
Frequent Epigenetic Suppression of Tumor Suppressor Gene Glutathione Peroxidase 3 by Promoter Hypermethylation and Its Clinical Implication in Clear Cell Renal Cell Carcinoma

Abstract
:1. Introduction
2. Results
2.1. Methylation of the GPX3 Promoter Correlates with Its Downregulation in ccRCC Cell Lines

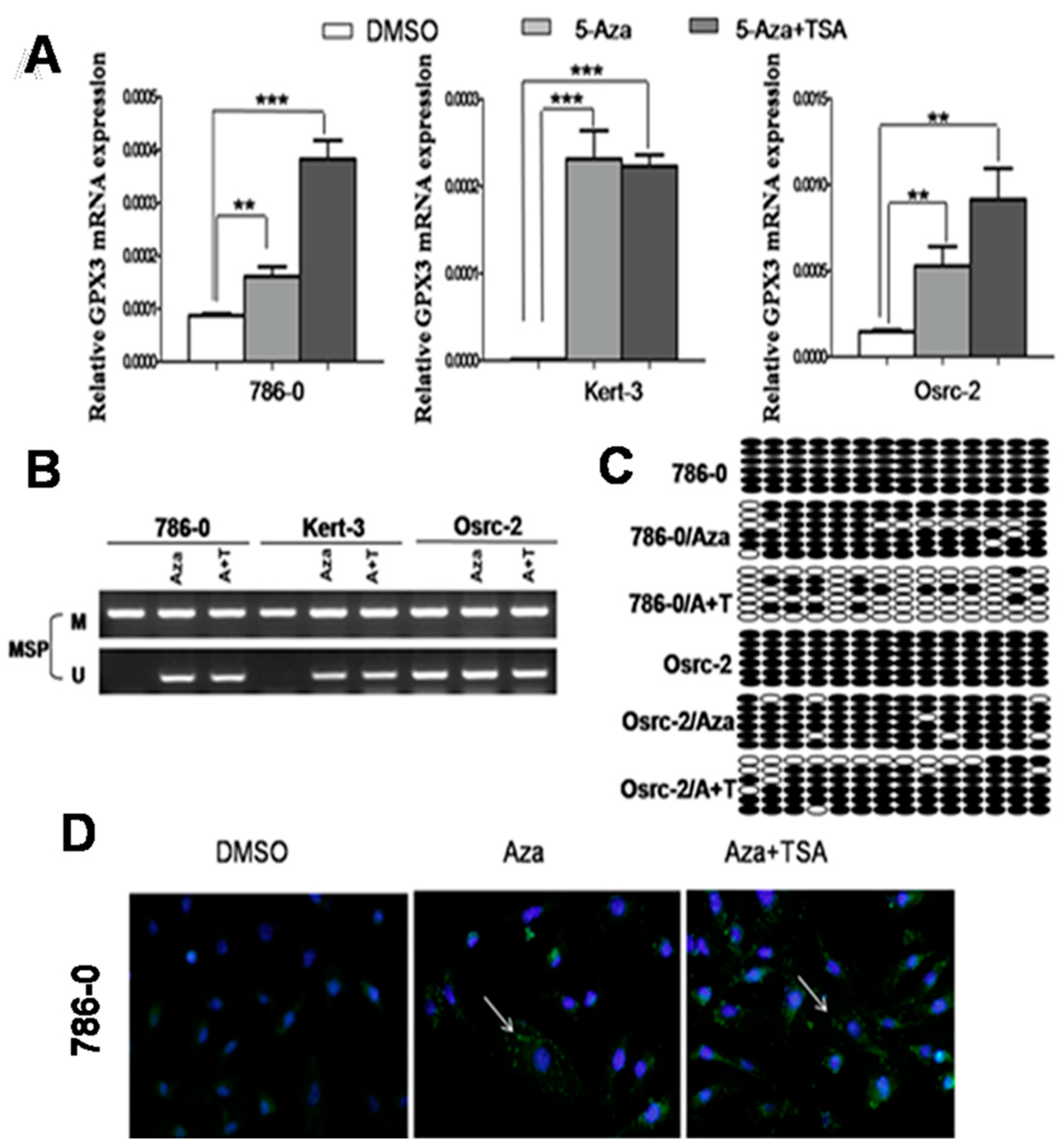
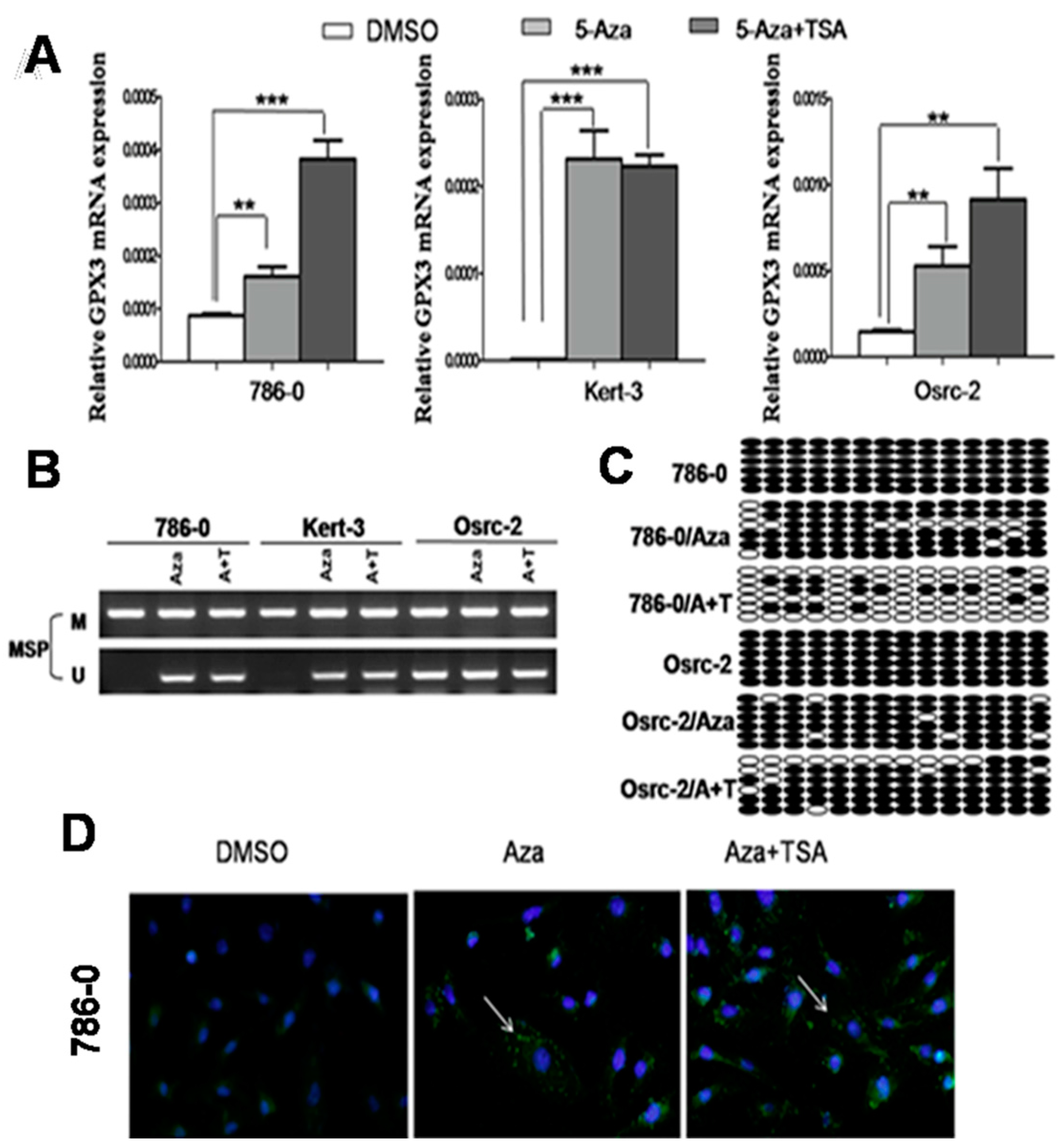
2.2. Pharmacological Demethylation Restored GPX3 Expression in ccRCC Cells
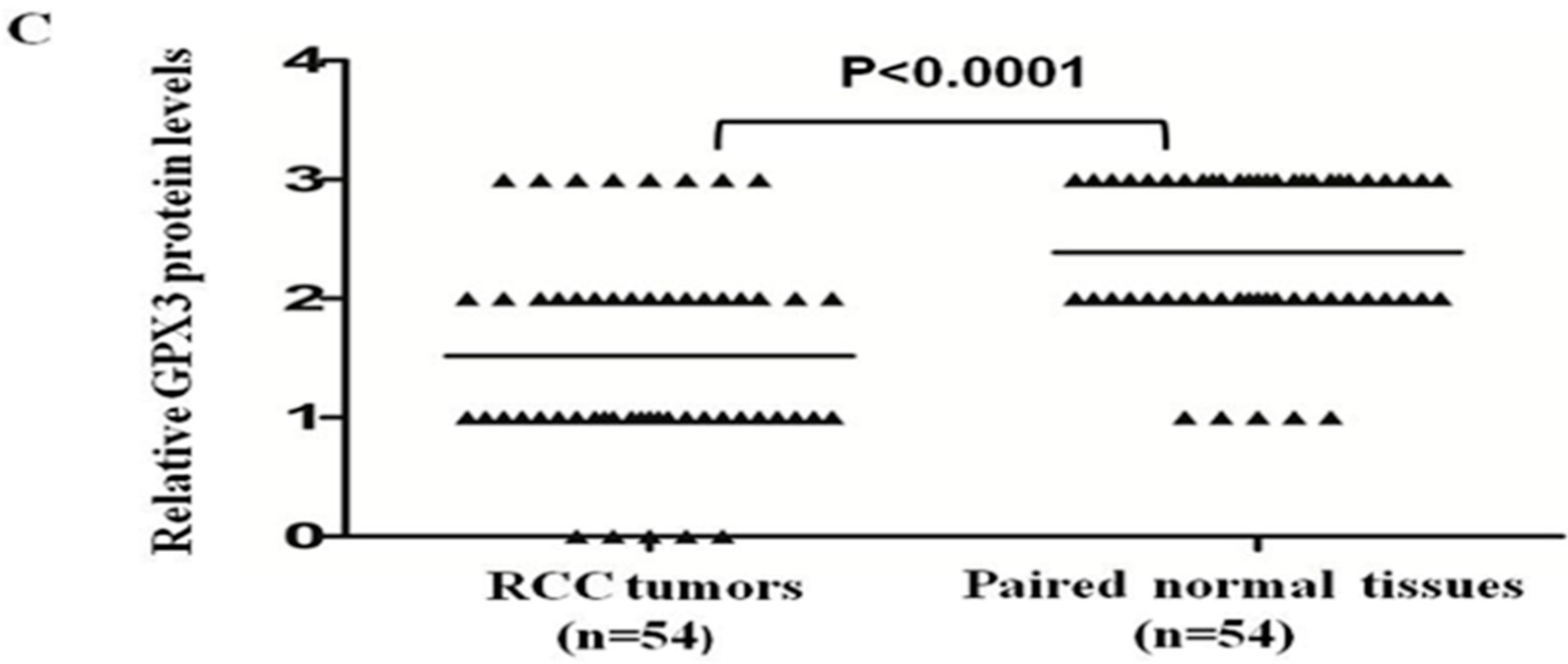
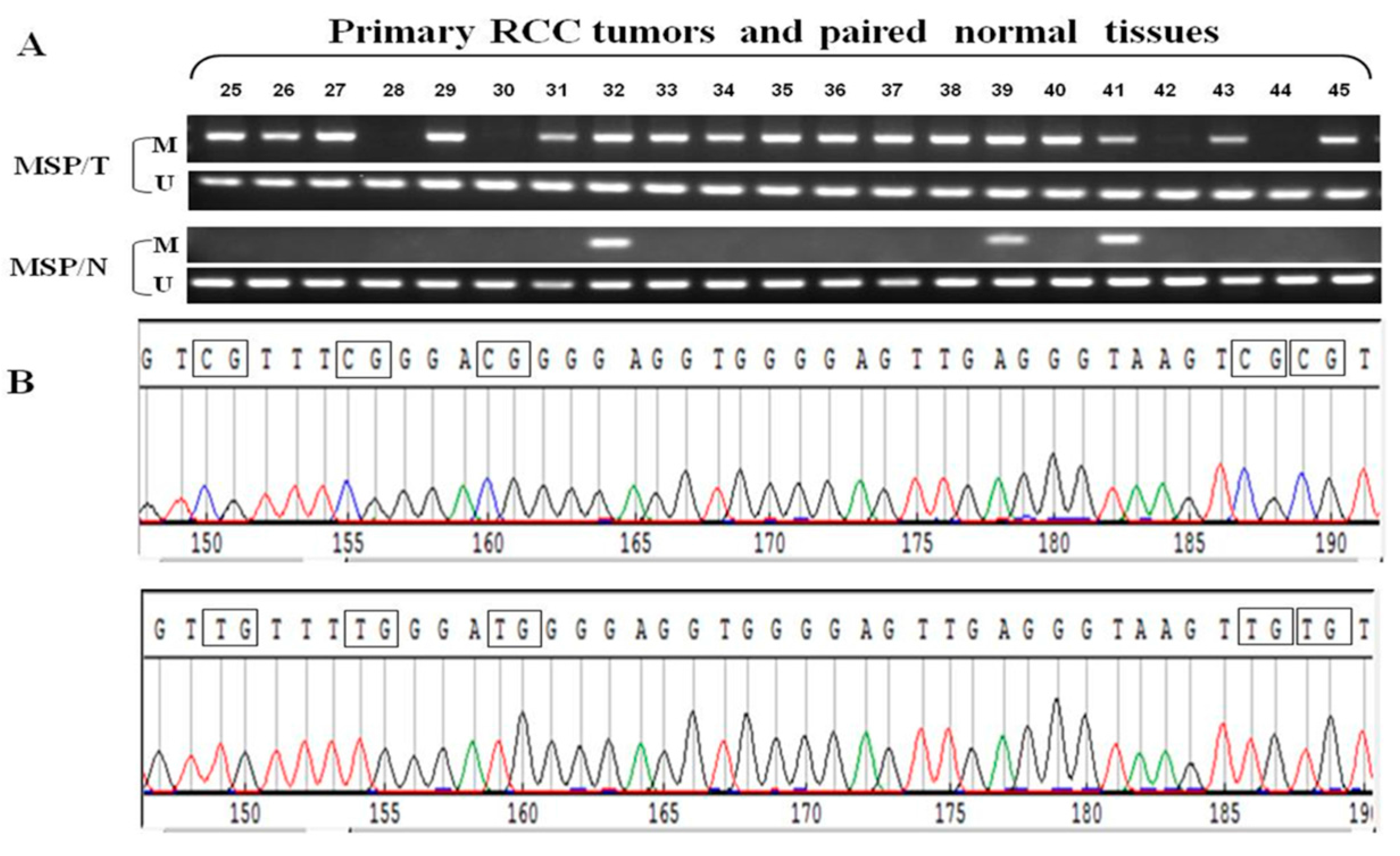
2.3. GPX3 mRNA and Protein Expression Was Frequently Downregulated or Silenced in Primary ccRCC Tumor Tissues

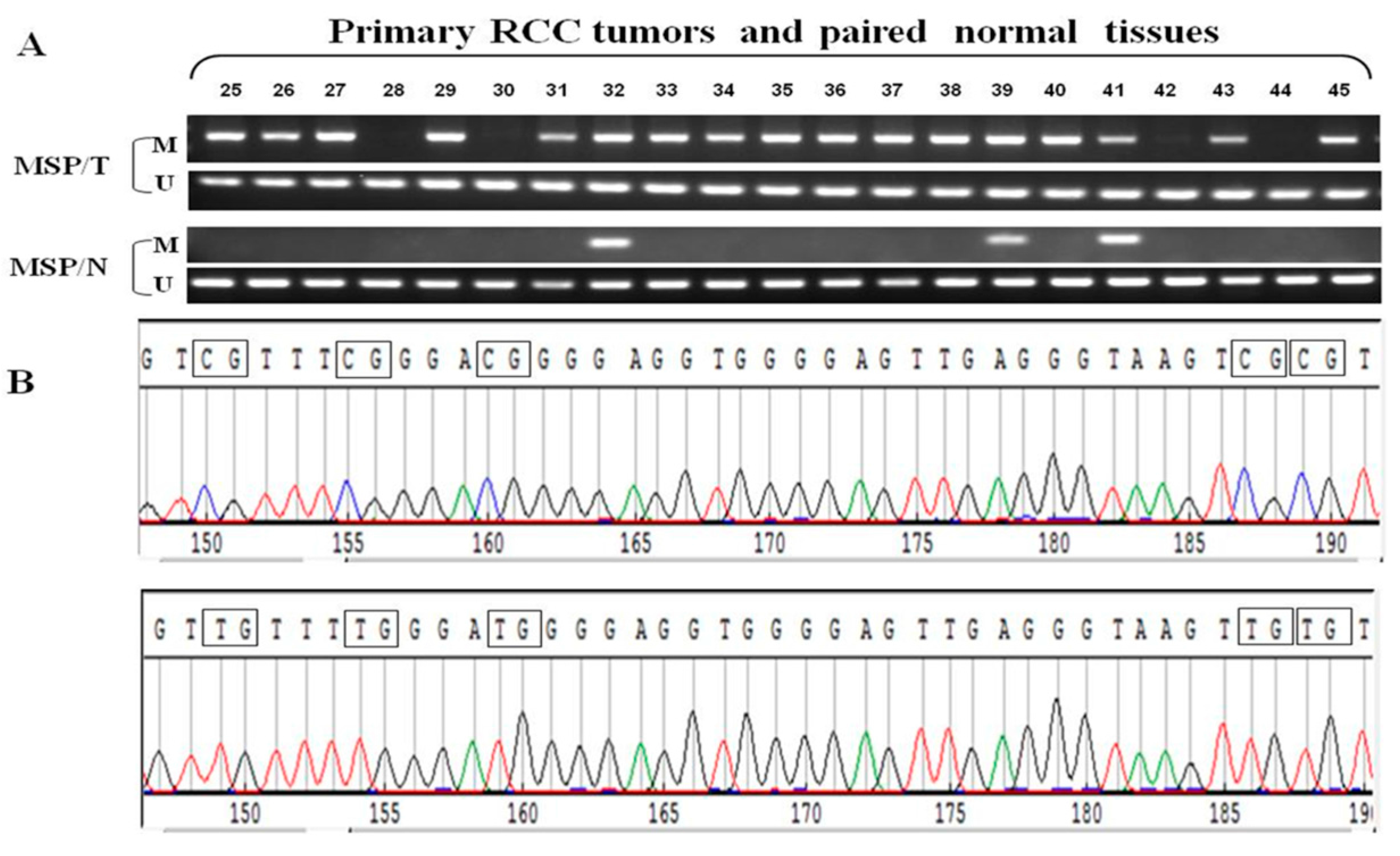
2.4. Frequent GPX3 Promoter Methylation in Primary RCC Tumors Is Associated with Poor Prognosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Features | Number ( n = 210) | GPX3 Methylation Status Methylated Unmethylated | p Value |
|---|---|---|---|
| Overall | 210 | 162 (77.1) 48 (22.9) | |
| Gender | |||
| M | 146 | 112 (76.7) 34 (23.3) | 0.860 |
| F | 64 | 50 (78.1) 14 (21.9) | (Fisher’s exact test) |
| Age | |||
| <60 (median) | 109 | 84 (77.1) 25 (22.9) | 1.000 |
| ≥60 | 101 | 78 (77.2) 23 (22.8) | (Fisher’s exact test) |
| Side | |||
| Rt | 113 | 82 (72.6) 31 (27.4) | 0.088 |
| Lt | 97 | 80 (82.5) 17 (17.5) | (Fisher’s exact test) |
| TNM Classification | |||
| pT1a | 79 | 56 (70.9) 23 (29.1) | 0.206 |
| pT1b | 72 | 60 (83.3) 12 (16.7) | (chi-square test) |
| pT2 | 17 | 15 (83.3) 2 (16.7) | |
| pT3 | 42 | 32 (76.2) 10 (23.8) | |
| Nuclear Grade | |||
| G1 | 48 | 30 (62.5) 18 (37.5) | 0.014 * |
| G2 | 135 | 108 (80.0) 27 (20.0) | (chi-square test) |
| G3 | 27 | 24 (88.9) 3 (11.1) | |
| Histological Type | |||
| Clear cell Rcc | 193 | 150 (77.7) 43 (23.3) | 0.734 |
| papillary Rcc | 9 | 6 (66.7) 3 (33.3) | (chi-square test) |
| chromophobe Rcc | 8 | 6 (75.0) 2 (25.0) |
3. Discussion
4. Experimental Section
4.1. Patients and Tissue Samples
4.2. Cell Culture
4.3. Drug Treatment
4.4. Quantitative Real-Time PCR
| Gene | Primer Sequence (5'-3') | Anneal. Temp. (°C) | No. of Cycles |
|---|---|---|---|
| Real Time | |||
| GPX3 F | CTTCCTACCCTCAAGTATGTCCG | 55 | 45 |
| GPX3 R | GAGGTGGGAGGACAGGAGTTCTT | ||
| GAPDH F | GGTGGTCTCCTCTGACTTCAACA | 55 | 45 |
| GAPDH R | GTTGCTGTAGCCAAATTCGTTGT | ||
| MSP | |||
| GPX3 m1 | TATGTTATTGTCGTTTCGGGAC | 59 | 40 |
| GPX3 m2 | GTCCGTCTAAAATATCCGACG | ||
| GPX3 U1 | TTTATGTTATTGTTGTTTTGGGATG | 59 | 40 |
| GPX3 U2 | ATCCATCTAAAATATCCAACACTCC | ||
| BGS | |||
| GPX3 BGS F | GGAGTTAAAAGAGGAAGGG | 58 | 40 |
| GPX3 BGS R | CCCAACCACCTTTCAAAC |
4.5. Immunohistochemistry
4.6. Immunofluorescence
4.7. Bisulfite Treatment and Methylation-Specific PCR
4.8. Bisulfite Genomic Sequencing
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Abbreviations
| Aza | 5-Aza-2'-deoxycytidine |
| BGS | bisulfite genomic sequencing |
| GPX3 | glutathione peroxidase 3 |
| LOH | loss of heterozygosity |
| MSP | methylation- specific PCR |
| PCR | polymerase chain reaction |
| RCC | renal cell carcinoma |
| ROS | reactive oxygen species |
| TSA | trichostatin A |
| TSG | tumor suppressor gene |
Conflicts of Interest
References
- Thillai, K.; Allan, S.; Powles, T.; Rudman, S.; Chowdhury, S. Neoadjuvant and adjuvant treatment of renal cell carcinoma. Expert Rev. Anticancer Ther. 2012, 12, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Casla, M.T.; Maestro, M.L.; Del, B.V.; Zanna, I.; Moreno, J.; Vidaurreta, M.; Almansa, I.; Fernandez, C.; Blanco, J.; Maestro, C.; et al. Loss of heterozygosity and methylation of p16 in renal cell carcinoma. Urol. Res. 2003, 31, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.R.; Gentle, D.; Abdulrahman, M.; Maina, E.N.; Gupta, K.; Banks, R.E.; Wiesener, M.S.; Kishida, T.; Yao, M.; Teh, B.; et al. Tumor suppressor activity and epigenetic inactivation of hepatocyte growth factor activator inhibitor type 2/SPINT2 in papillary and clear cell renal cell carcinoma. Cancer Res. 2005, 65, 4598–4606. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, C.; Martinez, A.; Zatyka, M.; Agathanggelou, A.; Honorio, S.; Astuti, D.; Morgan, N.V.; Moch, H.; Richards, F.M.; Kishida, T.; et al. Epigenetic inactivation of the RASSF1A 3p21.3 tumor suppressor gene in both clear cell and papillary renal cell carcinoma. Cancer Res. 2001, 61, 7277–7281. [Google Scholar] [PubMed]
- Morris, M.R.; Ricketts, C.J.; Gentle, D.; McRonald, F.; Carli, N.; Khalili, H.; Brown, M.; Kishida, T.; Yao, M.; Banks, R.E.; et al. Genome-wide methylation analysis identifies epigenetically inactivated candidate tumour suppressor genes in renal cell carcinoma. Oncogene 2011, 30, 1390–1401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ying, J.; Zhang, K.; Li, H.; Ng, K.M.; Zhao, Y.; He, Q.; Yang, X.; Xin, D.; Liao, S.K.; et al. Aberrant methylation of the 8p22 tumor suppressor gene DLC1 in renal cell carcinoma. Cancer Lett. 2007, 249, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ying, J.; Li, J.; Fan, Y.; Poon, F.F.; Ng, K.M.; Tao, Q.; Jin, J. Aberrant promoter methylation of DLEC1, a critical 3p22 tumor suppressor for renal cell carcinoma, is associated with more advanced tumor stage. J. Urol. 2010, 184, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, L.; Li, L.; Wang, Z.; Ying, J.; Fan, Y.; Xu, B.; Wang, L.; Liu, Q.; Chen, G.; et al. Interferon regulatory factor 8 functions as a tumor suppressor in renal cell carcinoma and its promoter methylation is associated with patient poor prognosis. Cancer Lett. 2014, 354, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Malins, D.C.; Polissar, N.L.; Gunselman, S.J. Progression of human breast cancers to the metastatic state is linked to hydroxyl radical-induced DNA damage. Proc. Natl. Acad. Sci. USA 1996, 93, 2557–2563. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Takebe, G.; Yarimizu, J.; Saito, Y.; Hayashi, T.; Nakamura, H.; Yodoi, J.; Nagasawa, S.; Takahashi, K. A comparative study on the hydroperoxide and thiol specificity of the glutathione peroxidase family and selenoprotein P. J. Biol. Chem. 2002, 277, 41254–41258. [Google Scholar] [CrossRef] [PubMed]
- Bjornstedt, M.; Xue, J.; Huang, W.; Akesson, B.; Holmgren, A. The thioredoxin and glutaredoxin systems are efficient electron donors to human plasma glutathione peroxidase. J. Biol. Chem. 1994, 269, 29382–29384. [Google Scholar] [PubMed]
- Tham, D.M.; Whitin, J.C.; Kim, K.K.; Zhu, S.X.; Cohen, H.J. Expression of extracellular glutathione peroxidase in human and mouse gastrointestinal tract. Am. J. Physiol. 1998, 275, G1463–G1471. [Google Scholar] [PubMed]
- Chu, F.F.; Esworthy, R.S.; Doroshow, J.H.; Doan, K.; Liu, X.F. Expression of plasma glutathione peroxidase in human liver in addition to kidney, heart, lung, and breast in humans and rodents. Blood 1992, 79, 3233–3238. [Google Scholar]
- Chen, B.; Rao, X.; House, M.G.; Nephew, K.P.; Cullen, K.J.; Guo, Z. GPx3 promoter hypermethylation is a frequent event in human cancer and is associated with tumorigenesis and chemotherapy response. Cancer Lett. 2011, 309, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.J.; Schneider-Stock, R.; McChesney, P.A.; Kuester, D.; Roessner, A.; Vieth, M.; Moskaluk, C.A.; El-Rifai, W. Hypermethylation and loss of expression of glutathione peroxidase-3 in Barrett’s tumorigenesis. Neoplasia 2005, 7, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.P.; Yu, G.; Tseng, G.; Cieply, K.; Nelson, J.; Defrances, M.; Zarnegar, R.; Michalopoulos, G.; Luo, J.H. Glutathione peroxidase 3, deleted or methylated in prostate cancer, suppresses prostate cancer growth and metastasis. Cancer Res. 2007, 67, 8043–8050. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef] [PubMed]
- Lo, I.C.; Shih, J.M.; Jiang, M.J. Reactive oxygen species and ERK 1/2 mediate monocyte chemotactic protein-1-stimulated smooth muscle cell migration. J. Biomed. Sci. 2005, 12, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Taulli, R.; Scuoppo, C.; Bersani, F.; Accornero, P.; Forni, P.E.; Miretti, S.; Grinza, A.; Allegra, P.; Schmitt-Ney, M.; Crepaldi, T.; et al. Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma. Cancer Res. 2006, 66, 4742–4749. [Google Scholar] [CrossRef] [PubMed]
- Lutterbach, B.; Zeng, Q.; Davis, L.J.; Hatch, H.; Hang, G.; Kohl, N.E.; Gibbs, J.B.; Pan, B.S. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res. 2007, 67, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Luo, K.; Tan, L.Z.; Ren, B.G.; Gu, L.Q.; Michalopoulos, G.; Luo, J.H.; Yu, Y.P. p53-induced gene 3 mediates cell death induced by glutathione peroxidase 3. J. Biol. Chem. 2012, 287, 16890–16902. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.W.; Ning, W.; Chen, X.; Smith, J.J.; Washington, M.K.; Hill, K.E.; Coburn, L.A.; Peek, R.M.; Chaturvedi, R.; Wilson, K.T.; et al. Tumor suppressor function of the plasma glutathione peroxidase GPX3 in colitis-associated carcinoma. Cancer Res. 2013, 73, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, Y.; Li, P.; Zhu, S.; Wang, J.; Zhang, S. Identification of GPX3 epigenetically silenced by CpG methylation in human esophageal squamous cell carcinoma. Dig. Dis. Sci. 2011, 56, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.F.; Hu, T.L.; Schneider, B.G.; Chen, Z.; Xu, Z.K.; El-Rifai, W. Silencing of glutathione peroxidase 3 through DNA hypermethylation is associated with lymph node metastasis in gastric carcinomas. PLoS ONE 2012, 7, e46214. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.O.; Begum, S.; Topaloglu, O.; Jeronimo, C.; Mambo, E.; Westra, W.H.; Califano, J.A.; Sidransky, D. Quantitative detection of promoter hypermethylation of multiple genes in the tumor, urine, and serum DNA of patients with renal cancer. Cancer Res. 2004, 64, 5511–5517. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.; Srivastava, G.; Hsieh, W.S.; Gao, Z.; Murray, P.; Liao, S.K.; Ambinder, R.; Tao, Q. The stress-responsive gene GADD45G is a functional tumor suppressor, with its response to environmental stresses frequently disrupted epigenetically in multiple tumors. Clin. Cancer Res. 2005, 11, 6442–6449. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Jin, J.; Ying, J.; Sun, M.; Cui, Y.; Zhang, L.; Xu, B.; Fan, Y.; Zhang, Q. Frequent Epigenetic Suppression of Tumor Suppressor Gene Glutathione Peroxidase 3 by Promoter Hypermethylation and Its Clinical Implication in Clear Cell Renal Cell Carcinoma. Int. J. Mol. Sci. 2015, 16, 10636-10649. https://doi.org/10.3390/ijms160510636
Liu Q, Jin J, Ying J, Sun M, Cui Y, Zhang L, Xu B, Fan Y, Zhang Q. Frequent Epigenetic Suppression of Tumor Suppressor Gene Glutathione Peroxidase 3 by Promoter Hypermethylation and Its Clinical Implication in Clear Cell Renal Cell Carcinoma. International Journal of Molecular Sciences. 2015; 16(5):10636-10649. https://doi.org/10.3390/ijms160510636
Chicago/Turabian StyleLiu, Qianling, Jie Jin, Jianming Ying, Mengkui Sun, Yun Cui, Lian Zhang, Ben Xu, Yu Fan, and Qian Zhang. 2015. "Frequent Epigenetic Suppression of Tumor Suppressor Gene Glutathione Peroxidase 3 by Promoter Hypermethylation and Its Clinical Implication in Clear Cell Renal Cell Carcinoma" International Journal of Molecular Sciences 16, no. 5: 10636-10649. https://doi.org/10.3390/ijms160510636
APA StyleLiu, Q., Jin, J., Ying, J., Sun, M., Cui, Y., Zhang, L., Xu, B., Fan, Y., & Zhang, Q. (2015). Frequent Epigenetic Suppression of Tumor Suppressor Gene Glutathione Peroxidase 3 by Promoter Hypermethylation and Its Clinical Implication in Clear Cell Renal Cell Carcinoma. International Journal of Molecular Sciences, 16(5), 10636-10649. https://doi.org/10.3390/ijms160510636