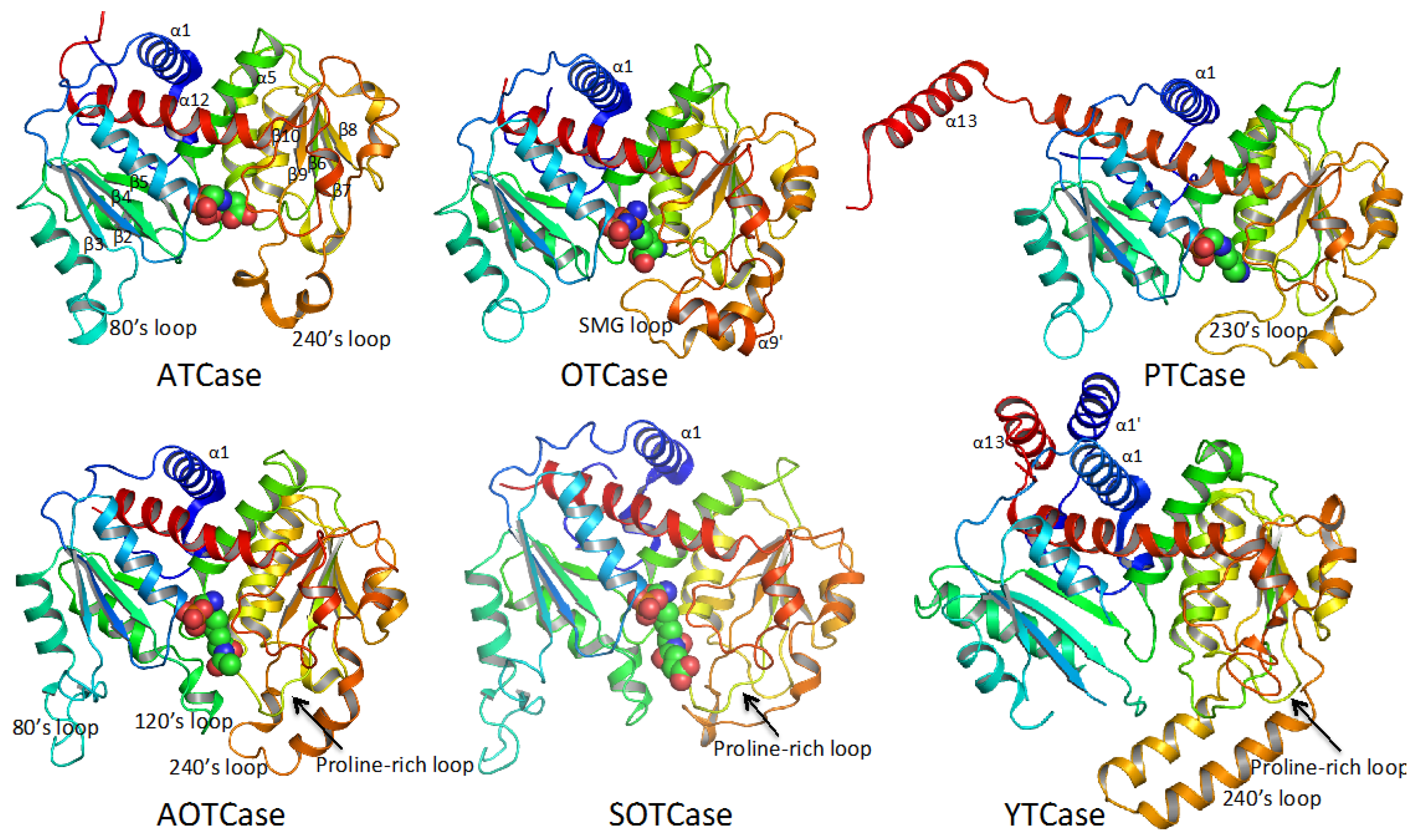
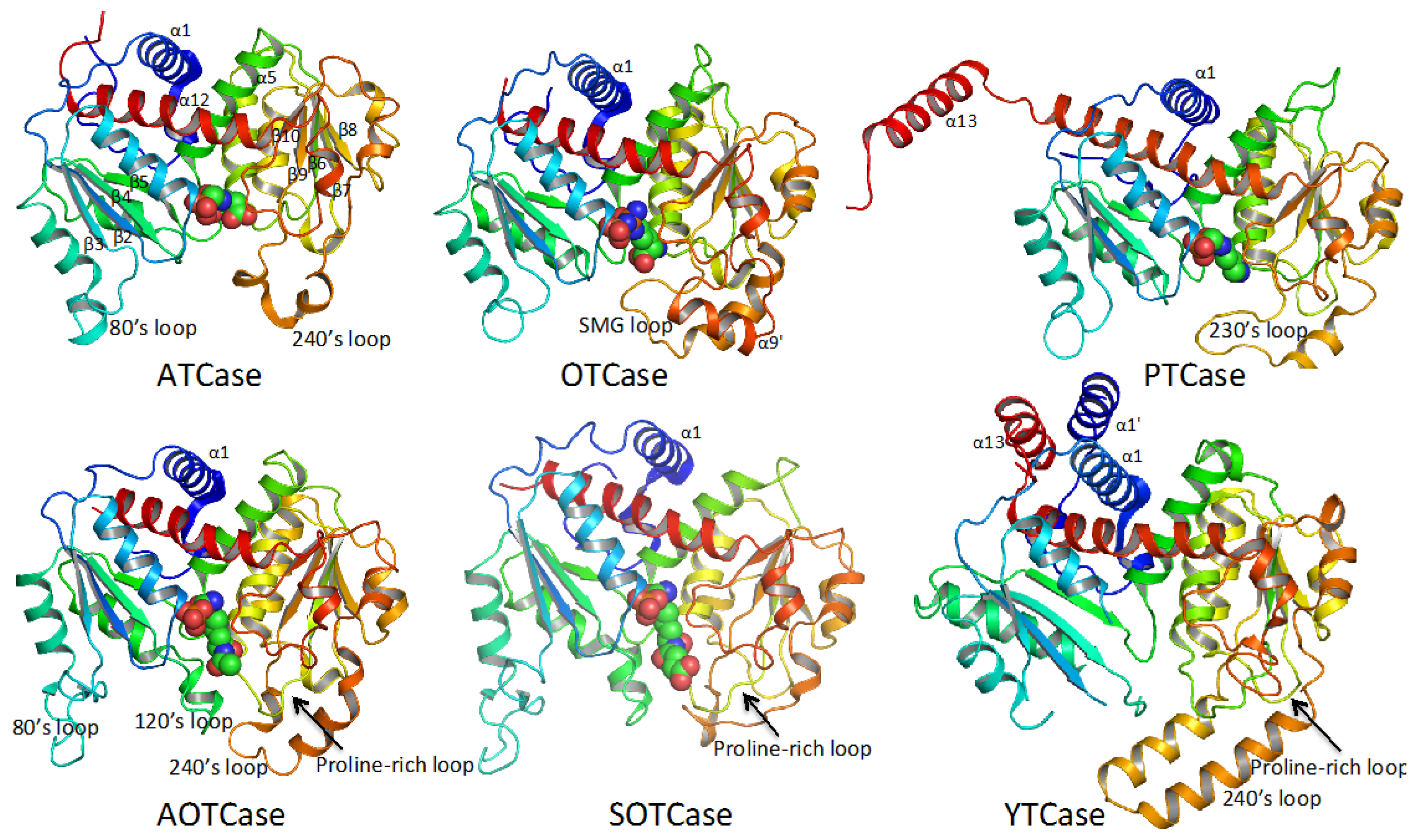
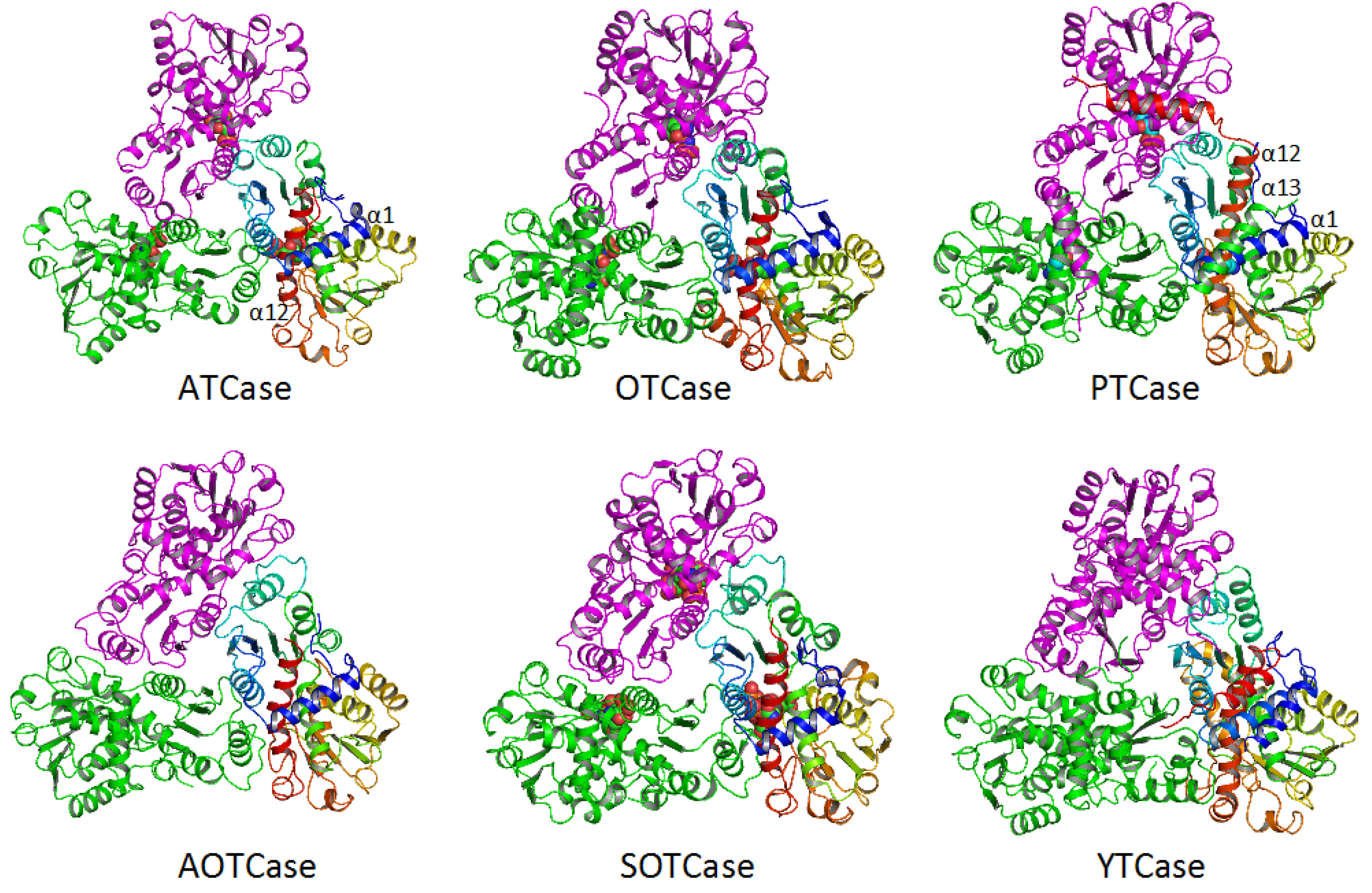
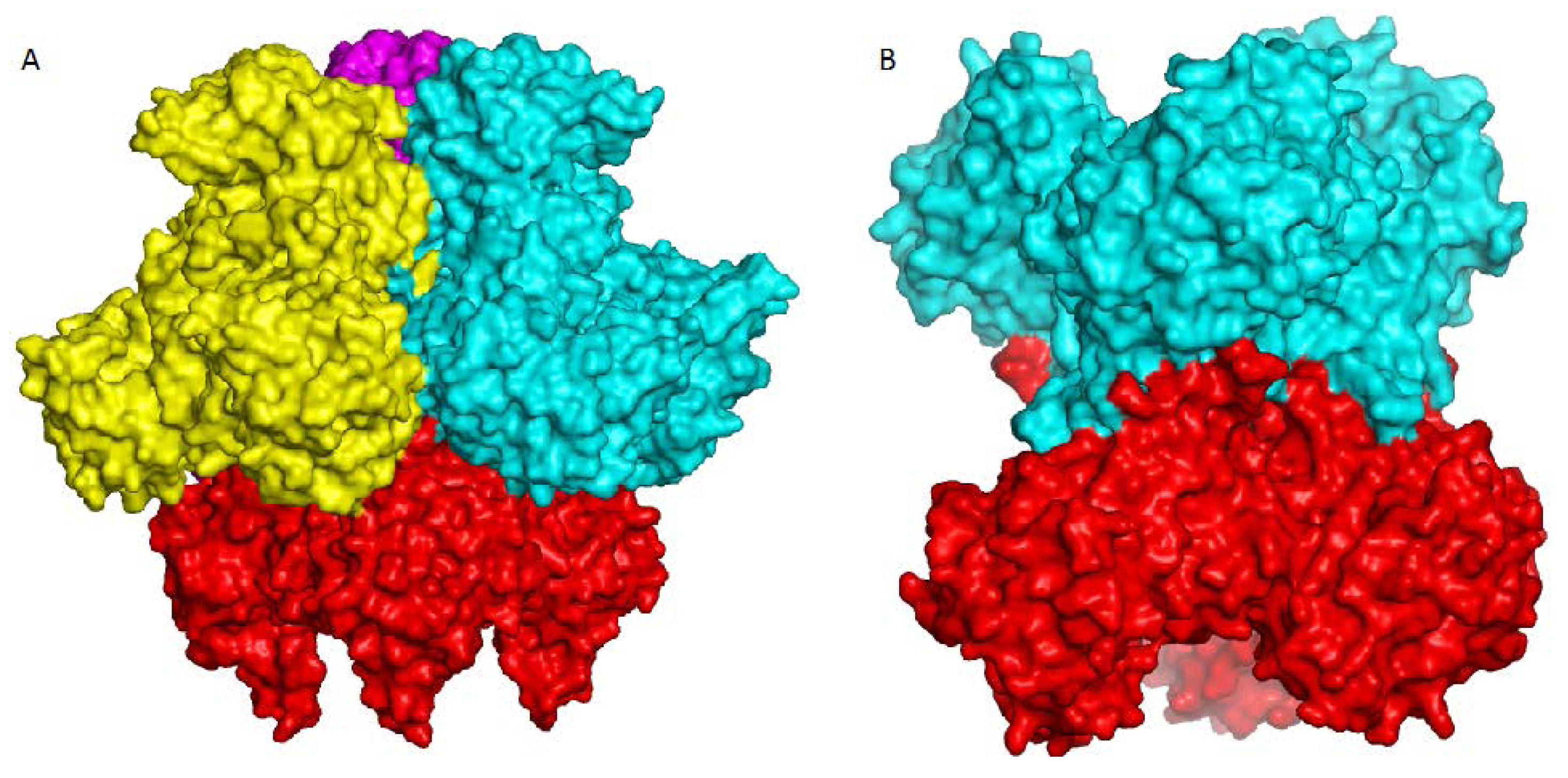
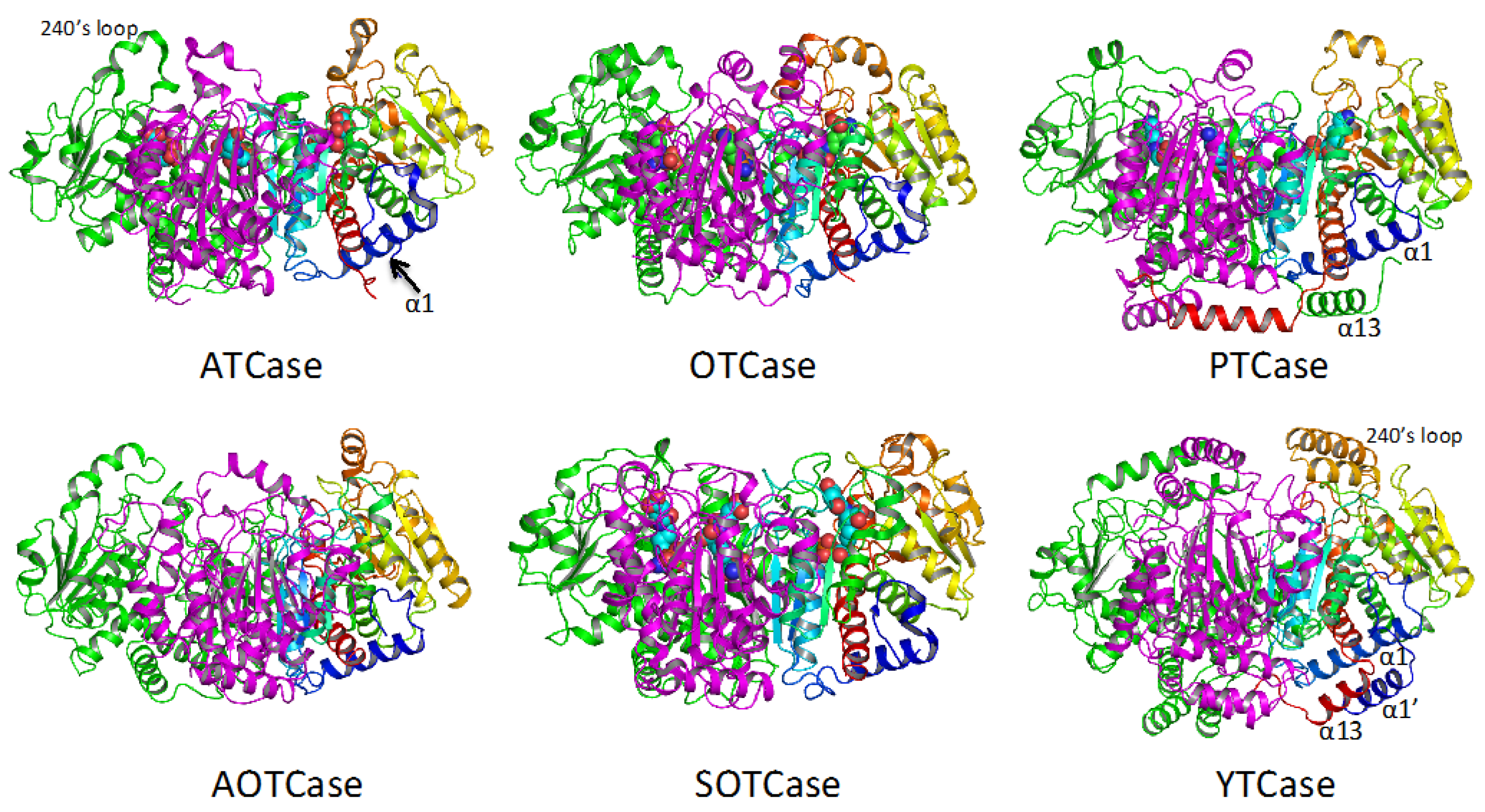
4. Overview of the Structural Fold
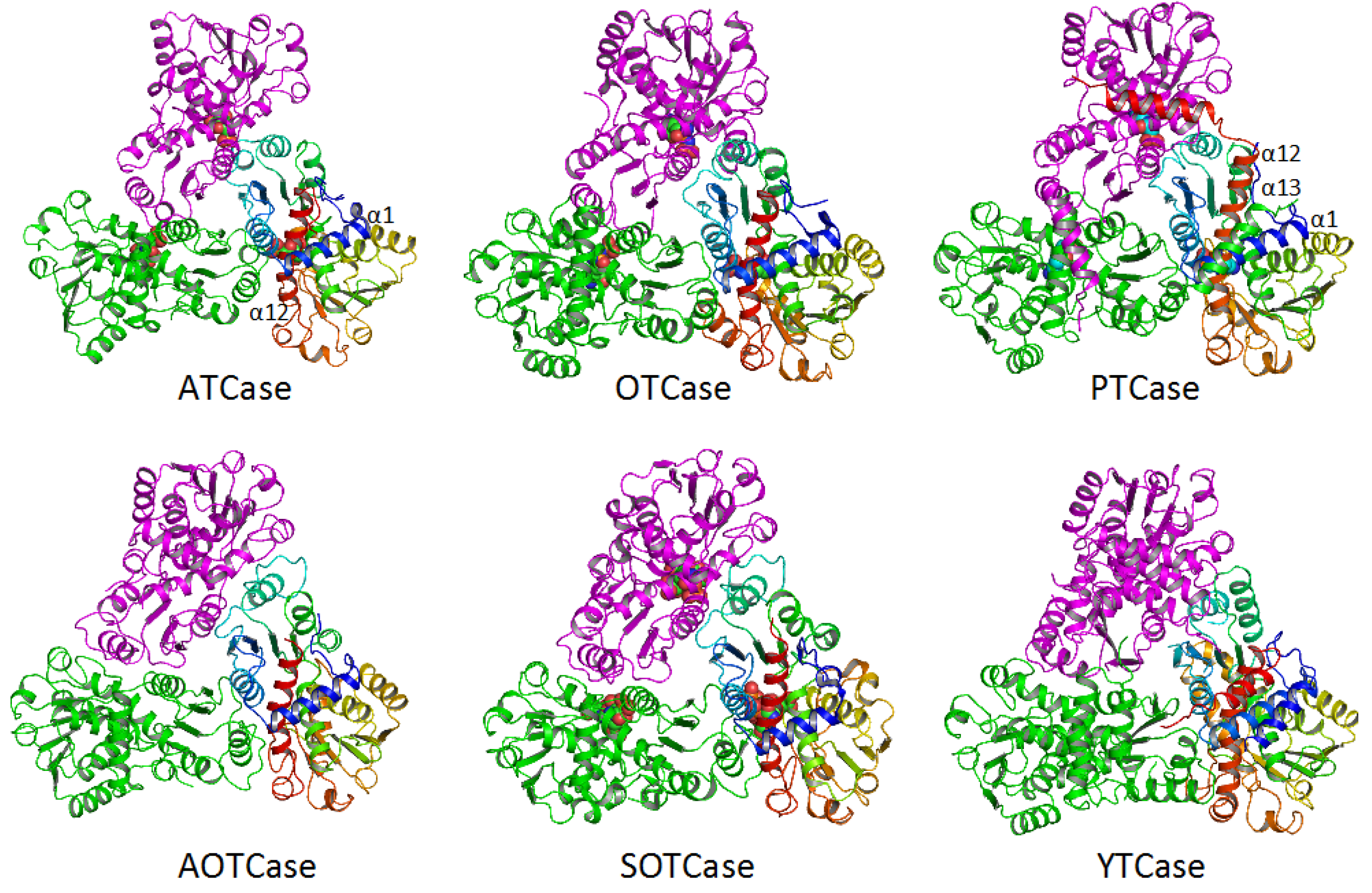
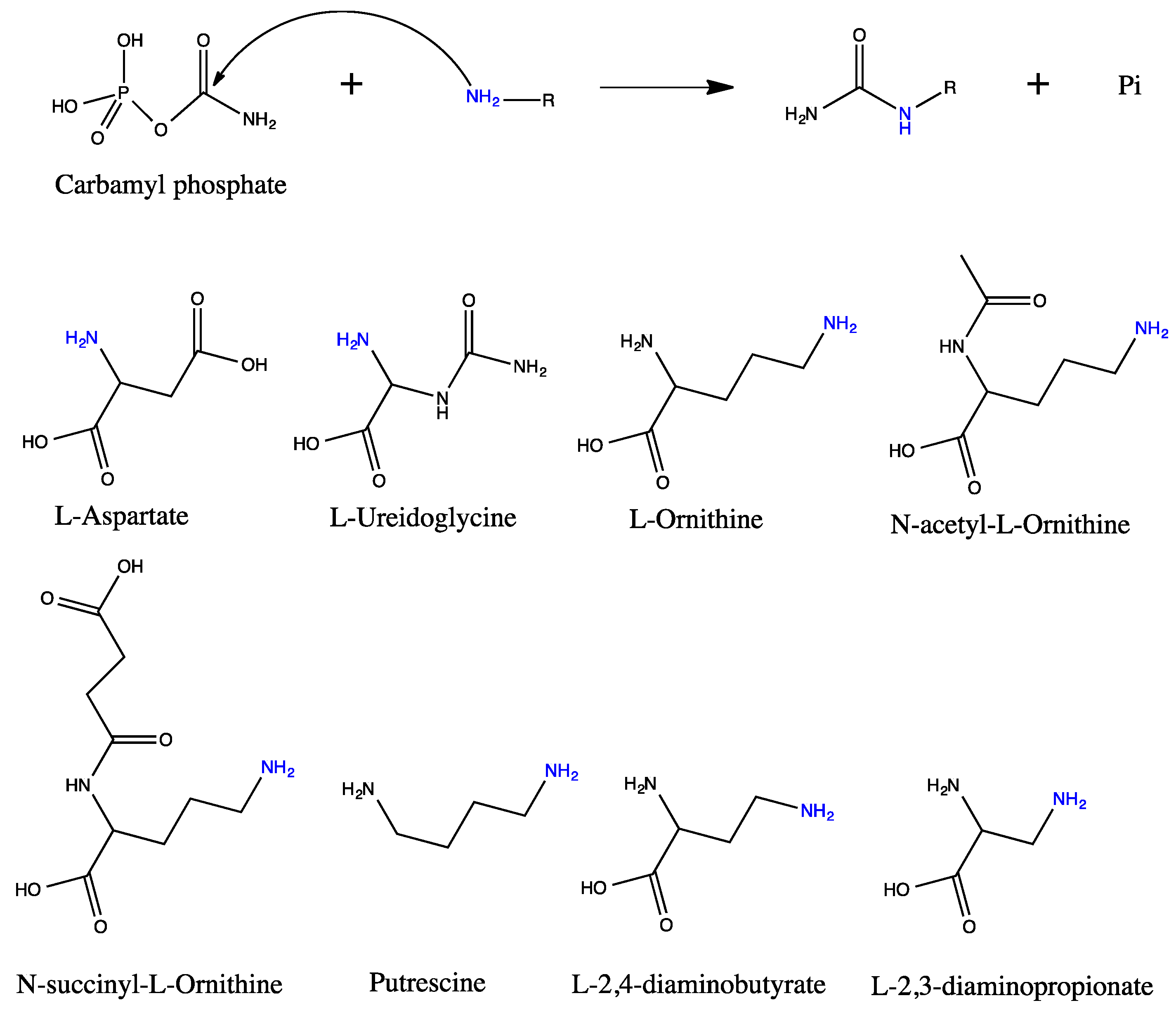
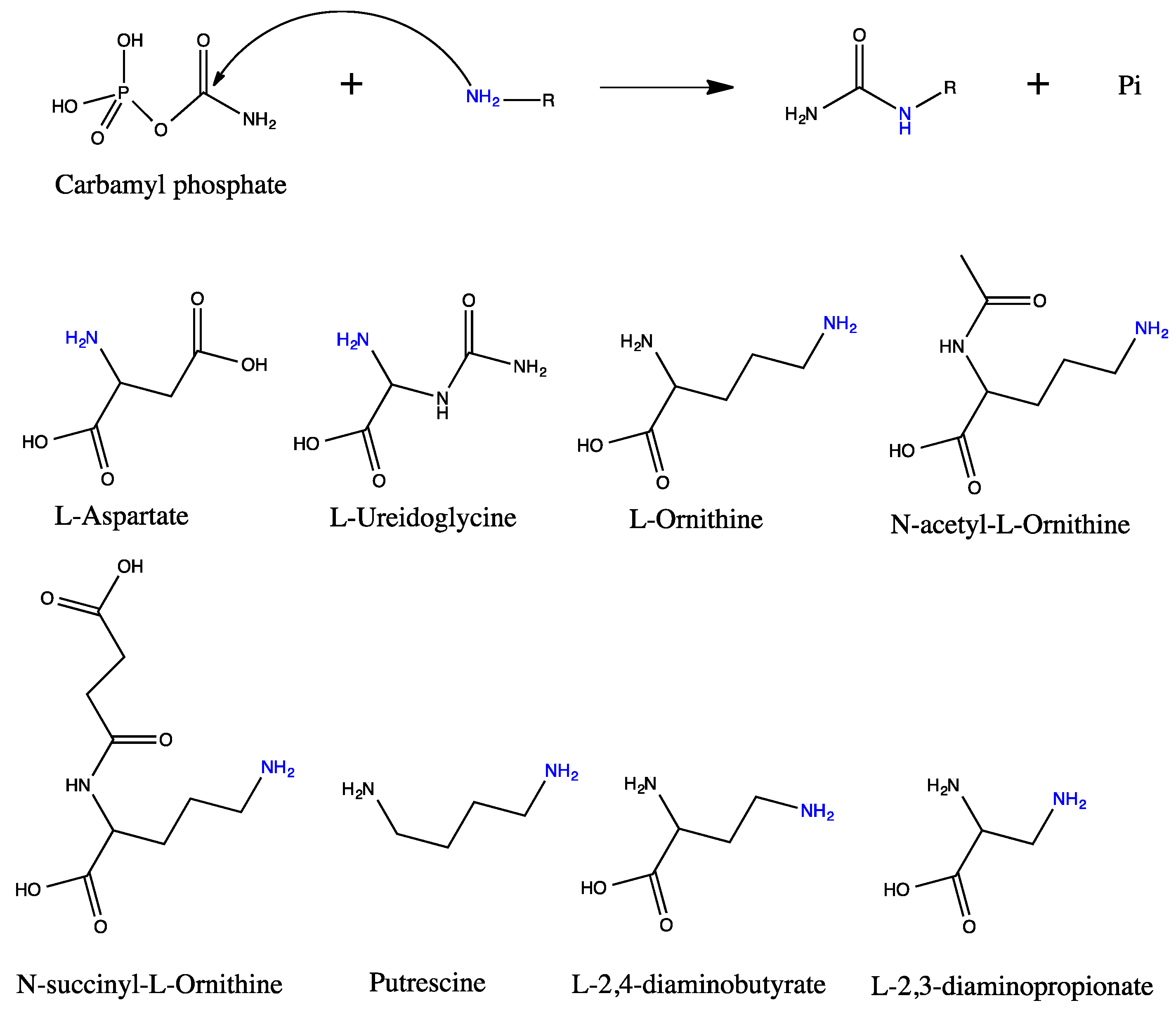
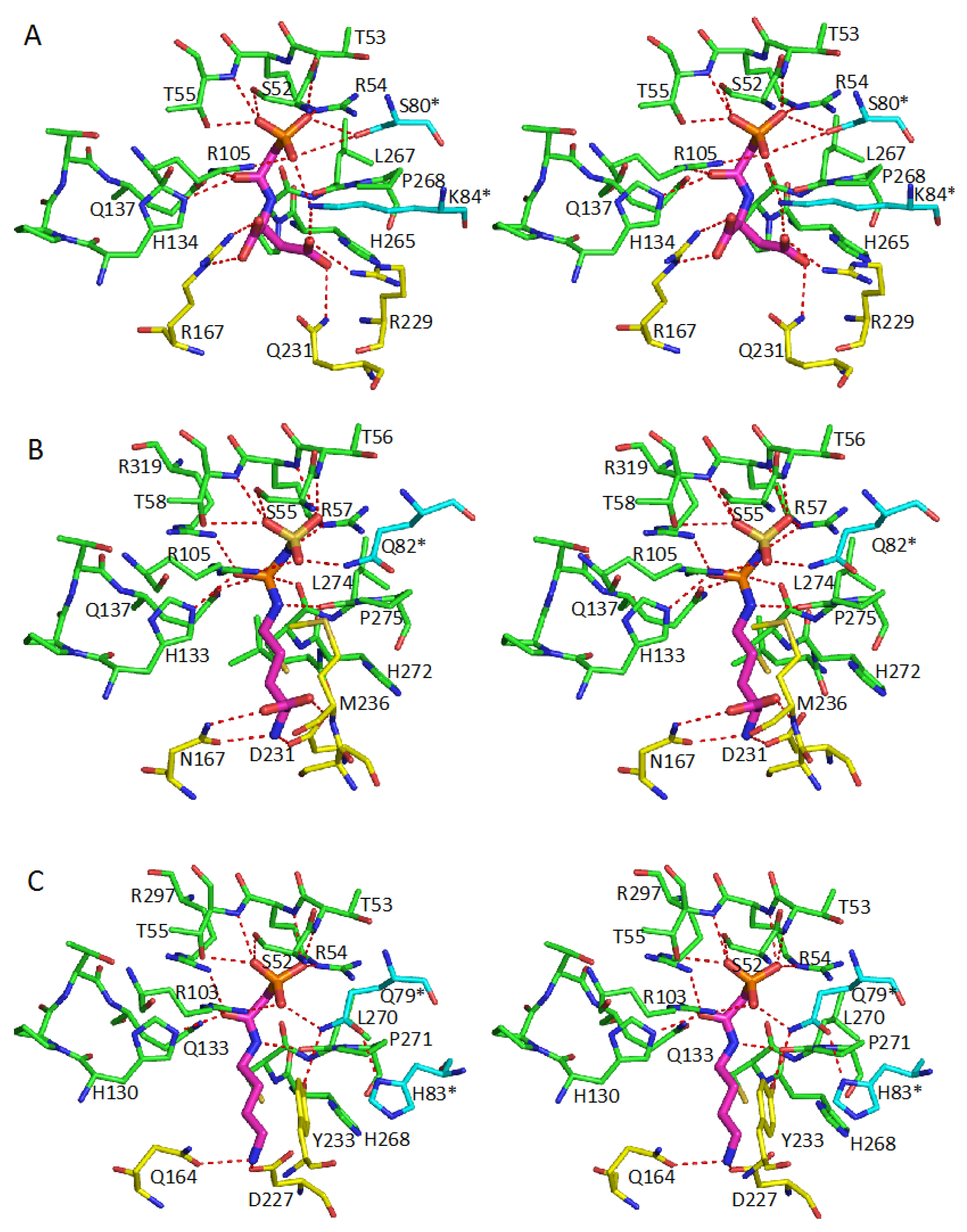
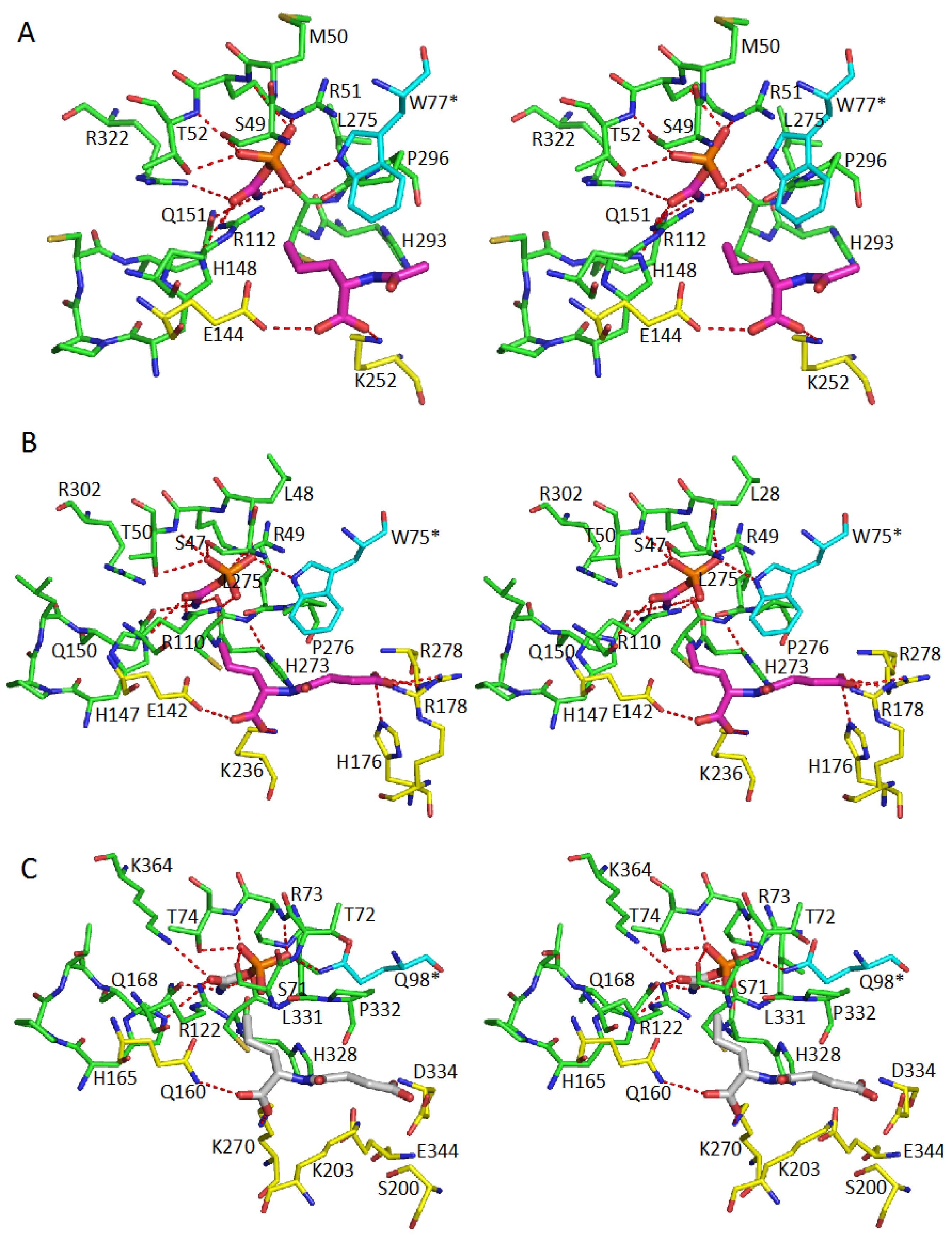
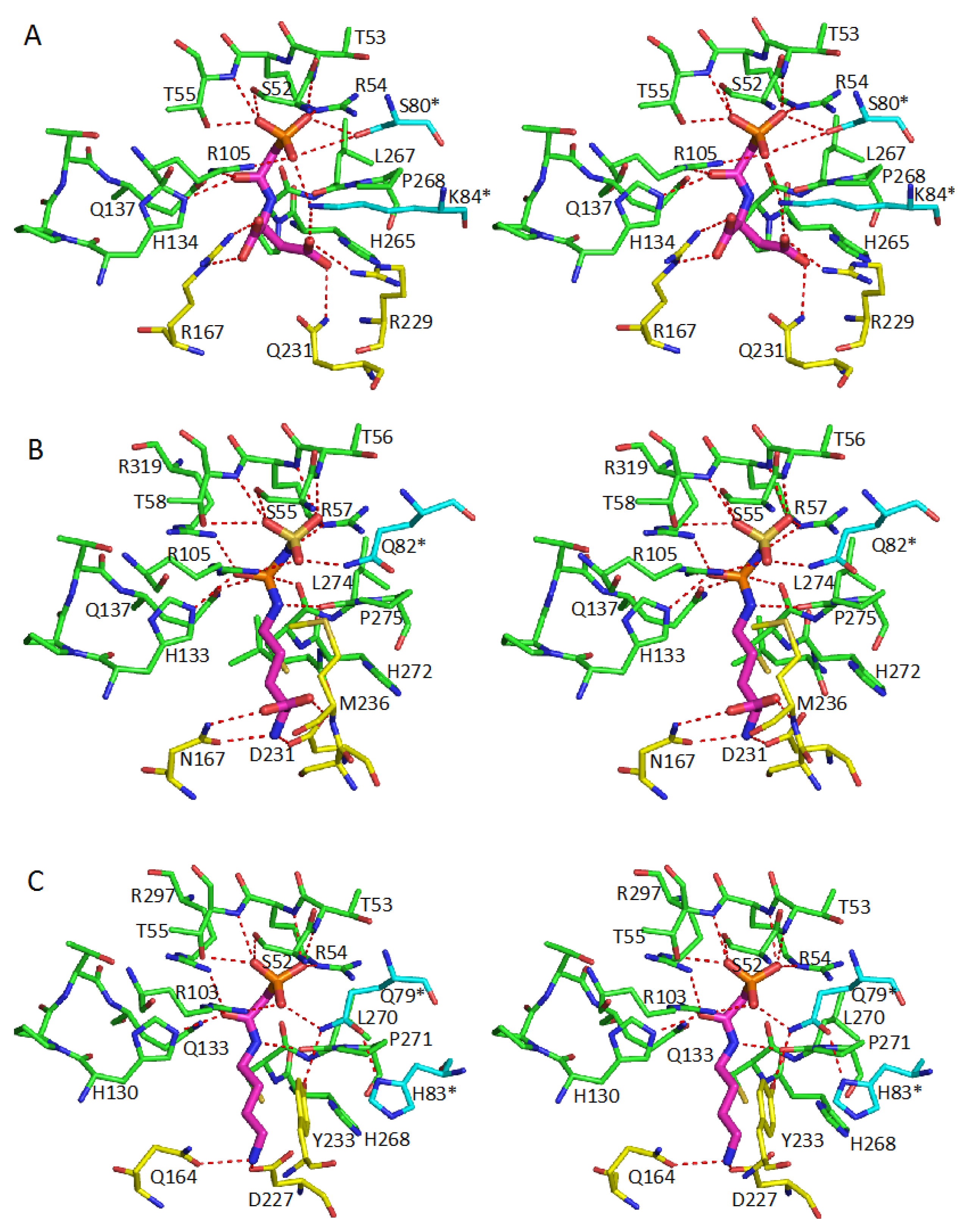
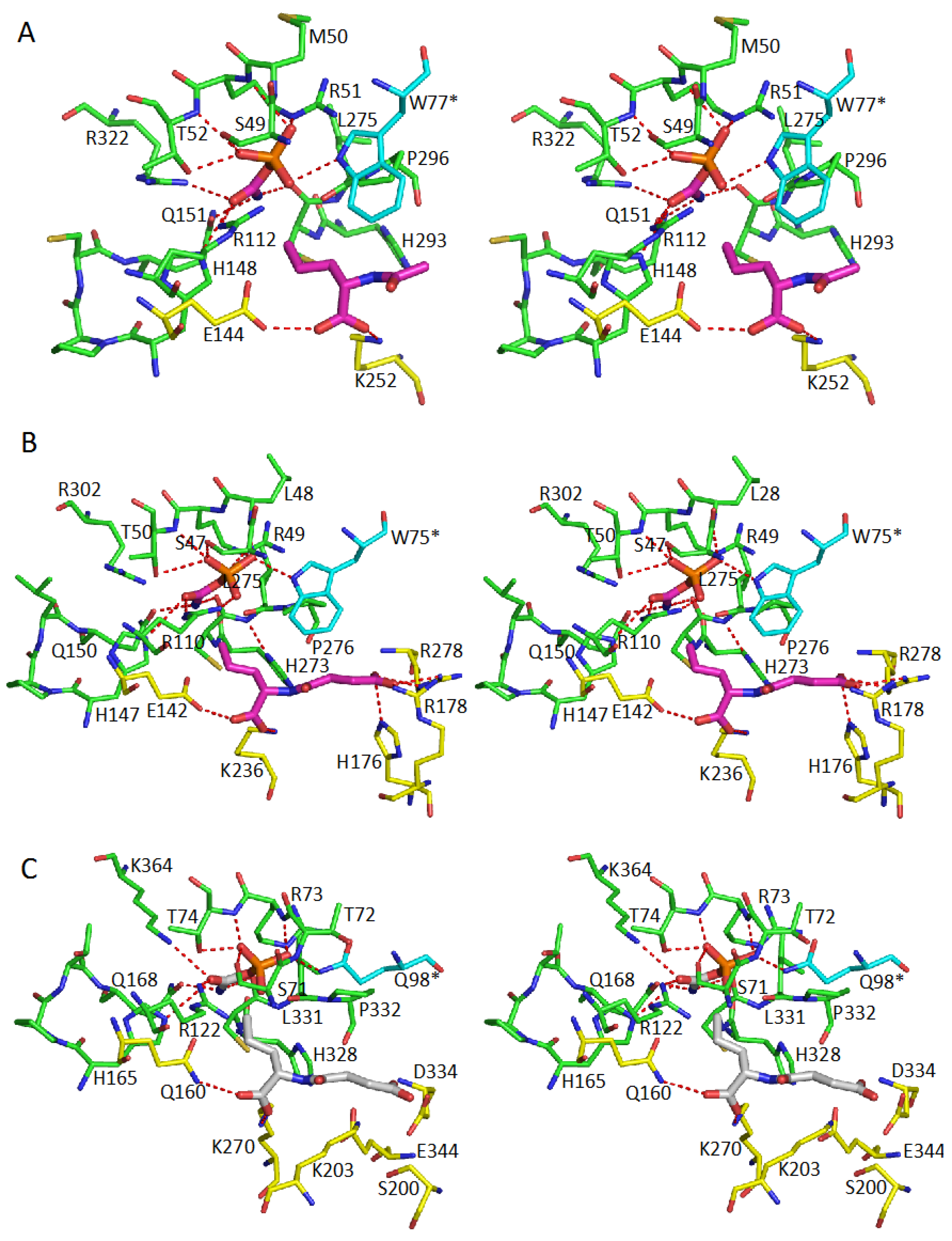
5. Active Site and Substrate Specificities

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | CP-Binding Site | The Second Substrate-Binding Site |
|---|---|---|
| ATCase | S52, T53, R54, T55, R105 H134, Q137, P266, L267 Ser80 *, Lys84 * | R167, Q231, R229, L267 |
| OTCase | S55, T56, R57, T58, R106 H133, Q136, C273, L274 R319, Q82 * | N167, D231, S235, M236, L274 |
| PTCase | S52, T53, R54, T55, R103 H130, Q133, C269, L240 R297, Q79 * | Q164, D227, Y233, L240 |
| AOTCase | S49, M50, R51, T52, R112 H148, Q151, C294, L295 R322, W77 * | E144, K252, L295 |
| SOTCase | S47, L48, R49, T50, R110 H147, Q150, C274, L275 R302, W75 | E142, K236, H176, R178, R278, L275 |
| YTCase | S71, T72, R73, T74, R122 H165, Q168, C330, L331 K363, Q98 * | Q160, K270, D124, S200, K203, L331 |
| DPTCase | S50, T51, R52, T53, R100 H128, Q131, D250, L251 K278, Q76 * | N159, T160, T211, R212, D250, L251 |
| DBTCase | S57, T58, R59, T60, R108 H135, Q138, D271, L272 K299, Q84 * | N166, T229, S233, M234, L272 |
| UGTCase | S74, T75, R76, T77, R126 H155, Q158, T298, L299 S102 *, K106 * | R189, S258, K261, T298, L299 |
6. Catalytic Mechanism and Domain Movement
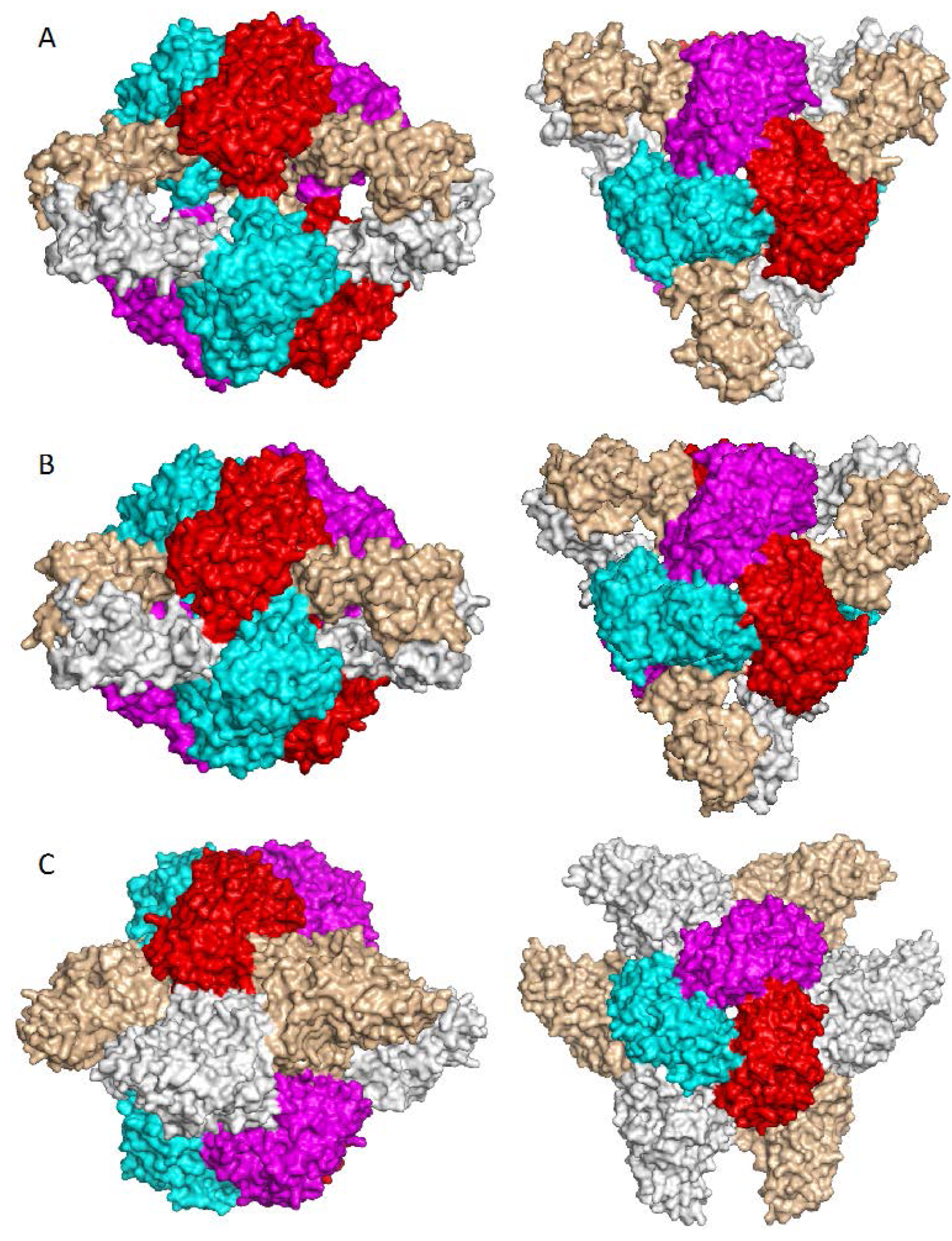
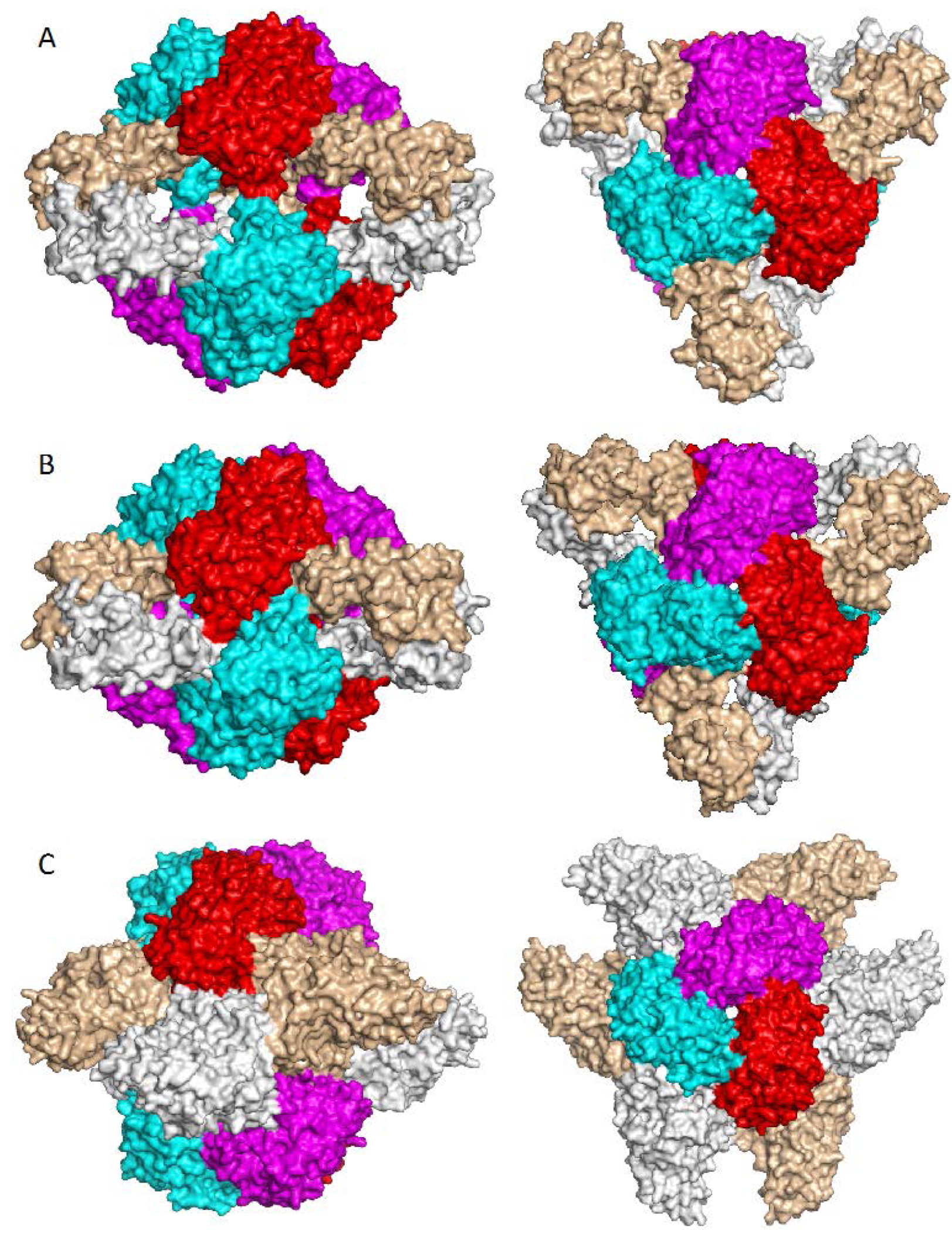
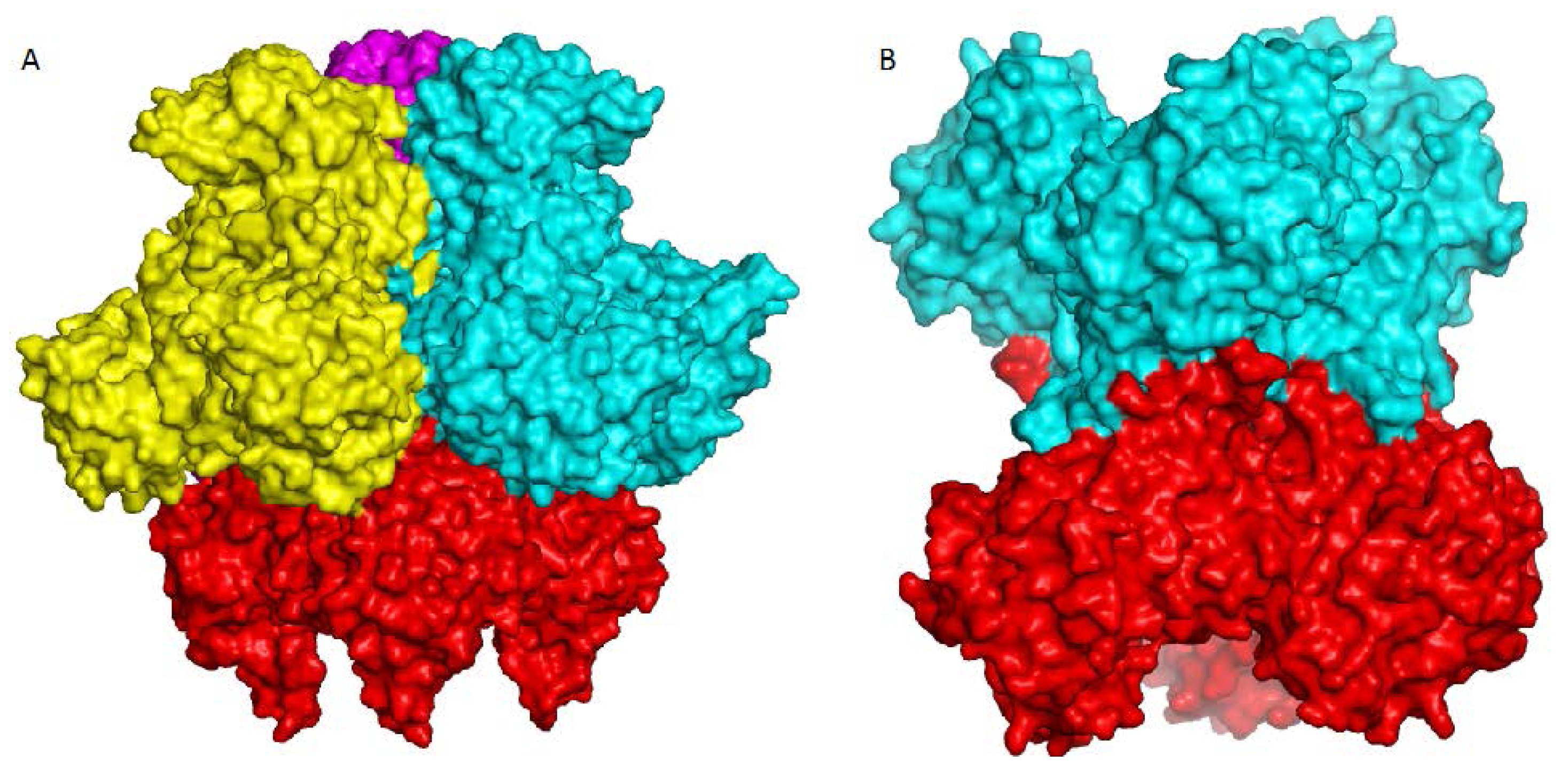
7. Higher Oligomer Structure and Biological Significance
8. Annotation of Transcarbamylases
9. Future Outlook
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jones, M.E.; Spector, L.; Lipmann, F. Carbamyl phosphate, the carbamyl donor in enzymatic citrulline synthesis. J. Am. Chem. Soc. 1955, 77, 819–820. [Google Scholar] [CrossRef]
- Bergh, S.T.; Evans, D.R. Subunit structure of a class a aspartate transcarbamoylase from Pseudomonas fluorescens. Proc. Natl. Acad. Sci. USA 1993, 90, 9818–9822. [Google Scholar] [CrossRef] [PubMed]
- Schurr, M.J.; Vickrey, J.F.; Kumar, A.P.; Campbell, A.L.; Cunin, R.; Benjamin, R.C.; Shanley, M.S.; O’Donovan, G.A. Aspartate transcarbamoylase genes of Pseudomonas putida: Requirement for an inactive dihydroorotase for assembly into the dodecameric holoenzyme. J. Bacteriol. 1995, 177, 1751–1759. [Google Scholar] [PubMed]
- Hughes, L.E.; Hooshdaran, M.Z.; O’Donovan, G.A. Streptomyces aspartate transcarbamoylase is a dodecamer with dihydroorotase activity. Curr. Microbiol. 1999, 39, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Lipscomb, W.N. Aspartate transcarbamylase from Escherichia coli: Activity and regulation. Adv. Enzymol. Relat. Areas Mol. Biol. 1994, 68, 67–151. [Google Scholar] [PubMed]
- Chen, P.; van Vliet, F.; van de Casteele, M.; Legrain, C.; Cunin, R.; Glansdorff, N. Aspartate transcarbamylase from the hyperthermophilic eubacterium Thermotoga maritima: Fused catalytic and regulatory polypeptides form an allosteric enzyme. J. Bacteriol. 1998, 180, 6389–6391. [Google Scholar] [PubMed]
- Stevens, R.C.; Reinisch, K.M.; Lipscomb, W.N. Molecular structure of Bacillus subtilis aspartate transcarbamoylase at 3.0 Å resolution. Proc. Natl. Acad. Sci. USA 1991, 88, 6087–6091. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.I.; Chowdhry, B.Z.; Yon, R.J. Wheat-germ aspartate transcarbamoylase: Revised purification, stability and re-evaluation of regulatory kinetics in terms of the monod-wyman-changeux model. Eur. J. Biochem. 1999, 259, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Coleman, P.F.; Suttle, D.P.; Stark, G.R. Purification of a multifunctional protein bearing carbamyl-phosphate synthase, aspartate transcarbamylase, and dihydroorotase enzyme activities from mutant hamster cells. Methods Enzymol. 1978, 51, 121–134. [Google Scholar] [PubMed]
- Souciet, J.L.; Nagy, M.; le Gouar, M.; Lacroute, F.; Potier, S. Organization of the yeast URA2 gene: Identification of a defective dihydroorotase-like domain in the multifunctional carbamoylphosphate synthetase-aspartate transcarbamylase complex. Gene 1989, 79, 59–70. [Google Scholar] [CrossRef]
- Snodgrass, P.J. The effects of pH on the kinetics of human liver ornithinecarbamyl phosphate transferase. Biochemistry 1968, 7, 3047–3051. [Google Scholar] [CrossRef] [PubMed]
- Legrain, C.; Stalon, V.; Noullez, J.P.; Mercenier, A.; Simon, J.P.; Broman, K.; Wiame, J.M. Structure and function of ornithine carbamoyltransferases. Eur. J. Biochem. 1977, 80, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Stalon, V. Regulation of the catabolic ornithine carbamoyltransferase of Pseudomonas fluorescens. A study of the allosteric interactions. Eur. J. Biochem. 1972, 29, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Haas, D.; Holloway, B.W.; Schambock, A.; Leisinger, T. The genetic organization of arginine biosynthesis in Pseudomonas aeruginosa. Mol. Gen. Genet. 1977, 154, 7–22. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, J.L.; Martin, J.F.; Liras, P. New type of hexameric ornithine carbamoyltransferase with arginase activity in the cephamycin producers Streptomyces clavuligerus and Nocardia lactamdurans. Biochem. J. 1996, 320 Pt 1, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Legrain, C.; Villeret, V.; Roovers, M.; Gigot, D.; Dideberg, O.; Pierard, A.; Glansdorff, N. Biochemical characterisation of ornithine carbamoyltransferase from Pyrococcus furiosus. Eur. J. Biochem. 1997, 247, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Baur, H.; Stalon, V.; Falmagne, P.; Luethi, E.; Haas, D. Primary and quaternary structure of the catabolic ornithine carbamoyltransferase from pseudomonas aeruginosa. Extensive sequence homology with the anabolic ornithine carbamoyltransferases of Escherichia coli. Eur. J. Biochem. 1987, 166, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Stalon, V.; Ramos, F.; Pierard, A.; Wiame, J.M. Regulation of the catabolic ornithine carbamoyltransferase of Pseudomonas fluorescens. A comparison with the anabolic transferase and with a mutationally modified catabolic transferase. Eur. J. Biochem. 1972, 29, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Gallego, P.; Planell, R.; Benach, J.; Querol, E.; Perez-Pons, J.A.; Reverter, D. Structural characterization of the enzymes composing the arginine deiminase pathway in Mycoplasma penetrans. PLoS ONE 2012, 7, e47886. [Google Scholar] [CrossRef] [PubMed]
- De Las Rivas, B.; Fox, G.C.; Angulo, I.; Ripoll, M.M.; Rodriguez, H.; Munoz, R.; Mancheno, J.M. Crystal structure of the hexameric catabolic ornithine transcarbamylase from Lactobacillus hilgardii: Structural insights into the oligomeric assembly and metal binding. J. Mol. Biol 2009, 393, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Morizono, H.; Yu, X.; Roth, L.; Caldovic, L.; Allewell, N.M.; Malamy, M.H.; Tuchman, M. Crystal structure of N-acetylornithine transcarbamylase from Xanthomonas campestris: A novel enzyme in a new arginine biosynthetic pathway found in several eubacteria. J. Biol. Chem. 2005, 280, 14366–14369. [Google Scholar] [CrossRef] [PubMed]
- Morizono, H.; Cabrera-Luque, J.; Shi, D.; Gallegos, R.; Yamaguchi, S.; Yu, X.; Allewell, N.M.; Malamy, M.H.; Tuchman, M. Acetylornithine transcarbamylase: A novel enzyme in arginine biosynthesis. J. Bacteriol. 2006, 188, 2974–2982. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Gallegos, R.; DePonte, J., 3rd; Morizono, H.; Yu, X.; Allewell, N.M.; Malamy, M.; Tuchman, M. Crystal structure of a transcarbamylase-like protein from the anaerobic bacterium Bacteroides fragilis at 2.0 Å resolution. J. Mol. Biol. 2002, 320, 899–908. [Google Scholar] [CrossRef]
- Shi, D.; Morizono, H.; Cabrera-Luque, J.; Yu, X.; Roth, L.; Malamy, M.H.; Allewell, N.M.; Tuchman, M. Structure and catalytic mechanism of a novel N-succinyl-l-ornithine transcarbamylase in arginine biosynthesis of Bacteroides fragilis. J. Biol. Chem. 2006, 281, 20623–20631. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Yu, X.; Cabrera-Luque, J.; Chen, T.Y.; Roth, L.; Morizono, H.; Allewell, N.M.; Tuchman, M. A single mutation in the active site swaps the substrate specificity of N-acetyl-l-ornithine transcarbamylase and N-succinyl-l-ornithine transcarbamylase. Protein Sci. 2007, 16, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Naumoff, D.G.; Xu, Y.; Glansdorff, N.; Labedan, B. Retrieving sequences of enzymes experimentally characterized but erroneously annotated: The case of the putrescine carbamoyltransferase. BMC Genom. 2004, 5, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, J.P.; Stalon, V. Enzymes of agmatine degradation and the control of their synthesis in Streptococcus faecalis. J. Bacteriol. 1982, 152, 676–681. [Google Scholar] [PubMed]
- Llacer, J.L.; Polo, L.M.; Tavarez, S.; Alarcon, B.; Hilario, R.; Rubio, V. The gene cluster for agmatine catabolism of Enterococcus faecalis: Study of recombinant putrescine transcarbamylase and agmatine deiminase and a snapshot of agmatine deiminase catalyzing its reaction. J. Bacteriol. 2007, 189, 1254–1265. [Google Scholar] [CrossRef] [PubMed]
- Polo, L.M.; Gil-Ortiz, F.; Cantin, A.; Rubio, V. New insight into the transcarbamylase family: The structure of putrescine transcarbamylase, a key catalyst for fermentative utilization of agmatine. PLoS ONE 2012, 7, e31528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, D.; Yu, X.; Zhao, G.; Ho, J.; Lu, S.; Allewell, N.M.; Tuchman, M. Crystal structure and biochemical properties of putrescine carbamoyltransferase from Enterococcus faecalis: Assembly, active site, and allosteric regulation. Proteins 2012, 80, 1436–1447. [Google Scholar] [CrossRef] [PubMed]
- Barba, M.; Dutoit, R.; Legrain, C.; Labedan, B. Identifying reaction modules in metabolic pathways: Bioinformatic deduction and experimental validation of a new putative route in purine catabolism. BMC Syst. Biol. 2013, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jin, Z.; Yu, X.; Allewell, N.M.; Tuchman, M.; Shi, D. The ygeW encoded protein from Escherichia coli is a knotted ancestral catabolic transcarbamylase. Proteins 2011, 79, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Barkei, J.J.; Kevany, B.M.; Felnagle, E.A.; Thomas, M.G. Investigations into viomycin biosynthesis by using heterologous production in streptomyces lividans. Chembiochem 2009, 10, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Kevany, B.M.; Rasko, D.A.; Thomas, M.G. Characterization of the complete zwittermicin a biosynthesis gene cluster from bacillus cereus. Appl. Environ. Microbiol. 2009, 75, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, E.R. Allostery and cooperativity in Escherichia coli aspartate transcarbamoylase. Arch. Biochem. Biophys. 2012, 519, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Lipscomb, W.N.; Kantrowitz, E.R. Structure and mechanisms of Escherichia coli aspartate transcarbamoylase. Acc. Chem. Res. 2012, 45, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Schachman, H.K. From allostery to mutagenesis: 20 years with aspartate transcarbamoylase. Biochem. Soc. Trans. 1987, 15, 772–775. [Google Scholar] [CrossRef] [PubMed]
- Allewell, N.M. Escherichia coli aspartate transcarbamoylase: Structure, energetics, and catalytic and regulatory mechanisms. Annu. Rev. Biophys. Biophys. Chem. 1989, 18, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, E.R.; Lipscomb, W.N. Escherichia coli aspartate transcarbamoylase: The molecular basis for a concerted allosteric transition. Trends Biochem. Sci. 1990, 15, 53–59. [Google Scholar] [CrossRef]
- Beernink, P.T.; Endrizzi, J.A.; Alber, T.; Schachman, H.K. Assessment of the allosteric mechanism of aspartate transcarbamoylase based on the crystalline structure of the unregulated catalytic subunit. Proc. Natl. Acad. Sci. USA 1999, 96, 5388–5393. [Google Scholar] [CrossRef] [PubMed]
- Endrizzi, J.A.; Beernink, P.T.; Alber, T.; Schachman, H.K. Binding of bisubstrate analog promotes large structural changes in the unregulated catalytic trimer of aspartate transcarbamoylase: Implications for allosteric regulation induced cell migration. Proc. Natl. Acad. Sci. USA 2000, 97, 5077–5082. [Google Scholar] [CrossRef] [PubMed]
- Van Boxstael, S.; Cunin, R.; Khan, S.; Maes, D. Aspartate transcarbamylase from the hyperthermophilic archaeon Pyrococcus abyssi: Thermostability and 1.8 Å resolution crystal structure of the catalytic subunit complexed with the bisubstrate analogue N-phosphonacetyl-l-aspartate. J. Mol. Biol. 2003, 326, 203–216. [Google Scholar] [CrossRef]
- De Vos, D.; van Petegem, F.; Remaut, H.; Legrain, C.; Glansdorff, N.; van Beeumen, J.J. Crystal structure of T state aspartate carbamoyltransferase of the hyperthermophilic archaeon Sulfolobus acidocaldarius. J. Mol. Biol. 2004, 339, 887–900. [Google Scholar] [CrossRef] [PubMed]
- De Vos, D.; Xu, Y.; Aerts, T.; van Petegem, F.; van Beeumen, J.J. Crystal structure of Sulfolobus acidocaldarius aspartate carbamoyltransferase in complex with its allosteric activator CTP. Biochem. Biophys. Res. Commun. 2008, 372, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Vitali, J.; Colaneri, M.J. Structure of the catalytic trimer of Methanococcus jannaschii aspartate transcarbamoylase in an orthorhombic crystal form. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2008, 64, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Vitali, J.; Colaneri, M.J.; Kantrowitz, E. Crystal structure of the catalytic trimer of Methanococcus jannaschii aspartate transcarbamoylase. Proteins 2008, 71, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Vitali, J.; Singh, A.K.; Soares, A.S.; Colaneri, M.J. Structure of the catalytic chain of Methanococcus jannaschii aspartate transcarbamoylase in a hexagonal crystal form: Insights into the path of carbamoyl phosphate to the active site of the enzyme. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 527–534. [Google Scholar] [CrossRef] [PubMed]
- De Vos, D.; Xu, Y.; Hulpiau, P.; Vergauwen, B.; van Beeumen, J.J. Structural investigation of cold activity and regulation of aspartate carbamoyltransferase from the extreme psychrophilic bacterium Moritella profunda. J. Mol. Biol. 2007, 365, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.M.; Cockrell, G.M.; Puleo, D.E.; Kantrowitz, E.R. Crystallographic snapshots of the complete catalytic cycle of the unregulated aspartate transcarbamoylase from Bacillus subtilis. J. Mol. Biol. 2011, 411, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Martin, P.D.; Purcarea, C.; Vaishnav, A.; Brunzelle, J.S.; Fernando, R.; Guy-Evans, H.I.; Evans, D.R.; Edwards, B.F. Dihydroorotase from the hyperthermophile Aquifex aeolicus is activated by stoichiometric association with aspartate transcarbamoylase and forms a one-pot reactor for pyrimidine biosynthesis. Biochemistry 2009, 48, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Edwards, B.F.; Fernando, R.; Martin, P.D.; Grimley, E.; Cordes, M.; Vaishnav, A.; Brunzelle, J.S.; Evans, H.G.; Evans, D.R. The mononuclear metal center of type-I dihydroorotase from Aquifex aeolicus. BMC Biochem. 2013, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.; McCann, M.T.; Tuchman, M.; Allewell, N.M. Substrate-induced conformational change in a trimeric ornithine transcarbamoylase. Proc. Natl. Acad. Sci. USA 1997, 94, 9550–9555. [Google Scholar] [CrossRef] [PubMed]
- Langley, D.B.; Templeton, M.D.; Fields, B.A.; Mitchell, R.E.; Collyer, C.A. Mechanism of inactivation of ornithine transcarbamoylase by Nδ-(N′-sulfodiaminophosphinyl)-l-ornithine, a true transition state analogue? Crystal structure and implications for catalytic mechanism. J. Biol. Chem. 2000, 275, 20012–20019. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Seaton, B.A.; Head, J.F. Crystal structure at 2.8 Å resolution of anabolic ornithine transcarbamylase from Escherichia coli. Nat. Struct. Biol. 1997, 4, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Villeret, V.; Tricot, C.; Stalon, V.; Dideberg, O. Crystal structure of Pseudomonas aeruginosa catabolic ornithine transcarbamoylase at 3.0-Å resolution: A different oligomeric organization in the transcarbamoylase family. Proc. Natl. Acad. Sci. USA 1995, 92, 10762–10766. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Cherney, M.M.; Garen, C.; Garen, G.; Niu, C.; Yuan, M.; James, M.N. The molecular structure of ornithine acetyltransferase from Mycobacterium tuberculosis bound to ornithine, a competitive inhibitor. J. Mol. Biol. 2010, 397, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Villeret, V.; Clantin, B.; Tricot, C.; Legrain, C.; Roovers, M.; Stalon, V.; Glansdorff, N.; van Beeumen, J. The crystal structure of Pyrococcus furiosus ornithine carbamoyltransferase reveals a key role for oligomerization in enzyme stability at extremely high temperatures. Proc. Natl. Acad. Sci. USA 1998, 95, 2801–2806. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Morizono, H.; Ha, Y.; Aoyagi, M.; Tuchman, M.; Allewell, N.M. 1. 85-Å resolution crystal structure of human ornithine transcarbamoylase complexed with N-phosphonacetyl-l-ornithine. Catalytic mechanism and correlation with inherited deficiency. J. Biol. Chem. 1998, 273, 34247–34254. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Morizono, H.; Yu, X.; Tong, L.; Allewell, N.M.; Tuchman, M. Crystallization and preliminary X-ray crystallographic studies of wild-type human ornithine transcarbamylase and two naturally occurring mutants at position 277. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Morizono, H.; Yu, X.; Tong, L.; Allewell, N.M.; Tuchman, M. Human ornithine transcarbamylase: Crystallographic insights into substrate recognition and conformational changes. Biochem. J. 2001, 354, 501–509. [Google Scholar] [CrossRef] [PubMed]
- De Gregorio, A.; Battistutta, R.; Arena, N.; Panzalorto, M.; Francescato, P.; Valentini, G.; Bruno, G.; Zanotti, G. Functional and structural characterization of ovine ornithine transcarbamoylase. Org. Biomol. Chem. 2003, 1, 3178–3185. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Yu, X.; Roth, L.; Morizono, H.; Tuchman, M.; Allewell, N.M. Structures of N-acetylornithine transcarbamoylase from Xanthomonas campestris complexed with substrates and substrate analogs imply mechanisms for substrate binding and catalysis. Proteins 2006, 64, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Morada, M.; Manzur, M.; Lam, B.; Tan, C.; Tachezy, J.; Rappelli, P.; Dessi, D.; Fiori, P.L.; Yarlett, N. Arginine metabolism in Trichomonas vaginalis infected with Mycoplasma hominis. Microbiology 2010, 156, 3734–3743. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kwon, Y.M. Identification of an isoform of ornithine carbamoyltransferase that can effectively utilize canaline as a substrate from the leaves of Canavalia lineata. Plant Sci. 2000, 151, 145–151. [Google Scholar] [CrossRef]
- Slocum, R.D. Genes, enzymes and regulation of arginine biosynthesis in plants. Plant. Physiol. Biochem. 2005, 43, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.L.; Slocum, R.D. Molecular cloning and characterization of the pyrB1 and pyrB2 genes encoding aspartate transcarbamoylase in pea (Pisum sativum L.). Plant Physiol. 1994, 105, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Serre, V.; Penverne, B.; Souciet, J.L.; Potier, S.; Guy, H.; Evans, D.; Vicart, P.; Herve, G. Integrated allosteric regulation in the S. cerevisiae carbamylphosphate synthetase—Aspartate transcarbamylase multifunctional protein. BMC Biochem. 2004, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lollier, M.; Jaquet, L.; Nedeva, T.; Lacroute, F.; Potier, S.; Souciet, J.L. As in Saccharomyces cerevisiae, aspartate transcarbamoylase is assembled on a multifunctional protein including a dihydroorotase-like cryptic domain in Schizosaccharomyces pombe. Curr. Genet. 1995, 28, 138–149. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Atkins, J.D.; Salehe, B.R.; Shuid, A.N.; Roche, D.B. IntFOLD: An integrated server for modelling protein structures and functions from amino acid sequences. Nucleic Acids Res. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.L.; Dalisay, D.S.; Andersen, R.J.; Ryan, K.S. N-carbamoylation of 2,4-diaminobutyrate reroutes the outcome in padanamide biosynthesis. Chem. Biol. 2013, 20, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Stieglitz, K.A.; Cardia, J.P.; Kantrowitz, E.R. Structural basis for ordered substrate binding and cooperativity in aspartate transcarbamoylase. Proc. Natl. Acad. Sci. USA 2005, 102, 8881–8886. [Google Scholar] [CrossRef] [PubMed]
- Gouaux, J.E.; Krause, K.L.; Lipscomb, W.N. The catalytic mechanism of Escherichia coli aspartate carbamoyltransferase: A molecular modelling study. Biochem. Biophys. Res. Commun. 1987, 142, 893–897. [Google Scholar] [CrossRef]
- Gerhart, J.C.; Schachman, H.K. Distinct subunits for the regulation and catalytic activity of aspartate transcarbamylase. Biochemistry 1965, 4, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.C.; Lipscomb, W.N. A molecular mechanism for pyrimidine and purine nucleotide control of aspartate transcarbamoylase. Proc. Natl. Acad. Sci. USA 1992, 89, 5281–5285. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.R.; Guy, H.I. Mammalian pyrimidine biosynthesis: Fresh insights into an ancient pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef] [PubMed]
- Grande-Garcia, A.; Lallous, N.; Diaz-Tejada, C.; Ramon-Maiques, S. Structure, functional characterization, and evolution of the dihydroorotase domain of human cad. Structure 2014, 22, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Massant, J.; Wouters, J.; Glansdorff, N. Refined structure of Pyrococcus furiosus ornithine carbamoyltransferase at 1.87 Å. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 2140–2149. [Google Scholar] [CrossRef] [PubMed]
- Tricot, C.; Villeret, V.; Sainz, G.; Dideberg, O.; Stalon, V. Allosteric regulation in Pseudomonas aeruginosa catabolic ornithine carbamoyltransferase revisited: Association of concerted homotropic cooperative interactions and local heterotropic effects. J. Mol. Biol. 1998, 283, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Labedan, B.; Boyen, A.; Baetens, M.; Charlier, D.; Chen, P.; Cunin, R.; Durbeco, V.; Glansdorff, N.; Herve, G.; Legrain, C.; et al. The evolutionary history of carbamoyltransferases: A complex set of paralogous genes was already present in the last universal common ancestor. J. Mol. Evol. 1999, 49, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Harp, D.F.; Chowdhury, I. Trichomoniasis: Evaluation to execution. Eur. J. Obstet. Gynecol. Reprod. Biol. 2011, 157, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Yarlett, N.; Martinez, M.P.; Moharrami, M.A.; Tachezy, J. The contribution of the arginine dihydrolase pathway to energy metabolism by trichomonas vaginalis. Mol. Biochem. Parasitol. 1996, 78, 117–125. [Google Scholar] [CrossRef]
- Felnagle, E.A.; Rondon, M.R.; Berti, A.D.; Crosby, H.A.; Thomas, M.G. Identification of the biosynthetic gene cluster and an additional gene for resistance to the antituberculosis drug capreomycin. Appl. Environ. Microbiol. 2007, 73, 4162–4170. [Google Scholar] [CrossRef] [PubMed]
- Caldovic, L.; Morizono, H.; Daikhin, Y.; Nissim, I.; McCarter, R.J.; Yudkoff, M.; Tuchman, M. Restoration of ureagenesis in N-acetylglutamate synthase deficiency by N-carbamylglutamate. J. Pediatr. 2004, 145, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Plecko, B.; Erwa, W.; Wermuth, B. Partial N-acetylglutamate synthetase deficiency in a 13-year-old girl: Diagnosis and response to treatment with N-carbamylglutamate. Eur. J. Pediatr. 1998, 157, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.A.; Richmond, S.W.; Oddie, S.J.; Pourfarzam, M.; Worthington, V.; Leonard, J.V. N-acetylglutamate synthetase deficiency: Favourable experience with carbamylglutamate. J. Inherit. Metab. Dis. 1998, 21, 867–868. [Google Scholar] [CrossRef] [PubMed]
- Hinnie, J.; Colombo, J.P.; Wermuth, B.; Dryburgh, F.J. N-acetylglutamate synthetase deficiency responding to carbamylglutamate. J. Inherit. Metab. Dis. 1997, 20, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Guffon, N.; Vianey-Saban, C.; Bourgeois, J.; Rabier, D.; Colombo, J.P.; Guibaud, P. A new neonatal case of N-acetylglutamate synthase deficiency treated by carbamylglutamate. J. Inherit. Metab. Dis. 1995, 18, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Li, S.; Gan, R.Y.; Zhou, T.; Xu, D.P.; Li, H.B. Impacts of gut bacteria on human health and diseases. Int. J. Mol. Sci. 2015, 16, 7493–7519. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, J.W.; Robertson, D.E.; Roberts, M.F.; Stevens, R.C.; Lipscomb, W.N.; Kantrowitz, E.R. Arginine 54 in the active site of Escherichia coli aspartate transcarbamoylase is critical for catalysis: A site-specific mutagenesis, NMR, and X-ray crystallographic study. Protein Sci. 1992, 1, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Gouaux, J.E.; Lipscomb, W.N. Crystal structures of phosphonoacetamide ligated T and phosphonoacetamide and malonate ligated R states of aspartate carbamoyltransferase at 2.8-Å resolution and neutral pH. Biochemistry 1990, 29, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Stec, B.; Lipscomb, W.N.; Kantrowitz, E.R. Insights into the mechanisms of catalysis and heterotropic regulation of Escherichia coli aspartate transcarbamoylase based upon a structure of the enzyme complexed with the bisubstrate analogue N-phosphonacetyl-l-aspartate at 2.1 Å. Proteins 1999, 37, 729–742. [Google Scholar] [CrossRef]
- Jin, L.; Stec, B.; Kantrowitz, E.R. A cis-proline to alanine mutant of E. coli aspartate transcarbamoylase: Kinetic studies and three-dimensional crystal structures. Biochemistry 2000, 39, 8058–8066. [Google Scholar] [CrossRef] [PubMed]
- Macol, C.P.; Tsuruta, H.; Stec, B.; Kantrowitz, E.R. Direct structural evidence for a concerted allosteric transition in Escherichia coli aspartate transcarbamoylase. Nat. Struct. Biol. 2001, 8, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.K.; Stec, B.; Kantrowitz, E.R. A single mutation in the regulatory chain of Escherichia coli aspartate transcarbamoylase results in an extreme T-state structure. J. Mol. Biol. 1998, 281, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lipscomb, W.N. Aspartate transcarbamylase (ATcase) of Escherichia coli: A new crystalline R-state bound to PALA, or to product analogues citrate and phosphate. Biochemistry 2004, 43, 6415–6421. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lipscomb, W.N. Products in the T-state of aspartate transcarbamylase: Crystal structure of the phosphate and N-carbamyl-l-aspartate ligated enzyme. Biochemistry 2004, 43, 6422–6426. [Google Scholar] [CrossRef] [PubMed]
- Kosman, R.P.; Gouaux, J.E.; Lipscomb, W.N. Crystal structure of CTP-ligated t state aspartate transcarbamoylase at 2.5 Å resolution: Implications for atcase mutants and the mechanism of negative cooperativity. Proteins 1993, 15, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Alam, N.; Stieglitz, K.A.; Caban, M.D.; Gourinath, S.; Tsuruta, H.; Kantrowitz, E.R. 240s loop interactions stabilize the T state of Escherichia coli aspartate transcarbamoylase. J. Biol. Chem. 2004, 279, 23302–23310. [Google Scholar] [CrossRef] [PubMed]
- Stieglitz, K.; Stec, B.; Baker, D.P.; Kantrowitz, E.R. Monitoring the transition from the T to the R state in E. coli aspartate transcarbamoylase by X-ray crystallography: Crystal structures of the e50a mutant enzyme in four distinct allosteric states. J. Mol. Biol. 2004, 341, 853–868. [Google Scholar] [CrossRef] [PubMed]
- Stieglitz, K.A.; Pastra-Landis, S.C.; Xia, J.; Tsuruta, H.; Kantrowitz, E.R. A single amino acid substitution in the active site of Escherichia coli aspartate transcarbamoylase prevents the allosteric transition. J. Mol. Biol. 2005, 349, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Stieglitz, K.A.; Dusinberre, K.J.; Cardia, J.P.; Tsuruta, H.; Kantrowitz, E.R. Structure of the E. coli aspartate transcarbamoylase trapped in the middle of the catalytic cycle. J. Mol. Biol. 2005, 352, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Honzatko, R.B.; Crawford, J.L.; Monaco, H.L.; Ladner, J.E.; Ewards, B.F.; Evans, D.R.; Warren, S.G.; Wiley, D.C.; Ladner, R.C.; Lipscomb, W.N. Crystal and molecular structures of native and CTP-liganded aspartate carbamoyltransferase from Escherichia coli. J. Mol. Biol. 1982, 160, 219–263. [Google Scholar] [CrossRef]
- Stevens, R.C.; Gouaux, J.E.; Lipscomb, W.N. Structural consequences of effector binding to the T state of aspartate carbamoyltransferase: Crystal structures of the unligated and ATP- and CTP-complexed enzymes at 2.6-Å resolution. Biochemistry 1990, 29, 7691–7701. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.M.; Lipscomb, W.N.; Cho, Y.J.; Honzatko, R.B. Complex of N-phosphonacetyl-l-aspartate with aspartate carbamoyltransferase. X-ray refinement, analysis of conformational changes and catalytic and allosteric mechanisms. J. Mol. Biol. 1988, 204, 725–747. [Google Scholar] [CrossRef]
- Ha, Y.; Allewell, N.M. Intersubunit hydrogen bond acts as a global molecular switch in Escherichia coli aspartate transcarbamoylase. Proteins 1998, 33, 430–443. [Google Scholar] [CrossRef]
- Huang, J.; Lipscomb, W.N. T-state active site of aspartate transcarbamylase: Crystal structure of the carbamyl phosphate and l-alanosine ligated enzyme. Biochemistry 2006, 45, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Heng, S.; Stieglitz, K.A.; Eldo, J.; Xia, J.; Cardia, J.P.; Kantrowitz, E.R. T-state inhibitors of E. coli aspartate transcarbamoylase that prevent the allosteric transition. Biochemistry 2006, 45, 10062–10071. [Google Scholar] [CrossRef] [PubMed]
- Cardia, J.P.; Eldo, J.; Xia, J.; O’Day, E.M.; Tsuruta, H.; Gryncel, K.R.; Kantrowitz, E.R. Use of l-asparagine and N-phosphonacetyl-l-asparagine to investigate the linkage of catalysis and homotropic cooperativity in E. coli aspartate transcarbomoylase. Proteins 2008, 71, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Eldo, J.; Cardia, J.P.; O’Day, E.M.; Xia, J.; Tsuruta, H.; Kantrowitz, E.R. N-phosphonacetyl-l-isoasparagine a potent and specific inhibitor of Escherichia coli aspartate transcarbamoylase. J. Med. Chem. 2006, 49, 5932–5938. [Google Scholar] [CrossRef] [PubMed]
- Stec, B.; Williams, M.K.; Stieglitz, K.A.; Kantrowitz, E.R. Comparison of two T-state structures of regulatory-chain mutants of Escherichia coli aspartate transcarbamoylase suggests that his20 and asp19 modulate the response to heterotropic effectors. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Stieglitz, K.A.; Xia, J.; Kantrowitz, E.R. The first high pH structure of Escherichia coli aspartate transcarbamoylase. Proteins 2009, 74, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Cockrell, G.M.; Zheng, Y.; Guo, W.; Peterson, A.W.; Truong, J.K.; Kantrowitz, E.R. New paradigm for allosteric regulation of Escherichia coli aspartate transcarbamoylase. Biochemistry 2013, 52, 8036–8047. [Google Scholar] [CrossRef] [PubMed]
- Cockrell, G.M.; Kantrowitz, E.R. Metal ion involvement in the allosteric mechanism of Escherichia coli aspartate transcarbamoylase. Biochemistry 2012, 51, 7128–7137. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.W.; Cockrell, G.M.; Kantrowitz, E.R. A second allosteric site in Escherichia coli aspartate transcarbamoylase. Biochemistry 2012, 51, 4776–4778. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; West, J.M.; Dutton, A.S.; Tsuruta, H.; Kantrowitz, E.R. Trapping and structure determination of an intermediate in the allosteric transition of aspartate transcarbamoylase. Proc. Natl. Acad. Sci. USA 2012, 109, 7741–7746. [Google Scholar] [CrossRef] [PubMed]
- Mendes, K.R.; Kantrowitz, E.R. A cooperative Escherichia coli aspartate transcarbamoylase without regulatory subunits. Biochemistry 2010, 49, 7694–7703. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Morizono, H.; Aoyagi, M.; Tuchman, M.; Allewell, N.M. Crystal structure of human ornithine transcarbamylase complexed with carbamoyl phosphate and l-norvaline at 1.9 Å resolution. Proteins 2000, 39, 271–277. [Google Scholar] [CrossRef]
- Baugh, L.; Gallagher, L.A.; Patrapuvich, R.; Clifton, M.C.; Gardberg, A.S.; Edwards, T.E.; Armour, B.; Begley, D.W.; Dieterich, S.H.; Dranow, D.M.; et al. Combining functional and structural genomics to sample the essential burkholderia structome. PLoS ONE 2013, 8, e53851. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Cherney, M.M.; Cherney, L.T.; Garen, C.R.; Moradian, F.; James, M.N. The crystal structures of ornithine carbamoyltransferase from mycobacterium tuberculosis and its ternary complex with carbamoyl phosphate and l-norvaline reveal the enzyme’s catalytic mechanism. J. Mol. Biol. 2008, 375, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Galkin, A.; Kulakova, L.; Wu, R.; Gong, M.; Dunaway-Mariano, D.; Herzberg, O. X-ray structure and kinetic properties of ornithine transcarbamoylase from the human parasite giardia lamblia. Proteins 2009, 76, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Shabalin, I.G.; Porebski, P.J.; Cooper, D.R.; Grabowski, M.; Onopriyenko, O.; Grimshaw, S.; Savchenko, A.; Chruszcz, M.; Minor, W. Structure of anabolic ornithine carbamoyltransferase from campylobacter jejuni at 2.7 Å resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.N.; Choi, R.; Kelley, A.; Crowther, G.J.; Napuli, A.J.; van Voorhis, W.C. Expression of proteins in Escherichia coli as fusions with maltose-binding protein to rescue non-expressed targets in a high-throughput protein-expression and purification pipeline. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, X.; Ho, J.; Fushman, D.; Allewell, N.M.; Tuchman, M.; Shi, D. Reversible post-translational carboxylation modulates the enzymatic activity of N-acetyl-l-ornithine transcarbamylase. Biochemistry 2010, 49, 6887–6895. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, D.; Allewell, N.M.; Tuchman, M. From Genome to Structure and Back Again: A Family Portrait of the Transcarbamylases. Int. J. Mol. Sci. 2015, 16, 18836-18864. https://doi.org/10.3390/ijms160818836
Shi D, Allewell NM, Tuchman M. From Genome to Structure and Back Again: A Family Portrait of the Transcarbamylases. International Journal of Molecular Sciences. 2015; 16(8):18836-18864. https://doi.org/10.3390/ijms160818836
Chicago/Turabian StyleShi, Dashuang, Norma M. Allewell, and Mendel Tuchman. 2015. "From Genome to Structure and Back Again: A Family Portrait of the Transcarbamylases" International Journal of Molecular Sciences 16, no. 8: 18836-18864. https://doi.org/10.3390/ijms160818836
APA StyleShi, D., Allewell, N. M., & Tuchman, M. (2015). From Genome to Structure and Back Again: A Family Portrait of the Transcarbamylases. International Journal of Molecular Sciences, 16(8), 18836-18864. https://doi.org/10.3390/ijms160818836