
Treatment Strategies that Enhance the Efficacy and Selectivity of Mitochondria-Targeted Anticancer Agents


Abstract
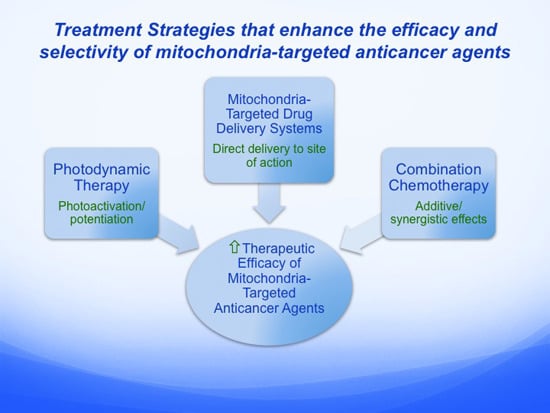
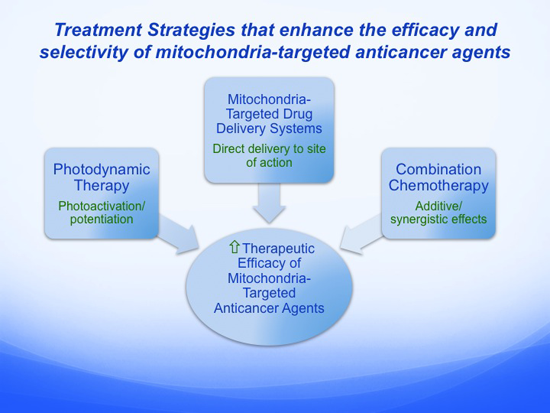
:
1. Introduction
2. Mitochondria Structure and Function
3. Some Notable Differences between Mitochondria of Cancer Cells and Normal Cells
4. Mitochondria-Targeted Drugs that Show Selective Cancer Cell Killing

{kind=link}
| Class | Compound | Mode of Action | Demonstrated Efficacy | References |
|---|---|---|---|---|
| OxPhos Inhibitors | Rhodamine 123 | ATP Synthase inhibitor | Preclinical (in vitro, in vivo) | [60,61,62] |
| Dequalinium Chloride | Complex I inhibitor | Preclinical (in vitro, in vivo) | [63,64] | |
| AA-1 | ATP Synthase inhibitor | Preclinical (in vitro, in vivo) | [65] | |
| MKT-077 | General inhibition of ETC enzymes | Preclinical (in vitro, in vivo) | [66,67,68,69] | |
| Clinical, Phase I | ||||
| Metformin | Complex I inhibitor | Preclinical (in vitro, in vivo) | [70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89] | |
| Clinical, Phase I | ||||
| ROS Regulators | Elesclomol | Enhanced ROS production | Preclinical (in vitro, in vivo) | [90,91,92] |
| Clinical, Phase I | ||||
| Bezielle | Enhanced ROS production | Preclinical (in vitro, in vivo) | [93,94,95,96,97,98,99] | |
| Clinical, Phase I | ||||
| Intrinsic Apoptosis Inducers | ABT-737 | BH3 mimetic | Preclinical (in vitro, in vivo) | [100,101,102] |
| ABT-263 (Navitoclax) | BH3 mimetic | Preclinical (in vitro, in vivo) | [103,104,105] | |
| Clinical, Phase I/II | ||||
| Gossypol | BH3 mimetic | Preclinical (in vitro, in vivo) | [106,107] | |
| GX15-070 (Obatoclax) | BH3 mimetic | Preclinical (in vitro, in vivo) | [108,109] | |
| HA14-1 | BH3 mimetic | Preclinical (in vitro, in vivo) | [110,111] |
5. Alternative Treatment Strategies that Enhance the Efficacy and Selectivity of Mitochondria-Targeted Anticancer Agents
| Strategy | Carrier/Class | Anticancer Agent | References |
|---|---|---|---|
| Mitochondria-Targeted Drug Delivery Systems | TPP+-conjugated molecules | Vitamin E succinate | [122,123] |
| Coenzyme Q | [124] | ||
| DQAsomes | Paclitaxel | [125,126,127] | |
| Curcumin | [128] | ||
| Resveratrol | [129] | ||
| STPP+ liposomes | Paclitaxel | [130,131] | |
| Doxorubicin | [132] | ||
| Mito-targeted nanontubes | Platinum (IV) | [133] | |
| Photodynamic Therapy | Cationic photosensitizers | EDKC | [134] |
| Rh123 | [135] | ||
| MKT-077 | [136] | ||
| Non-cationic photosensitizers | Pba | [137,138,139,140,141,142,143] | |
| BBr2 | [144] | ||
| Combination Chemotherapy | Inhibitors of glycolysis and oxidative phosphorylation | 2-DG plus metformin | [145,146] |
| Inhibitors of two or more mitochondrial target sites | AZT plus MKT-077 | [147] |
5.1. Mitochondria-Targeted Drug Delivery Systems
5.2. Photodynamic Therapy
5.3. Combination Chemotherapy
6. Summary and Concluding Remarks
Author Contributions
Conflicts of Interest
References
- American Cancer Society. Lifetime Risk of Developing or Dying from Cancer. Available online: http://www.cancer.org/cancer/cancerbasics/lifetime-probability-of-developing-or-dying-from-cancer (accessed on 18 June 2015).
- Chen, H.; Chan, D.C. Emerging functions of mammalian mitochondrial fusion and fission. Hum. Mol. Genet. 2005, 14, R283–R289. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Kukat, C.; Larsson, N.G. mtDNA makes a U-turn for the mitochondrial nucleoid. Trends Cell Biol. 2013, 23, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Shagger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial Complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [PubMed]
- Drose, S. Differential effects of Complex II on mitochondrial ROS production and their relation to cardioprotective pre- and post-conditioning. Biochim. Biophys. Acta 2013, 1827, 578–587. [Google Scholar]
- Weinberg, F.; Chandel, N.S. Reactive oxygen species-dependent signaling regulates cancer. Cell. Mol. Life Sci. 2009, 66, 3663–3673. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Hirata, H. Redox regulation of cellular signalling. Cell Signal. 1999, 11, 1–14. [Google Scholar] [CrossRef]
- Lee, Y.J.; Shacter, E. Oxidative stress inhibits apoptosis in human lymphoma cells. J. Biol. Chem. 1999, 274, 19792–19798. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Dickens, F. The Metabolism of Tumors; Arnold Constable: London, UK, 1930. [Google Scholar]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman-Kocher, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1 mediates mitochondrial biogenesis and oxidative phosphorylation to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1015. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.L.; Qin, L.; Liu, H.C.; Candas, D.; Fan, M.; Li, J.J. Tumor cells switch to mitochondrial oxidative phosphorylation under radiation via mTOR-mediated hexokinase II inhibition—A Warburg-reversing effect. PLoS ONE 2015, 10, e0121046. [Google Scholar] [CrossRef] [PubMed]
- Guppy, M.; Leedman, P.; Zu, X.L.; Russell, V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem. J. 2002, 364, 309–315. [Google Scholar] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. Bcl-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Lunt, S.Y.; Dayton, T.L.; Fiske, B.P.; Israelsen, W.J.; Mattaini, K.R.; Vokes, N.I.; Stephanopoulos, G.; Cantley, L.C.; Metallo, C.M.; et al. Metabolic pathway alterations that support cell proliferation. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.L. Tumor mitochondria and the bioenergetics of cancer cells. Prog. Exp. Tumor Res. 1978, 22, 190–274. [Google Scholar] [PubMed]
- Modica-Napolitano, J.S.; Singh, K.K. Mitochondria as targets for detection and treatment of cancer. Expert Rev. Mol. Med. 2002, 4, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Singh, K.K. Mitochondrial dysfunction in cancer. Mitochondrion 2004, 4, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Kulawiec, M.; Singh, K.K. Mitochondria and human cancer. Curr. Mol. Med. 2007, 7, 121–31. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Mitochondria in cancer. Oncogene 2006, 25, 4630–4632. [Google Scholar] [CrossRef] [PubMed]
- Fogg, V.C.; Lanning, N.J.; MacKeigan, J.P. Mitochondria in cancer: At the crossroads of life and death. Chin. J. Cancer 2011, 30, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Touma, S.E. Functional differences in mitochondrial enzymes from normal epithelial and carcinoma cells. In Mitochondrial Dysfunction in Pathogenesis; Keystone Symposia: Silverthorne, CO, USA, 2000. [Google Scholar]
- Sun, A.S.; Sepkowitz, K.; Geller, S.A. A study of some mitochondrial and peroxisomal enzymes in human colonic adenocarcinoma. Lab. Investig. 1981, 44, 13–17. [Google Scholar] [PubMed]
- Chan, S.H.; Barbour, R.L. Adenine nucleotide transport in hepatoma mitochondria. Characterization of factors influencing the kinetics of ADP and ATP uptake. Biochim. Biophys. Acta 1983, 723, 104–113. [Google Scholar] [CrossRef]
- Sul, H.S.; Shrago, E.; Goldfarb, S.; Rose, F. Comparison of the adenine nucleotide translocase in hepatomas and rat liver mitochondria. Biochim. Biophys. Acta 1979, 551, 148–155. [Google Scholar] [CrossRef]
- Woldegiorgis, G.; Shrago, E. Adenine nucleotide translocase activity and sensitivity to inhibitors in hepatomas. Comparison of the ADP/ATP carrier in mitochondria and in a purified reconstituted liposome system. J. Biol. Chem. 1985, 260, 7585–7590. [Google Scholar] [PubMed]
- Pedersen, P.L.; Morris, H.P. Uncoupler-stimulated adenosine triphosphatase activity. Deficiency in intact mitochondria from Morris hepatomas and ascites tumor cells. J. Biol. Chem. 1974, 249, 3327–3334. [Google Scholar] [PubMed]
- Johnson, L.V.; Walsh, M.L.; Bockus, B.J.; Chen, L.B. Monitoring of relative mitochondrial membrane potential in living cells by fluorescence microscopy. J. Cell Biol. 1981, 88, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Weiss, M.J.; Wong, J.R.; Lampidis, T.J.; Chen, L.B. Mitochondrial and plasma membrane potentials cause unusual accumulation and retention of rhodamine 123 by human breast adenocarcinoma-derived MCF-7 cells. J. Biol. Chem. 1985, 260, 13844–13850. [Google Scholar] [PubMed]
- Modica-Napolitano, J.S.; Aprille, J.R. Basis for the selective cytotoxicity of rhodamine 123. Cancer Res. 1987, 47, 4361–4365. [Google Scholar] [PubMed]
- Canter, J.A.; Kallianpur, A.R.; Parl, F.F.; Millikan, R.C. Mitochondrial DNA G10398A polymorphism and invasive breast cancer in African–American women. Cancer Res. 2005, 65, 8028–8033. [Google Scholar] [PubMed]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial mutations in cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar] [CrossRef] [PubMed]
- Kulawiec, M.; Owens, K.M.; Singh, K.K. mtDNA G10398A variant in African–American women with breast cancer provides resistance to apoptosis and promotes metastasis in mice. J. Hum. Genet. 2009, 54, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Booker, L.M.; Habermacher, G.M.; Jessie, B.C.; Sun, Q.C.; Baumann, A.K.; Amin, M.; Lim, S.D.; Fernandez-Golarz, C.; Lyles, R.H.; Brown, M.D.; et al. North American white mitochondrial haplogroups in prostate and renal cancer. J. Urol. 2006, 175, 468–472. [Google Scholar] [CrossRef]
- Liu, V.W.; Wang, Y.; Yang, H.J.; Tsang, P.C.; Ng, T.Y.; Wong, L.C.; Nagley, P.; Ngan, H.Y. Mitochondrial DNA variant 16189T>C is associated with susceptibility to endometrial cancer. Hum. Mutat. 2003, 22, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Fruehauf, J.P.; Meyskens, F.L., Jr. Reactive oxygen species: A breath of life or death? Clin. Cancer Res. 2007, 13, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at Complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Cannito, S.; Novo, E.; di Bonzo, L.V.; Busletta, C.; Colombatto, S.; Parola, M. Epithelial-mesenchymal transition: From molecular mechanisms, redox regulation to implications in human health and disease. Antioxid. Redox Signal. 2010, 12, 1383–1430. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Dysregulation of apoptosis in cancer. J. Clin. Oncol. 1999, 17, 2941–2953. [Google Scholar] [PubMed]
- Wong, R. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed]
- Pepper, C.; Hoy, T.; Bentley, D.P. Bcl-2/Bax ratios in chronic lymphocytic leukaemia and their correlation with in vitro apoptosis and clinical resistance. Br. J. Cancer 1997, 76, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Goolsby, C.; Paniagua, M.; Tallman, M.; Gartenhaus, R.B. Bcl-2 regulatory pathway is functional in chronic lymphocytic leukaemia. Cytom. Part B Clin. Cytom. 2005, 63, 36–46. [Google Scholar]
- Raffo, A.J.; Perlman, H.; Chen, M.W.; Day, M.L.; Streitman, J.S.; Buttyan, R. Overexpression of Bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. 1995, 55, 4438–4445. [Google Scholar] [PubMed]
- Kitada, S.; Pedersen, I.M.; Schimmer, A.D.; Reed, J.C. Dysregulation of apoptosis genes in hematopoietic malignancies. Oncogene 2002, 21, 3459–3474. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; Joos, S.; Zornig, M. The role of Bcl-2 family members in tumorigenesis. Biochim. Biophys. Acta 2004, 1644, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Meyer, E.; Debatin, K.M. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene 2000, 21, 2283–2294. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J.; Rudin, C.M.; Boise, L.H.; Thompson, C.B. Expression of Bcl-XL can confer a multidrug resistance phenotype. Blood 1995, 86, 1903–1910. [Google Scholar] [PubMed]
- Neuzil, J.; Dong, L.F.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 2013, 13, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Bernal, S.D.; Lampidis, T.J.; Summerhayes, I.C.; Chen, L.B. Rhodamine-123 selectively reduces clonal growth of carcinoma cells in vitro. Science 1982, 218, 1117–1119. [Google Scholar] [CrossRef] [PubMed]
- Bernal, S.D.; Lampidis, T.J.; McIsaac, R.M.; Chen, L.B. Anticarcinoma activity in vivo of rhodamine 123, a mitochondrial-specific dye. Science 1983, 222, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Weiss, M.J.; Chen, L.B.; Aprille, J.R. Rhodamine 123 inhibits bioenergetic function in isolated rat liver mitochondria. Biochem. Biophys. Res. Commun. 1984, 118, 717–723. [Google Scholar] [CrossRef]
- Bleday, R.; Weiss, M.J.; Salem, R.R.; Wilson, R.E.; Chen, L.B.; Steele, G., Jr. Inhibition of rat colon tumor isograft growth with dequalinium chloride. Arch. Surg. 1986, 121, 1272–1275. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.J.; Wong, J.R.; Ha, C.S.; Bleday, R.; Salem, R.R.; Steele, G.D., Jr.; Chen, L.B. Dequalinium, a topical antimicrobial agent, displays anticarcinoma activity based on selective mitochondrial accumulation. Proc. Natl. Acad. Sci. USA 1987, 84, 5444–5448. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wong, J.R.; Song, K.; Hu, J.; Garlid, K.D.; Chen, L.B. AA1, a newly synthesized monovalent lipophilic cation, expresses potent in vivo antitumor activity. Cancer Res. 1994, 54, 1465–1471. [Google Scholar] [PubMed]
- Koya, K.; Li, Y.; Wang, H.; Ukai, T.; Tatsuta, N.; Kawakami, M.; Shishido, T.; Chen, L.B. MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996, 56, 538–543. [Google Scholar] [PubMed]
- Modica-Napolitano, J.S.; Koya, K.; Weisberg, E.; Brunelli, B.T.; Li, Y.; Chen, L.B. Selective damage to carcinoma mitochondria by the rhodacyanine MKT-077. Cancer Res. 1996, 56, 544–550. [Google Scholar] [PubMed]
- Britten, C.D.; Rowinsky, E.K.; Baker, S.D.; Weiss, G.R.; Smith, L.; Stephenson, J.; Rothenberg, M.; Smetzer, L.; Cramer, J.; Collins, W.; et al. A phase I and pharmacokinetic study of the mitochondrial-specific rhodacyanine dye analog MKT 077. Clin. Cancer Res. 2000, 6, 42–49. [Google Scholar] [PubMed]
- Propper, D.J.; Braybrooke, J.P.; Taylor, D.J.; Lodi, R.; Styles, P.; Cramer, J.A.; Collins, W.C.J.; Levitt, N.C.; Talbot, D.C.; Ganesan, T.S.; et al. Phase I trial of the selective mitochondrial toxin MKT 077 in chemo-resistant solid tumours. Ann. Oncol. 1999, 10, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Libby, G.; Donnelly, L.A.; Donnan, P.T.; Alessi, D.R.; Morris, A.D.; Evans, J.M. New users of metformin are at low risk of incident cancer: A cohort study among people with type 2 diabetes. Diabetes Care 2009, 32, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Murtola, T.J.; Tammela, T.L.; Lahtela, J.; Auvinen, A. Antidiabetic medication and prostate cancer risk: A population-based case-control study. Am. J. Epidemiol. 2008, 168, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Jiralerspong, S.; Angulo, A.M.; Hung, M.C. Expanding the arsenal: Metformin for the treatment of triple-negative breast cancer? Cell Cycle 2009, 8, 2681. [Google Scholar] [CrossRef] [PubMed]
- Dowling, R.J.; Niraula, S.; Stambolic, V.; Goodwin, P.J. Metformin in cancer: Translational challenges. J. Mol. Endocrinol. 2012, 48, R31–R43. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; Le Marchand-Brustel, Y.; Bost, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef] [PubMed]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [PubMed]
- Gotlieb, W.H.; Saumet, J.; Beauchamp, M.C.; Gu, J.; Lau, S.; Pollak, M.N.; Bruchim, I. In vitro metformin antineoplastic activity in epithelial ovarian cancer. Gynecol. Oncol. 2008, 110, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.W.; Li, Z.S.; Zou, D.W.; Jin, Z.D.; Gao, J.; Xu, G.M. Metformin induces apoptosis of pancreatic cancer cells. World J. Gastroenterol. 2008, 14, 7192–7198. [Google Scholar] [CrossRef] [PubMed]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Berstein, L.M.; Egormin, P.A.; Piskunova, T.S.; Popovich, I.G.; Zabezhinski, M.A.; Kovalenko, I.G.; Poroshina, T.E.; Semenchenko, A.V.; Provinciali, M.; et al. 2005 Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp. Gerontol. 2005, 40, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wullschleger, S.; Shpiro, N.; McGuire, V.A.; Sakamoto, K.; Woods, Y.L.; McBurnie, W.; Fleming, S.; Alessi, D.R. Important role of the LKB1–AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem. J. 2008, 412, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Memmott, R.M.; Mercado, J.R.; Maier, C.R.; Kawabata, S.; Fox, S.D.; Dennis, P.A. Metformin prevents tobacco carcinogen–induced lung tumorigenesis. Cancer Prev. Res. 2010, 3, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Algire, C.; Zakikhani, M.; Blouin, M.J.; Shuai, J.H.; Pollak, M. 2008 Metformin attenuates the stimulatory effect of a high-energy diet on in vivo LLC1 carcinoma growth. Endocr. Relat. Cancer 2008, 15, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, K.N.; Vumbaca, F.; Fox, M.M.; Evans, R.; Claffey, K.P. Dietary energy availability affects primary and metastatic breast cancer and metformin efficacy. Breast Cancer Res. Treat. 2010, 123, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Hosono, K.; Endo, H.; Takahashi, H.; Sugiyama, M.; Sakai, E.; Uchiyama, T.; Suzuki, K.; Iida, H.; Sakamoto, Y.; Yoneda, K.; et al. Metformin suppresses colorectal aberrant crypt foci in a short-term clinical trial. Cancer Prev. Res. 2010, 3, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Niraula, S.; Dowling, R.J.; Ennis, M.; Chang, M.C.; Done, S.J.; Hood, N.; Escallon, J.; Leong, W.L.; McCready, D.R.; Reedijk, M.; et al. Metformin in early breast cancer: A prospective window of opportunity neoadjuvant study. Breast Cancer Res. Treat. 2012, 135, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Hadad, S.; Iwamoto, T.; Jordan, L.; Purdie, C.; Bray, S.; Baker, L.; Jellema, G.; Deharo, S.; Hardie, D.G.; Pusztai, L.; et al. 2011 Evidence for biological effects of metformin in operable breast cancer: A pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res. Treat. 2011, 128, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.R.S.; et al. Metformin inhibits mitochondrial Complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Blackman, R.K.; Cheung-Ong, K.; Gebbia, M.; Proia, D.A.; He, S.; Kepros, J.; Jonneaux, A.; Marchetti, P.; Kluza, J.; Rao, P.E.; et al. Mitochondrial electron transport is the cellular target of the oncology drug elesclomol. PLoS ONE 2012, 7, e29798. [Google Scholar] [CrossRef] [PubMed]
- O’Day, S.J.; Eggermont, A.M.; Chiarion-Sileni, V.; Kefford, R.; Grob, J.J.; Mortier, L.; Robert, C.; Schachter, J.; Testori, A.; Mackiewicz, J.; et al. Final results of phase III SYMMETRY study: Randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?term=elesclomol&Search=Search (accessed on 19 May 2015).
- Dai, Z.J.; Wang, X.J.; Li, Z.F.; Ji, Z.Z.; Ren, H.T.; Tang, W.; Liu, X.X.; Kang, H.F.; Guan, H.T.; Song, L.Q. Scutellaria barbate extract induces apoptosis of hepatoma H22 cells via the mitochondrial pathway involving caspase-3. World J. Gastroenterol. 2008, 14, 7321–7328. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Kwon, K.B.; Han, M.J.; Song, M.Y.; Lee, J.H.; Ko, Y.S.; Shin, B.C.; Yu, J.; Lee, Y.R.; Ryu, D.G.; et al. Induction of G1 arrest and apoptosis by Scutellaria barbata in the human promyelocytic leukemia HL-60 cell line. Int. J. Mol. Med. 2007, 20, 123–128. [Google Scholar] [PubMed]
- Marconett, C.N.; Morgenstern, T.J.; san Roman, A.K.; Sundar, S.N.; Singhal, A.K.; Firestone, G.L. BZL101, a phytochemical extract from the Scutellaria barbata plant, disrupts proliferation of human breast and prostate cancer cells through distinct mechanisms dependent on the cancer cell phenotype. Cancer Biol. Ther. 2010, 10, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Fong, S.; Shoemaker, M.; Cadaoas, J.; Lo, A.; Liao, W.; Tagliaferri, M.; Cohen, I.; Shtivelman, E. Molecular mechanisms underlying selective cytotoxic activity of BZL101, an extract of Scutellaria barbata, towards breast cancer cells. Cancer Biol. Ther. 2008, 7, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.; Staub, R.E.; Fong, S.; Tagliaferri, M.; Cohen, I.; Shtivelman, E. Bezielle selectively targets mitochondria of cancer cells to inhibit glycolysis and OXPHOS. PLoS ONE 2012, 7, e30300. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.; Shtivelman, E.; Perez, A.; Vogel, C.; Franco, S.; Chiu, E.T.; Melisko, M.; Tagliaferri, M.; Cohen, I.; Shoemaker, M.; et al. Phase I trial and antitumor effects of BZL101 for patients with advanced breast cancer. Breast Cancer Res. Treat. 2007, 105, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Perez, A.T.; Arun, B.; Tripathy, D.; Tagliaferri, M.A.; Shaw, H.S.; Kimmick, G.G.; Cohen, I.; Shtivelman, E.; Caygill, K.A.; Grady, D.; et al. A phase 1B dose escalation trial of Scutellaria barbata (BZL101) for patients with metastatic breast cancer. Breast Cancer Res. Treat. 2010, 120, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.X.; Sneed, T.; et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Hann, C.L.; Daniel, V.C.; Sugar, E.A.; Dobromilskaya, I.; Murphy, S.C.; Cope, L.; Lin, X.; Hierman, J.S.; Wilburn, D.L.; Neil Watkins, D.; et al. Therapeutic efficacy of ABT-737, a selective inhibitor of Bcl-2, in small cell lung cancer. Cancer Res. 2008, 68, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Camidge, D.R.; de Oliveira, M.R.; Bonomi, P.; Gandara, D.; Khaira, D.; Hann, C.L.; McKeegan, E.M.; Litvinovich, E.; Hemken, P.M.; et al. Phase I study of navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J. Clin. Oncol. 2011, 29, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; de Oliveira, M.R.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. 2012, 18, 3163–3169. [Google Scholar] [CrossRef] [PubMed]
- Sadahira, K.; Sagawa, M.; Nakazato, T.; Uchida, H.; Ikeda, Y.; Okamoto, S.; Nakajima, H.; Kizaki, M. Gossypol induces apoptosis in multiple myeloma cells by inhibition of interleukin-6 signaling and Bcl-2/Mcl-1 pathway. Int. J. Oncol. 2014, 45, 2278–2286. [Google Scholar] [CrossRef] [PubMed]
- Kline, M.P.; Rajkumar, S.V.; Timm, M.M.; Kimlinger, T.K.; Haug, J.L.; Lust, J.A.; Greipp, P.R.; Kumar, S. R-(−)-gossypol (AT-101) activates programmed cell death in multiple myeloma cells. Exp. Hematol. 2008, 36, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Watt, J.; Contractor, R.; Tsao, T.; Harris, D.; Estrov, Z.; Bornmann, W.; Kantarjian, H.; Viallet, J.; Samudio, I.; et al. Mechanisms of antileukemic activity of the novel BH3 mimetic GX15–070 (obatoclax). Cancer Res. 2008, 68, 3413–3420. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Marcellus, R.C.; Roulston, A.; Watson, M.; Serfass, L.; Madiraju, S.R.M.; Goulet, D.; Viallet, J.; Bélec, L.; Billot, X.; et al. Small molecule obatoclax (GX15–070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 19512–19517. [Google Scholar] [CrossRef] [PubMed]
- Heikaus, S.; van den Berg, L.; Kempf, T.; Mahotka, C.; Gabbert, H.E.; Ramp, U. HA14–1 is able to reconstitute the impaired mitochondrial pathway of apoptosis in renal cell carcinoma cell lines. Cell Oncol. 2008, 30, 419–433. [Google Scholar] [PubMed]
- Rehman, K.; Tariq, M.; Akash, M.S.; Gillani, Z.; Qazi, M.H. Effect of HA14–1 on apoptosis-regulating proteins in HeLa cells. Chem. Biol. Drug Des. 2014, 83, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.M.; Wood, J.M.; Anderson, A.C. Inhibition of mitochondrial and Paracoccus denitrificans NADH-ubiquinone reductase by oxacarbocyanine dyes. A structure-activity study. Biochem. Pharmacol. 1993, 45, 691–696. [Google Scholar] [CrossRef]
- Rideout, D.; Bustamante, A.; Patel, J. Mechanism of inhibition of FaDu hypopharyngeal carcinoma cell growth by tetraphenylphosphonium chloride. Int. J. Cancer 1994, 57, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Beel, J.A.; Lillehei, K.O. A threshold concept for cancer therapy. Med. Hypotheses 2000, 55, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Billard, C. BH3 mimetics: Status of the field and new developments. Mol. Cancer Ther. 2013, 12, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, A.R.; Oleksijew, A.; Bauch, J.; Belli, B.A.; Borre, T.; Bruncko, M.; Deckwirth, T.; Frost, D.J.; Jarvis, K.; Joseph, M.K.; et al. A small-molecule inhibitor of Bcl-XL potentiates the activity of cytotoxic drugs in vitro and in vivo. Cancer Res. 2006, 66, 8731–8739. [Google Scholar] [CrossRef] [PubMed]
- Hikita, H.; Takehara, T.; Shimizu, S.; Kodama, T.; Shigekawa, M.; Iwase, K.; Hosui, A.; Miyagi, T.; Tatsumi, T.; Ishida, H.; et al. The Bcl-xL inhibitor, ABT-737, efficiently induces apoptosis and suppresses growth of hepatoma cells in combination with sorafenib. Hepatology 2010, 52, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Jain, H.V.; Meyer-Hermann, M. The molecular basis of synergism between carboplatin and ABT-737 therapy targeting ovarian carcinomas. Cancer Res. 2011, 71, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Zall, H.; Weber, A.; Besch, R.; Zantl, N.; Hacker, G. Chemotherapeutic drugs sensitize human renal cell carcinoma cells to ABT-737 by a mechanism involving the Noxa-dependent inactivation of Mcl-1 or A1. Mol. Cancer 2010, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.; Malek, M.; Zha, J.; Yue, P.; Kassees, R.; Berry, L.; Fairbrother, W.J.; Sampath, D.; Belmont, L.D. Navitoclax enhances the efficacy of taxanes in non-small cell lung cancer models. Clin. Cancer Res. 2011, 17, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Truksa, J.; Dong, L.F.; Rohlena, J.; Stursa, J.; Vondrusova, M.; Goodwin, J.; Nguyen, M.; Kluckova, K.; Rychtarcikova, Z.; Lettlova, S.; et al. Mitochondrially targeted vitamin E succinate modulates expression of mitochondrial DNA transcripts and mitochondrial biogenesis. Antiox. Redox Signal. 2015, 22, 883–900. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.F.; Jameson, V.J.A.; Tilly, D.; Cerny, J.; Mahdavian, E.; Marín-Hernandez, A.; Hernandez-Esquivel, L.; Rodríguez-Enríquez, S.; Stursa, J.; Witting, P.K.; et al. Mitochondrial targeting of vitamin E succinate enhances its pro-apoptotic and anti-cancer activity via mitochondrial Complex II. J. Biol. Chem. 2011, 286, 3717–3728. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.A.; Klein, S.R.; Bonar, S.J.; Zielonka, J.; Mizuno, N.; Dickey, J.S.; Keller, P.W.; Joseph, J.; Kalyanaraman, B.; Shacter, E. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J. Biol. Chem. 2010, 285, 34447–34459. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, G.G.M.; Cheng, S.M.; Boddapati, S.V.; Horobin, R.W.; Weissig, V. Nanocarrier-assisted sub-cellular targeting to the site of mitochondria improves the pro-apoptotic activity of paclitaxel. J. Drug Target. 2008, 16, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, R.; Rai, S.; Vaidya, B.; Gupta, P.N.; Mahor, S.; Khatri, K.; Goyal, A.K.; Rawat, A.; Vyas, S.P. Cell-selective mitochondrial targeting: Progress in mitochondrial medicine. Curr. Drug Deliv. 2007, 4, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Dodwadkar, N.S.; Deshpande, P.P.; Torchilin, V.P. Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium-PEG-PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo. J. Control. Release 2012, 159, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Zupancic, S.; Kocbek, P.; Zariwala, M.G.; Renshaw, D.; Gul, M.O.; Elsaid, Z.; Taylor, K.M.; Somavarapu, S. Design and development of novel mitochondrial targeted nanocarriers, DQAsomes for curcumin inhalation. Mol. Pharm. 2014, 11, 2334–2345. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Li, Y.B.; Yao, H.J.; Ju, R.J.; Zhang, Y.; Li, R.J.; Yu, Y.; Zhang, L.; Lu, W.L. The use of mitochondrial targeting resveratrol liposomes modified with a dequalinium polyethylene glycol-distearoylphosphatidyl ethanolamine conjugate to induce apoptosis in resistant lung cancer cells. Biomaterials 2011, 32, 5673–5687. [Google Scholar] [CrossRef] [PubMed]
- Solomon, M.A.; Shah, A.A.; D’Souza, G.G. In vitro assessment of the utility of stearyl triphenyl phosphonium modified liposomes in overcoming the resistance of ovarian carcinoma Ovcar-3 cells to paclitaxel. Mitochondrion 2013, 13, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhao, W.Y.; Ma, X.; Ju, R.J.; Li, X.Y.; Li, N.; Sun, M.G.; Shi, J.F.; Zhnag, C.X.; Lu, W.L. The anticancer efficacy of paclitaxel liposomes modified with mitochondrial targeting conjugate in resistant lung cancer. Biomaterials 2013, 34, 3626–3638. [Google Scholar] [CrossRef] [PubMed]
- Malhi, S.S.; Budhiraja, A.; Arora, S.; Chaudhari, K.R.; Nepali, K.; Kumar, R.; Sohi, H.; Murthy, R.S. Intracellular delivery of redox cycler-doxorubicin to the mitochondria of cancer cell by folate receptor targeted mitocancerotropic liposomes. Int. J. Pharm. 2012, 432, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Yoong, S.L.; Wong, B.S.; Zhou, Q.L.; Chin, C.F.; Li, J.; Venkatesan, T.; Ho, H.K.; Yu, V.; Ang, W.H.; Pastorin, G. Enhanced cytotoxicity to cancer cells by mitochondria-targeting MWCNTs containing platinum(IV) prodrug of cisplatin. Biomaterials 2014, 35, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Ara, G.; Aprille, J.R.; Malis, C.D.; Kane, S.B.; Cincotta, L.; Foley, J.; Bonventre, J.V.; Oseroff, A.R. Mechanisms of mitochondrial photosensitization by the cationic dye, N,Nʹ-bis(2-ethyl-l,3-dioxylene)kryptocyanine (EDKC): Preferential inactivation of Complex I in the electron transport chain. Cancer Res. 1987, 47, 6580–6585. [Google Scholar] [PubMed]
- Powers, S.K.; Pribil, S.; Gillespie, G.Y.; Watkins, P.J. Laser photochemotherapy of rhodamine-123 sensitized human glioma cells in vitro. J. Neurosurg. 1986, 64, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Brunelli, B.T.; Koya, K.; Chen, L.B. Photoactivation enhances the mitochondrial toxicity of the cationic rhodacyanine MKT-077. Cancer Res. 1998, 58, 71–75. [Google Scholar] [PubMed]
- Chan, J.Y.; Tang, P.M.; Hon, P.M.; Au, S.W.; Tsui, S.K.; Waye, M.M.; Kong, S.K.; Mak, T.C.; Fung, K.P. Pheophorbide a, a major antitumor component purified from Scutellaria barbata, induces apoptosis in human hepatocellular carcinoma cells. Planta Med. 2006, 72, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Li, W.T.; Tsao, H.W.; Chen, Y.Y.; Cheng, S.W.; Hsu, Y.C. A study on the photodynamic properties of chlorophyll derivatives using human hepatocellular carcinoma cells. Photochem. Photobiol. Sci. 2007, 6, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Hajri, A.; Coffy, S.; Vallat, F.; Evrard, S.; Marescaux, J.; Aprahamian, M. Human pancreatic carcinoma cells are sensitive to photodynamic therapy in vitro and in vivo. Br. J. Surg. 1999, 86, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Hibasami, H.; Kyohkon, M.; Ohwaki, S.; Katsuzaki, H.; Imai, K.; Nakagawa, M.; Ishi, Y.; Komiya, T. Pheophorbide a, a moiety of chlorophyll a, induces apoptosis in human lymphoid leukemia molt 4B cells. Int. J. Mol. Med. 2000, 6, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.M.; Liu, X.Z.; Zhang, D.M.; Fong, W.P.; Fung, K.P. Pheophorbide a based photodynamic therapy induces apoptosis via mitochondrial-mediated pathway in human uterine carcinosarcoma. Cancer Biol. Ther. 2009, 8, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Hoi, S.W.; Wong, H.M.; Chan, J.Y.; Yue, G.G.; Tse, G.M.; Law, B.K.; Fong, W.P.; Fung, K.P. Photodynamic therapy of Pheophorbide a inhibits the proliferation of human breast tumour via both caspase-dependent and -independent apoptotic pathways in in vitro and in vivo models. Phytother. Res. 2012, 26, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Hajri, A.; Wack, S.; Meyer, C.; Smith, M.K.; Leberquier, C.; Kedinger, M.; Aprahamian, M. In vitro and in vivo efficacy of Photofrin® and pheophorbide a, a bacteriochlorin, in photodynamic therapy of colonic cancer cells. Photochem. Photobiol. 2002, 75, 140–148. [Google Scholar] [CrossRef]
- Laranjo, M.; Serra, A.C.; Abrantes, M.; Piñeiro, M.; Gonçalves, A.C.; Casalta-Lopes, J.; Carvalho, L.; Sarmento-Ribeiro, A.B.; Rocha-Gonsalves, A.; Botelho, F. 2-Bromo-5-hydroxyphenylporphyrins for photodynamic therapy: Photosensitization efficiency, subcellular localization and in vivo studies. Photodiagn. Photodyn. Ther. 2013, 10, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.H.; Park, E.S.; Liang, J.; Dennison, J.B.; Tsavachidou, D.; Nguyen-Charles, C.; Cheng, K.W.; Hall, H.; Zhang, D.; Lu, Y.; et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol. Cancer Ther. 2011, 10, 2350–2362. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Nalbandian, R.; Kidd, M.E.; Nalbandian, A.; Nguyen, C.C. The selective in vitro cytotoxicity of carcinoma cells by AZT is enhanced by concurrent treatment with delocalized lipophilic cations. Cancer Lett. 2003, 19, 859–868. [Google Scholar] [CrossRef]
- Westermann, B.; Neupert, W. Mitochondria-targeted green fluorescent proteins: Convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast 2000, 16, 1421–1427. [Google Scholar] [CrossRef]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef] [PubMed]
- Horobin, R.W.; Trapp, S.; Weissig, V. Mitochondriotropics: A review of their mode of action, and their applications for drug and DNA delivery to mammalian mitochondria. J. Control. Release 2007, 121, 125–136. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, G.G.M.; Weissig, V. An introduction to subcellular and nanomedicine: Current trends and future developments. In Organelle-Specific Pharmaceutical Nanotechnology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 1–13. [Google Scholar]
- Weissig, V.; Lasch, J.; Erdos, G.; Meyer, H.W.; Rowe, T.C.; Hughes, J. DQAsomes: A novel potential drug and gene delivery system made from dequalinium. Pharm. Res. 1998, 15, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V.; Torchilin, V.P. Towards mitochondrial gene therapy: DQAsomes as a strategy. J. Drug Target. 2001, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V.; Torchilin, V.P. Cationic bolasomes with delocalized charge centers as mitochondria-specific DNA delivery systems. Adv. Drug Deliv. Rev. 2001, 49, 127–149. [Google Scholar] [CrossRef]
- Weissig, V. DQAsomes as the prototype of mitochondria-targeted pharmaceutical nanocarriers: Preparation, characterization, and use. Methods Mol. Biol. 2015, 1265, 1–11. [Google Scholar] [PubMed]
- Lyrawati, D.; Trounson, A.; Cram, D. Expression of GFP in the mitochondrial compartment using DQAsome-mediated delivery of an artificial mini-mitochondrial genome. Pharm. Res. 2011, 28, 2848–2862. [Google Scholar] [CrossRef] [PubMed]
- Boddapati, S.V.; Tongcharoensirikul, P.; Hanson, R.N.; D’Souza, G.G.; Torchilin, V.P.; Weissig, V. Mitochondriotropic liposomes. J. Liposome Res. 2005, 15, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Boddapati, S.V.; D’Souza, G.G.; Erdogan, S.; Torchilin, V.P.; Weissig, V. Organelle-targeted nanocarriers: Specific delivery of liposomal ceramide to mitochondria enhances its cytotoxicity in vitro and in vivo. Nano Lett. 2008, 8, 2559–2263. [Google Scholar] [CrossRef] [PubMed]
- Boddapati, S.V.; D’Souza, G.G.; Weissig, V. Liposomes for drug delivery to mitochondria. Methods Mol. Biol. 2010, 605, 295–303. [Google Scholar] [PubMed]
- Weissig, V.; Boddapati, S.V.; Cheng, S.M.; D’Souza, G.G. Liposomes and liposome-like vesicles for drug and DNA delivery to mitochondria. J. Liposome Res. 2006, 16, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V.; Lasch, J.; Klibanov, A.L.; Torchilin, V.P. A new hydrophobic anchor for the attachment of proteins to liposomal membranes. FEBS Lett. 1986, 202, 86–90. [Google Scholar] [CrossRef]
- Weissig, V.; Lasch, J.; Gregoriadis, G. Covalent coupling of sugars to liposomes. Biochim. Biophys. Acta 1989, 1003, 54–57. [Google Scholar] [CrossRef]
- Theodossiou, T.A.; Sideratou, Z.; Tsiourvas, D.; Paleos, C.M. A novel mitotropic oligolysine nanocarrier: Targeted delivery of covalently bound D-Luciferin to cell mitochondria. Mitochondrion 2011, 11, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Dodwadkar, N.S.; Piroyan, A.; Torchilin, V.P. Surface conjugation of triphenylphosphonium to target poly(amidoamine) dendrimers to mitochondria. Biomaterials 2012, 33, 4773–4782. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Soliman, G.M.; Al-Hajaj, N.; Sharma, R.; Maysinger, D.; Kakkar, A. Design and evaluation of multifunctional nanocarriers for selective delivery of coenzyme Q10 to mitochondria. Biomacromolecules 2012, 13, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Marrache, S.; Dhar, S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc. Natl. Acad. Sci. USA 2012, 109, 16288–16293. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.K.; Kolishetti, N.; Dhar, S. Targeted nanoparticles in mitochondrial medicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Theodossiou, T.A.; Sideratou, Z.; Katsarou, M.E.; Tsiourvas, D. Mitochondrial delivery of doxorubicin by triphenylphosphonium-functionalized hyperbranched nanocarriers results in rapid and severe cytotoxicity. Pharm. Res. 2013, 30, 2832–2842. [Google Scholar] [CrossRef] [PubMed]
- Battigelli, A.; Russier, J.; Venturelli, E.; Fabbro, C.; Petronilli, V.; Bernardi, P.; Da Ros, T.; Prato, M.; Bianco, A. Peptide-based carbon nanotubes for mitochondrial targeting. Nanoscale 2013, 5, 9110–9117. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.S.; Turchiello, R.; Kowaltowski, A.J.; Indig, G.L.; Baptista, M.S. Major determinants of photoinduced cell death: Subcellular localization versus photosensitization efficiency. Free Radic. Biol. Med. 2011, 51, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Joyal, J.L.; Ara, G.; Oseroff, A.R.; Aprille, J.R. Mitochondrial toxicity of cationic photosensitizers for photochemotherapy. Cancer Res. 1990, 50, 7876–7881. [Google Scholar] [PubMed]
- Powers, S.K.; Walstad, D.L.; Brown, J.T.; Detty, M.; Watkins, P.J. Photosensitization of human glioma cells by chalcogenapyrylium dyes. J. Neurooncol. 1989, 7, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Lee, W.Y.; Hahn, B.S.; Han, M.J.; Yang, W.I.; Kim, B.S. Chlorophyll derivatives—A new photosensitizer for photodynamic therapy of cancer in mice. Yonsei Med. J. 1989, 30, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Deslandes, E.; Villedieu, M.; Poulain, L.; Duval, M.; Gauduchon, P.; Scwartz, L.; Icard, P. Effect of 2-deoxy-d-glucose on various malignant cell lines in vitro. Anticancer Res. 2006, 26, 3561–3566. [Google Scholar] [PubMed]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-d-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Pedersen, P.L.; Geschwind, J.F. Glucose catabolism in the rabbit VX2 tumor model for liver cancer: Characterization and targeting hexokinase. Cancer Lett. 2001, 173, 83–91. [Google Scholar] [CrossRef]
- Pedersen, P.L. 3-Bromopyruvate (3BP) a fast acting, promising, powerful, specific, and effective “small molecule” anti-cancer agent taken from labside to bedside: Introduction to a special issue. J. Bioenerg. Biomembr. 2012, 44, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Oudard, S.; Poirson, F.; Miccoli, L.; Bourgeois, Y.; Vassault, A.; Poisson, M.; Magdelénat, H.; Dutrillaux, B.; Poupon, M.F. Mitochondria-bound hexokinase as target for therapy of malignant gliomas. Int. J. Cancer 1995, 62, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Pulselli, R.; Amadio, L.; Fanciulli, M.; Floridi, A. Effect of lonidamine on the mitochondrial potential in situ in Ehrlich ascites tumor cells. Anticancer Res. 1996, 16, 419–423. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Modica-Napolitano, J.S.; Weissig, V. Treatment Strategies that Enhance the Efficacy and Selectivity of Mitochondria-Targeted Anticancer Agents. Int. J. Mol. Sci. 2015, 16, 17394-17421. https://doi.org/10.3390/ijms160817394
Modica-Napolitano JS, Weissig V. Treatment Strategies that Enhance the Efficacy and Selectivity of Mitochondria-Targeted Anticancer Agents. International Journal of Molecular Sciences. 2015; 16(8):17394-17421. https://doi.org/10.3390/ijms160817394
Chicago/Turabian StyleModica-Napolitano, Josephine S., and Volkmar Weissig. 2015. "Treatment Strategies that Enhance the Efficacy and Selectivity of Mitochondria-Targeted Anticancer Agents" International Journal of Molecular Sciences 16, no. 8: 17394-17421. https://doi.org/10.3390/ijms160817394
APA StyleModica-Napolitano, J. S., & Weissig, V. (2015). Treatment Strategies that Enhance the Efficacy and Selectivity of Mitochondria-Targeted Anticancer Agents. International Journal of Molecular Sciences, 16(8), 17394-17421. https://doi.org/10.3390/ijms160817394