
Mitochondrial Mechanisms in Septic Cardiomyopathy

Abstract
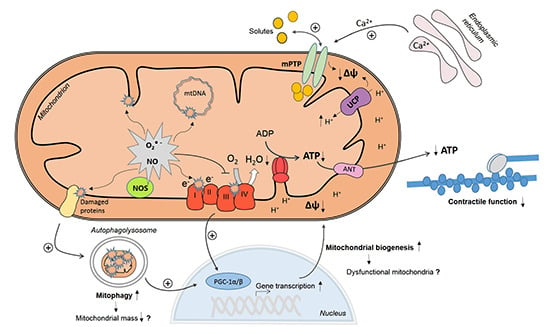
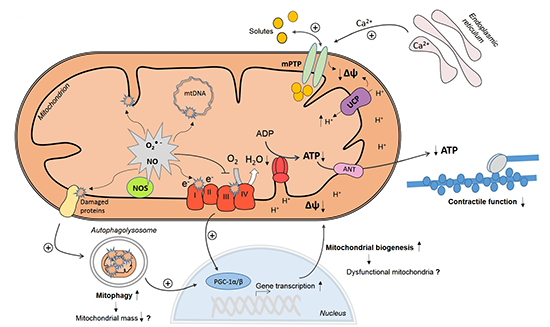
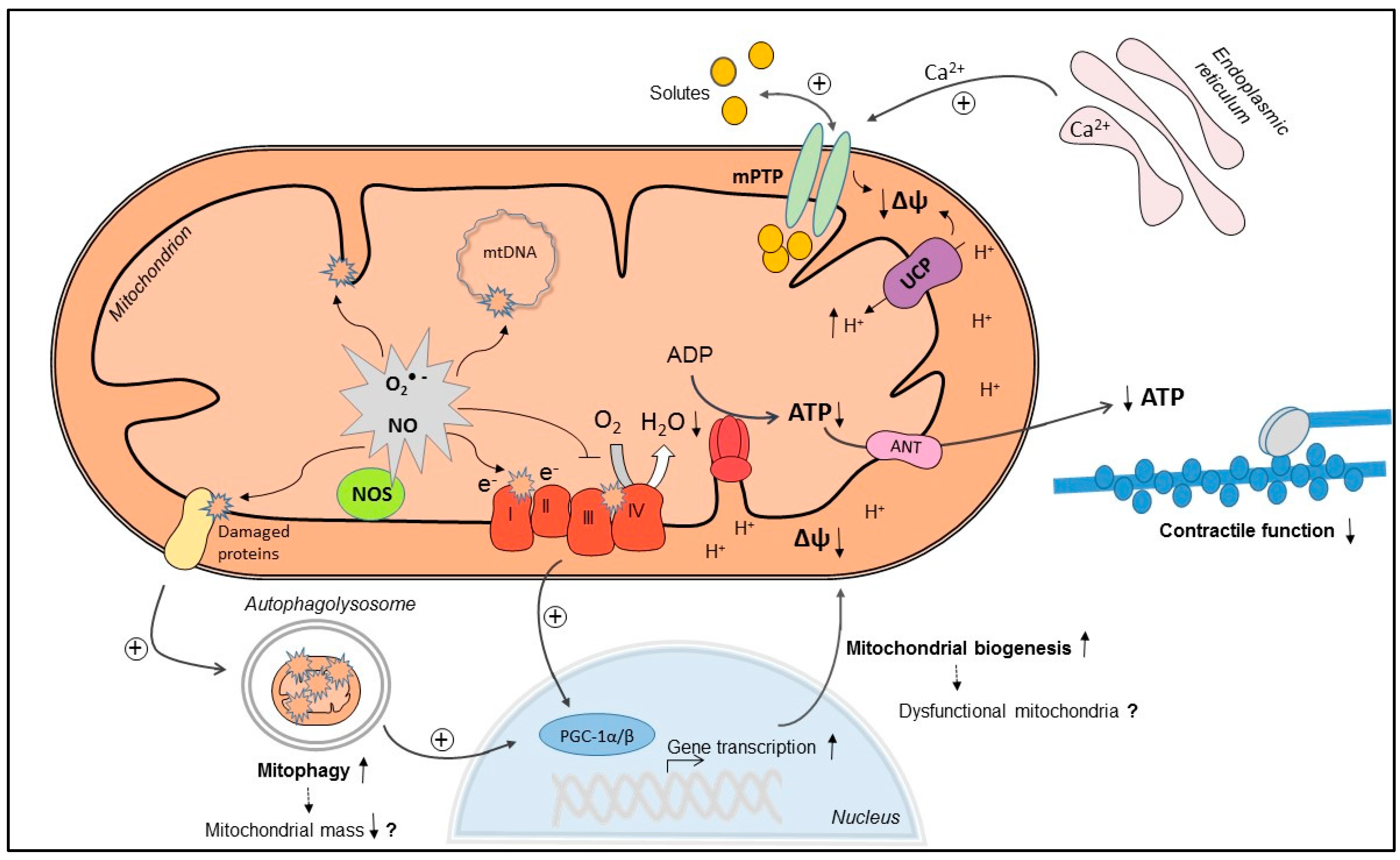
:
{kind=link}
{kind=link}
1. Introduction
2. Myocardial Mitochondrial Dysfunction in Sepsis
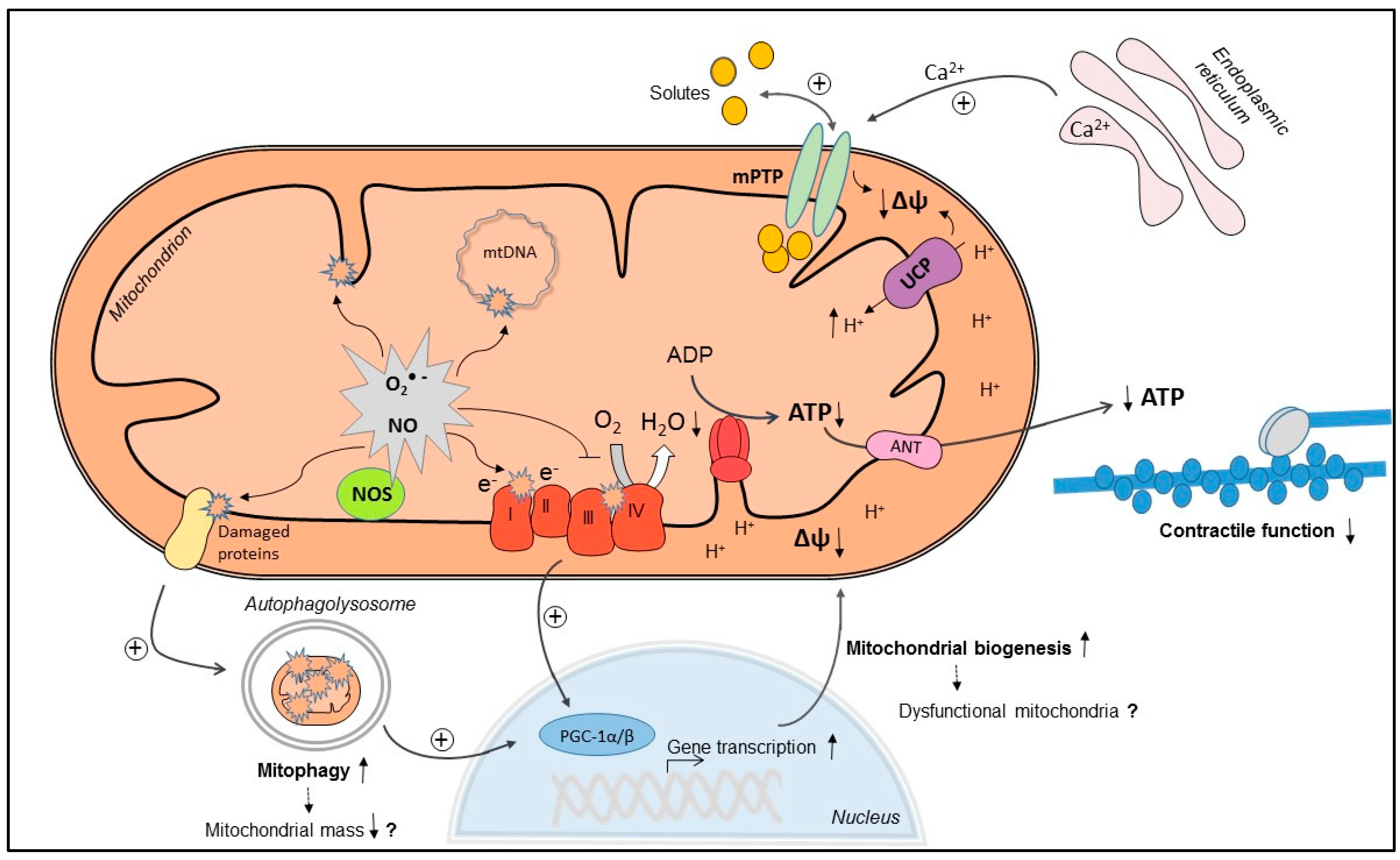
3. Mechanisms of Myocardial Mitochondrial Dysfunction in Sepsis
3.1. Mitochondrial NO Production and Oxidative Stress
3.2. Ca2+ Handling and Mitochondrial Permeability Transition
3.3. Mitochondrial Uncoupling
3.4. Mitochondrial Biogenesis
3.5. Mitophagy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Deutschman, C.S.; Tracey, K.J. Sepsis: Current dogma and new perspectives. Immunity 2014, 40, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Soong, J.; Soni, N. Sepsis: Recognition and treatment. Clin. Med. 2012, 12, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Sagy, M.; Al-Qaqaa, Y.; Kim, P. Definitions and pathophysiology of sepsis. Curr. Prob. Pediatr. Adolesc. Health Care 2013, 43, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Court, O.; Kumar, A.; Parrillo, J.E.; Kumar, A. Clinical review: Myocardial depression in sepsis and septic shock. Crit. Care 2002, 6, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Cunnion, R.E.; Schaer, G.L.; Parker, M.M.; Natanson, C.; Parrillo, J.E. The coronary circulation in human septic shock. Circulation 1986, 73, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Romero-Bermejo, F.J.; Ruiz-Bailen, M.; Gil-Cebrian, J.; Huertos-Ranchal, M.J. Sepsis-induced cardiomyopathy. Curr. Cardiol. Rev. 2011, 7, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Rudiger, A.; Singer, M. Mechanisms of sepsis-induced cardiac dysfunction. Crit. Care Med. 2007, 35, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Vallet, B.; Lund, N.; Curtis, S.E.; Kelly, D.; Cain, S.M. Gut and muscle tissue PO2 in endotoxemic dogs during shock and resuscitation. J. Appl. Physiol. 1994, 76, 793–800. [Google Scholar] [CrossRef]
- Joseph, B. On anoxaemia. Lancet 1920, 485–489. [Google Scholar]
- Sair, M.; Etherington, P.J.; Curzen, N.P.; Winlove, C.P.; Evans, T.W. Tissue oxygenation and perfusion in endotoxemia. Am. J. Physiol. 1996, 271, H1620–H1625. [Google Scholar] [PubMed]
- Hotchkiss, R.S.; Rust, R.S.; Dence, C.S.; Wasserman, T.H.; Song, S.K.; Hwang, D.R.; Karl, I.E.; Welch, M.J. Evaluation of the role of cellular hypoxia in sepsis by the hypoxic marker [18F] fluoromisonidazole. Am. J. Physiol. 1991, 261, R965–R972. [Google Scholar] [PubMed]
- Fink, M.P. Cytopathic hypoxia. Mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit. Care Clin. 2001, 17, 219–237. [Google Scholar] [CrossRef]
- Soriano, F.G.; Nogueira, A.C.; Caldini, E.G.; Lins, M.H.; Teixeira, A.C.; Cappi, S.B.; Lotufo, P.A.; Bernik, M.M.; Zsengeller, Z.; Chen, M.; et al. Potential role of poly(adenosine 5′-diphosphate-ribose) polymerase activation in the pathogenesis of myocardial contractile dysfunction associated with human septic shock. Crit. Care Med. 2006, 34, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Vanasco, V.; Saez, T.; Magnani, N.D.; Pereyra, L.; Marchini, T.; Corach, A.; Vaccaro, M.I.; Corach, D.; Evelson, P.; Alvarez, S. Cardiac mitochondrial biogenesis in endotoxemia is not accompanied by mitochondrial function recovery. Free Radic. Biol. Med. 2014, 77, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tavener, S.A.; Long, E.M.; Robbins, S.M.; McRae, K.M.; van Remmen, H.; Kubes, P. Immune cell toll-like receptor 4 is required for cardiac myocyte impairment during endotoxemia. Circ. Res. 2004, 95, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Drosatos, K.; Khan, R.S.; Trent, C.M.; Jiang, H.; Son, N.H.; Blaner, W.S.; Homma, S.; Schulze, P.C.; Goldberg, I.J. Peroxisome proliferator-activated receptor-γ activation prevents sepsis-related cardiac dysfunction and mortality in mice. Circ. Heart Fail. 2013, 6, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Escames, G.; Leon, J.; Macias, M.; Khaldy, H.; Acuna-Castroviejo, D. Melatonin counteracts lipopolysaccharide-induced expression and activity of mitochondrial nitric oxide synthase in rats. FASEB J. 2003, 17, 932–934. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.; Boveris, A. Mitochondrial nitric oxide metabolism in rat muscle during endotoxemia. Free Radic. Biol. Med. 2004, 37, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Abel, E.D. Mitochondria in the diabetic heart. Cardiovasc. Res. 2010, 88, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Boudina, S.; Hu, X.X.; Tuinei, J.; Zaha, V.G.; Theobald, H.A.; Yun, U.J.; McQueen, A.P.; Wayment, B.; Litwin, S.E.; et al. Type 1 diabetic akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3. Diabetes 2008, 57, 2924–2932. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Chen, Q.; Slabe, T.J.; Stoll, M.S.; Minkler, P.E.; Hassan, M.O.; Tandler, B.; Hoppel, C.L. Ischemia, rather than reperfusion, inhibits respiration through cytochrome oxidase in the isolated, perfused rabbit heart: Role of cardiolipin. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H258–H267. [Google Scholar] [CrossRef] [PubMed]
- Takasu, O.; Gaut, J.P.; Watanabe, E.; To, K.; Fagley, R.E.; Sato, B.; Jarman, S.; Efimov, I.R.; Janks, D.L.; Srivastava, A.; et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am. J. Respir. Crit. Care Med. 2013, 187, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Leelahavanichkul, A.; Yuen, P.S.; Star, R.A. Animal models of sepsis and sepsis-induced kidney injury. J. Clin. Investig. 2009, 119, 2868–2878. [Google Scholar] [CrossRef] [PubMed]
- Smeding, L.; Plotz, F.B.; Groeneveld, A.B.; Kneyber, M.C. Structural changes of the heart during severe sepsis or septic shock. Shock 2012, 37, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.M.; Suliman, H.B.; Hollingsworth, J.W.; Welty-Wolf, K.E.; Carraway, M.S.; Piantadosi, C.A. Nitric oxide synthase-2 induction optimizes cardiac mitochondrial biogenesis after endotoxemia. Free Radic. Biol. Med. 2009, 46, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.S.; Julian, M.W.; Huff, J.E.; Bauer, J.A.; Xia, Y.; Crouser, E.D. Calcineurin regulates myocardial function during acute endotoxemia. Am. J. Respir. Crit. Care Med. 2006, 173, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Du, J.; Wei, N.; Guan, T.; Camara, A.K.; Shi, Y. Differential sensitivity to LPS-induced myocardial dysfunction in the isolated brown norway and Dahl S rat hearts: Roles of mitochondrial function, NF-κB activation, and TNF-α production. Shock 2012, 37, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Chopra, M.; Golden, H.B.; Mullapudi, S.; Dowhan, W.; Dostal, D.E.; Sharma, A.C. Modulation of myocardial mitochondrial mechanisms during severe polymicrobial sepsis in the rat. PLoS ONE 2011, 6, e21285. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.V.; Staniek, K.; Haindl, S.; Piskernik, C.; Ohlinger, W.; Gille, L.; Nohl, H.; Bahrami, S.; Redl, H. Different effects of endotoxic shock on the respiratory function of liver and heart mitochondria in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G543–G549. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, S.M.; Marechal, X.; Montaigne, D.; Bouazza, Y.; Decoster, B.; Lancel, S.; Neviere, R. Prevention of endotoxin-induced sarcoplasmic reticulum calcium leak improves mitochondrial and myocardial dysfunction. Crit. Care Med. 2008, 36, 2590–2596. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Godin, R.; Deschenes, S.; Bessi, V.L.; Mofarrahi, M.; Hussain, S.N.; Burelle, Y. Protective role of PARK2/Parkin in sepsis-induced cardiac contractile and mitochondrial dysfunction. Autophagy 2013, 9, 1837–1851. [Google Scholar] [CrossRef] [PubMed]
- Zang, Q.S.; Sadek, H.; Maass, D.L.; Martinez, B.; Ma, L.; Kilgore, J.A.; Williams, N.S.; Frantz, D.E.; Wigginton, J.G.; Nwariaku, F.E.; et al. Specific inhibition of mitochondrial oxidative stress suppresses inflammation and improves cardiac function in a rat pneumonia-related sepsis model. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1847–H1859. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.; Evelson, P.; Cimolai, M.C. Oxygen and nitric oxide metabolism in sepsi. In Free Radical Pathophysiology; Alvarez, S., Ed.; Transworld Research Network: Kerala, India, 2008; pp. 223–236. [Google Scholar]
- Wallace, D.C.; Shoffner, J.M.; Watts, R.L.; Juncos, J.L.; Torroni, A. Mitochondrial oxidative phosphorylation defects in Parkinson’s disease. Ann. Neurol. 1992, 32, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Kanai, A.J.; Pearce, L.L.; Clemens, P.R.; Birder, L.A.; VanBibber, M.M.; Choi, S.Y.; de Groat, W.C.; Peterson, J. Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc. Natl. Acad. Sci. USA 2001, 98, 14126–14131. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F. Oxidative stress and mitochondrial dysfunction in sepsis. Br. J. Anaesth. 2011, 107, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Van de Sandt, A.M.; Windler, R.; Godecke, A.; Ohlig, J.; Zander, S.; Reinartz, M.; Graf, J.; van Faassen, E.E.; Rassaf, T.; Schrader, J.; et al. Endothelial nos (NOS3) impairs myocardial function in developing sepsis. Basic Res. Cardiol. 2013, 108, 330. [Google Scholar] [CrossRef] [PubMed]
- Vanasco, V.; Magnani, N.D.; Cimolai, M.C.; Valdez, L.B.; Evelson, P.; Boveris, A.; Alvarez, S. Endotoxemia impairs heart mitochondrial function by decreasing electron transfer, ATP synthesis and atp content without affecting membrane potential. J. Bioenerg. Biomembr. 2012, 44, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, J.R.; Rudyk, O.; Mayr, M.; Eaton, P. Nitrosative protein oxidation is modulated during early endotoxemia. Nitric Oxide 2011, 25, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Zang, Q.; Maass, D.L.; Tsai, S.J.; Horton, J.W. Cardiac mitochondrial damage and inflammation responses in sepsis. Surg. Infect. 2007, 8, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Welty-Wolf, K.E.; Carraway, M.; Tatro, L.; Piantadosi, C.A. Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc. Res. 2004, 64, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A.; Alvarez, S.; Navarro, A. The role of mitochondrial nitric oxide synthase in inflammation and septic shock. Free Radic. Biol. Med. 2002, 33, 1186–1193. [Google Scholar] [CrossRef]
- Escames, G.; Lopez, L.C.; Ortiz, F.; Lopez, A.; Garcia, J.A.; Ros, E.; Acuna-Castroviejo, D. Attenuation of cardiac mitochondrial dysfunction by melatonin in septic mice. FEBS J. 2007, 274, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Yi, C.; Wang, H.; Bruce, I.C.; Xia, Q. Mitochondrial nitric oxide synthase participates in septic shock myocardial depression by nitric oxide overproduction and mitochondrial permeability transition pore opening. Shock 2012, 37, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Vanasco, V.; Cimolai, M.C.; Evelson, P.; Alvarez, S. The oxidative stress and the mitochondrial dysfunction caused by endotoxemia are prevented by α-lipoic acid. Free Radic. Res. 2008, 42, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Supinski, G.S.; Murphy, M.P.; Callahan, L.A. MitoQ administration prevents endotoxin-induced cardiac dysfunction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1095–R1102. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; McStay, G.P.; Clarke, S.J. The permeability transition pore complex: Another view. Biochimie 2002, 84, 153–166. [Google Scholar] [CrossRef]
- Larche, J.; Lancel, S.; Hassoun, S.M.; Favory, R.; Decoster, B.; Marchetti, P.; Chopin, C.; Neviere, R. Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardial dysfunction and mortality. J. Am. Coll. Cardiol. 2006, 48, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Fauvel, H.; Marchetti, P.; Obert, G.; Joulain, O.; Chopin, C.; Formstecher, P.; Neviere, R. Protective effects of cyclosporin a from endotoxin-induced myocardial dysfunction and apoptosis in rats. Am. J. Respir. Crit. Rare Red. 2002, 165, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.D.; Quinlan, C.L.; Andrukhiv, A.; West, I.C.; Jaburek, M.; Garlid, K.D. The direct physiological effects of mitoKATP opening on heart mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H406–H415. [Google Scholar] [CrossRef] [PubMed]
- Bougaki, M.; Searles, R.J.; Kida, K.; Yu, J.; Buys, E.S.; Ichinose, F. Nos3 protects against systemic inflammation and myocardial dysfunction in murine polymicrobial sepsis. Shock 2010, 34, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Ricquier, D.; Bouillaud, F. Mitochondrial uncoupling proteins: From mitochondria to the regulation of energy balance. J. Physiol. 2000, 1, 3–10. [Google Scholar] [CrossRef]
- Boudina, S.; Sena, S.; Theobald, H.; Sheng, X.; Wright, J.J.; Hu, X.X.; Aziz, S.; Johnson, J.I.; Bugger, H.; Zaha, V.G.; et al. Mitochondrial energetics in the heart in obesity-related diabetes: Direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 2007, 56, 2457–2466. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Konig, A.; Bode, C.; Bugger, H. Diabetes mellitus and myocardial mitochondrial dysfunction: Bench to bedside. Heart Fail. Clin. 2012, 8, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.J.; Cole, M.A.; Lygate, C.A.; Carr, C.A.; Stuckey, D.J.; Little, S.E.; Neubauer, S.; Clarke, K. Increased mitochondrial uncoupling proteins, respiratory uncoupling and decreased efficiency in the chronically infarcted rat heart. J. Mol. Cell. Cardiol. 2008, 44, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Roshon, M.J.; Kline, J.A.; Thornton, L.R.; Watts, J.A. Cardiac UCP2 expression and myocardial oxidative metabolism during acute septic shock in the rat. Shock 2003, 19, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, D.; Chai, W.; Long, Y.; Su, L.; Yang, R. The role of uncoupling protein-2 (UCP2) during myocardial dysfunction in a canine model of endotoxin shock. Shock 2014, 3, 292–297. [Google Scholar]
- Zheng, G.; Lyu, J.; Liu, S.; Huang, J.; Liu, C.; Xiang, D.; Xie, M.; Zeng, Q. Silencing of uncoupling protein 2 by small interfering rna aggravates mitochondrial dysfunction in cardiomyocytes under septic conditions. Int. J. Mol. Med. 2015, 35, 1525–1536. [Google Scholar] [PubMed]
- Aguirre, E.; Cadenas, S. GDP and carboxyatractylate inhibit 4-hydroxynonenal-activated proton conductance to differing degrees in mitochondria from skeletal muscle and heart. Biochim. Biophys. Acta 2010, 1797, 1716–1726. [Google Scholar] [CrossRef] [PubMed]
- Hickson-Bick, D.L.; Jones, C.; Buja, L.M. Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. J. Mol. Cell. Cardiol. 2008, 44, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A.; Gustafsson, A.B. Mitochondrial turnover in the heart. Biochim. Biophys. Acta 2011, 1813, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Wenz, T. Regulation of mitochondrial biogenesis and PGC-1α under cellular stress. Mitochondrion 2013, 13, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Lancel, S.; Hassoun, S.M.; Favory, R.; Decoster, B.; Motterlini, R.; Neviere, R. Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J. Pharmacol. Exp. Ther. 2009, 329, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Russell, L.K.; Mansfield, C.M.; Lehman, J.J.; Kovacs, A.; Courtois, M.; Saffitz, J.E.; Medeiros, D.M.; Valencik, M.L.; McDonald, J.A.; Kelly, D.P. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator-1α promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ. Res. 2004, 94, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Schilling, J.; Lai, L.; Sambandam, N.; Dey, C.E.; Leone, T.C.; Kelly, D.P. Toll-like receptor-mediated inflammatory signaling reprograms cardiac energy metabolism by repressing peroxisome proliferator-activated receptor gamma coactivator-1 signaling. Circ. Heart Fail. 2011, 4, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Gustafsson, A.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Carchman, E.H.; Rao, J.; Loughran, P.A.; Rosengart, M.R.; Zuckerbraun, B.S. Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology 2011, 53, 2053–2062. [Google Scholar] [CrossRef] [PubMed]
- Carchman, E.H.; Whelan, S.; Loughran, P.; Mollen, K.; Stratamirovic, S.; Shiva, S.; Rosengart, M.R.; Zuckerbraun, B.S. Experimental sepsis-induced mitochondrial biogenesis is dependent on autophagy, TLR4, and TLR9 signaling in liver. FASEB J. 2013, 27, 4703–4711. [Google Scholar] [CrossRef] [PubMed]
- Ceylan-Isik, A.F.; Zhao, P.; Zhang, B.; Xiao, X.; Su, G.; Ren, J. Cardiac overexpression of metallothionein rescues cardiac contractile dysfunction and endoplasmic reticulum stress but not autophagy in sepsis. J. Mol. Cell. Cardiol. 2010, 48, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Welty-Wolf, K.E.; Carraway, M.S.; Schwartz, D.A.; Hollingsworth, J.W.; Piantadosi, C.A. Toll-like receptor 4 mediates mitochondrial DNA damage and biogenic responses after heat-inactivated. E. coli. FASEB J. 2005, 19, 1531–1533. [Google Scholar] [CrossRef] [PubMed]
- Crouser, E.D.; Julian, M.W.; Huff, J.E.; Struck, J.; Cook, C.H. Carbamoyl phosphate synthase-1: A marker of mitochondrial damage and depletion in the liver during sepsis. Crit. Care Med. 2006, 34, 2439–2446. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Perry, C.N.; Huang, C.; Iwai-Kanai, E.; Carreira, R.S.; Glembotski, C.C.; Gottlieb, R.A. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H470–H479. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cimolai, M.C.; Alvarez, S.; Bode, C.; Bugger, H. Mitochondrial Mechanisms in Septic Cardiomyopathy. Int. J. Mol. Sci. 2015, 16, 17763-17778. https://doi.org/10.3390/ijms160817763
Cimolai MC, Alvarez S, Bode C, Bugger H. Mitochondrial Mechanisms in Septic Cardiomyopathy. International Journal of Molecular Sciences. 2015; 16(8):17763-17778. https://doi.org/10.3390/ijms160817763
Chicago/Turabian StyleCimolai, María Cecilia, Silvia Alvarez, Christoph Bode, and Heiko Bugger. 2015. "Mitochondrial Mechanisms in Septic Cardiomyopathy" International Journal of Molecular Sciences 16, no. 8: 17763-17778. https://doi.org/10.3390/ijms160817763
APA StyleCimolai, M. C., Alvarez, S., Bode, C., & Bugger, H. (2015). Mitochondrial Mechanisms in Septic Cardiomyopathy. International Journal of Molecular Sciences, 16(8), 17763-17778. https://doi.org/10.3390/ijms160817763