
Cyanobacterial Hydrogenases and Hydrogen Metabolism Revisited: Recent Progress and Future Prospects

Abstract
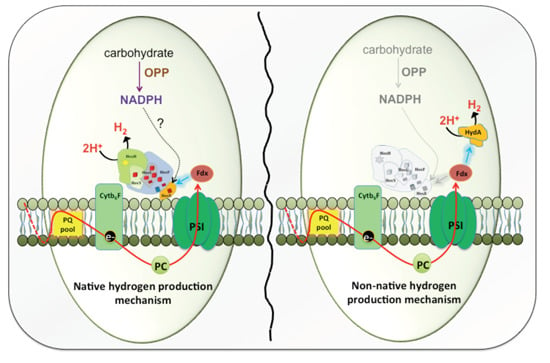
:
1. Introduction
2. Key Advances in Cyanobacterial Research
2.1. Hox Reduced by NAD(P)H/NADH in Synechocystis—A Myth Busted
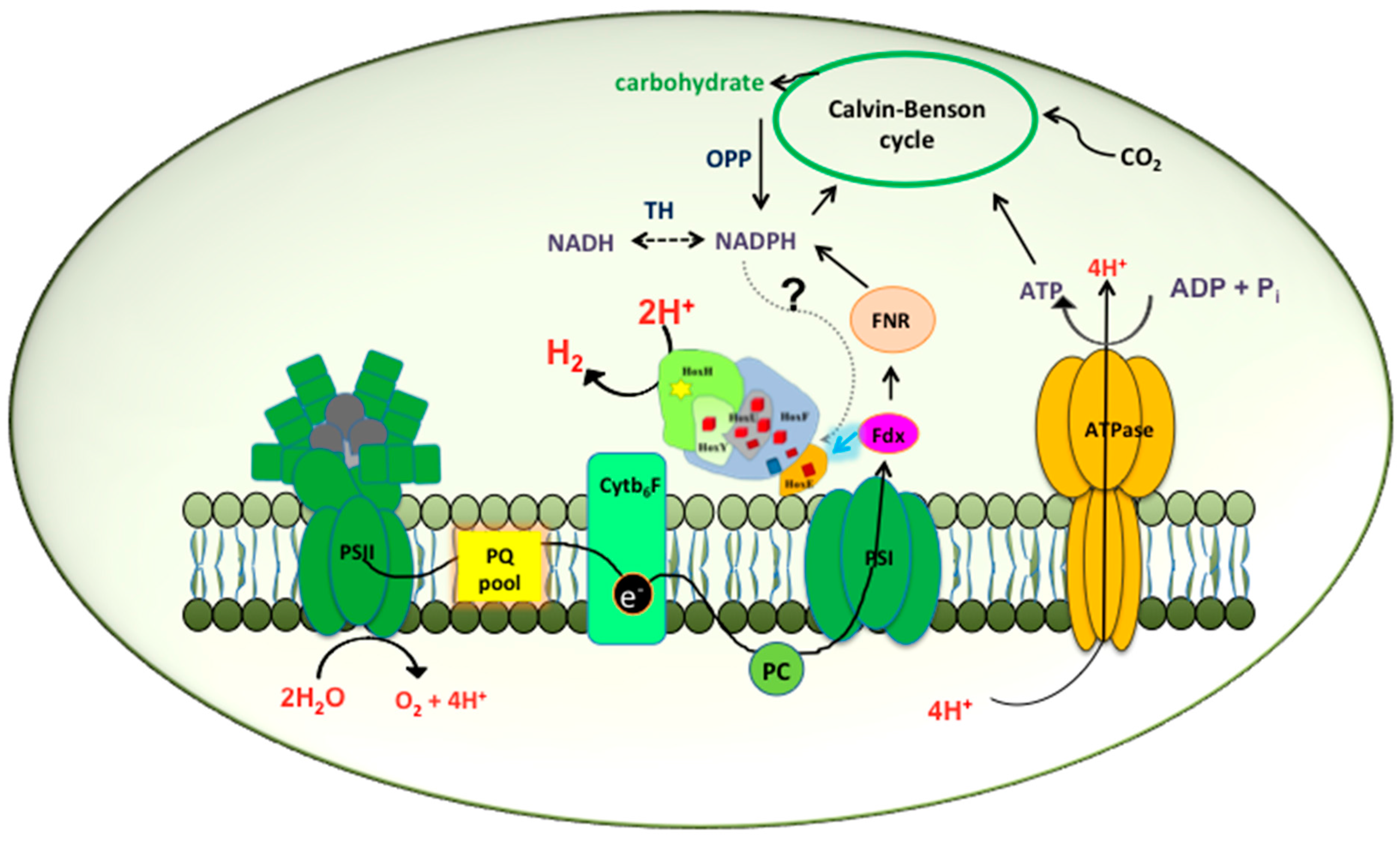
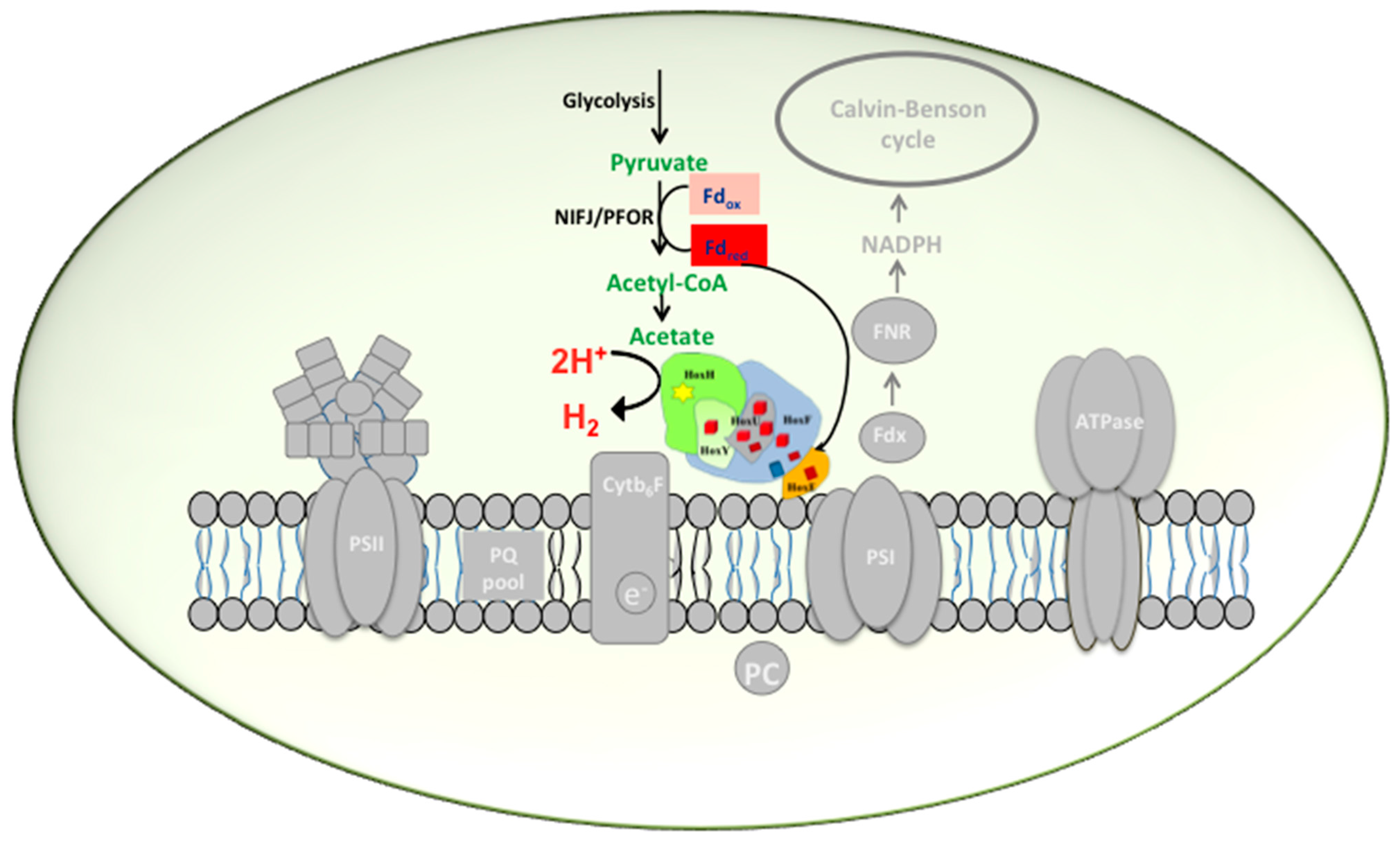
2.2. Introduction of Non-Native Hydrogenases in Cyanobacteria

{kind=link}
{kind=link}
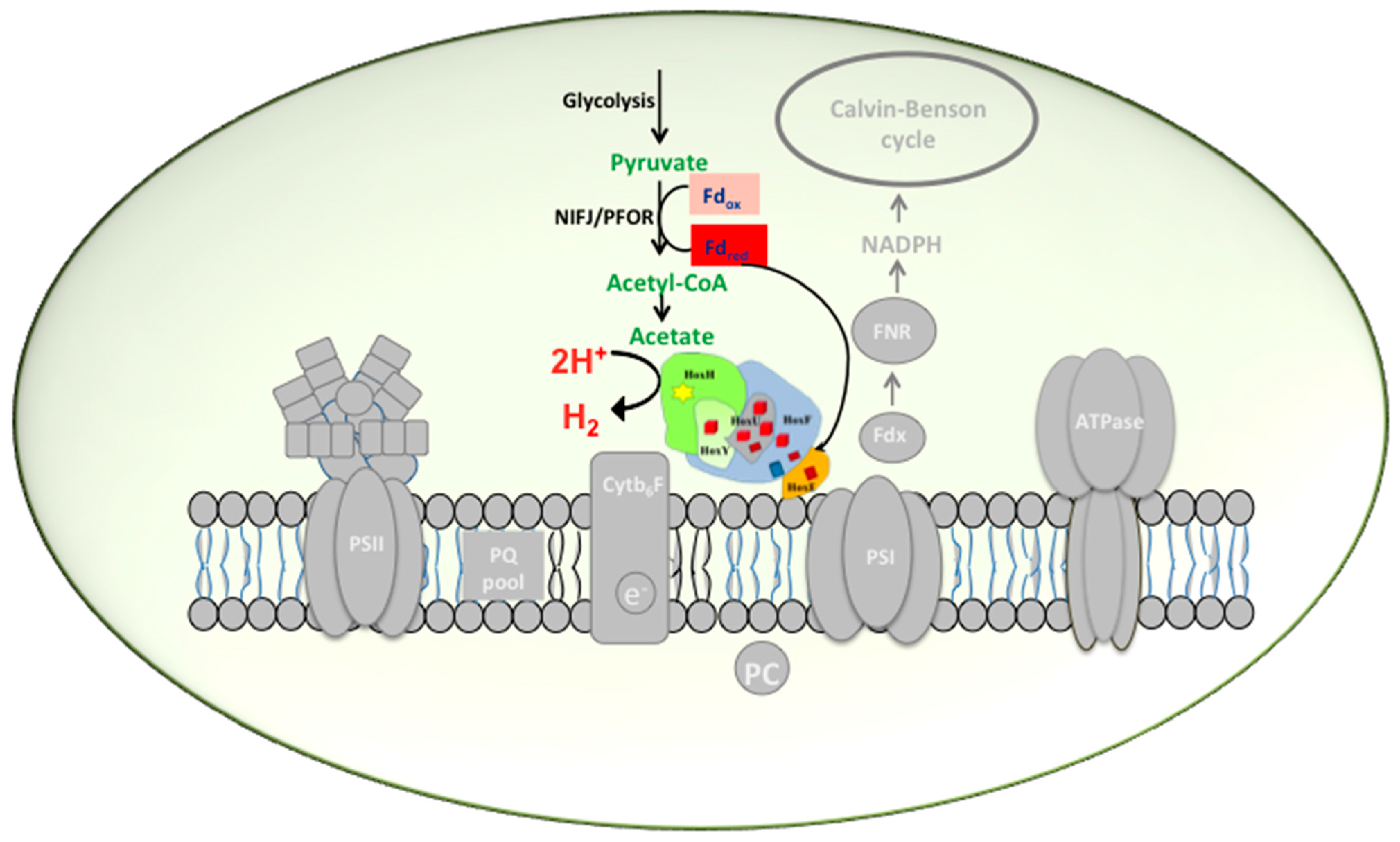{kind=link}
| Organism | Genes Expressed | Comments | Reference |
|---|---|---|---|
| Synechococcus elongatus | Expression of [FeFe] hydrogenase and the accessory HydEFG from Clostridium acetobutylicum in neutral site 3 and 1 respectively. | The first report on expression of non-native [FeFe] hydrogenase along with their accessory genes in cyanobacteria. The article clearly demonstrated in vitro and in vivo hydrogenase activity. The authors established that the in vivo hydrogenase activity was connected to the light-dependent reactions of the electron transport chain. | [88] |
| Synechocystis PCC 6803 | Expression of [FeFe] hydA from Chlamydomonas reinhardtii in a non-coding region of the Synechocystis chromosome(s) identified between the sll1865 and sll1864 genes, which encode for the peptide chain release factor 2 and for a chloride channel protein, respectively. | The report suggested that foreign [FeFe] hydrogenase could be matured in absence of auxiliary proteins, HydEFG. The report showed five times higher hydrogen production from the recombinant cells. This however, needs further confirmation. | [89] |
| Anabaena PCC 7120 | Expression of Shewanella oneidensis MR-1 [FeFe] hydrogenase and accessory genes on a pK3 self-replicating vector. | The first publication that showed the expression of an [FeFe] hydrogenase under a heterocyst specific promoter, in a filamentous organism. [FeFe] hydrogenase expression was detected and the protein was purified from aerobically grown filaments cultivated under nitrate-depleted conditions. Activity assays confirmed in vitro and in situ activities, however, in vivo activities could not be detected. This perhaps demonstrated substrate limitation for the successful endogenous activity of the introduced hydrogenase. | [90] |
| Synechococcus elongatus | Expression of [NiFe] hydrogenase from Alteromonas macleodii and Thiocapsa roseopersici. | The first report that demonstrated expression of non-native [NiFe] hydrogenase in cyanobacteria. Thiocapsa roseopersicina (hynSL) and the 11 accessory genes from Alteromonas macleodii were co-expressed. The article showed in vitro activity of the expressed protein. | [91] |
2.3. Introduction of “Improved” Non-Native Pathways into Cyanobacteria
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Eberle, U.; Mueller, B.; von Helmolt, R. Fuel cell electric vehicles and hydrogen infrastructure: Status 2012. Energy Environ. Sci. 2012, 5, 8780–8798. [Google Scholar] [CrossRef]
- Vignais, P.M.; Billoud, B. Occurrence, classification, and biological function of hydrogenases: An overview. Chem. Rev. 2007, 107, 4206–4272. [Google Scholar] [CrossRef] [PubMed]
- Björn, L.O.; Govindjee, G. The Evolution of Photosynthesis and Chloroplasts. Curr. Sci. 2009, 96, 1466–1474. [Google Scholar]
- Rexroth, S.; Weigand, K.; Rögner, M. Cyanobacterial design cell for the production of hydrogen from water. In Biohydrogen; Rögner, M., Ed.; Walter de Gruyter GmbH: Berlin, Germany, 2015; pp. 1–14. [Google Scholar]
- McNeely, K.; Xu, Y.; Bennette, N.; Bryant, D.A.; Dismukes, G.C. Redirecting reductant flux into hydrogen production via metabolic engineering of fermentative carbon metabolism in a cyanobacterium. Appl. Environ. Microbiol. 2010, 76, 5032–5038. [Google Scholar] [CrossRef] [PubMed]
- Cassier-Chauvat, C.; Veaudor, T.; Chauvat, F. Advances in the Function and Regulation of Hydrogenase in the Cyanobacterium Synechocystis PCC6803. Int. J. Mol. Sci. 2014, 15, 19938–19951. [Google Scholar] [CrossRef] [PubMed]
- Kothe, T.; Schuhmann, W.; Rögner, M.; Pluemere, N. Semi artificial photosynthetic Z-scheme for hydrogen production for water. In Biohydrogen; Rögner, M., Ed.; Walter de Gruyter GmbH: Berlin, Germany, 2015; pp. 189–206. [Google Scholar]
- Rousset, M.; Montet, Y.; Guigliarelli, B.; Forget, N.; Asso, M.; Bertrand, P.; Fontecilla-Camps, J.C.; Hatchikian, E.C. [3Fe-4S] to [4Fe-4S] cluster conversion in Desulfovibrio fructosovorans [NiFe] hydrogenase by site-directed mutagenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 11625–11630. [Google Scholar] [CrossRef] [PubMed]
- Dementin, S.; Belle, V.; Bertrand, P.; Guigliarelli, B.; Adryanczyk-Perrier, G.; de Lacey, A.L.; Fernandez, V.M.; Rousset, M.; Leger, C. Changing the ligation of the distal [4Fe4S] cluster in NiFe hydrogenase impairs inter and intramolecular electron transfers. J. Am. Chem. Soc. 2006, 128, 5209–5218. [Google Scholar] [CrossRef] [PubMed]
- Yonemoto, I.T.; Matteri, C.W.; Nguyen, T.M.; Smith, H.M.; Weyman, P.D. Dual organism design cycle reveals small subunit substitutions that improve [NiFe] hydrogenase hydrogen evolution. J. Biol. Eng. 2013, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Yonemoto, I.T.; Clarkson, B.R.; Smith, H.M.; Weyman, P.D. A broad survey reveals substitution tolerance of residues ligating FeS clusters in [NiFe] hydrogenase. BMC Biochem. 2014, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.J.; Sargent, F.; Armstrong, F.A. Transforming an oxygen-tolerant [NiFe] uptake hydrogenase into a proficient, reversible hydrogen producer. Energy Environ. Sci. 2014, 7, 1426–1433. [Google Scholar] [CrossRef]
- Hamdan, A.A.; Dementin, S.; Liebgott, P.P.; Gutierrez-Sanz, O.; Richaud, P.; de Lacey, A.L.; Rousset, M.; Bertrand, P.; Cournac, L.; Léger, C. Understanding and tuning the catalytic bias of hydrogenase. J. Am. Chem. Soc. 2012, 134, 8368–8371. [Google Scholar] [CrossRef] [PubMed]
- Bingemann, R.; Klein, A. Conversion of the central [4Fe–4S] cluster into a [3Fe–4S] cluster leads to reduced hydrogen-uptake activity of the F420-reducing hydrogenase of Methanococcus voltae. Eur. J. Biochem. 2000, 267, 6612–6618. [Google Scholar] [CrossRef] [PubMed]
- Berggren, G.; Adamska, A.; Lambertz, C.; Simmons, T.R.; Esselborn, J.; Atta, M.; Gambarelli, S.; Mouesca, J.-M.; Reijerse, E.; Lubitz, W.; et al. Biomimetic assembly and activation of [FeFe]-hydrogenases. Nature 2013, 499, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Gutekunst, K.; Chen, X.; Schreiber, K.; Kaspar, U.; Makam, S.; Appel, J. The bidirectional NiFe-hydrogenase in Synechocystis sp. PCC 6803 is reduced by flavodoxin and ferredoxin and is essential under mixotrophic, nitrate-limiting conditions. J. Biol. Chem. 2014, 289, 1930–1937. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, C.L.; Germer, F.; Schulz, R.; Appel, J.; Jones, A.K. The [NiFe]-hydrogenase of the cyanobacterium Synechocystis sp. PCC 6803 works bidirectionally with a bias to H2 production. J. Am. Chem. Soc. 2011, 133, 11308–11310. [Google Scholar] [CrossRef] [PubMed]
- Adamska-Venkatesh, A.; Simmons, T.R.; Siebel, J.F.; Artero, V.; Fontecave, M.; Reijerse, E.; Lubitz, W. Artificially maturated [FeFe] hydrogenase from Chlamydomonas reinhardtii: A HYSCORE and ENDOR study of a non-natural H-cluster. Phys. Chem. Chem. Phys. 2015, 17, 5421–5430. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, P.; Khanna, N. Engineering of cyanobacteria for increased hydrogen production. In Biohydrogen; Rögner, M., Ed.; Walter de Gruyter GmbH: Berlin, Germany, 2015; pp. 175–190. [Google Scholar]
- Lindblad, P.; Lindberg, P.; Oliveira, P.; Stensjö, K.; Heidorn, T. Design, engineering, and construction of photosynthetic microbial cell factories for renewable solar fuel production. Ambio 2012, 41, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.W.; Schut, G.J.; Boyd, E.S.; Mulder, D.W.; Shepard, E.M.; Broderick, J.B.; King, P.W.; Adams, M.W. [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim. Biophys. Acta 2014, 1853, 1350–1369. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, A.; Martin, L.; Liebgott, P.P.; de Lacey, A.L.; Fontecilla-Camps, J.C. [NiFe]-hydrogenases revisited: Nickel-carboxamido bond formation in a variant with accrued O2 tolerance and a tentative re-interpretation of Ni-SI states. Metallomics 2015, 7, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Pandelia, M.E.; Nitschke, W.; Infossi, P.; Giudici-Orticoni, M.T.; Bill, E.; Lubitz, W. Characterization of a unique [FeS] cluster in the electron transfer chain of the oxygen tolerant [NiFe] hydrogenase from Aquifex aeolicus. Proc. Natl. Acad. Sci. USA 2011, 108, 6097–6102. [Google Scholar] [CrossRef] [PubMed]
- Tamagnini, P.; Axelsson, R.; Lindberg, P.; Oxelfelt, F.; Wunschiers, R.; Lindblad, P. Hydrogenases and hydrogen metabolism of cyanobacteria. Microbiol. Mol. Biol. Rev. 2002, 66, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Tamagnini, P.; Leita, E.; Oliveira, P.; Ferreira, D.; Pinto, F.A.L.; Harris, D.J.; Heidorn, T.; Lindblad, P. Cyanobacterial hydrogenases diversity, regulation and application. FEMS Microbiol. Rev. 2007, 31, 692–720. [Google Scholar] [CrossRef] [PubMed]
- Bothe, H.; Schmitz, O.; Yates, M.G.; Newton, W.E. Nitrogen Fixation and Hydrogen Metabolism in Cyanobacteria. Microbiol. Mol. Biol. Rev. 2010, 74, 529–551. [Google Scholar] [CrossRef] [PubMed]
- Rögner, M. Metabolic engineering of cyanobacteria for the production of hydrogen from water. Biochem. Soc. Trans. 2013, 41, 1254–1259. [Google Scholar] [CrossRef] [PubMed]
- Ananyev, G.; Carrieri, D.; Dismukes, G.C. Optimization of metabolic capacity and flux through environmental cues to maximize hydrogen production by the cyanobacterium “Arthrospira (Spirulina) maxima”. Appl. Environ. Microbiol. 2008, 74, 6102–6113. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, O.; Boison, G.; Bothe, H. Quantitative analysis of expression of two circadian clock-controlled gene clusters coding for the bidirectional hydrogenase in the cyanobacterium Synechococcus sp. PCC7942. Mol. Microbiol. 2001, 41, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Ohki, K.; Taniuchi, Y. Detection of nitrogenase in individual cells of a natural population of trichodesmium using immunocytochemical methods for fluorescent cells. J. Oceanogr. 2009, 65, 427–432. [Google Scholar] [CrossRef]
- Sandh, G.; Ramström, M.; Stensjö, K. Analysis of the early heterocyst Cys-proteome in the multicellular cyanobacterium Nostoc punctiforme reveals novel insights into the division of labor within diazotrophic filaments. BMC Genomics 2014, 4, 1064. [Google Scholar] [CrossRef]
- Khetkorn, W.; Khanna, N.; Incharoensakdi, A.; Lindblad, P. Metabolic and genetic engineering of cyanobacteria for enhanced hydrogen production. Biofuels 2013, 4, 535–561. [Google Scholar] [CrossRef]
- Carrieri, D.; Wawrousek, K.; Eckert, C.; Yu, J.; Maness, P.C. The role of the bidirectional hydrogenase in cyanobacteria. Bioresour. Technol. 2011, 102, 8368–8377. [Google Scholar] [CrossRef] [PubMed]
- Appel, J.; Phunpruch, S.; Steinmüller, K.; Schulz, R. The bidirectional hydrogenase of Synechocystis sp. PCC 6803 works as an electron valve during photosynthesis. Arch. Microbiol. 2000, 173, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Cournac, L.; Guedeney, G.; Peltier, G.; Vignais, P.M. Sustained photoevolution of molecular hydrogen in a mutant of Synechocystis sp. strain PCC 6803 deficient in the type I NADPH-dehydrogenase complex. J. Bacteriol. 2004, 186, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Gutthann, F.; Egert, M.; Marques, A.; Appel, J. Inhibition of respiration and nitrate assimilation enhances photohydrogen evolution under low oxygen concentration in Synechocystis sp. PCC 6803. Biochim. Biophys. Acta 2007, 1767, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, M.; Schulz-Friedrich, R.; Appel, J. Occurrence of hydrogenases in cyanobacteria and anoxygenic photosynthetic bacteria: Implications for the phylogenetic origin of cyanobacterial and algal hydrogenases. J. Mol. Evol. 2006, 63, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Eckert, C.; Boehm, M.; Carrieri, D.; Yu, J.; Dubini, A.; Nixon, P.J.; Maness, P.C. Genetic analysis of the Hox hydrogenase in the cyanobacterium Synechocystis sp. PCC 6803 reveals subunit roles in association, assembly, maturation, and function. J. Biol. Chem. 2012, 287, 43502–43515. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.; Kós, P.B.; Vass, I. Transcriptional regulation of the bidirectional hydrogenase in the cyanobacterium Synechocystis 6803. J. Biotechnol. 2009, 142, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Shestakov, S.V.; Mikheeva, L.E. Genetic control of hydrogen metabolism in cyanobacteria. Russ. J. Genet. 2006, 42, 1272–1284. [Google Scholar] [CrossRef]
- Boison, G.; Schmitz, O.; Schmitz, B.; Bothe, H. Unusual gene arrangement of the bidirectional hydrogenase and functional analysis of its diaphorase subunit HoxU in respiration of the unicellular cyanobacterium Anacystis nidulans. Curr. Microbiol. 1998, 36, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Boer, J.L.; Mulrooney, S.B.; Hausinger, R.P. Nickel-dependent metalloenzymes. Arch. Biochem. Biophys. 2014, 544, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Shafaat, H.S.; Rüdiger, O.; Ogata, H.; Lubitz, W. [NiFe] hydrogenases: A common active site for hydrogen metabolism under diverse conditions. Biochim. Biophys. Acta 2013, 1827, 986–1002. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Higuchi, Y. Studies on hydrogenase. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2013, 89, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Horch, M.; Lauterbach, L.; Lenz, O.; Hildebrandt, P.; Zebger, I. NAD(H)-coupled hydrogen cycling—Structure-function relationships of bidirectional [NiFe] hydrogenases. FEBS Lett. 2012, 586, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Bock, A.; King, P.W.; Blokesch, M.; Posewitz, M.C. Maturation of hydrogenases. Adv. Microb. Physiol. 2006, 51, 1–71. [Google Scholar] [PubMed]
- Broderick, J.B.; Byer, A.S.; Duschene, K.S.; Duffus, B.R.; Betz, J.N.; Shepard, E.M.; Peters, J.W. H-Cluster assembly during maturation of the [FeFe] hydrogenase. J. Biol. Inorg. Chem. 2014, 19, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Wunschiers, R.; Batur, M.; Lindblad, P. Presence and expression of hydrogenase specific C-terminal endopeptidases in cyanobacteria. BMC Microbiol. 2003, 3, 8–20. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Soboh, B.; Lindenstrauss, U.; Granich, C.; Javed, M.; Herzberg, M.; Thomas, C.; Stripp, S.T. [NiFe]-hydrogenase maturation in vitro: Analysis of the roles of the HybG and HypD accessory proteins1. Biochem. J. 2014, 464, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Hansel, A.; Axelsson, R.; Lindberg, P.; Troshina, O.Y.; Wunschiers, R.; Lindblad, P. Cloning and characterization of a hyp gene cluster in the filamentous cyanobacterium Nostoc sp. strain. FEMS Microbiol. Lett. 2001, 201, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Sato, S.; Kotani, H.; Tanaka, A.; Asamizu, E.; Nakamura, Y.; Miyajima, N.; Hirosawa, M.; Sugiura, M.; Sasamoto, S.; et al. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC 6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 1996, 3, 185–209. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Gutekunst, K.; Klissenbauer, M.; Schulz-Friedrich, R.; Appel, J. Mutagenesis of hydrogenase accessory genes of Synechocystis sp. PCC 6803. Additional homologues of hypA and hypB are not active in hydrogenase maturation. FEBS J. 2006, 273, 4516–4527. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.; Lindblad, P. LexA, a transcriptional regulator binding in the promoter region of the bidirectional hydrogenase in the cyanobacterium Synechocystis sp. strain PCC6803. FEMS Microbiol. Lett. 2005, 251, 59–66. [Google Scholar] [CrossRef]
- Oliveira, P.; Lindblad, P. An AbrB-like protein regulates the expression of the bidirectional hydrogenase in Synechocystis sp. strain 6803. J. Bacteriol. 2008, 190, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.; Lindblad, P. Transcriptional regulation of the cyanobacterial Hox-hydrogenase. Dalton Trans. 2009, 45, 9990–9996. [Google Scholar] [CrossRef] [PubMed]
- Gutekunst, K.; Phunpruch, S.; Schwarz, C.; Schuchardt, S.; Schulz-Friedrich, R.; Appel, J. LexA regulates the bidirectional hydrogenase in the cyanobacterium Synechocystis sp. PCC 6803 as a transcription activator. Mol. Microbiol. 2005, 58, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Masukawa, H.; Kitashima, M.; Inoue, K. How close we are to achieving commercially viable large-scale photobiological hydrogen production by cyanobacteria: A review of the biological aspects. Life 2015, 5, 997–1018. [Google Scholar] [CrossRef] [PubMed]
- Tye, J.W.; Hall, M.B.; Darensbourg, M.Y. Better than platinum? Fuel cells energized by enzymes. Proc. Natl. Acad. Sci. USA 2005, 102, 16911–16912. [Google Scholar] [CrossRef] [PubMed]
- Lojou, E.; Luo, X.; Brugna, M.; Candoni, N.; Dementin, S.; Giudici-Orticoni, M.T. Biocatalysts for fuel cells: Efficient hydrogenase orientation for H2 oxidation at electrodes modified with carbon nanotubes. J. Biol. Inorg. Chem. 2008, 13, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Houchins, J.P.; Burris, R.H. Occurrence and localization of two distinct hydrogenases in the heterocystous cyanobacterium Anabaena sp. strain 7120. J. Bacteriol. 1981, 146, 209–214. [Google Scholar] [PubMed]
- Houchins, J.P.; Burris, R.H. Comparative characterization of two distinct hydrogenases from Anabaena sp. strain 7120. J. Bacteriol. 1981, 146, 215–221. [Google Scholar] [PubMed]
- Schmitz, O.; Boison, G.; Hilscher, R.; Hundeshangen, B.; Zimmer, W.; Lottspeich, F.; Bothe, H. Molecular biological analysis of a directional hydrogenase from cyanobacteria. Eur. J. Biochem. 1995, 233, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Sigfridsson, K.G.; Leidel, N.; Sanganas, O.; Chernev, P.; Lenz, O.; Yoon, K.S.; Nishihara, H.; Parkin, A.; Armstrong, F.A.; Dementin, S.; et al. Structural differences of oxidized iron-sulfur and nickel-iron cofactors in O2-tolerant and O2-sensitive hydrogenases studied by X-ray absorption spectroscopy. Biochim. Biophys. Acta 2015, 1847, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Stripp, S.T.; Lindenstrauss, U.; Granich, C.; Sawers, R.G.; Soboh, B. The influence of oxygen on [NiFe]-hydrogenase cofactor biosynthesis and how ligation of carbon monoxide precedes cyanation. PLoS ONE 2014, 9, e107488. [Google Scholar] [CrossRef] [PubMed]
- Pinto, F.; Elburg, K.; Pacheco, C.C.; Lopo, M.; Noirel, J.; Montagud, A.; Urcueguía, J.F.; Wright, P.C.; Tamagnini, P. Construction of a chassis for hydrogen production: Physiological and molecular characterization of a Synechocystis sp. PCC 6803 mutant lacking a functional bidirectional hydrogenase. Microbiology 2012, 158, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, N.J.; Boehm, M.; Eckert, C.; Mastroianni, G.; Spence, E.M.; Yu, J.; Nixon, P.J.; Appel, J.; Mullineaux, C.W.; Bryan, S.J.; et al. Solar powered biohydrogen production requires specific localization of the hydrogenase. Energy Environ. Sci. 2014, 7, 3791–3800. [Google Scholar] [CrossRef]
- Liu, L-N.; Bryan, S.J.; Huang, F.; Yu, J.; Nixon, P.J.; Rich, P.R.; Mullineaux, W.C. Control of electron transport routes through redox-regulated redistribution of respiratory complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 11431–11436. [Google Scholar] [CrossRef] [PubMed]
- Appel, J.; Schulz, R. Hydrogen metabolism in organisms with oxygenic photosynthesis: Hydrogenases as important regulatory devices for a proper redox poising? J. Photochem. Photobiol. B Biol. 1998, 47, 1–11. [Google Scholar] [CrossRef]
- Schmitz, O.; Bothe, H. NAD(P)+ Dependent Hydrogenase Activity in Extracts from the Cyanobacterium Anacystis nidulans. FEMS Microbiol. Lett. 1996, 35, 97–101. [Google Scholar]
- Schmitz, O.; Boison, G.; Salzmann, H.; Bothe, H.; Schultz, K.; Wang, S.H.; Happe, T. HoxE—A subunit specific for the pentameric bidirectional hydrogenase complex (HoxEFUYH) of cyanobacteria. Biochim. Biophys. Acta 2002, 1554, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Battchikova, N.; Eisenhut, M.; Aro, E.M. Cyanobacterial NDH-1 complexes: Novel insights and remaining puzzles. Biochim. Biophys. Acta 2011, 1807, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Battchikova, N.; Paakkarinen, V.; Katoh, H.; Iwai, M.; Ikeuchi, M.; Battchikova, N.; Zhang, P.; Rudd, S.; Ogawa, T.; et al. Identification of NdhL and Ssl1690 (NdhO) in NDH-1L and NDH-1M complexes of Synechocystis sp. PCC 6803. J. Biol. Chem. 2005, 280, 2587–2595. [Google Scholar] [CrossRef] [PubMed]
- Prommeenate, P.; Lennon, A.M.; Markert, C.; Hippler, M.; Nixon, P.J. Subunit composition of NDH-1 complexes of Synechocystis sp. PCC 6803—Identification of two new ndh gene products with nuclear-encoded homologues in the chloroplast Ndh complex. J. Biol. Chem. 2004, 279, 28165–28173. [Google Scholar]
- Appel, J.; Schulz, R. Sequence analysis of an operon of a NAD(P)-reducing nickel hydrogenase from the cyanobacterium Synechocystis sp. PCC6803 gives additional evidence for direct coupling of the enzyme to NAD(P)H-dehydrogenase (complex I). Biochim. Biophys. Acta 1996, 1298, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Boison, G.; Bothe, H.; Hansel, A.; Lindblad, P. Evidence against a common use of the diaphorase subunits by the bidirectional hydrogenase and by the respiratory complex I in cyanobacteria. FEMS Microbiol. Lett. 1999, 174, 159–165. [Google Scholar] [CrossRef]
- Howitt, C.A.; Vermaas, W.F.J. Subunits of the NAD(P)-reducing nickel-containing hydrogenase do not act as part of the type-1 NAD(P)H-dehydrogenase in the cyanobacterium Synechocystis sp. PCC 6803. In The Phototrophic Prokaryotes; Peschek, G.A., Löffelhardt, W., Schmetterer, G., Eds.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 1999; pp. 595–601. [Google Scholar]
- Marreiros, B.C.; Batista, A.P.; Duarte, A.M.S.; Pereira, M.M. A missing link between complex I and group 4 membrane-bound [NiFe] hydrogenases. Biochim. Biophys. Acta 2013, 1827, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Aubert-Jousset, E.; Cano, M.; Guedeney, G.; Richaud, P.; Cournac, L. Role of HoxE subunit in Synechocystis PCC6803 hydrogenase. FEBS J. 2011, 278, 4035–4043. [Google Scholar] [CrossRef] [PubMed]
- McNeely, K.; Xu, Y.; Ananyev, G.; Bennette, N.; Bryant, D.A.; Dismukes, C.G. Characterization of a nifJ mutant of Synechococcus sp. strain PCC 7002 lacking pyruvate:ferredoxin oxidoreductase. Appl. Environ. Microbiol. 2011, 77, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Guerra, T.L.; Li, Z.; Ludwig, M.; Dismukes, C.G.; Bryant, D.A. Altered carbohydrate metabolism in glycogen synthase mutants of Synechococcus sp. strain PCC 7002: Cell factories for soluble sugars. Metab. Eng. 2013, 16, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Skizim, N.J.; Ananyev, G.M.; Krishnan, A.; Dismukes, C.G. Metabolic pathways for photobiological hydrogen production by nitrogenase- and hydrogenase-containing unicellular cyanobacteria Cyanothece. J. Biol. Chem. 2012, 287, 2777–2786. [Google Scholar] [CrossRef] [PubMed]
- Kothari, A.; Potrafka, R.; Garcia-Pichel, F. Diversity in hydrogen evolution from bidirectional hydrogenases in cyanobacteria from terrestrial, freshwater and marine intertidal environments. J. Biotechnol. 2012, 162, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.D.; Kimball, E.H.; Gao, M.; Osterhout, R.; van Dien, S.J.; Rabinowitz, J.D. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 2009, 5, 593–599. [Google Scholar] [CrossRef] [PubMed]
- De Graef, M.R.; Alexeeva, S.; Snoep, J.L.; Teixeira de Mattos, M.J. The steady-state internal redox state (NADH/NAD) reflects the external redox state and is correlated with catabolic adaptation in Escherichia coli. J. Bacteriol. 1999, 181, 2351–2357. [Google Scholar] [PubMed]
- Germer, F.; Zebger, I.; Saggu, M.; Lendzian, F.; Schulz, R. Overexpression, Isolation, and Spectroscopic Characterization of the Bidirectional [NiFe] Hydrogenase from Synechocystis sp. PCC 6803. J. Biol. Chem. 2009, 284, 36462–36472. [Google Scholar] [CrossRef] [PubMed]
- Goss, T.; Hanke, G. The end of the line: Can ferredoxin and ferredoxin NADP(H) oxidoreductase determine the fate of photosynthetic electrons? Curr. Protein Pept. Sci. 2014, 15, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Rosner, V.; Wagner, H.-J. Analysis and assessment of current photobioreactor systems for photobiological hydrogen production. In Biohydrogen; Rögner, M., Ed.; Walter de Gruyter GmbH: Berlin, Germany, 2015; pp. 19–37. [Google Scholar]
- Ducat, D.C.; Sachdeva, G.; Silver, P.A. Rewiring hydrogenase-dependent redox circuits in cyanobacteria. Proc. Natl. Acad. Sci. USA 2011, 108, 3941–3946. [Google Scholar] [CrossRef] [PubMed]
- Berto, P.; Adamo, S.D.; Bergantino, E.; Vallese, F.; Giacometti, G.M.; Costantini, P. The Cyanobacterium Synechocystis sp. PCC 6803 is able to express an active [FeFe] hydrogenase without additional maturation proteins. Biochem. Biophys. Res. Commun. 2011, 405, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Gartner, K.; Lechno-Yossef, S.; Cornish, A.J.; Wolk, P.C.; Hegg, E.C. Expression of Shewanella Oneidensis MR-1 [FeFe] hydrogenase genes in Anabaena Sp. strain PCC 7120. Appl. Environ. Microbiol. 2012, 78, 8579–8586. [Google Scholar] [CrossRef] [PubMed]
- Weyman, P.D.; Vargas, W.; Tong, Y.; Yu, J.; Maness, P.C.; Smith, H.O.; Xu, Q. Heterologous expression of Alteromonas macleodii and Thiocapsa roseopersicina [NiFe] hydrogenases in Synechococcus elongatus. PLoS ONE 2011, 6, e20126. [Google Scholar] [CrossRef] [PubMed]
- Posewitz, M.C.; Mulder, D.W.; Peters, J.W. New frontiers in hydrogenase structure and biosynthesis. Curr. Chem. Biol. 2008, 2, 178–199. [Google Scholar]
- Mulder, D.W.; Shepard, E.M.; Meuser, J.E.; Joshi, N.; King, P.W.; Posewitz, M.C.; Broderick, J.B.; Peters, J.W. Insights into [FeFe] hydrogenase structure, mechanism, and maturation. Structure 2011, 19, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Baebpraserta, W.; Jantaroa, S.; Khetkorna, W.; Lindblad, P.; Incharoensakdia, A. Increased H2 production in the cyanobacterium Synechocystis sp. strain PCC 6803 by redirecting the electron supply via genetic engineering of the nitrate assimilation pathway. Metab. Eng. 2011, 13, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Stripp, S.T.; Goldetb, G.B.; Brandmayrb, C.; Sanganasc, O.; Vincent, K.A.; Haumannc, M. How oxygen attacks [FeFe] hydrogenases from photosynthetic organisms. Proc. Natl. Acad. Sci. USA 2009, 106, 17331–17336. [Google Scholar] [CrossRef] [PubMed]
- Wolk, C.P.; Ernest, A.; Elhai, J. Heterocyst metabolism and development. In The Molecular Biology of Cyanobacteria; Bryant, A., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1994; pp. 769–823. [Google Scholar]
- Fay, P. Oxygen relations of nitrogen fixation in cyanobacteria. Microbiol. Rev. 1992, 56, 340–373. [Google Scholar] [PubMed]
- Masukawa, H.; Inoue, K.; Sakurai, H.; Wolk, C.P.; Hausinger, R.P. Site-directed mutagenesis of the Anabaena sp. strain PCC 7120 nitrogenase active site to increase photobiological hydrogen production. Appl. Environ. Microbiol. 2010, 76, 6741–6750. [Google Scholar] [CrossRef] [PubMed]
- Vargas, W.A.; Weyman, P.D.; Tong, Y.; Smith, H.O.; Xu, Q. A [NiFe]-hydrogenase from Alteromonas macleodii with unusual stability in the presence of oxygen and high temperature. Appl. Environ. Microbiol. 2011, 77, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Weyman, P.D.; Vargas, W.; Chuang, R.Y.; Chang, Y.; Smith, H.O.; Xu, Q. Heterologous expression of Alteromonas macleodii and Thiocapsa roseopersicina [NiFe] hydrogenases in Escherichia coli. Microbiology 2011, 157, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Weyman, P.D.; Umetani, M.; Xiong, J.; Qi, X.; Xu, Q.; Iwasaki, H.; Johnson, C.H. Circadian Yin-Yang regulation and its manipulation to globally reprogram gene expression. Curr. Biol. 2013, 23, 2365–2374. [Google Scholar] [CrossRef] [PubMed]
- Maróti, G.; Tong, Y.; Yooseph, S.; Baden-Tillson, H.; Smith, H.O.; Kovács, K.L.; Frazier, M.; Venter, J.C.; Xu, Q. Discovery of [NiFe] hydrogenase genes in metagenomic DNA: Cloning and heterologous expression in Thiocapsa roseopersicina. Appl. Environ. Microbiol. 2009, 75, 5821–5830. [Google Scholar] [CrossRef] [PubMed]
- Lukey, M.J.; Parkin, A.; Roessler, M.M.; Murphy, B.J.; Harmer, J.; Palmer, T.; Sargent, F.; Armstrong, F.A. How Escherichia coli is equipped to oxidize hydrogen under different redox conditions. J. Biol. Chem. 2010, 285, 3928–3938. [Google Scholar] [CrossRef] [PubMed]
- Leger, C.; Dementin, S.; Bertrand, P.; Rousset, M.; Guigliarelli, B. Inhibition and anaerobic inactivation kinetics of Desulfovibrio fructosovorans NiFe hydrogenase studied by protein film voltammetry. J. Am. Chem. Soc. 2004, 126, 12162–12172. [Google Scholar] [CrossRef] [PubMed]
- Leger, C.; Bertrand, P. Direct Electrochemistry of redox enzymes as a tool for mechanistic studies. Chem. Rev. 2008, 108, 2379–2438. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, A.; Charon, M.H.; Piras, C.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 1995, 373, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Page, C.C.; Moser, C.C.; Chen, X.; Dutton, P.L. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature 1999, 402, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Page, C.C.; Moser, C.C.; Dutton, P.L. Mechanism for electron transfer within and between proteins. Curr. Opin. Chem. Biol. 2003, 7, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.M.; Heffron, K.; Kotlyar, V.; Sher, Y.; Maklashina, E.; Cecchini, G.; Armstrong, F.A. Electron transfer and catalytic control by the iron-sulfur clusters in a respiratory enzyme, E. coli fumarate reductase. J. Am. Chem. Soc. 2005, 127, 6977–6989. [Google Scholar] [CrossRef] [PubMed]
- Raleiras, P.; Kellers, P.; Lindblad, P.; Styring, S.; Magnuson, A. Isolation and characterization of the small subunit of the uptake hydrogenase from the cyanobacterium Nostoc punctiforme. J. Biol. Chem. 2013, 288, 18345–18352. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khanna, N.; Lindblad, P. Cyanobacterial Hydrogenases and Hydrogen Metabolism Revisited: Recent Progress and Future Prospects. Int. J. Mol. Sci. 2015, 16, 10537-10561. https://doi.org/10.3390/ijms160510537
Khanna N, Lindblad P. Cyanobacterial Hydrogenases and Hydrogen Metabolism Revisited: Recent Progress and Future Prospects. International Journal of Molecular Sciences. 2015; 16(5):10537-10561. https://doi.org/10.3390/ijms160510537
Chicago/Turabian StyleKhanna, Namita, and Peter Lindblad. 2015. "Cyanobacterial Hydrogenases and Hydrogen Metabolism Revisited: Recent Progress and Future Prospects" International Journal of Molecular Sciences 16, no. 5: 10537-10561. https://doi.org/10.3390/ijms160510537
APA StyleKhanna, N., & Lindblad, P. (2015). Cyanobacterial Hydrogenases and Hydrogen Metabolism Revisited: Recent Progress and Future Prospects. International Journal of Molecular Sciences, 16(5), 10537-10561. https://doi.org/10.3390/ijms160510537