Abstract
In normal cells, mitochondria are the primary organelles that generate energy, which is critical for cellular metabolism. Mitochondrial dysfunction, caused by mitochondrial DNA (mtDNA) mutations or an abnormal mtDNA copy number, is linked to a range of human diseases, including Alzheimer’s disease, premature aging and cancer. mtDNA resides in the mitochondrial lumen, and its duplication requires the mtDNA replicative helicase, Twinkle. In addition to Twinkle, many DNA helicases, which are encoded by the nuclear genome and are crucial for nuclear genome integrity, are transported into the mitochondrion to also function in mtDNA replication and repair. To date, these helicases include RecQ-like helicase 4 (RECQ4), petite integration frequency 1 (PIF1), DNA replication helicase/nuclease 2 (DNA2) and suppressor of var1 3-like protein 1 (SUV3). Although the nuclear functions of some of these DNA helicases have been extensively studied, the regulation of their mitochondrial transport and the mechanisms by which they contribute to mtDNA synthesis and maintenance remain largely unknown. In this review, we attempt to summarize recent research progress on the role of mammalian DNA helicases in mitochondrial genome maintenance and the effects on mitochondria-associated diseases.
1. Introduction
The mitochondrion, once an autonomous free-living Proteobacterium, became a part of the eukaryotic cell through endosymbiosis approximately two billion years ago [1]. A symbiotic relationship was established, and now, mitochondria not only serve as the powerhouses of the cell by generating adenosine triphosphate (ATP) via oxidative phosphorylation, but also regulate cellular metabolism through synthesizing heme and steroids, supplying reactive oxygen species (ROS), establishing the membrane potential and controlling calcium and apoptotic signaling [2]. Human mitochondria are maternally inherited organelles, which reside in the cytoplasm. The mitochondrial architecture consists of an outer membrane, an inner membrane, an intermembrane space and the matrix or lumen (Figure 1). The mitochondrial number per cell differs from one cell type to another, and each mitochondrion contains multiple copies of the mitochondrial DNA (mtDNA), ranging from one to 15 copies per mitochondrion [3,4]. mtDNA copy number per cell also varies among different tissues due to the tissue-specific epigenetic regulation of the expression of mtDNA replication polymerase γ (Pol γ) [5]. The human mtDNA resides in the lumen and attaches to the inner membrane [6]. The mtDNA forms a small circle, which consists of 16,569 base pairs that encode two rRNA genes, 22 tRNA genes and 13 protein-encoding genes that produce parts of the electron transport chain and ATP Synthase complexes.
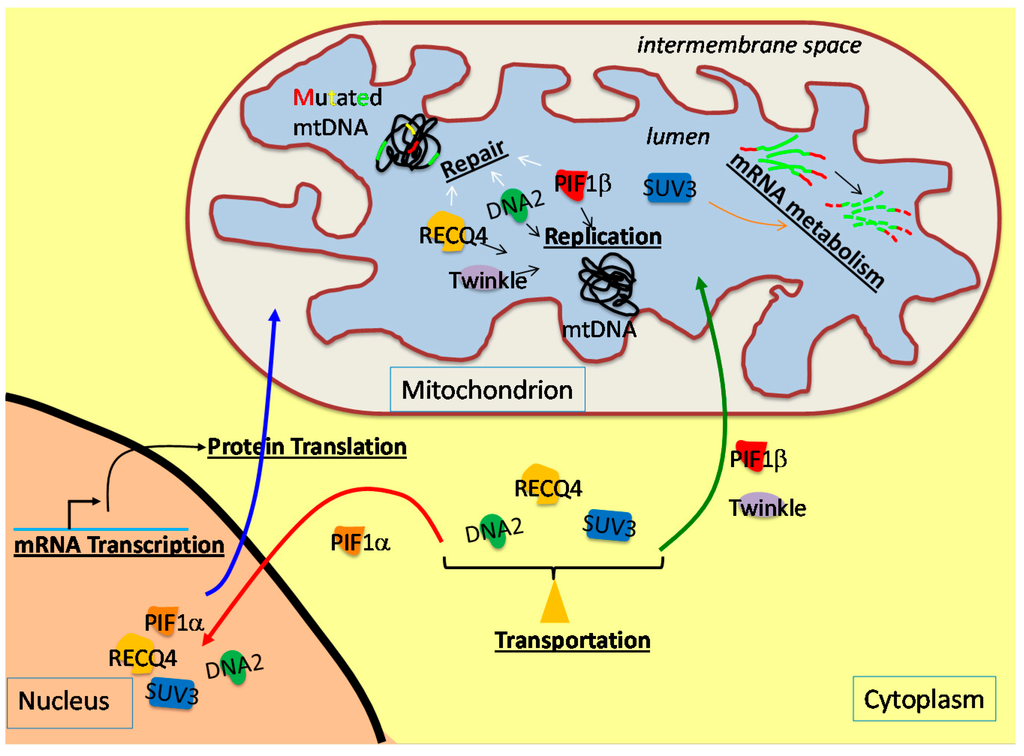
Figure 1.
Schematic diagram of the production and the cellular localization of the DNA helicases (Twinkle, purple; RecQ-like helicase 4 (RECQ4), yellow; DNA replication helicase/nuclease 2 (DNA2), green; petite integration frequency 1 (PIF1), red; suppressor of var1 3-like protein 1 (SUV3), blue) that function in the mitochondrion. These DNA helicases are encoded in the nuclear genome, produced in the cytoplasm and transported into the mitochondrial lumen. With the exception of Twinkle, other DNA helicases, including RECQ4, DNA2, PIF1 and SUV3, are transported into the mitochondrial lumen or nucleus depending on the molecular cue. In the mitochondrion, these helicases participate in DNA replication and repair, as well as mRNA metabolism, in order to maintain mtDNA stability.
mtDNA is thought to be duplicated through either strand-displacement replication or RNA incorporation throughout the lagging strand [7]. Interestingly, mtDNA sequences are highly polymorphic, even within an individual. This is due to the fact that somatic mutations in mtDNA, as a result of replication errors, ROS exposure and aging, make mtDNA sequences different from each other, even within the same cell (heteroplasmic), rather than genetically identical (homoplasmic). To safeguard a healthy population of mitochondria in a cell, mitochondria are constantly dividing (fission) and rejoining (fusion). However, should a pathogenic somatic mutation be introduced into the mtDNA genome, the entire mitochondrial population could be affected. Therefore, to maintain mtDNA stability, it is crucial to ensure faithful mtDNA synthesis. In addition, mitochondria employ several DNA repair pathways to restore DNA integrity in response to damage or replication errors [8]. Failure to do so causes mitochondrial morphological changes [9], which may lead to mitochondrial dysfunction, a phenomenon that has been linked to Alzheimer’s disease and premature aging [10]. Moreover, recent studies have shown that changes in mtDNA copy number are often associated with human cancers [11,12].
DNA synthesis and DNA repair are sophisticated processes that involve multi-protein complexes. Due to the small size of the mtDNA genome and the limited number of genes it encodes, mitochondria have adapted a mechanism to “borrow” enzymes encoded in the nuclear genome for many of its functions, including mtDNA synthesis and repair. For example, DNA helicases are ATPases that break the hydrogen bonds between DNA base pairs and transiently convert double-stranded DNA (dsDNA) into single-stranded DNA (ssDNA), the latter of which can serve as the template for DNA synthesis or allow the repair of damaged bases or nucleotides. These DNA helicases are transcribed in the nucleus, synthesized in the cytoplasm and imported into the mitochondrial compartment. Mitochondrial transport occurs primarily through either the presequence pathway or the carrier pathway. Both pathways involve interactions with the translocase of the outer membrane (TOM) and the translocase of the inner membrane (TIM) protein complexes, though the protein subunits are different for each pathway [13,14,15]. The presequence pathway targets the precursor protein to the lumen, where a mitochondrial targeting signal (MTS) located at the N-terminus of the precursor protein is then cleaved by a mitochondrial processing peptidase. The precursor proteins that also express hydrophobic sorting signals are either inserted into the inner membrane or released into the inter membrane space. The carrier pathway usually targets the mitochondrial proteins to the inner membrane, and these precursors have a non-cleavable internal targeting signal (ITS) and form complexes with cytosolic chaperones to prevent aggregation. Nonetheless, there are exceptions to these rules; some proteins, such as the tumor suppressor p53, can also be targeted to the mitochondrion via protein-protein interactions [16].
In mammalian cells, mtDNA replication is promoted by the replicative DNA helicase Twinkle, which is encoded by the C10orf2 gene in the nucleus. In addition to Twinkle, there are many DNA helicases that contribute to mammalian mtDNA integrity. Interestingly, unlike Twinkle, which is known to exclusively function in the mitochondrion, many of these DNA helicases not only are expressed from the nuclear genome, but also are involved in nuclear DNA replication and repair (Figure 1). This raises several questions. How do these DNA helicases balance their distribution and function in the nucleus and mitochondrion? What triggers the translocation of these helicases between different cellular compartments? How do Twinkle and other helicases collaborate in mtDNA replication and repair? In this review, we summarize recent findings on how these nuclear-encoding DNA helicases contribute to mtDNA integrity and associated diseases, and we will try to shine light on future studies in this active field.
2. Mitochondrial Replicative Helicase, Twinkle
Twinkle (for the T7 gp4-like protein with intramitochondrial nucleoid localization or PEO1) was first identified based on a sequence homology search as T7 gene 4 primase/helicase in 2001 [17]. Although Twinkle is conserved in many eukaryotes, such as the mouse, Drosophila and zebra fish, it has no orthologs in yeast [18]. It is possible that other yeast helicases compensate for its role in mtDNA replication. Twinkle is essential for embryonic development in mammalian systems, and it is known to unwind mtDNA for mtDNA synthesis by Pol γ [19]. Immunofluorescence microscopy has revealed that Twinkle proteins form punctate foci within mitochondria and colocalize with mitochondrial nucleoids [17], which are aggregates containing mtDNA and proteins that enact mitochondrial genome maintenance and transcription [20,21]. These foci resemble twinkling stars [17]. Human Twinkle, a 684 amino-acid (aa)-long polypeptide with a molecular weight of 77 kDa, oligomerizes to form a hexamer and exhibits 5'–3' helicase activity due to the conserved superfamily 4 (SF4) helicase domain located at its C-terminus (Figure 2) [22]. In addition to the conserved SF4 domain, Twinkle also contains a 42-aa MTS for mitochondrial targeting and a non-functional N-terminal primase-like domain that connects to the SF4 domain by a linker domain (Figure 2) [22,23]. The linker region is important for the hexamerization of Twinkle and its DNA helicase activity [24]. Recent studies have also found that Twinkle exhibits DNA annealing activity, indicating a possible involvement of Twinkle in recombination-mediated replication initiation or the fork regression pathway of DNA repair [25]. Interestingly, an alternatively-spliced product, Twinky, lacks part of the C-terminus, exists as monomers and has no enzymatic activity [23]. The function of Twinky remains unclear, as it cannot localize to the mitochondrial nucleoids [17] nor associate with Twinkle [23], despite the fact that Twinky contains the proposed MTS at the N-terminus (Figure 2). This suggests that the unique C-terminus of Twinkle may contain an additional sequence that is also important for its mitochondrial localization. Recombinant human Twinkle, combined with Pol γ purified from insect cells, is sufficient to form the minimal mammalian mtDNA replisome [26]. The hexameric Twinkle ring can efficiently bind to the single-stranded region of a closed circular DNA without a helicase loader and support DNA synthesis by Pol γ through the duplex region [26]. The helicase activity of Twinkle is stimulated by mitochondrial single-stranded DNA-binding protein (mtSSB) [22,27].
Given the essential role of Twinkle in mtDNA synthesis, mtDNA stability is greatly influenced by the Twinkle expression level in cells. For example, overexpression of the wild-type Twinkle is associated with increased mtDNA copy number in skeletal muscle in mice and reduced ROS-induced mtDNA mutations [28,29], whereas depletion of the Twinkle protein by small interfering RNA (siRNA) leads to a significant decrease in mtDNA copy number [30]. Furthermore, increasing evidence has linked a set of mutations, which change the stability and enzymatic activity of Twinkle [31], to a wide range of diseases [32], such as mitochondrial myopathy [33] and autosomal dominant progressive external ophthalmoplegia (adPEO) [34]. Individuals suffering from adPEO bear multiple deletions in their mitochondrial genome and exhibit multiple symptoms, including muscle weakening, hearing loss, nerve damage and Parkinsonism [35,36].
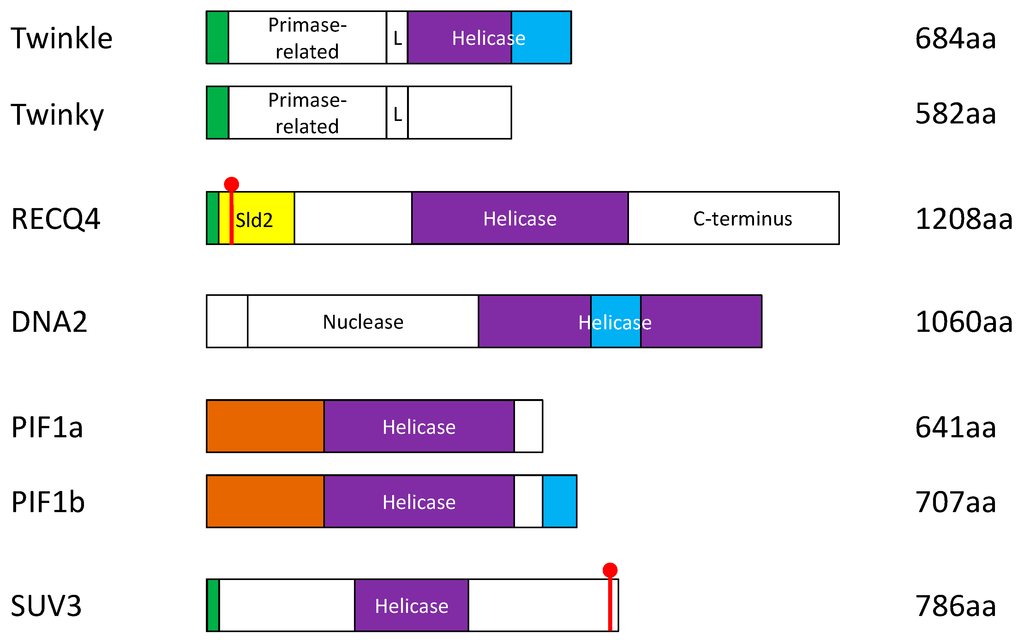
Figure 2.
Schematic diagram of the protein domains and alternatively-spliced variants of the human DNA helicases that have known functions in the mitochondrion. Green: mitochondrial targeting sequence (MTS). Red: nuclear localization signal (NLS). Purple: helicase domain. Blue: non-MTS sequence required for mitochondrial localization. Brown: arginine-rich region where potential NLSs reside. Yellow: unique sld2-like domain. L = linker region.
3. The Involvement of the Nuclear DNA Helicases
3.1. RecQ-Like Helicase 4 (RECQ4)
The gene that encodes RecQ-like helicase 4 (RECQ4) was first identified and cloned based on its limited sequence homology to the highly-conserved RECQ family of superfamily 2 (SF2) DNA helicases [37]. RECQ4 mutations were later identified in patients suffering from Rothmund-Thomson syndrome (RTS), Baller-Gerold syndrome and RAPADILINO (RAdial hypo-/aplasia, PAtellae hypo-/aplasia and cleft or highly arched PAlate, DIarrhea and DIslocated joints, LIttle size and LImb malformation, NOse slender and NOrmal intelligence) syndrome, with phenotypes ranging from premature aging to cancer predisposition [38]. In vitro, purified recombinant RECQ4 proteins exist as multimeric proteins and unwind DNA in a 3'–5' direction [39]. Interestingly, RECQ4 not only unwinds DNA, but also exhibits strong DNA annealing activity [40]. In addition to the conserved SF2 helicase domain, the vertebrate RECQ4 contains a unique Sld2-like N-terminus (Figure 2) that resembles the essential yeast DNA replication initiation factor Sld2 [41]. Researchers have shown that RECQ4 forms a chromatin-specific complex via this Sld2-like N-terminal domain with the MCM2-7 replicative helicase complex and participates in nuclear DNA replication initiation [42,43,44,45,46,47]. This function explains why recq4 knockout results in embryonic lethality in mice. Furthermore, the expression of a RECQ4 fragment containing only the Sld2-like N-terminal domain is sufficient to support embryonic development [48,49]. The C-terminus of RECQ4, which is highly conserved among vertebrates, contains a putative RecQ-C-terminal domain (RQC) [50]. Although this C-terminal domain is not required for unperturbed DNA replication, a recent study suggests that it is crucial for replication elongation when cells are exposed to ionizing radiation [51]. It has been demonstrated with other members of the RECQ family helicases that the RQC domain is important for the DNA unwinding activity [52]. Therefore, it is possible that the helicase activity of RECQ4 is involved in stabilizing or repairing the damaged replication forks. Because many disease-associated RECQ4 mutations disrupt the conserved C-terminal domain [38], understanding the potential function of RECQ4 in replication fork stability in response to ionizing radiation may provide important insight into the pathogenicity of these diseases.
In addition to affecting nuclear DNA replication, RECQ4 expression level also affects mtDNA copy number [53]. Consistent with this, RECQ4 also localizes to the mitochondrion [16,53,54,55,56], and the existence of a MTS within the first 20 aa has been proposed [16]. That said, whether RECQ4 is targeted to the mitochondrion via the conventional MTS remains to be validated. Given that RECQ4 interacts with the nuclear DNA replicative helicase complex and plays a critical role in nuclear DNA replication [42,43,44,45,46,47], it is possible that RECQ4 might have a similar role in mtDNA synthesis. Indeed, in a recent study from our laboratory, we reported a weak interaction between RECQ4 and the mitochondrial replicative helicase Twinkle that can be detected in human whole-cell extracts [56]. Perhaps most surprisingly, we found that this interaction between RECQ4 and Twinkle was significantly enhanced in human cells carrying the most common lymphoma-prone RECQ4 mutation: c.1390+2 delT. This mutation produces RECQ4 polypeptides lacking Ala420-Ala463 residues immediately upstream of the conserved helicase domain [56]. As a consequence, there is increased mtDNA synthesis, leading to an increase in the mtDNA copy number and mitochondrial dysfunction in these cells. Clearly, residues Ala420–Ala463, which are missing in this cancer-prone RECQ4 mutant, have an important inhibitory role in mtDNA synthesis, and we further elucidated how this regulation works. We found that residues Ala420–Ala463 of RECQ4 are required for the interaction with p32, and this interaction negatively regulates RECQ4 mitochondrial localization [56]. p32, which resides in both the mitochondrion and the nucleus [56,57], is involved in regulating mitochondrial innate immunity [58], energy production [57,59] and mitochondrial protein translocation [60]. Cells expressing RECQ4 mutants that lack these 44 aa show defective RECQ4-p32 interactions, increased RECQ4 mutant proteins in the mitochondrion and decreased nuclear RECQ4, suggesting that the excess of RECQ4 molecules in the mitochondrion likely results from increased nuclear-mitochondrial transport [56]. Therefore, this work presents a model for the mechanism used by cells to balance the distribution of RECQ4 in the nucleus and mitochondrion via direct protein-protein interaction.
Although mitochondrial localization of RECQ4 is restricted by p32, RECQ4 itself has also been suggested to function as a positive regulator of the mitochondrial transport of the p53 tumor suppressor via a direct protein–protein interaction [16]. Interestingly, mitochondrial localization of p53 can be blocked by the chaperone protein nucleophosmin (NPM) [61], which was found to also interact with RECQ4 in the nucleoplasm [56]. Although the domain of RECQ4 that interacts with NPM remains to be determined, it is tempting to speculate that NPM inhibits the mitochondrial transport of p53 via its interaction with RECQ4 in the nucleus. In summary, RECQ4 is a dynamic interacting protein, and its protein-protein interactions not only govern the rate of nuclear and mitochondrial DNA synthesis, but also regulate its cellular localization.
3.2. DNA Replication Helicase/Nuclease 2 (DNA2)
The DNA replication helicase/nuclease 2 (DNA2) gene was first isolated from a genetic screen in budding yeast [62], and the human DNA2 gene was later identified based on its sequence homology to the yeast counterpart [63]. Human DNA2, a 120-kDa polypeptide, has two independent functional domains: the N-terminal nuclease and the C-terminal helicase domain (Figure 2). DNA2 is highly conserved among the eukaryotes. As such, expression of either the human or Xenopus laevis DNA2 complements the temperature sensitivity of a DNA2 (DNA2-1) mutation in budding yeast [64]. However, the specificity of DNA2 enzymatic activity may vary across species due to the widely divergent sequences of the distal N-terminal regions [65,66]. DNA2 proteins purified from human cells and insect cells show that the nuclease domain has both 5'–3' and 3'–5' nuclease activities [65,67], whereas the helicase unwinds dsDNA that contains a 5' flap as a tail [67]. Data from yeast studies suggest that the 5'–3' helicase activity of DNA2 facilitates the production of a 5' flap structure, a substrate of DNA2 nuclease activity in vitro [68]. Nonetheless, the yeast helicase activity in vivo is dispensable for cell growth under normal conditions [69,70]. It remains to be determined if this is also the case in higher eukaryotes and if other DNA helicases can compensate for the DNA2 helicase activity.
In the nucleus, DNA2 interacts with proliferating cell nuclear antigen, also known as PCNA, a protein that is important for replication processivity and prevents the accumulation of DNA double-strand breaks (DSBs) during replication [71]. In addition, DNA2 interacts with the Fanconi anemia complementation group D2 (FANCD2) protein and functions in the FANCD2-dependent interstrand crosslink repair pathway [72]. Cells with depleted DNA2 show increased DSBs [71], internuclear chromatin bridges [73] and increased sensitivity to interstrand crosslinking agents due to a reduced homologous recombination frequency [72]. Furthermore, DNA2 participates in long-range DNA resection, in concert with the Werner syndrome ATP-dependent helicase (WRN) and the Bloom syndrome protein (BLM), in DSB repair [74,75,76]. DNA2 also stimulates BLM helicase activity [75]. Recently, DNA2 was also implicated in telomere maintenance based on its ability to cleave G-quadruplex DNA, and heterozygous DNA2 knockout mice were found to be prone to telomeric DNA damage and aneuploidy [77].
Although DNA2 can localize to the nucleus and play a role in nuclear DNA repair, immunofluorescence microscopy data suggest that the majority of the DNA2 molecules are found in the mitochondrion [73,78]. DNA2 does not contain a classical MTS/ITS, but its localization to the mitochondrion requires the sequence located within 734 and 829 aa [78]. It remains unclear how cells regulate the distribution of DNA2 in the mitochondrion and the nucleus in response to either the cell cycle or DNA damage. DNA2 interacts with and stimulates Pol γ in the mitochondrion and is thought to also function in concert with flap structure-specific endonuclease 1 (FEN1) to process 5'-flap intermediates and participate in repairing oxidative lesions in mtDNA by long-range base excision repair [78]. Indeed, DNA2 proteins colocalize with mtDNA nucleoids and Twinkle and through an unknown mechanism; this localization increases in cells carrying some of the adPEO-associated Twinkle mutations [73]. Interestingly, point mutations in the DNA2 gene itself have also been linked to adPEO, and these patients show progressive myopathy with mitochondrial dysfunction [79]. Importantly, one mutation located within the helicase domain altered the DNA unwinding efficiency [79], suggesting that the helicase activity of DNA2 has an important role in mtDNA maintenance in humans. Therefore, similar to Twinkle, DNA2 is important for maintaining healthy mitochondrial DNA and preventing related diseases.
3.3. Petite Integration Frequency 1 (PIF1)
PIF1, which stands for petite integration frequency 1, is conserved in both budding yeast and humans [80,81,82,83]. PIF1 is a member of the superfamily 1 (SF1) helicase family and has 5'–3' DNA unwinding activity (Figure 2) [84,85,86]. Similar to RECQ4 and DNA2, PIF1 localizes to both the nucleus and the mitochondrion [83]. However, unlike RECQ4 and DNA2, PIF1 mitochondrial localization in human cells is regulated by alternative splicing, which produces α and β isoforms [83]. Both the α and β isoforms contain the intact helicase domain and the N-terminus (Figure 2), which has arginine-rich nuclear localization signals [83] and is important for the interaction with ssDNA [84].
The PIF1 α isoform consists of 641 aa and has a short distal C-terminus. This isoform localizes to the nucleus [83], and PIF1 function in the nucleus has been extensively demonstrated [83,85,87,88]. The expression of PIF1 is cell cycle regulated, and the downregulation of PIF1 leads to cell cycle delay [81,83]. Both yeast and human PIF1 bind DNA and promote DNA replication through interaction with G-quadruplex DNA regions [86,87,89,90,91,92]. This activity is important for maintaining telomere integrity and for resolving stalled replication forks [85]. Reduction of PIF1α by siRNA knockdown decreases cancer cell survival, but has no impact on non-malignant cells [93], and this is likely due to its role in restarting stalled replication forks [85,94].
The PIF1 β isoform (707 aa) has a long distal C-terminus with a lipocalin motif (protein secretion signal; Figure 2). This C-terminal region results from alternative splicing and is not present in the α isoform. PIF1β is expected to have similar biochemical properties, compared to PIF1α, as they contain the same helicase domain. However, unlike the α isoform, the majority of this β isoform localizes to the mitochondrion, with some residual nuclear signal [83]. Evidence from yeast studies suggests that PIF1 may associate with mtDNA and mitochondrial inner membranes [95] and contribute to reducing DSBs in mtDNA [96]. Furthermore, it is required for repairing UV- and ethidium bromide-damaged mtDNA [80]. In addition, Twinkle, which cannot efficiently unwind G-quadruplex DNA [97], may rely on PIF1 helicase activity to remove G-quadruplexes, which could potentially lead to mtDNA deletions. Nonetheless, it is unknown how the distal C-terminus, which is unique to PIF1β, promotes its mitochondrial localization and how PIF1β protects mtDNA from DSBs. Interestingly, deletion of PIF1 rescued the lethal phenotype of DNA2 in budding yeast, suggesting that PIF1 and DNA2 may be involved in similar, but non-redundant pathways in the mitochondrion [98].
3.4. Suppressor of Var1 3-Like Protein 1 (SUV3)
SUV3, a member of the DExH-box helicase family, was first identified in budding yeast as the suppressor of var1 (the small subunit of mitochondrial ribosomal protein) [99], and the gene was later found to be conserved in humans [100]. SUV3 knockout mice are embryonic lethal, whereas heterozygous mice have shortened lifespan and develop tumors at multiple sites, due to a reduced mtDNA copy number and an elevated number of mtDNA mutations [101]. Reduced SUV3 expression was observed in human breast tumor samples [101]. Nonetheless, unlike RECQ4, PIF1 and DNA2 helicases, the effect of SUV3 deficiency on mtDNA copy number and stability is likely indirect. For example, in the mitochondrion, SUV3 forms a complex with polynucleotide phosphorylase (PNPase) to function in mtRNA degradation [102]. Indeed, analysis using purified recombinant human SUV3 proteins demonstrated that SUV3 is an active ATPase and capable of unwinding not only DNA, but also RNA in a 3'–5' direction [102,103,104]. This SUV3-PNPase complex transiently associates with the mitochondrial polyadenylation polymerase when the inorganic phosphate level is low in the mitochondrial lumen [105]. The three-component complex is capable of regulating the length of the RNA poly(A) tail. Consistent with this, siRNA knockdown leads to an increase in the amount of mtRNA with shorter poly(A) tails, a reduction in mtDNA copy number [106] and an increase in the rate of apoptosis [107]. In addition, expression of a mutant defective in the ATPase function leads to an abnormally high level of mtRNA, due to the slow mRNA turnover rate [108]. Although it remains unclear how a defect in mtRNA degradation contributes to mtDNA instability in SUV3-deficient cells, it is possible that the abnormal level of mtRNA imposes cellular stress, leading to overproduction of ROS and mtDNA damage.
Early studies suggest that SUV3 localizes to the lumen of the mitochondrion, presumably through cleavage of an MTS localized at the distal N-terminus (Figure 2) [103,107]. However, recent studies provide evidence that SUV3 also localizes to the nucleus with a potential nuclear localization signal located between residues 777 and 781 at the C-terminus [104,107]. In the nucleus, SUV3 interacts with nuclear DNA replication and repair factors, such as the RECQ helicases BLM and WRN [109], as well as replication protein A (RPA) and FEN1 [104]. Therefore, it is possible that, at least in humans, SUV3 is a key player in nuclear genome maintenance due to its participation in DNA damage repair, whereas it maintains mitochondrial genome integrity by participating in mtRNA metabolism. The reason why cells utilize an mtRNA helicase in nuclear DNA damage repair remains unknown. Interestingly, in mammalian cells, there is an increase in the degradation of mtRNA, but not cytoplasmic RNAs, to protect cells in response to oxidative stress [110]. It is possible that the involvement of SUV3 in nuclear DNA repair provides a mechanism for cells to “sense” oxidative DNA damage and induce mtRNA degradation. Therefore, identifying the molecular switch that balances the localization and the two distinct functions of SUV3 might reveal a novel crosstalk between the nucleus and the mitochondrion in response to DNA damage.
4. Conclusions
Given that mitochondria provide the vital ATP energy source needed by diverse cellular processes that support the development of an organism, it is not surprising that abnormal mtDNA copy number and mitochondrial dysfunction have been correlated with a decline in tissue maintenance and regeneration. Tissue degeneration may contribute to some of the symptoms, such as muscle weakening, hearing loss, nerve damage and Parkinsonism observed in the adPEO1 patients [35,36]. Growing evidence also suggests a close association between mitochondrial dysfunction and age-related bone diseases. For example, osteoporosis is a result of the loss of bone mass and is one of the common symptoms associated with aging [12,111]. Studies in mice indicate that increased apoptosis in osteoblasts, due to the accumulation of ROS generated by damaged mitochondria, is one of the main causes of bone loss [112]. Interestingly, RTS patients with RECQ4 mutations show abnormal bone development and osteoporosis at an early age [113]. Therefore, it is possible that these RTS-associated RECQ4 mutations lead to mitochondrial dysfunction and contribute to the premature aging phenotypes.
In addition to their association with tissue degeneration and developmental defects, mitochondria have recently gained attention for their potential use both as diagnostic tools and as therapeutic targets for cancer treatment [11]. Variations in mtDNA copy number are observed in many cancers and correlate with tumor aggressiveness and survival outcome. For example, mtDNA copy number is significantly elevated in various types of lymphoma, including Burkitt lymphoma and non-Hodgkin lymphoma [114,115,116,117]. In addition, highly invasive osteosarcoma cells contain enlarged mitochondria and larger amounts of mtDNA, and inhibiting replication of mtDNA in these cells also effectively slows down tumor growth [118,119,120]. Because mtDNA copy number correlates with cell growth [121], deregulated mtDNA synthesis could be a risk factor that contributes to cancer pathogenesis or that sustains cancer cell growth. Therefore, reducing aberrant mtDNA synthesis in cancer by targeting enzymes involved in mtDNA synthesis or mtDNA repair may be an effective strategy for controlling tumor progression. It would be of great interest for future studies to explore the possibility that the DNA helicases we have summarized here may be cancer drug targets or biomarkers for cancer diagnosis and prevention.
Acknowledgments
We thank Nancy Linford and Keely Walker for their comments and expert editing of this manuscript. Yilun Liu was supported by funding from the National Cancer Institute (R01 CA151245).
Author Contributions
Lin Ding and Yilun Liu wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kurland, C.G.; Andersson, S.G. Origin and evolution of the mitochondrial proteome. Microb. Mol. Biol. Rev. 2000, 64, 786–820. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Iborra, F.J.; Kimura, H.; Cook, P.R. The functional organization of mitochondrial genomes in human cells. BMC Biol. 2004, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Kuroiwa, T. Organization of multiple nucleoids and DNA molecules in mitochondria of a human cell. Exp. Cell Res. 1991, 196, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.D.; Mahmud, A.; McKenzie, M.; Trounce, I.A.; St John, J.C. Mitochondrial DNA copy number is regulated in a tissue specific manner by DNA methylation of the nuclear-encoded DNA polymerase gamma A. Nucleic Acids Res. 2012, 40, 10124–10138. [Google Scholar] [CrossRef] [PubMed]
- Albring, M.; Griffith, J.; Attardi, G. Association of a protein structure of probable membrane derivation with HeLa cell mitochondrial DNA near its origin of replication. Proc. Natl. Acad. Sci. USA 1977, 74, 1348–1352. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Reyes, A. Human mitochondrial DNA replication. Cold Spring Harb. Perspect. Biol. 2012, 4, a012971. [Google Scholar] [CrossRef] [PubMed]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.W.; Margineantu, D.H.; Capaldi, R.A.; Selker, J.M. Mitochondrial DNA depletion causes morphological changes in the mitochondrial reticulum of cultured human cells. FEBS Lett. 2000, 474, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondrial disease. Lancet 2006, 368, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Yu, M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci. 2011, 89, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Clay Montier, L.L.; Deng, J.J.; Bai, Y. Number matters: Control of mammalian mitochondrial DNA copy number. J. Genet. Genomics 2009, 36, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.; Bottinger, L.; Pfanner, N. Mitochondrial protein import: From transport pathways to an integrated network. Trends Biochem. Sci. 2012, 37, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Gebert, N.; Ryan, M.T.; Pfanner, N.; Wiedemann, N.; Stojanovski, D. Mitochondrial protein import machineries and lipids: A functional connection. Biochim. Biophys. Acta 2011, 1808, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Kumari, J.; Mudgal, R.; Modi, P.; Gupta, S.; Futami, K.; Goto, H.; Lindor, N.M.; Furuichi, Y.; Mohanty, D.; et al. RECQL4 is essential for the transport of p53 to mitochondria in normal human cells in the absence of exogenous stress. J. Cell Sci. 2012, 125, 2509–2522. [Google Scholar] [CrossRef] [PubMed]
- Spelbrink, J.N.; Li, F.Y.; Tiranti, V.; Nikali, K.; Yuan, Q.P.; Tariq, M.; Wanrooij, S.; Garrido, N.; Comi, G.; Morandi, L.; et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 2001, 28, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial inheritance in yeast. Biochim. Biophys. Acta 2014, 1837, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, D.; Matic, S.; Kuhl, I.; Ruzzenente, B.; Freyer, C.; Jemt, E.; Park, C.B.; Falkenberg, M.; Larsson, N.G. TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D-loop strands and complete mtDNA replication. Hum. Mol. Genet. 2013, 22, 1983–1993. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.; Bravo, L.; Garcia, I.; Gaytan, N.; Herrera, A.; Maldonado, A.; Quintanilla, B. The mitochondrial nucleoid: Integrating mitochondrial DNA into cellular homeostasis. Cold Spring Harb. Perspect. Biol. 2013, 5, a011080. [Google Scholar] [CrossRef] [PubMed]
- Bogenhagen, D.F.; Rousseau, D.; Burke, S. The layered structure of human mitochondrial DNA nucleoids. J. Biol. Chem. 2008, 283, 3665–3675. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, J.A.; Gaspari, M.; Falkenberg, M. TWINKLE has 5'→3' DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J. Biol. Chem. 2003, 278, 48627–48632. [Google Scholar] [CrossRef] [PubMed]
- Farge, G.; Holmlund, T.; Khvorostova, J.; Rofougaran, R.; Hofer, A.; Falkenberg, M. The N-terminal domain of TWINKLE contributes to single-stranded DNA binding and DNA helicase activities. Nucleic Acids Res. 2008, 36, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, J.A.; Pande, V.; Holmlund, T.; Farge, G.; Pham, X.H.; Nilsson, L.; Falkenberg, M. Structure–function defects of the TWINKLE linker region in progressive external ophthalmoplegia. J. Mol. Biol. 2008, 377, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Nandakumar, D.; Tang, G.Q.; Patel, S.S. Human mitochondrial DNA helicase TWINKLE is both an unwinding and annealing helicase. J. Biol. Chem. 2012, 287, 14545–14556. [Google Scholar] [CrossRef] [PubMed]
- Jemt, E.; Farge, G.; Backstrom, S.; Holmlund, T.; Gustafsson, C.M.; Falkenberg, M. The mitochondrial DNA helicase TWINKLE can assemble on a closed circular template and support initiation of DNA synthesis. Nucleic Acids Res. 2011, 39, 9238–9249. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, J.A.; Pham, X.H.; Pellegrini, M.; Falkenberg, M. Reconstitution of a minimal mtDNA replisome in vitro. EMBO J. 2004, 23, 2423–2429. [Google Scholar] [CrossRef] [PubMed]
- Ylikallio, E.; Tyynismaa, H.; Tsutsui, H.; Ide, T.; Suomalainen, A. High mitochondrial DNA copy number has detrimental effects in mice. Hum. Mol. Genet. 2010, 19, 2695–2705. [Google Scholar] [CrossRef] [PubMed]
- Pohjoismaki, J.L.; Williams, S.L.; Boettger, T.; Goffart, S.; Kim, J.; Suomalainen, A.; Moraes, C.T.; Braun, T. Over-expression of Twinkle-helicase protects cardiomyocytes from genotoxic stress caused by reactive oxygen species. Proc. Natl. Acad. Sci. USA 2013, 110, 19408–19013. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Sembongi, H.; Bokori-Brown, M.; Granycome, C.; Ashley, N.; Poulton, J.; Jalanko, A.; Spelbrink, J.N.; Holt, I.J.; Suomalainen, A. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum. Mol. Genet. 2004, 13, 3219–3227. [Google Scholar] [CrossRef] [PubMed]
- Longley, M.J.; Humble, M.M.; Sharief, F.S.; Copeland, W.C. Disease variants of the human mitochondrial DNA helicase encoded by C10orf2 differentially alter protein stability, nucleotide hydrolysis, and helicase activity. J. Biol. Chem. 2010, 285, 29690–29702. [Google Scholar] [CrossRef] [PubMed]
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and disease. J. Pathol. 2012, 226, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Arenas, J.; Briem, E.; Dahl, H.; Hutchison, W.; Lewis, S.; Martin, M.A.; Spelbrink, H.; Tiranti, V.; Jacobs, H.; Zeviani, M. The V368i mutation in Twinkle does not segregate with adPEO. Ann. Neurol. 2003, 53, 278. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Y.; Tariq, M.; Croxen, R.; Morten, K.; Squier, W.; Newsom-Davis, J.; Beeson, D.; Larsson, C. Mapping of autosomal dominant progressive external ophthalmoplegia to a 7-cM critical region on 10q24. Neurology 1999, 53, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Servidei, S.; Gellera, C.; Bertini, E.; DiMauro, S.; DiDonato, S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature 1989, 339, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Moslemi, A.R.; Melberg, A.; Holme, E.; Oldfors, A. Autosomal dominant progressive external ophthalmoplegia: Distribution of multiple mitochondrial DNA deletions. Neurology 1999, 53, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Kitao, S.; Ohsugi, I.; Ichikawa, K.; Goto, M.; Furuichi, Y.; Shimamoto, A. Cloning of two new human helicase genes of the RecQ family: Biological significance of multiple species in higher eukaryotes. Genomics 1998, 54, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Rothmund-Thomson syndrome helicase, RECQ4: On the crossroad between DNA replication and repair. DNA Repair. (Amst.) 2010, 9, 325–330. [Google Scholar] [CrossRef]
- Suzuki, T.; Kohno, T.; Ishimi, Y. DNA helicase activity in purified human RECQL4 protein. J. Biochem. 2009, 146, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, Y. Dual DNA unwinding activities of the Rothmund-Thomson syndrome protein, RECQ4. EMBO J. 2009, 28, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, Y.; Masumoto, H.; Sugino, A.; Araki, H. Sld2, which interacts with Dpb11 in Saccharomyces cerevisiae, is required for chromosomal DNA replication. Mol. Cell. Biol. 1998, 18, 6102–6109. [Google Scholar] [PubMed]
- Im, J.S.; Ki, S.H.; Farina, A.; Jung, D.S.; Hurwitz, J.; Lee, J.K. Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 15628–15632. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Rochette, P.J.; Feyissa, E.A.; Su, T.V.; Liu, Y. MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO J. 2009, 28, 3005–3014. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, S.; Mendoza-Maldonado, R.; Tissino, E.; Sidorova, J.M.; Yin, J.; Wang, W.; Monnat, R.J., Jr.; Falaschi, A.; Vindigni, A. Human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Mol. Cell. Biol. 2010, 30, 1382–1396. [Google Scholar] [CrossRef] [PubMed]
- Im, J.S.; Park, S.Y.; Cho, W.H.; Bae, S.H.; Hurwitz, J.; Lee, J.K. RecQL4 is required for the association of Mcm10 and Ctf4 with replication origins in human cells. Cell Cycle 2015, 14, 1001–1009. [Google Scholar] [PubMed]
- Sangrithi, M.N.; Bernal, J.A.; Madine, M.; Philpott, A.; Lee, J.; Dunphy, W.G.; Venkitaraman, A.R. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell 2005, 121, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Kumano, M.; Kubota, Y.; Hashimoto, Y.; Takisawa, H. The N-terminal noncatalytic region of Xenopus RecQ4 is required for chromatin binding of DNA polymerase α in the initiation of DNA replication. Mol. Cell. Biol. 2006, 26, 4843–4852. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Noda, T.; Furuichi, Y. Preparation of the gene targeted knockout mice for human premature aging diseases, Werner syndrome, and Rothmund-Thomson syndrome caused by the mutation of DNA helicases. Nihon Yakurigaku Zasshi 2002, 119, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.B.; Hodges, C.A.; Barnes, E.; Vogel, H.; Hassold, T.J.; Luo, G. Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund-Thomson syndrome. Hum. Mol. Genet. 2005, 14, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Marino, F.; Vindigni, A.; Onesti, S. Bioinformatic analysis of RecQ4 helicases reveals the presence of a RQC domain and a Zn knuckle. Biophys. Chem. 2013, 177, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Kohzaki, M.; Chiourea, M.; Versini, G.; Adachi, N.; Takeda, S.; Gagos, S.; Halazonetis, T.D. The helicase domain and C-terminus of human RecQL4 facilitate replication elongation on DNA templates damaged by ionizing radiation. Carcinogenesis 2012, 33, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Lucic, B.; Zhang, Y.; King, O.; Mendoza-Maldonado, R.; Berti, M.; Niesen, F.H.; Burgess-Brown, N.A.; Pike, A.C.; Cooper, C.D.; Gileadi, O.; et al. A prominent beta-hairpin structure in the winged-helix domain of RECQ1 is required for DNA unwinding and oligomer formation. Nucleic Acids Res. 2011, 39, 1703–1717. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.; Nie, L.; Peng, Z.; Yang, Q.; Yang, K.; Tao, J.; Mi, Y.; Fang, X.; Balajee, A.S.; Zhao, Y. RecQL4 cytoplasmic localization: Implications in mitochondrial DNA oxidative damage repair. Int. J. Biochem. Cell Biol. 2012, 44, 1942–1951. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.L.; Rossi, M.L.; Canugovi, C.; Tian, J.; Sykora, P.; Ramamoorthy, M.; Wang, Z.M.; Singh, D.K.; Akbari, M.; Kasiviswanathan, R.; et al. RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell 2012, 11, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; De, S.; Srivastava, V.; Hussain, M.; Kumari, J.; Muniyappa, K.; Sengupta, S. RECQL4 and p53 potentiate the activity of polymerase gamma and maintain the integrity of the human mitochondrial genome. Carcinogenesis 2014, 35, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Xu, X.; Alontaga, A.Y.; Chen, Y.; Liu, Y. Impaired p32 regulation caused by the lymphoma-prone RECQ4 mutation drives mitochondrial dysfunction. Cell Rep. 2014, 7, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Muta, T.; Kang, D.; Kitajima, S.; Fujiwara, T.; Hamasaki, N. p32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. J. Biol. Chem. 1997, 272, 24363–24370. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Fogal, V.; Richardson, A.D.; Karmali, P.P.; Scheffler, I.E.; Smith, J.W.; Ruoslahti, E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol. Cell. Biol. 2010, 30, 1303–1318. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Zhang, Y. Mitochondrial p32 is a critical mediator of ARF-induced apoptosis. Cancer Cell 2008, 13, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.K.; St Clair, D.K. Nucleophosmin blocks mitochondrial localization of p53 and apoptosis. J. Biol. Chem. 2009, 284, 16409–16418. [Google Scholar] [CrossRef] [PubMed]
- Dumas, L.B.; Lussky, J.P.; McFarland, E.J.; Shampay, J. New temperature-sensitive mutants of Saccharomyces cerevisiae affecting DNA replication. Mol. Gen. Genet. 1982, 187, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Eki, T.; Okumura, K.; Shiratori, A.; Abe, M.; Nogami, M.; Taguchi, H.; Shibata, T.; Murakami, Y.; Hanaoka, F. Assignment of the closest human homologue (DNA2L:KIAA0083) of the yeast DNA2 helicase gene to chromosome band 10q21.3-q22.1. Genomics 1996, 37, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Imamura, O.; Campbell, J.L. The human Bloom syndrome gene suppresses the DNA replication and repair defects of yeast DNA2 mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 8193–8198. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.D.; Ryu, G.H.; Kim, D.H.; Hurwitz, J.; Seo, Y.S. Isolation of human DNA2 endonuclease and characterization of its enzymatic properties. Nucleic Acids Res. 2006, 34, 1854–1864. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lee, M.; Kang, H.J.; Kim, D.H.; Kang, Y.H.; Bae, S.H.; Seo, Y.S. The N-terminal 45-kDa domain of DNA2 endonuclease/helicase targets the enzyme to secondary structure DNA. J. Biol. Chem. 2013, 288, 9468–9481. [Google Scholar] [CrossRef] [PubMed]
- Masuda-Sasa, T.; Imamura, O.; Campbell, J.L. Biochemical analysis of human DNA2. Nucleic Acids Res. 2006, 34, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Kim, D.W.; Kim, J.; Kim, J.H.; Kim, D.H.; Kim, H.D.; Kang, H.Y.; Seo, Y.S. Coupling of DNA helicase and endonuclease activities of yeast DNA2 facilitates Okazaki fragment processing. J. Biol. Chem. 2002, 277, 26632–26641. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Sun, L.; Shen, F.; Chen, Y.; Hua, Y.; Liu, Y.; Zhang, M.; Hu, Y.; Wang, Q.; Xu, W.; et al. The intra-S phase checkpoint targets DNA2 to prevent stalled replication forks from reversing. Cell 2012, 149, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Formosa, T.; Nittis, T. DNA2 mutants reveal interactions with DNA polymerase α and Ctf4, a Pol α accessory factor, and show that full DNA2 helicase activity is not essential for growth. Genetics 1999, 151, 1459–1470. [Google Scholar] [PubMed]
- Peng, G.; Dai, H.; Zhang, W.; Hsieh, H.J.; Pan, M.R.; Park, Y.Y.; Tsai, R.Y.; Bedrosian, I.; Lee, J.S.; Ira, G.; et al. Human nuclease/helicase DNA2 alleviates replication stress by promoting DNA end resection. Cancer Res. 2012, 72, 2802–2813. [Google Scholar] [CrossRef] [PubMed]
- Karanja, K.K.; Cox, S.W.; Duxin, J.P.; Stewart, S.A.; Campbell, J.L. DNA2 and EXO1 in replication-coupled, homology-directed repair and in the interplay between HDR and the FA/BRCA network. Cell Cycle 2012, 11, 3983–3996. [Google Scholar] [CrossRef] [PubMed]
- Duxin, J.P.; Dao, B.; Martinsson, P.; Rajala, N.; Guittat, L.; Campbell, J.L.; Spelbrink, J.N.; Stewart, S.A. Human DNA2 is a nuclear and mitochondrial DNA maintenance protein. Mol. Cell. Biol. 2009, 29, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Chiba, T.; Xue, X.; Niu, H.; Sung, P. Multifaceted role of the Topo IIIα-RMI1-RMI2 complex and DNA2 in the BLM-dependent pathway of DNA break end resection. Nucleic Acids Res. 2014, 42, 11083–11091. [Google Scholar] [CrossRef] [PubMed]
- Sturzenegger, A.; Burdova, K.; Kanagaraj, R.; Levikova, M.; Pinto, C.; Cejka, P.; Janscak, P. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J. Biol. Chem. 2014, 289, 27314–27326. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Sampathi, S.; Dai, H.; Liu, C.; Zhou, M.; Hu, J.; Huang, Q.; Campbell, J.; Shin-Ya, K.; Zheng, L.; et al. Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 2013, 32, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhou, M.; Guo, Z.; Lu, H.; Qian, L.; Dai, H.; Qiu, J.; Yakubovskaya, E.; Bogenhagen, D.F.; Demple, B.; et al. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol. Cell 2008, 32, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, D.; Di Fonzo, A.; Lin, W.; Bordoni, A.; Liu, C.; Fassone, E.; Pagliarani, S.; Rizzuti, M.; Zheng, L.; Filosto, M.; et al. Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am. J. Hum. Genet. 2013, 92, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Foury, F.; Kolodynski, J. PIF mutation blocks recombination between mitochondrial ρ+ and ρ− genomes having tandemly arrayed repeat units in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1983, 80, 5345–5349. [Google Scholar] [CrossRef] [PubMed]
- Mateyak, M.K.; Zakian, V.A. Human PIF helicase is cell cycle regulated and associates with telomerase. Cell Cycle 2006, 5, 2796–2804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.H.; Zhou, B.; Huang, Y.; Xu, L.X.; Zhou, J.Q. The human PIF1 helicase, a potential Escherichia coli RecD homologue, inhibits telomerase activity. Nucleic Acids Res. 2006, 34, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Futami, K.; Shimamoto, A.; Furuichi, Y. Mitochondrial and nuclear localization of human PIF1 helicase. Biol. Pharm. Bull. 2007, 30, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Masuda, Y.; Kamiya, K. Biochemical analysis of human PIF1 helicase and functions of its N-terminal domain. Nucleic Acids Res. 2008, 36, 6295–6308. [Google Scholar] [CrossRef] [PubMed]
- George, T.; Wen, Q.; Griffiths, R.; Ganesh, A.; Meuth, M.; Sanders, C.M. Human PIF1 helicase unwinds synthetic DNA structures resembling stalled DNA replication forks. Nucleic Acids Res. 2009, 37, 6491–6502. [Google Scholar] [CrossRef] [PubMed]
- Sanders, C.M. Human PIF1 helicase is a G-quadruplex DNA-binding protein with G-quadruplex DNA-unwinding activity. Biochem. J. 2010, 430, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Sabouri, N.; Zakian, V.A. Unwinding the functions of the PIF1 family helicases. DNA Repair (Amst.) 2010, 9, 237–249. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, J.; Li, S.; Kamiya, K.; Chen, X.; Zhou, P. Determination of the biochemical properties of full-length human PIF1 ATPase. Prion 2013, 7, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.L.; Liu, N.N.; Yang, Y.T.; Li, H.H.; Li, M.; Dou, S.X.; Xi, X. G-quadruplexes significantly stimulate PIF1 helicase-catalyzed duplex DNA unwinding. J. Biol. Chem. 2015, 290, 7722–7735. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.M.; Wu, W.Q.; Duan, X.L.; Liu, N.N.; Li, H.H.; Fu, J.; Dou, S.X.; Li, M.; Xi, X.G. Molecular mechanism of G-quadruplex unwinding helicase: Sequential and repetitive unfolding of G-quadruplex by PIF1 helicase. Biochem. J. 2015, 466, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zhang, J.; Bochman, M.L.; Zakian, V.A.; Ha, T. Periodic DNA patrolling underlies diverse functions of PIF1 on R-loops and G-rich DNA. Elife 2014, 3, e02190. [Google Scholar] [PubMed]
- Byrd, A.K.; Raney, K.D. A parallel quadruplex DNA is bound. Tightly but unfolded slowly by PIF1 helicase. J. Biol. Chem. 2015, 290, 6482–6494. [Google Scholar] [CrossRef] [PubMed]
- Gagou, M.E.; Ganesh, A.; Thompson, R.; Phear, G.; Sanders, C.; Meuth, M. Suppression of apoptosis by PIF1 helicase in human tumor cells. Cancer Res. 2011, 71, 4998–5008. [Google Scholar] [CrossRef] [PubMed]
- Gagou, M.E.; Ganesh, A.; Phear, G.; Robinson, D.; Petermann, E.; Cox, A.; Meuth, M. Human PIF1 helicase supports DNA replication and cell growth under oncogenic-stress. Oncotarget 2014, 5, 11381–11398. [Google Scholar] [PubMed]
- Cheng, X.; Ivessa, A.S. Association of the yeast DNA helicase PIF1p with mitochondrial membranes and mitochondrial DNA. Eur. J. Cell Biol. 2010, 89, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Dunaway, S.; Ivessa, A.S. The role of Pif1p, a DNA helicase in Saccharomyces cerevisiae, in maintaining mitochondrial DNA. Mitochondrion 2007, 7, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Sommers, J.A.; Zhou, J.; Kaplan, D.L.; Spelbrink, J.N.; Mergny, J.L.; Brosh, R.M., Jr. DNA sequences proximal to human mitochondrial DNA deletion breakpoints prevalent in human disease form G-quadruplexes, a class of DNA structures inefficiently unwound by the mitochondrial replicative Twinkle helicase. J. Biol. Chem. 2014, 289, 29975–29993. [Google Scholar] [CrossRef] [PubMed]
- Budd, M.E.; Reis, C.C.; Smith, S.; Myung, K.; Campbell, J.L. Evidence suggesting that PIF1 helicase functions in DNA replication with the DNA2 helicase/nuclease and DNA polymerase delta. Mol. Cell. Biol. 2006, 26, 2490–2500. [Google Scholar] [CrossRef] [PubMed]
- Butow, R.A.; Zhu, H.; Perlman, P.; Conrad-Webb, H. The role of a conserved dodecamer sequence in yeast mitochondrial gene expression. Genome 1989, 31, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Dmochowska, A.; Kalita, K.; Krawczyk, M.; Golik, P.; Mroczek, K.; Lazowska, J.; Stepien, P.P.; Bartnik, E. A human putative SUV3-like RNA helicase is conserved between Rhodobacter and all eukaryotes. Acta Biochim. Pol. 1999, 46, 155–162. [Google Scholar] [PubMed]
- Chen, P.L.; Chen, C.F.; Chen, Y.; Guo, X.E.; Huang, C.K.; Shew, J.Y.; Reddick, R.L.; Wallace, D.C.; Lee, W.H. Mitochondrial genome instability resulting from SUV3 haploinsufficiency leads to tumorigenesis and shortened lifespan. Oncogene 2013, 32, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Shu, Z.; Lieser, S.A.; Chen, P.L.; Lee, W.H. Human mitochondrial SUV3 and polynucleotide phosphorylase form a 330-kDa heteropentamer to cooperatively degrade double-stranded RNA with a 3'-to-5' directionality. J. Biol. Chem. 2009, 284, 20812–20821. [Google Scholar] [CrossRef] [PubMed]
- Minczuk, M.; Piwowarski, J.; Papworth, M.A.; Awiszus, K.; Schalinski, S.; Dziembowski, A.; Dmochowska, A.; Bartnik, E.; Tokatlidis, K.; Stepien, P.P.; et al. Localisation of the human hSUV3p helicase in the mitochondrial matrix and its preferential unwinding of dsDNA. Nucleic Acids Res. 2002, 30, 5074–5086. [Google Scholar] [CrossRef] [PubMed]
- Veno, S.T.; Kulikowicz, T.; Pestana, C.; Stepien, P.P.; Stevnsner, T.; Bohr, V.A. The human SUV3 helicase interacts with replication protein A and flap endonuclease 1 in the nucleus. Biochem. J. 2011, 440, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Guo, X.E.; Modrek, A.S.; Chen, C.F.; Chen, P.L.; Lee, W.H. Helicase SUV3, polynucleotide phosphorylase, and mitochondrial polyadenylation polymerase form a transient complex to modulate mitochondrial mRNA polyadenylated tail lengths in response to energetic changes. J. Biol. Chem. 2014, 289, 16727–16735. [Google Scholar] [CrossRef] [PubMed]
- Khidr, L.; Wu, G.; Davila, A.; Procaccio, V.; Wallace, D.; Lee, W.H. Role of SUV3 helicase in maintaining mitochondrial homeostasis in human cells. J. Biol. Chem. 2008, 283, 27064–27073. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, R.J.; Obriot, H.; Paczkowska, A.; Jedrzejczak, R.; Dmochowska, A.; Bartnik, E.; Formstecher, P.; Polakowska, R.; Stepien, P.P. Down-regulation of human RNA/DNA helicase SUV3 induces apoptosis by a caspase- and AIF-dependent pathway. Biol. Cell 2007, 99, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, R.J.; Borowski, L.S.; Brzezniak, L.K.; Dmochowska, A.; Gewartowski, K.; Bartnik, E.; Stepien, P.P. Human mitochondrial RNA turnover caught in flagranti: Involvement of hSUV3p helicase in RNA surveillance. Nucleic Acids Res. 2010, 38, 279–298. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.; Mason, P.; Szczesny, R.J.; Maddukuri, L.; Dziwura, S.; Jedrzejczak, R.; Paul, E.; Wojcik, A.; Dybczynska, L.; Tudek, B.; et al. Interaction of human SUV3 RNA/DNA helicase with BLM helicase: Loss of the SUV3 gene results in mouse embryonic lethality. Mech. Ageing Dev. 2007, 128, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.R.; Wang, Y.; Schools, G.P.; Kochheiser, J.; Davies, K.J. Down-regulation of mammalian mitochondrial RNAs during oxidative stress. Free Radic. Biol. Med. 1997, 22, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Desler, C.; Marcker, M.L.; Singh, K.K.; Rasmussen, L.J. The importance of mitochondrial DNA in aging and cancer. J. Aging Res. 2011, 2011, 407536. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M. Aging and oxidative stress: A new look at old bone. IBMS BoneKEy 2010, 7, 340–352. [Google Scholar] [CrossRef]
- Siitonen, H.A.; Sotkasiira, J.; Biervliet, M.; Benmansour, A.; Capri, Y.; Cormier-Daire, V.; Crandall, B.; Hannula-Jouppi, K.; Hennekam, R.; Herzog, D.; et al. The mutation spectrum in RECQL4 diseases. Eur. J. Hum. Genet. 2009, 17, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Nawrocki, S.T.; Xu, R.H.; Dunner, K.; McConkey, D.J.; Wierda, W.G.; Keating, M.J.; Huang, P. Increased mitochondrial biogenesis in primary leukemia cells: The role of endogenous nitric oxide and impact on sensitivity to fludarabine. Leukemia 2004, 18, 1934–1940. [Google Scholar] [CrossRef] [PubMed]
- Lan, Q.; Lim, U.; Liu, C.S.; Weinstein, S.J.; Chanock, S.; Bonner, M.R.; Virtamo, J.; Albanes, D.; Rothman, N. A prospective study of mitochondrial DNA copy number and risk of non-Hodgkin lymphoma. Blood 2008, 112, 4247–4249. [Google Scholar] [CrossRef] [PubMed]
- DʼSouza, A.D.; Parikh, N.; Kaech, S.M.; Shadel, G.S. Convergence of multiple signaling pathways is required to coordinately up-regulate mtDNA and mitochondrial biogenesis during T cell activation. Mitochondrion 2007, 7, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.P.; Shim, S.M.; Nam, H.Y.; Baik, S.Y.; Kim, J.W.; Han, B.G. Copy number increase of 1p36.33 and mitochondrial genome amplification in Epstein-Barr virus-transformed lymphoblastoid cell lines. Cancer Genet. Cytogenet. 2007, 173, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Shapovalov, Y.; Hoffman, D.; Zuch, D.; de Mesy Bentley, K.L.; Eliseev, R.A. Mitochondrial dysfunction in cancer cells due to aberrant mitochondrial replication. J. Biol. Chem. 2011, 286, 22331–22338. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Dass, C.R.; Choong, P.F. Novel therapeutic strategy for osteosarcoma targeting osteoclast differentiation, bone-resorbing activity, and apoptosis pathway. Mol. Cancer Ther. 2008, 7, 3461–3469. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Mori, S.; Shigemoto, K.; Larsson, N.; Nakamura, T.; Kato, S.; Nakashima, T.; Takayanagi, H.; Tanaka, S. Maintenance of mitochondrial DNA copy number is essential for osteoclast survival. Arthritis Res. Ther. 2012, 14, 40. [Google Scholar] [CrossRef]
- Jeng, J.Y.; Yeh, T.S.; Lee, J.W.; Lin, S.H.; Fong, T.H.; Hsieh, R.H. Maintenance of mitochondrial DNA copy number and expression are essential for preservation of mitochondrial function and cell growth. J. Cell. Biochem. 2008, 103, 347–357. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).