
Borrowing Nuclear DNA Helicases to Protect Mitochondrial DNA

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondrial Replicative Helicase, Twinkle
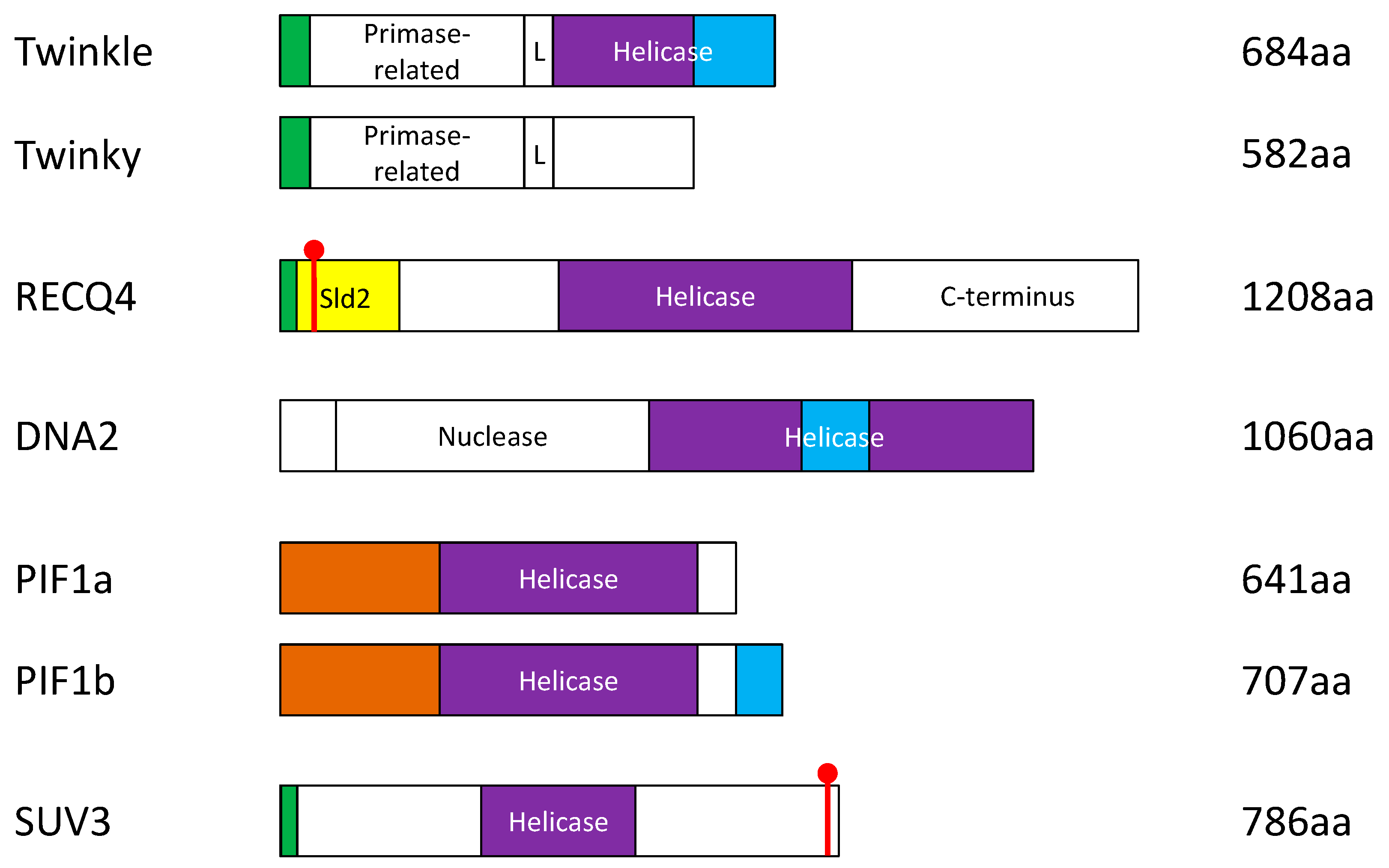
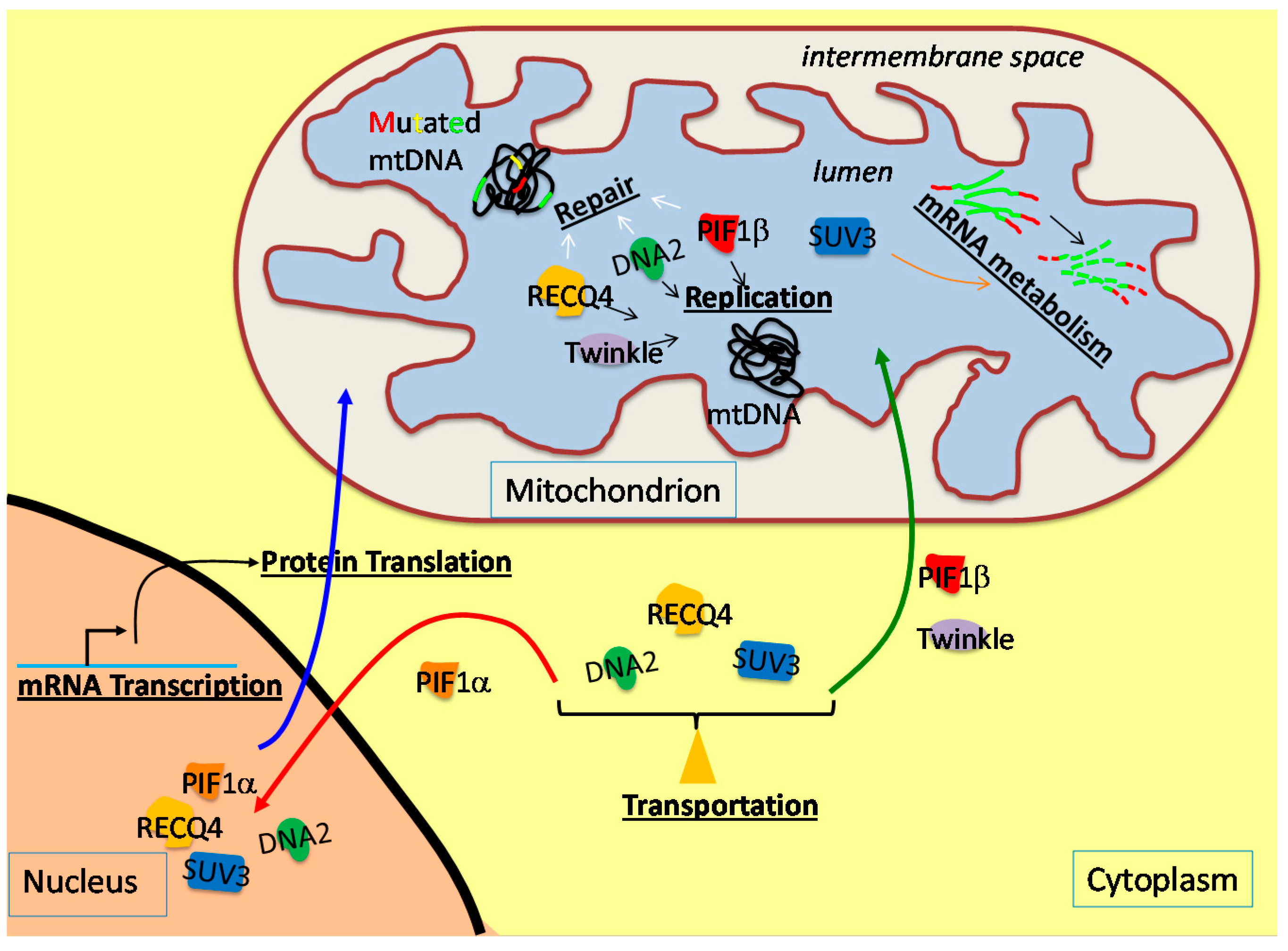
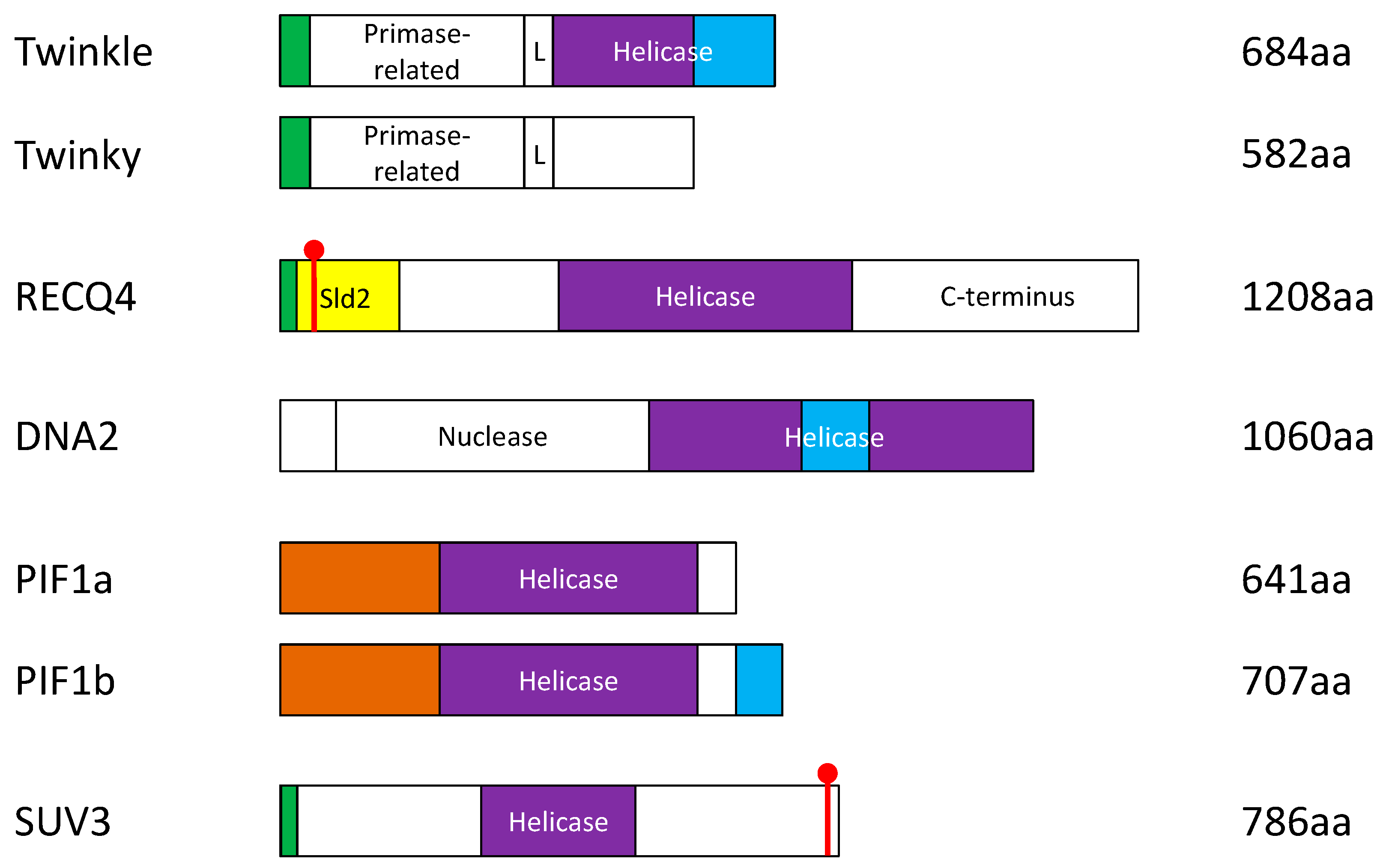
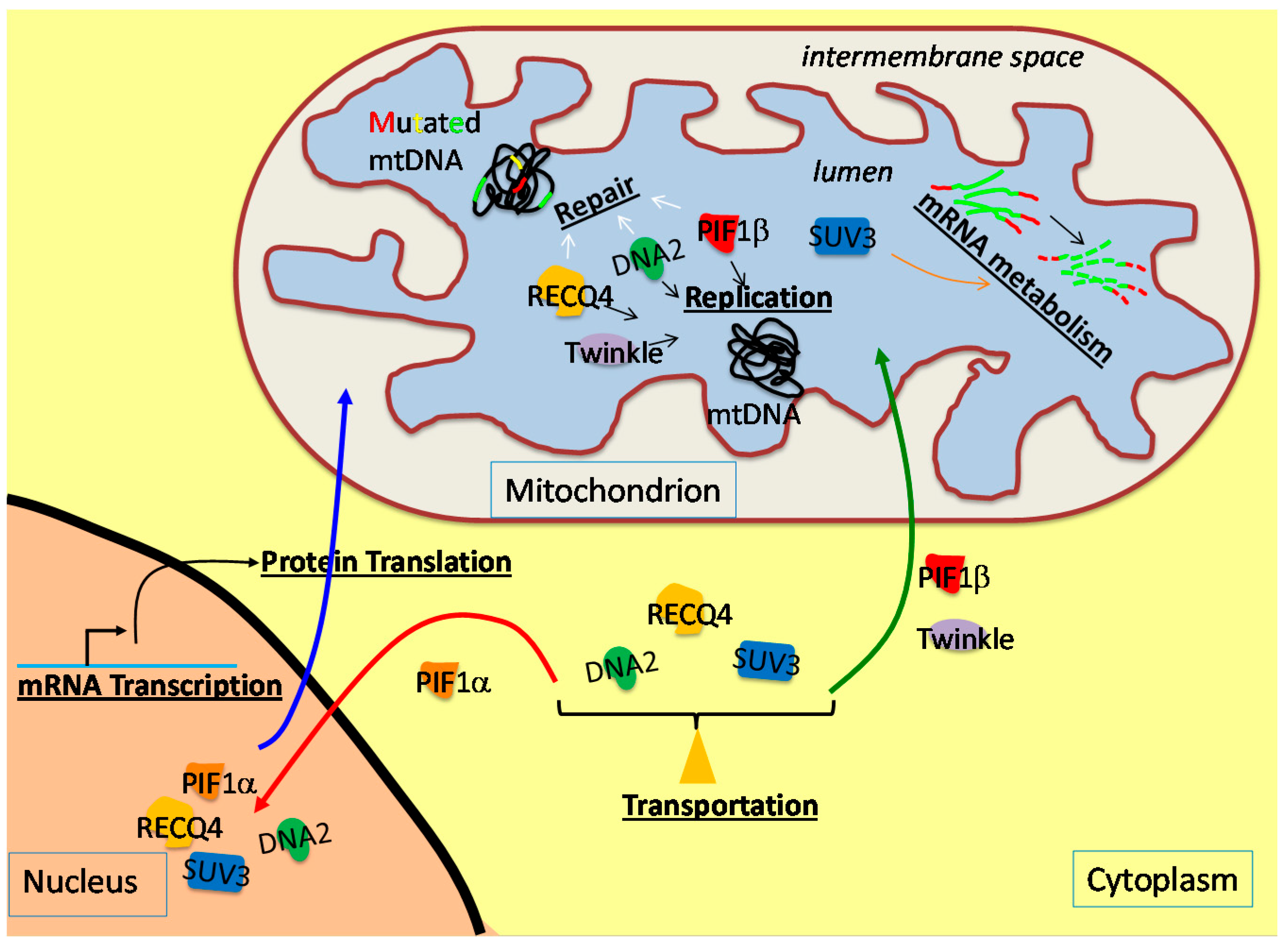
3. The Involvement of the Nuclear DNA Helicases
3.1. RecQ-Like Helicase 4 (RECQ4)
3.2. DNA Replication Helicase/Nuclease 2 (DNA2)
3.3. Petite Integration Frequency 1 (PIF1)
3.4. Suppressor of Var1 3-Like Protein 1 (SUV3)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kurland, C.G.; Andersson, S.G. Origin and evolution of the mitochondrial proteome. Microb. Mol. Biol. Rev. 2000, 64, 786–820. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Iborra, F.J.; Kimura, H.; Cook, P.R. The functional organization of mitochondrial genomes in human cells. BMC Biol. 2004, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Kuroiwa, T. Organization of multiple nucleoids and DNA molecules in mitochondria of a human cell. Exp. Cell Res. 1991, 196, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.D.; Mahmud, A.; McKenzie, M.; Trounce, I.A.; St John, J.C. Mitochondrial DNA copy number is regulated in a tissue specific manner by DNA methylation of the nuclear-encoded DNA polymerase gamma A. Nucleic Acids Res. 2012, 40, 10124–10138. [Google Scholar] [CrossRef] [PubMed]
- Albring, M.; Griffith, J.; Attardi, G. Association of a protein structure of probable membrane derivation with HeLa cell mitochondrial DNA near its origin of replication. Proc. Natl. Acad. Sci. USA 1977, 74, 1348–1352. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Reyes, A. Human mitochondrial DNA replication. Cold Spring Harb. Perspect. Biol. 2012, 4, a012971. [Google Scholar] [CrossRef] [PubMed]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.W.; Margineantu, D.H.; Capaldi, R.A.; Selker, J.M. Mitochondrial DNA depletion causes morphological changes in the mitochondrial reticulum of cultured human cells. FEBS Lett. 2000, 474, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondrial disease. Lancet 2006, 368, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Yu, M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci. 2011, 89, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Clay Montier, L.L.; Deng, J.J.; Bai, Y. Number matters: Control of mammalian mitochondrial DNA copy number. J. Genet. Genomics 2009, 36, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.; Bottinger, L.; Pfanner, N. Mitochondrial protein import: From transport pathways to an integrated network. Trends Biochem. Sci. 2012, 37, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Gebert, N.; Ryan, M.T.; Pfanner, N.; Wiedemann, N.; Stojanovski, D. Mitochondrial protein import machineries and lipids: A functional connection. Biochim. Biophys. Acta 2011, 1808, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Kumari, J.; Mudgal, R.; Modi, P.; Gupta, S.; Futami, K.; Goto, H.; Lindor, N.M.; Furuichi, Y.; Mohanty, D.; et al. RECQL4 is essential for the transport of p53 to mitochondria in normal human cells in the absence of exogenous stress. J. Cell Sci. 2012, 125, 2509–2522. [Google Scholar] [CrossRef] [PubMed]
- Spelbrink, J.N.; Li, F.Y.; Tiranti, V.; Nikali, K.; Yuan, Q.P.; Tariq, M.; Wanrooij, S.; Garrido, N.; Comi, G.; Morandi, L.; et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 2001, 28, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial inheritance in yeast. Biochim. Biophys. Acta 2014, 1837, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, D.; Matic, S.; Kuhl, I.; Ruzzenente, B.; Freyer, C.; Jemt, E.; Park, C.B.; Falkenberg, M.; Larsson, N.G. TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D-loop strands and complete mtDNA replication. Hum. Mol. Genet. 2013, 22, 1983–1993. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.; Bravo, L.; Garcia, I.; Gaytan, N.; Herrera, A.; Maldonado, A.; Quintanilla, B. The mitochondrial nucleoid: Integrating mitochondrial DNA into cellular homeostasis. Cold Spring Harb. Perspect. Biol. 2013, 5, a011080. [Google Scholar] [CrossRef] [PubMed]
- Bogenhagen, D.F.; Rousseau, D.; Burke, S. The layered structure of human mitochondrial DNA nucleoids. J. Biol. Chem. 2008, 283, 3665–3675. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, J.A.; Gaspari, M.; Falkenberg, M. TWINKLE has 5'→3' DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J. Biol. Chem. 2003, 278, 48627–48632. [Google Scholar] [CrossRef] [PubMed]
- Farge, G.; Holmlund, T.; Khvorostova, J.; Rofougaran, R.; Hofer, A.; Falkenberg, M. The N-terminal domain of TWINKLE contributes to single-stranded DNA binding and DNA helicase activities. Nucleic Acids Res. 2008, 36, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, J.A.; Pande, V.; Holmlund, T.; Farge, G.; Pham, X.H.; Nilsson, L.; Falkenberg, M. Structure–function defects of the TWINKLE linker region in progressive external ophthalmoplegia. J. Mol. Biol. 2008, 377, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Nandakumar, D.; Tang, G.Q.; Patel, S.S. Human mitochondrial DNA helicase TWINKLE is both an unwinding and annealing helicase. J. Biol. Chem. 2012, 287, 14545–14556. [Google Scholar] [CrossRef] [PubMed]
- Jemt, E.; Farge, G.; Backstrom, S.; Holmlund, T.; Gustafsson, C.M.; Falkenberg, M. The mitochondrial DNA helicase TWINKLE can assemble on a closed circular template and support initiation of DNA synthesis. Nucleic Acids Res. 2011, 39, 9238–9249. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, J.A.; Pham, X.H.; Pellegrini, M.; Falkenberg, M. Reconstitution of a minimal mtDNA replisome in vitro. EMBO J. 2004, 23, 2423–2429. [Google Scholar] [CrossRef] [PubMed]
- Ylikallio, E.; Tyynismaa, H.; Tsutsui, H.; Ide, T.; Suomalainen, A. High mitochondrial DNA copy number has detrimental effects in mice. Hum. Mol. Genet. 2010, 19, 2695–2705. [Google Scholar] [CrossRef] [PubMed]
- Pohjoismaki, J.L.; Williams, S.L.; Boettger, T.; Goffart, S.; Kim, J.; Suomalainen, A.; Moraes, C.T.; Braun, T. Over-expression of Twinkle-helicase protects cardiomyocytes from genotoxic stress caused by reactive oxygen species. Proc. Natl. Acad. Sci. USA 2013, 110, 19408–19013. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Sembongi, H.; Bokori-Brown, M.; Granycome, C.; Ashley, N.; Poulton, J.; Jalanko, A.; Spelbrink, J.N.; Holt, I.J.; Suomalainen, A. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum. Mol. Genet. 2004, 13, 3219–3227. [Google Scholar] [CrossRef] [PubMed]
- Longley, M.J.; Humble, M.M.; Sharief, F.S.; Copeland, W.C. Disease variants of the human mitochondrial DNA helicase encoded by C10orf2 differentially alter protein stability, nucleotide hydrolysis, and helicase activity. J. Biol. Chem. 2010, 285, 29690–29702. [Google Scholar] [CrossRef] [PubMed]
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and disease. J. Pathol. 2012, 226, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Arenas, J.; Briem, E.; Dahl, H.; Hutchison, W.; Lewis, S.; Martin, M.A.; Spelbrink, H.; Tiranti, V.; Jacobs, H.; Zeviani, M. The V368i mutation in Twinkle does not segregate with adPEO. Ann. Neurol. 2003, 53, 278. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Y.; Tariq, M.; Croxen, R.; Morten, K.; Squier, W.; Newsom-Davis, J.; Beeson, D.; Larsson, C. Mapping of autosomal dominant progressive external ophthalmoplegia to a 7-cM critical region on 10q24. Neurology 1999, 53, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Servidei, S.; Gellera, C.; Bertini, E.; DiMauro, S.; DiDonato, S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature 1989, 339, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Moslemi, A.R.; Melberg, A.; Holme, E.; Oldfors, A. Autosomal dominant progressive external ophthalmoplegia: Distribution of multiple mitochondrial DNA deletions. Neurology 1999, 53, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Kitao, S.; Ohsugi, I.; Ichikawa, K.; Goto, M.; Furuichi, Y.; Shimamoto, A. Cloning of two new human helicase genes of the RecQ family: Biological significance of multiple species in higher eukaryotes. Genomics 1998, 54, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Rothmund-Thomson syndrome helicase, RECQ4: On the crossroad between DNA replication and repair. DNA Repair. (Amst.) 2010, 9, 325–330. [Google Scholar] [CrossRef]
- Suzuki, T.; Kohno, T.; Ishimi, Y. DNA helicase activity in purified human RECQL4 protein. J. Biochem. 2009, 146, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, Y. Dual DNA unwinding activities of the Rothmund-Thomson syndrome protein, RECQ4. EMBO J. 2009, 28, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, Y.; Masumoto, H.; Sugino, A.; Araki, H. Sld2, which interacts with Dpb11 in Saccharomyces cerevisiae, is required for chromosomal DNA replication. Mol. Cell. Biol. 1998, 18, 6102–6109. [Google Scholar] [PubMed]
- Im, J.S.; Ki, S.H.; Farina, A.; Jung, D.S.; Hurwitz, J.; Lee, J.K. Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 15628–15632. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Rochette, P.J.; Feyissa, E.A.; Su, T.V.; Liu, Y. MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO J. 2009, 28, 3005–3014. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, S.; Mendoza-Maldonado, R.; Tissino, E.; Sidorova, J.M.; Yin, J.; Wang, W.; Monnat, R.J., Jr.; Falaschi, A.; Vindigni, A. Human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Mol. Cell. Biol. 2010, 30, 1382–1396. [Google Scholar] [CrossRef] [PubMed]
- Im, J.S.; Park, S.Y.; Cho, W.H.; Bae, S.H.; Hurwitz, J.; Lee, J.K. RecQL4 is required for the association of Mcm10 and Ctf4 with replication origins in human cells. Cell Cycle 2015, 14, 1001–1009. [Google Scholar] [PubMed]
- Sangrithi, M.N.; Bernal, J.A.; Madine, M.; Philpott, A.; Lee, J.; Dunphy, W.G.; Venkitaraman, A.R. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell 2005, 121, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Kumano, M.; Kubota, Y.; Hashimoto, Y.; Takisawa, H. The N-terminal noncatalytic region of Xenopus RecQ4 is required for chromatin binding of DNA polymerase α in the initiation of DNA replication. Mol. Cell. Biol. 2006, 26, 4843–4852. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Noda, T.; Furuichi, Y. Preparation of the gene targeted knockout mice for human premature aging diseases, Werner syndrome, and Rothmund-Thomson syndrome caused by the mutation of DNA helicases. Nihon Yakurigaku Zasshi 2002, 119, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.B.; Hodges, C.A.; Barnes, E.; Vogel, H.; Hassold, T.J.; Luo, G. Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund-Thomson syndrome. Hum. Mol. Genet. 2005, 14, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Marino, F.; Vindigni, A.; Onesti, S. Bioinformatic analysis of RecQ4 helicases reveals the presence of a RQC domain and a Zn knuckle. Biophys. Chem. 2013, 177, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Kohzaki, M.; Chiourea, M.; Versini, G.; Adachi, N.; Takeda, S.; Gagos, S.; Halazonetis, T.D. The helicase domain and C-terminus of human RecQL4 facilitate replication elongation on DNA templates damaged by ionizing radiation. Carcinogenesis 2012, 33, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Lucic, B.; Zhang, Y.; King, O.; Mendoza-Maldonado, R.; Berti, M.; Niesen, F.H.; Burgess-Brown, N.A.; Pike, A.C.; Cooper, C.D.; Gileadi, O.; et al. A prominent beta-hairpin structure in the winged-helix domain of RECQ1 is required for DNA unwinding and oligomer formation. Nucleic Acids Res. 2011, 39, 1703–1717. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.; Nie, L.; Peng, Z.; Yang, Q.; Yang, K.; Tao, J.; Mi, Y.; Fang, X.; Balajee, A.S.; Zhao, Y. RecQL4 cytoplasmic localization: Implications in mitochondrial DNA oxidative damage repair. Int. J. Biochem. Cell Biol. 2012, 44, 1942–1951. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.L.; Rossi, M.L.; Canugovi, C.; Tian, J.; Sykora, P.; Ramamoorthy, M.; Wang, Z.M.; Singh, D.K.; Akbari, M.; Kasiviswanathan, R.; et al. RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell 2012, 11, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; De, S.; Srivastava, V.; Hussain, M.; Kumari, J.; Muniyappa, K.; Sengupta, S. RECQL4 and p53 potentiate the activity of polymerase gamma and maintain the integrity of the human mitochondrial genome. Carcinogenesis 2014, 35, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Xu, X.; Alontaga, A.Y.; Chen, Y.; Liu, Y. Impaired p32 regulation caused by the lymphoma-prone RECQ4 mutation drives mitochondrial dysfunction. Cell Rep. 2014, 7, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Muta, T.; Kang, D.; Kitajima, S.; Fujiwara, T.; Hamasaki, N. p32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. J. Biol. Chem. 1997, 272, 24363–24370. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Fogal, V.; Richardson, A.D.; Karmali, P.P.; Scheffler, I.E.; Smith, J.W.; Ruoslahti, E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol. Cell. Biol. 2010, 30, 1303–1318. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Zhang, Y. Mitochondrial p32 is a critical mediator of ARF-induced apoptosis. Cancer Cell 2008, 13, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.K.; St Clair, D.K. Nucleophosmin blocks mitochondrial localization of p53 and apoptosis. J. Biol. Chem. 2009, 284, 16409–16418. [Google Scholar] [CrossRef] [PubMed]
- Dumas, L.B.; Lussky, J.P.; McFarland, E.J.; Shampay, J. New temperature-sensitive mutants of Saccharomyces cerevisiae affecting DNA replication. Mol. Gen. Genet. 1982, 187, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Eki, T.; Okumura, K.; Shiratori, A.; Abe, M.; Nogami, M.; Taguchi, H.; Shibata, T.; Murakami, Y.; Hanaoka, F. Assignment of the closest human homologue (DNA2L:KIAA0083) of the yeast DNA2 helicase gene to chromosome band 10q21.3-q22.1. Genomics 1996, 37, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Imamura, O.; Campbell, J.L. The human Bloom syndrome gene suppresses the DNA replication and repair defects of yeast DNA2 mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 8193–8198. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.D.; Ryu, G.H.; Kim, D.H.; Hurwitz, J.; Seo, Y.S. Isolation of human DNA2 endonuclease and characterization of its enzymatic properties. Nucleic Acids Res. 2006, 34, 1854–1864. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lee, M.; Kang, H.J.; Kim, D.H.; Kang, Y.H.; Bae, S.H.; Seo, Y.S. The N-terminal 45-kDa domain of DNA2 endonuclease/helicase targets the enzyme to secondary structure DNA. J. Biol. Chem. 2013, 288, 9468–9481. [Google Scholar] [CrossRef] [PubMed]
- Masuda-Sasa, T.; Imamura, O.; Campbell, J.L. Biochemical analysis of human DNA2. Nucleic Acids Res. 2006, 34, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Kim, D.W.; Kim, J.; Kim, J.H.; Kim, D.H.; Kim, H.D.; Kang, H.Y.; Seo, Y.S. Coupling of DNA helicase and endonuclease activities of yeast DNA2 facilitates Okazaki fragment processing. J. Biol. Chem. 2002, 277, 26632–26641. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Sun, L.; Shen, F.; Chen, Y.; Hua, Y.; Liu, Y.; Zhang, M.; Hu, Y.; Wang, Q.; Xu, W.; et al. The intra-S phase checkpoint targets DNA2 to prevent stalled replication forks from reversing. Cell 2012, 149, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Formosa, T.; Nittis, T. DNA2 mutants reveal interactions with DNA polymerase α and Ctf4, a Pol α accessory factor, and show that full DNA2 helicase activity is not essential for growth. Genetics 1999, 151, 1459–1470. [Google Scholar] [PubMed]
- Peng, G.; Dai, H.; Zhang, W.; Hsieh, H.J.; Pan, M.R.; Park, Y.Y.; Tsai, R.Y.; Bedrosian, I.; Lee, J.S.; Ira, G.; et al. Human nuclease/helicase DNA2 alleviates replication stress by promoting DNA end resection. Cancer Res. 2012, 72, 2802–2813. [Google Scholar] [CrossRef] [PubMed]
- Karanja, K.K.; Cox, S.W.; Duxin, J.P.; Stewart, S.A.; Campbell, J.L. DNA2 and EXO1 in replication-coupled, homology-directed repair and in the interplay between HDR and the FA/BRCA network. Cell Cycle 2012, 11, 3983–3996. [Google Scholar] [CrossRef] [PubMed]
- Duxin, J.P.; Dao, B.; Martinsson, P.; Rajala, N.; Guittat, L.; Campbell, J.L.; Spelbrink, J.N.; Stewart, S.A. Human DNA2 is a nuclear and mitochondrial DNA maintenance protein. Mol. Cell. Biol. 2009, 29, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Chiba, T.; Xue, X.; Niu, H.; Sung, P. Multifaceted role of the Topo IIIα-RMI1-RMI2 complex and DNA2 in the BLM-dependent pathway of DNA break end resection. Nucleic Acids Res. 2014, 42, 11083–11091. [Google Scholar] [CrossRef] [PubMed]
- Sturzenegger, A.; Burdova, K.; Kanagaraj, R.; Levikova, M.; Pinto, C.; Cejka, P.; Janscak, P. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J. Biol. Chem. 2014, 289, 27314–27326. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Sampathi, S.; Dai, H.; Liu, C.; Zhou, M.; Hu, J.; Huang, Q.; Campbell, J.; Shin-Ya, K.; Zheng, L.; et al. Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 2013, 32, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhou, M.; Guo, Z.; Lu, H.; Qian, L.; Dai, H.; Qiu, J.; Yakubovskaya, E.; Bogenhagen, D.F.; Demple, B.; et al. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol. Cell 2008, 32, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, D.; Di Fonzo, A.; Lin, W.; Bordoni, A.; Liu, C.; Fassone, E.; Pagliarani, S.; Rizzuti, M.; Zheng, L.; Filosto, M.; et al. Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am. J. Hum. Genet. 2013, 92, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Foury, F.; Kolodynski, J. PIF mutation blocks recombination between mitochondrial ρ+ and ρ− genomes having tandemly arrayed repeat units in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1983, 80, 5345–5349. [Google Scholar] [CrossRef] [PubMed]
- Mateyak, M.K.; Zakian, V.A. Human PIF helicase is cell cycle regulated and associates with telomerase. Cell Cycle 2006, 5, 2796–2804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.H.; Zhou, B.; Huang, Y.; Xu, L.X.; Zhou, J.Q. The human PIF1 helicase, a potential Escherichia coli RecD homologue, inhibits telomerase activity. Nucleic Acids Res. 2006, 34, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Futami, K.; Shimamoto, A.; Furuichi, Y. Mitochondrial and nuclear localization of human PIF1 helicase. Biol. Pharm. Bull. 2007, 30, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Masuda, Y.; Kamiya, K. Biochemical analysis of human PIF1 helicase and functions of its N-terminal domain. Nucleic Acids Res. 2008, 36, 6295–6308. [Google Scholar] [CrossRef] [PubMed]
- George, T.; Wen, Q.; Griffiths, R.; Ganesh, A.; Meuth, M.; Sanders, C.M. Human PIF1 helicase unwinds synthetic DNA structures resembling stalled DNA replication forks. Nucleic Acids Res. 2009, 37, 6491–6502. [Google Scholar] [CrossRef] [PubMed]
- Sanders, C.M. Human PIF1 helicase is a G-quadruplex DNA-binding protein with G-quadruplex DNA-unwinding activity. Biochem. J. 2010, 430, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Sabouri, N.; Zakian, V.A. Unwinding the functions of the PIF1 family helicases. DNA Repair (Amst.) 2010, 9, 237–249. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, J.; Li, S.; Kamiya, K.; Chen, X.; Zhou, P. Determination of the biochemical properties of full-length human PIF1 ATPase. Prion 2013, 7, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.L.; Liu, N.N.; Yang, Y.T.; Li, H.H.; Li, M.; Dou, S.X.; Xi, X. G-quadruplexes significantly stimulate PIF1 helicase-catalyzed duplex DNA unwinding. J. Biol. Chem. 2015, 290, 7722–7735. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.M.; Wu, W.Q.; Duan, X.L.; Liu, N.N.; Li, H.H.; Fu, J.; Dou, S.X.; Li, M.; Xi, X.G. Molecular mechanism of G-quadruplex unwinding helicase: Sequential and repetitive unfolding of G-quadruplex by PIF1 helicase. Biochem. J. 2015, 466, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zhang, J.; Bochman, M.L.; Zakian, V.A.; Ha, T. Periodic DNA patrolling underlies diverse functions of PIF1 on R-loops and G-rich DNA. Elife 2014, 3, e02190. [Google Scholar] [PubMed]
- Byrd, A.K.; Raney, K.D. A parallel quadruplex DNA is bound. Tightly but unfolded slowly by PIF1 helicase. J. Biol. Chem. 2015, 290, 6482–6494. [Google Scholar] [CrossRef] [PubMed]
- Gagou, M.E.; Ganesh, A.; Thompson, R.; Phear, G.; Sanders, C.; Meuth, M. Suppression of apoptosis by PIF1 helicase in human tumor cells. Cancer Res. 2011, 71, 4998–5008. [Google Scholar] [CrossRef] [PubMed]
- Gagou, M.E.; Ganesh, A.; Phear, G.; Robinson, D.; Petermann, E.; Cox, A.; Meuth, M. Human PIF1 helicase supports DNA replication and cell growth under oncogenic-stress. Oncotarget 2014, 5, 11381–11398. [Google Scholar] [PubMed]
- Cheng, X.; Ivessa, A.S. Association of the yeast DNA helicase PIF1p with mitochondrial membranes and mitochondrial DNA. Eur. J. Cell Biol. 2010, 89, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Dunaway, S.; Ivessa, A.S. The role of Pif1p, a DNA helicase in Saccharomyces cerevisiae, in maintaining mitochondrial DNA. Mitochondrion 2007, 7, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Sommers, J.A.; Zhou, J.; Kaplan, D.L.; Spelbrink, J.N.; Mergny, J.L.; Brosh, R.M., Jr. DNA sequences proximal to human mitochondrial DNA deletion breakpoints prevalent in human disease form G-quadruplexes, a class of DNA structures inefficiently unwound by the mitochondrial replicative Twinkle helicase. J. Biol. Chem. 2014, 289, 29975–29993. [Google Scholar] [CrossRef] [PubMed]
- Budd, M.E.; Reis, C.C.; Smith, S.; Myung, K.; Campbell, J.L. Evidence suggesting that PIF1 helicase functions in DNA replication with the DNA2 helicase/nuclease and DNA polymerase delta. Mol. Cell. Biol. 2006, 26, 2490–2500. [Google Scholar] [CrossRef] [PubMed]
- Butow, R.A.; Zhu, H.; Perlman, P.; Conrad-Webb, H. The role of a conserved dodecamer sequence in yeast mitochondrial gene expression. Genome 1989, 31, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Dmochowska, A.; Kalita, K.; Krawczyk, M.; Golik, P.; Mroczek, K.; Lazowska, J.; Stepien, P.P.; Bartnik, E. A human putative SUV3-like RNA helicase is conserved between Rhodobacter and all eukaryotes. Acta Biochim. Pol. 1999, 46, 155–162. [Google Scholar] [PubMed]
- Chen, P.L.; Chen, C.F.; Chen, Y.; Guo, X.E.; Huang, C.K.; Shew, J.Y.; Reddick, R.L.; Wallace, D.C.; Lee, W.H. Mitochondrial genome instability resulting from SUV3 haploinsufficiency leads to tumorigenesis and shortened lifespan. Oncogene 2013, 32, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Shu, Z.; Lieser, S.A.; Chen, P.L.; Lee, W.H. Human mitochondrial SUV3 and polynucleotide phosphorylase form a 330-kDa heteropentamer to cooperatively degrade double-stranded RNA with a 3'-to-5' directionality. J. Biol. Chem. 2009, 284, 20812–20821. [Google Scholar] [CrossRef] [PubMed]
- Minczuk, M.; Piwowarski, J.; Papworth, M.A.; Awiszus, K.; Schalinski, S.; Dziembowski, A.; Dmochowska, A.; Bartnik, E.; Tokatlidis, K.; Stepien, P.P.; et al. Localisation of the human hSUV3p helicase in the mitochondrial matrix and its preferential unwinding of dsDNA. Nucleic Acids Res. 2002, 30, 5074–5086. [Google Scholar] [CrossRef] [PubMed]
- Veno, S.T.; Kulikowicz, T.; Pestana, C.; Stepien, P.P.; Stevnsner, T.; Bohr, V.A. The human SUV3 helicase interacts with replication protein A and flap endonuclease 1 in the nucleus. Biochem. J. 2011, 440, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Guo, X.E.; Modrek, A.S.; Chen, C.F.; Chen, P.L.; Lee, W.H. Helicase SUV3, polynucleotide phosphorylase, and mitochondrial polyadenylation polymerase form a transient complex to modulate mitochondrial mRNA polyadenylated tail lengths in response to energetic changes. J. Biol. Chem. 2014, 289, 16727–16735. [Google Scholar] [CrossRef] [PubMed]
- Khidr, L.; Wu, G.; Davila, A.; Procaccio, V.; Wallace, D.; Lee, W.H. Role of SUV3 helicase in maintaining mitochondrial homeostasis in human cells. J. Biol. Chem. 2008, 283, 27064–27073. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, R.J.; Obriot, H.; Paczkowska, A.; Jedrzejczak, R.; Dmochowska, A.; Bartnik, E.; Formstecher, P.; Polakowska, R.; Stepien, P.P. Down-regulation of human RNA/DNA helicase SUV3 induces apoptosis by a caspase- and AIF-dependent pathway. Biol. Cell 2007, 99, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, R.J.; Borowski, L.S.; Brzezniak, L.K.; Dmochowska, A.; Gewartowski, K.; Bartnik, E.; Stepien, P.P. Human mitochondrial RNA turnover caught in flagranti: Involvement of hSUV3p helicase in RNA surveillance. Nucleic Acids Res. 2010, 38, 279–298. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.; Mason, P.; Szczesny, R.J.; Maddukuri, L.; Dziwura, S.; Jedrzejczak, R.; Paul, E.; Wojcik, A.; Dybczynska, L.; Tudek, B.; et al. Interaction of human SUV3 RNA/DNA helicase with BLM helicase: Loss of the SUV3 gene results in mouse embryonic lethality. Mech. Ageing Dev. 2007, 128, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.R.; Wang, Y.; Schools, G.P.; Kochheiser, J.; Davies, K.J. Down-regulation of mammalian mitochondrial RNAs during oxidative stress. Free Radic. Biol. Med. 1997, 22, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Desler, C.; Marcker, M.L.; Singh, K.K.; Rasmussen, L.J. The importance of mitochondrial DNA in aging and cancer. J. Aging Res. 2011, 2011, 407536. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M. Aging and oxidative stress: A new look at old bone. IBMS BoneKEy 2010, 7, 340–352. [Google Scholar] [CrossRef]
- Siitonen, H.A.; Sotkasiira, J.; Biervliet, M.; Benmansour, A.; Capri, Y.; Cormier-Daire, V.; Crandall, B.; Hannula-Jouppi, K.; Hennekam, R.; Herzog, D.; et al. The mutation spectrum in RECQL4 diseases. Eur. J. Hum. Genet. 2009, 17, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Nawrocki, S.T.; Xu, R.H.; Dunner, K.; McConkey, D.J.; Wierda, W.G.; Keating, M.J.; Huang, P. Increased mitochondrial biogenesis in primary leukemia cells: The role of endogenous nitric oxide and impact on sensitivity to fludarabine. Leukemia 2004, 18, 1934–1940. [Google Scholar] [CrossRef] [PubMed]
- Lan, Q.; Lim, U.; Liu, C.S.; Weinstein, S.J.; Chanock, S.; Bonner, M.R.; Virtamo, J.; Albanes, D.; Rothman, N. A prospective study of mitochondrial DNA copy number and risk of non-Hodgkin lymphoma. Blood 2008, 112, 4247–4249. [Google Scholar] [CrossRef] [PubMed]
- DʼSouza, A.D.; Parikh, N.; Kaech, S.M.; Shadel, G.S. Convergence of multiple signaling pathways is required to coordinately up-regulate mtDNA and mitochondrial biogenesis during T cell activation. Mitochondrion 2007, 7, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.P.; Shim, S.M.; Nam, H.Y.; Baik, S.Y.; Kim, J.W.; Han, B.G. Copy number increase of 1p36.33 and mitochondrial genome amplification in Epstein-Barr virus-transformed lymphoblastoid cell lines. Cancer Genet. Cytogenet. 2007, 173, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Shapovalov, Y.; Hoffman, D.; Zuch, D.; de Mesy Bentley, K.L.; Eliseev, R.A. Mitochondrial dysfunction in cancer cells due to aberrant mitochondrial replication. J. Biol. Chem. 2011, 286, 22331–22338. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Dass, C.R.; Choong, P.F. Novel therapeutic strategy for osteosarcoma targeting osteoclast differentiation, bone-resorbing activity, and apoptosis pathway. Mol. Cancer Ther. 2008, 7, 3461–3469. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Mori, S.; Shigemoto, K.; Larsson, N.; Nakamura, T.; Kato, S.; Nakashima, T.; Takayanagi, H.; Tanaka, S. Maintenance of mitochondrial DNA copy number is essential for osteoclast survival. Arthritis Res. Ther. 2012, 14, 40. [Google Scholar] [CrossRef]
- Jeng, J.Y.; Yeh, T.S.; Lee, J.W.; Lin, S.H.; Fong, T.H.; Hsieh, R.H. Maintenance of mitochondrial DNA copy number and expression are essential for preservation of mitochondrial function and cell growth. J. Cell. Biochem. 2008, 103, 347–357. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, L.; Liu, Y. Borrowing Nuclear DNA Helicases to Protect Mitochondrial DNA. Int. J. Mol. Sci. 2015, 16, 10870-10887. https://doi.org/10.3390/ijms160510870
Ding L, Liu Y. Borrowing Nuclear DNA Helicases to Protect Mitochondrial DNA. International Journal of Molecular Sciences. 2015; 16(5):10870-10887. https://doi.org/10.3390/ijms160510870
Chicago/Turabian StyleDing, Lin, and Yilun Liu. 2015. "Borrowing Nuclear DNA Helicases to Protect Mitochondrial DNA" International Journal of Molecular Sciences 16, no. 5: 10870-10887. https://doi.org/10.3390/ijms160510870
APA StyleDing, L., & Liu, Y. (2015). Borrowing Nuclear DNA Helicases to Protect Mitochondrial DNA. International Journal of Molecular Sciences, 16(5), 10870-10887. https://doi.org/10.3390/ijms160510870