
Identification and Validation of Evolutionarily Conserved Unusually Short Pre-mRNA Introns in the Human Genome


Abstract
:1. Introduction
2. Results and Discussion
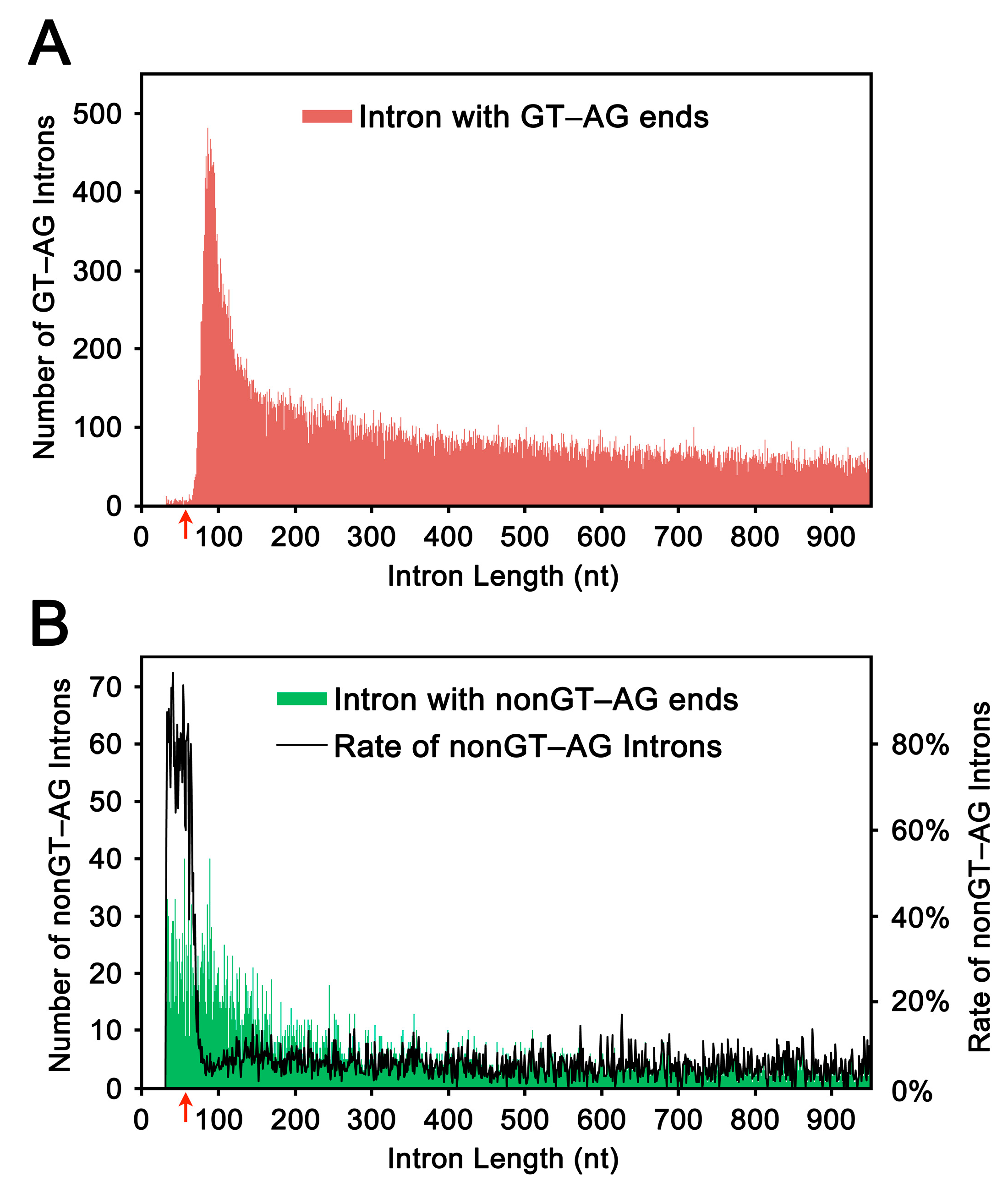
2.1. Length Distribution of Human Introns
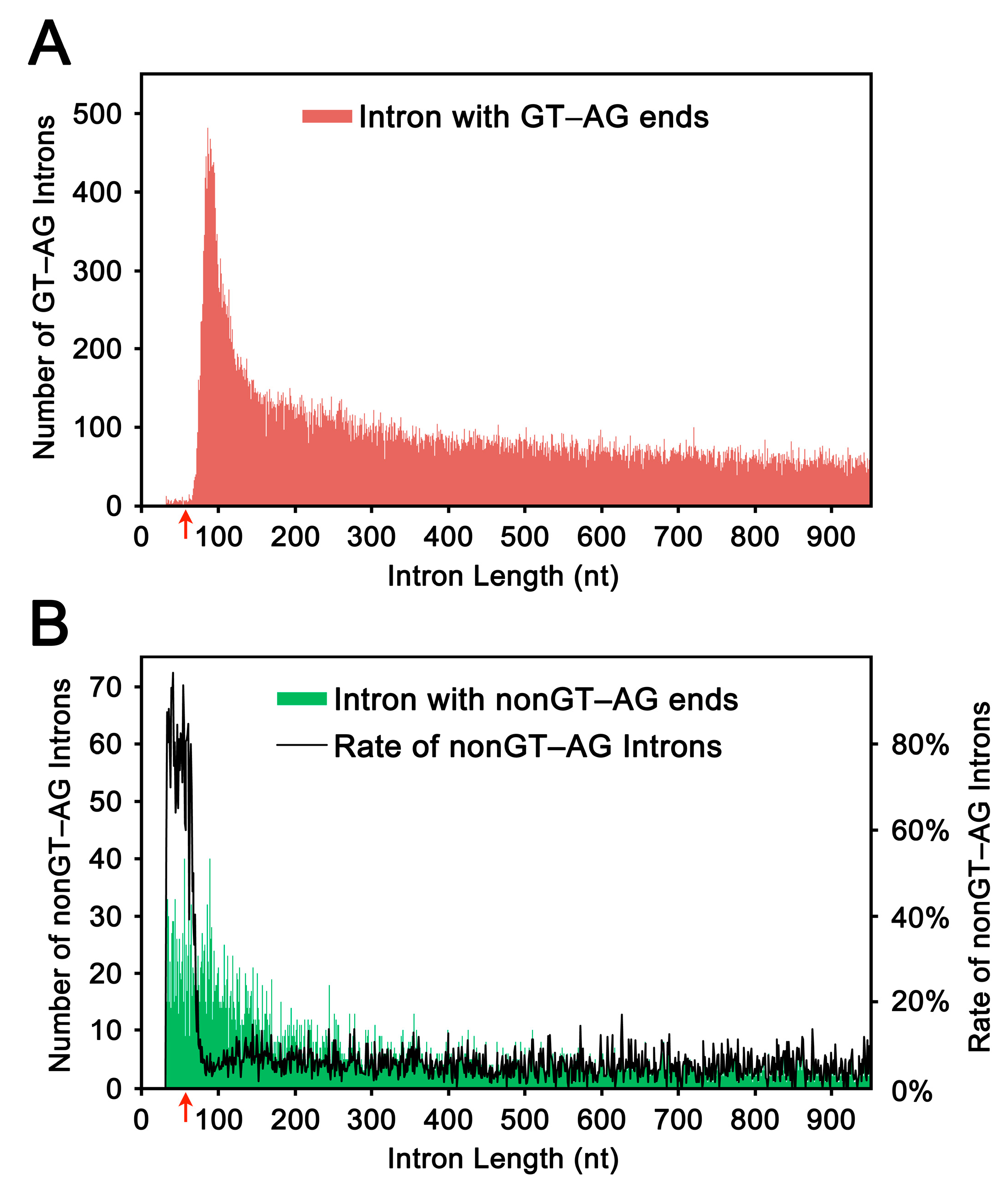
2.2. Selection and Validation of the Ultra-Short Introns

{kind=link}
{kind=link}
| SN a | Length (nt) b | ID number of HIT c | Intron number d | Total no. of introns e | Site of intron | Data in Ensembl f | AA-seq g | Intron frequency h | RT–PCR analysis * i | RNA-Seq data * j | Individually sequenced * k | Confirm. l | ID number of HIX m | Host gene (HGNC) n |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 37 | HIT000059291 | 1 | 3 | CDS | Yes | I | 1/2 | Expressed | No | Yes | Yes | HIX0029777 | AQP12A |
| 2 | 41 | HIT000276161 | 4 | 4 | CDS | Yes | II | 1/23 | Expressed | No | RM | No | HIX0001032 | ENSA |
| 3 | 43 | HIT000008845 | 6 | 14 | CDS | Yes | I | 1/4 | Spliced | Yes | No | Yes | HIX0013170 | ESRP2 |
| 4 | 47 | HIT000325704 | 2 | 15 | CDS | Yes | II | 1/1 | No-Exp | No | Yes | Yes | HIX0003317 | IFRD2 |
| 5 | 49 | HIT000009363 | 12 | 13 | CDS | Yes | I | 3/11 | Spliced | Yes | No | Yes | HIX0023123 | NDOR1 |
| 6 | 50 | HIT000084762 | 8 | 10 | CDS | Yes | III | 1/13 | Expressed | No | No | No | HIX0022245 | SAMD14 |
| 7 | 54 | HIT000325704 | 3 | 15 | CDS | Yes | II | 1/1 | Expressed | No | Yes | Yes | HIX0003317 | IFRD2 |
| 8 | 54 | HIT000333308 | 1 | 2 | CDS | No | VII | 1/1 | Expressed | No | No | No | HIX0059400 | HSP90B2P |
| 9 | 55 | HIT000278575 | 1 | 5 | CDS | No | IV | 1/7 | Expressed | No | RM | No | HIX0006057 | AKIRIN2 |
| 10 | 56 | HIT000192494 | 7 | 13 | CDS | Yes | I | 9/10 | Spliced | Yes | Yes | Yes | HIX0005482 | HNRNPH1 |
| 11 | 61 | HIT000302202 | 1 | 13 | 5′ UTR | Yes | I | 1/15 | Expressed | No | RM | No | HIX0001133 | MSTO1 |
| 12 | 62 | HIT000279220 | 1 | 7 | CDS | Yes | I | 9/11 | Spliced | Yes | Yes | Yes | HIX0027515 | SIGLEC6 |
| 13 | 62 | HIT000333305 | 1 | 2 | CDS | Yes | II | 1/1 | Expressed | No | No | No | HIX0202199 | HSP90AB4P |
| 14 | 62 | HIT000495960 | 1 | 6 | CDS | Yes | II | 5/5 | Spliced | Yes | No | Yes | HIX0202884 | SIGLECP3 |
| 15 | 63 | HIT000191419 | 3 | 4 | CDS | Yes | I | 3/3 | n/a | Yes | No | Yes | HIX0079411 | PRH1 |
| 16 | 63 | HIT000091849 | 1 | 2 | 5′ UTR | Yes | VI | 1/1 | Expressed | No | No | No | HIX0036362 | – |
| 17 | 65 | HIT000324311 | 10 | 28 | CDS | Yes | II | 1/1 | Spliced | Yes | Yes | Yes | HIX0003640 | PLXNA1 |
| 18 | 65 | HIT000058074 | 1 | 20 | CDS | Yes | I | 1/4 | No-PCR | Yes | Yes | Yes | HIX0034231 | RECQL4 |
| 19 | 65 | HIT000052133 | 11 | 13 | CDS | Yes | IV | 2/2 | No-PCR | Yes | No | Yes | HIX0026183 | C11orf35 |
| 20 | 65 | HIT000082518 | 3 | 11 | CDS | Yes | I | 1/9 | Spliced | Yes | No | Yes | HIX0202311 | PDIA2 |
| 21 | 65 | HIT000252921 | 4 | 4 | CDS | Yes | I | 6/8 | Spliced | Yes | UC | Yes | HIX0028549 | TNFRSF18 |
| 22 | 65 | HIT000058190 | 7 | 26 | CDS | Yes | I | 4/4 | Spliced | Yes | Yes | Yes | HIX0039022 | ADAM11 |
2.3. Confirmation of the Genuine Ultra-Short Introns by Database Analyses and Experiments
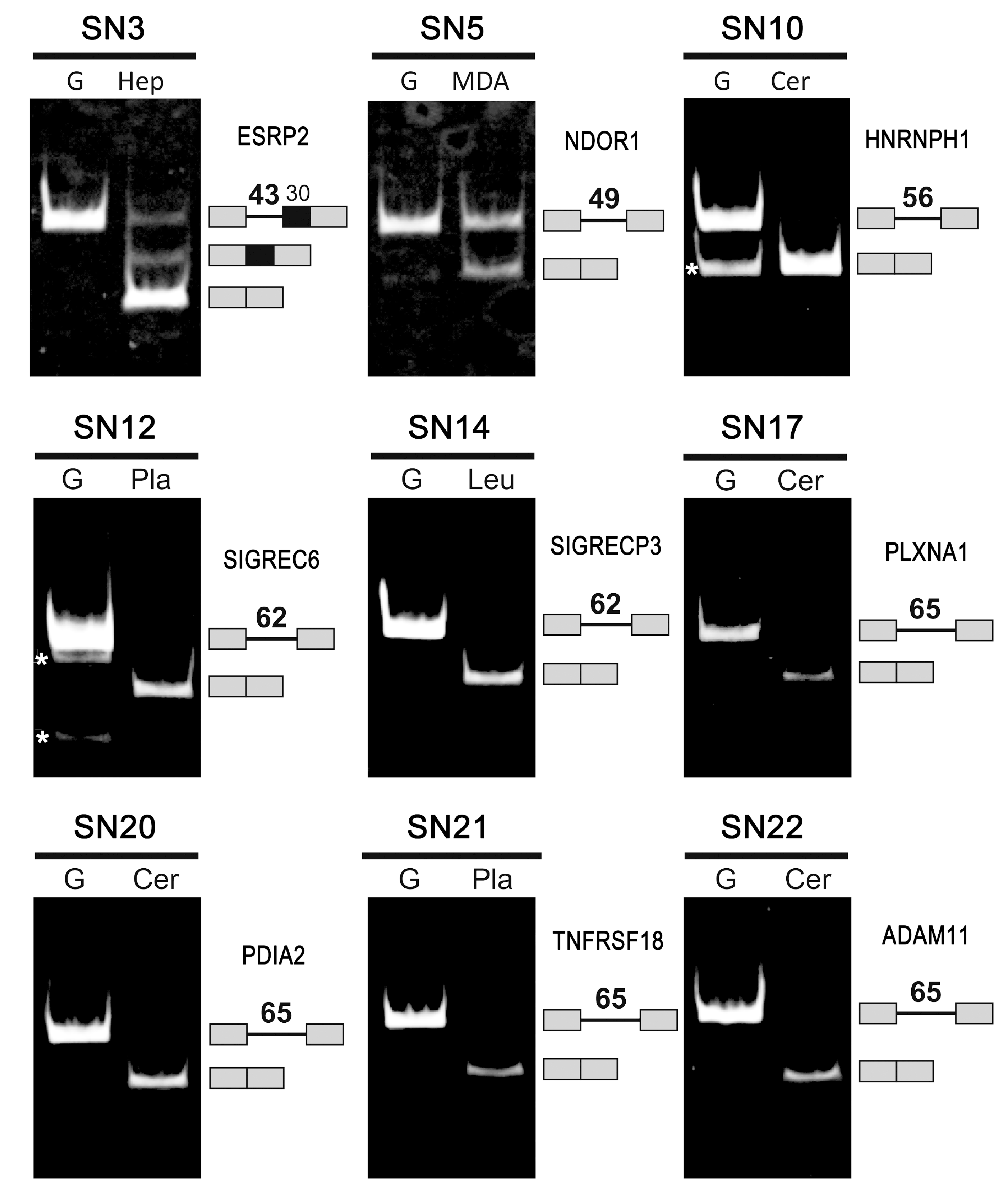
2.4. Evidence to Support the Existence of Ultra-Short Introns
2.5. Unorthodox Sequence Features in the Ultra-Short Introns
 |
2.6. Splicing Mechanism of the Ultra-Short Introns
3. Experimental Procedures
3.1. Extraction of Human Introns and Calculation of Intron Lengths
3.2. Selection of Ultra-Short Introns According to the Length and Conservation Status
3.3. Validation of the Ultra-Short Introns by Database Searches and Experiments
3.4. Collection of Evidence to Support the Authenticity of the Ultra-Short Introns
3.5. Analyses of Sequence Features in the Ultra-Short Introns
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lim, L.P.; Burge, C.B. A computational analysis of sequence features involved in recognition of short introns. Proc. Natl. Acad. Sci. USA 2001, 98, 11193–11198. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhang, Y.; Zhang, W.; Yang, S.; Chen, J.Q.; Tian, D. Patterns of exon-intron architecture variation of genes in eukaryotic genomes. BMC Genomics 2009, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Gotoh, O. Comparative analysis of information contents relevant to recognition of introns in many species. BMC Genomics 2011, 12, 45. [Google Scholar] [CrossRef] [PubMed]
- Matlin, A.J.; Moore, M.J. Spliceosome assembly and composition. Adv. Exp. Med. Biol. 2007, 623, 14–35. [Google Scholar] [PubMed]
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar]
- Behzadnia, N.; Golas, M.M.; Hartmuth, K.; Sander, B.; Kastner, B.; Deckert, J.; Dube, P.; Will, C.L.; Urlaub, H.; Stark, H.; et al. Composition and three-dimensional EM structure of double affinity-purified, human prespliceosomal A complexes. EMBO J. 2007, 26, 1737–1748. [Google Scholar]
- Glass, J.; Wertz, G.W. Different base per unit length ratios exist in single-stranded RNA and single-stranded DNA. Nucleic Acids Res. 1980, 8, 5739–5751. [Google Scholar] [CrossRef] [PubMed]
- Sasaki-Haraguchi, N.; Shimada, M.K.; Taniguchi, I.; Ohno, M.; Mayeda, A. Mechanistic insights into human pre-mRNA splicing of human ultra-short introns: Potential unusual mechanism identifies G-rich introns. Biochem. Biophys. Res. Commun. 2012, 423, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yang, Z.; Kibukawa, M.; Paddock, M.; Passey, D.A.; Wong, G.K. Minimal introns are not “junk”. Genome Res. 2002, 12, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Turunen, J.J.; Niemela, E.H.; Verma, B.; Frilander, M.J. The significant other: Splicing by the minor spliceosome. WIREs RNA 2013, 4, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Maquat, L.E. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem. Sci. 1998, 23, 198–199. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Bateman, J.F.; Mercer, J.F.; Lamande, S.R. Nonsense-mediated mRNA decay of collagen-emerging complexity in RNA surveillance mechanisms. J. Cell Sci. 2013, 126, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Gazave, E.; Marques-Bonet, T.; Fernando, O.; Charlesworth, B.; Navarro, A. Patterns and rates of intron divergence between humans and chimpanzees. Genome Biol. 2007, 8, R21. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yu, J. Both size and GC-content of minimal introns are selected in human populations. PLoS ONE 2011, 6, e17945. [Google Scholar] [CrossRef] [PubMed]
- Burge, C.B.; Tuschl, T.; Sharp, P.A. Splicing of precusors to mRNAs by the spliceosomes. In The RNA World, 2nd ed.; Gesteland, R.F., Cech, T.R., Atkins, J.F., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, New York, NY, USA, 1999; pp. 525–560. [Google Scholar]
- Gao, K.; Masuda, A.; Matsuura, T.; Ohno, K. Human branch point consensus sequence is yUnAy. Nucleic Acids Res. 2008, 36, 2257–2267. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Hall, E.; Ast, G. SROOGLE: webserver for integrative, user-friendly visualization of splicing signals. Nucleic Acids Res. 2009, 37, W189–W192. [Google Scholar] [CrossRef] [PubMed]
- Roca, X.; Sachidanandam, R.; Krainer, A.R. Determinants of the inherent strength of human 5′ splice sites. RNA 2005, 11, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, L.; Theiss, S.; Niederacher, D.; Schaal, H. Diagnostics of pathogenic splicing mutations: Does bioinformatics cover all bases? Front. Biosci. 2008, 13, 3252–3272. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Chasin, L.A. Searching for splicing motifs. Adv. Exp. Med. Biol. 2007, 623, 85–106. [Google Scholar] [PubMed]
- Crispino, J.D.; Blencowe, B.J.; Sharp, P.A. Complementation by SR proteins of pre-mRNA splicing reactions depleted of U1 snRNP. Science 1994, 265, 1866–1869. [Google Scholar] [CrossRef] [PubMed]
- Tarn, W.Y.; Steitz, J.A. SR proteins can compensate for the loss of U1 snRNP functions in vitro. Genes Dev. 1994, 8, 2704–2717. [Google Scholar] [CrossRef] [PubMed]
- Crispino, J.D.; Mermoud, J.E.; Lamond, A.I.; Sharp, P.A. Cis-acting elements distinct from the 5′ splice site promote U1- independent pre-mRNA splicing. RNA 1996, 2, 664–673. [Google Scholar] [PubMed]
- Fukumura, K.; Taniguchi, I.; Sakamoto, H.; Ohno, M.; Inoue, K. U1-independent pre-mRNA splicing contributes to the regulation of alternative splicing. Nucleic Acids Res. 2009, 37, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Herold, N.; Will, C.L.; Wolf, E.; Kastner, B.; Urlaub, H.; Lührmann, R. Conservation of the protein composition and electron microscopy structure of Drosophila melanogaster and human spliceosomal complexes. Mol. Cell. Biol. 2009, 29, 281–301. [Google Scholar] [CrossRef] [PubMed]
- Sirand-Pugnet, P.; Durosay, P.; Brody, E.; Marie, J. An intronic (A/U)GGG repeat enhances the splicing of an alternative intron of the chicken β-tropomyosin pre-mRNA. Nucleic Acids Res. 1995, 23, 3501–3507. [Google Scholar] [CrossRef] [PubMed]
- Carlo, T.; Sterner, D.A.; Berget, S.M. An intron splicing enhancer containing a G-rich repeat facilitates inclusion of a vertebrate micro-exon. RNA 1996, 2, 342–353. [Google Scholar] [PubMed]
- McCullough, A.J.; Berget, S.M. G triplets located throughout a class of small vertebrate introns enforce intron borders and regulate splice site selection. Mol. Cell. Biol. 1997, 17, 4562–4571. [Google Scholar] [PubMed]
- Haut, D.D.; Pintel, D.J. Intron definition is required for excision of the minute virus of mice small intron and definition of the upstream exon. J. Virol. 1998, 72, 1834–1843. [Google Scholar] [PubMed]
- McCarthy, E.M.; Phillips, J.A., III. Characterization of an intron splice enhancer that regulates alternative splicing of human GH pre-mRNA. Hum. Mol. Genet. 1998, 7, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- McCullough, A.J.; Berget, S.M. An intronic splicing enhancer binds U1 snRNPs to enhance splicing and select 5′ splice sites. Mol. Cell. Biol. 2000, 20, 9225–9235. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, C.; Murakami, K.; Fujii, Y.; Sato, Y.; Harada, E.; Takeda, J.; Taniya, T.; Sakate, R.; Kikugawa, S.; Shimada, M.; et al. The H-Invitational Database (H-InvDB), a comprehensive annotation resource for human genes and transcripts. Nucleic Acids Res. 2008, 36, D793–D799. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimada, M.K.; Sasaki-Haraguchi, N.; Mayeda, A. Identification and Validation of Evolutionarily Conserved Unusually Short Pre-mRNA Introns in the Human Genome. Int. J. Mol. Sci. 2015, 16, 10376-10388. https://doi.org/10.3390/ijms160510376
Shimada MK, Sasaki-Haraguchi N, Mayeda A. Identification and Validation of Evolutionarily Conserved Unusually Short Pre-mRNA Introns in the Human Genome. International Journal of Molecular Sciences. 2015; 16(5):10376-10388. https://doi.org/10.3390/ijms160510376
Chicago/Turabian StyleShimada, Makoto K., Noriko Sasaki-Haraguchi, and Akila Mayeda. 2015. "Identification and Validation of Evolutionarily Conserved Unusually Short Pre-mRNA Introns in the Human Genome" International Journal of Molecular Sciences 16, no. 5: 10376-10388. https://doi.org/10.3390/ijms160510376
APA StyleShimada, M. K., Sasaki-Haraguchi, N., & Mayeda, A. (2015). Identification and Validation of Evolutionarily Conserved Unusually Short Pre-mRNA Introns in the Human Genome. International Journal of Molecular Sciences, 16(5), 10376-10388. https://doi.org/10.3390/ijms160510376