
Mitochondria as Key Targets of Cardioprotection in Cardiac Ischemic Disease: Role of Thyroid Hormone Triiodothyronine



Abstract
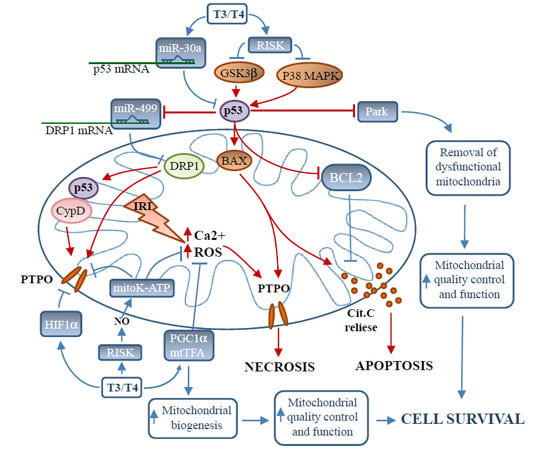
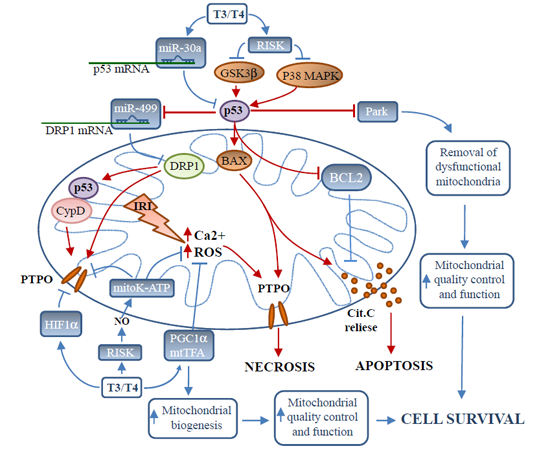
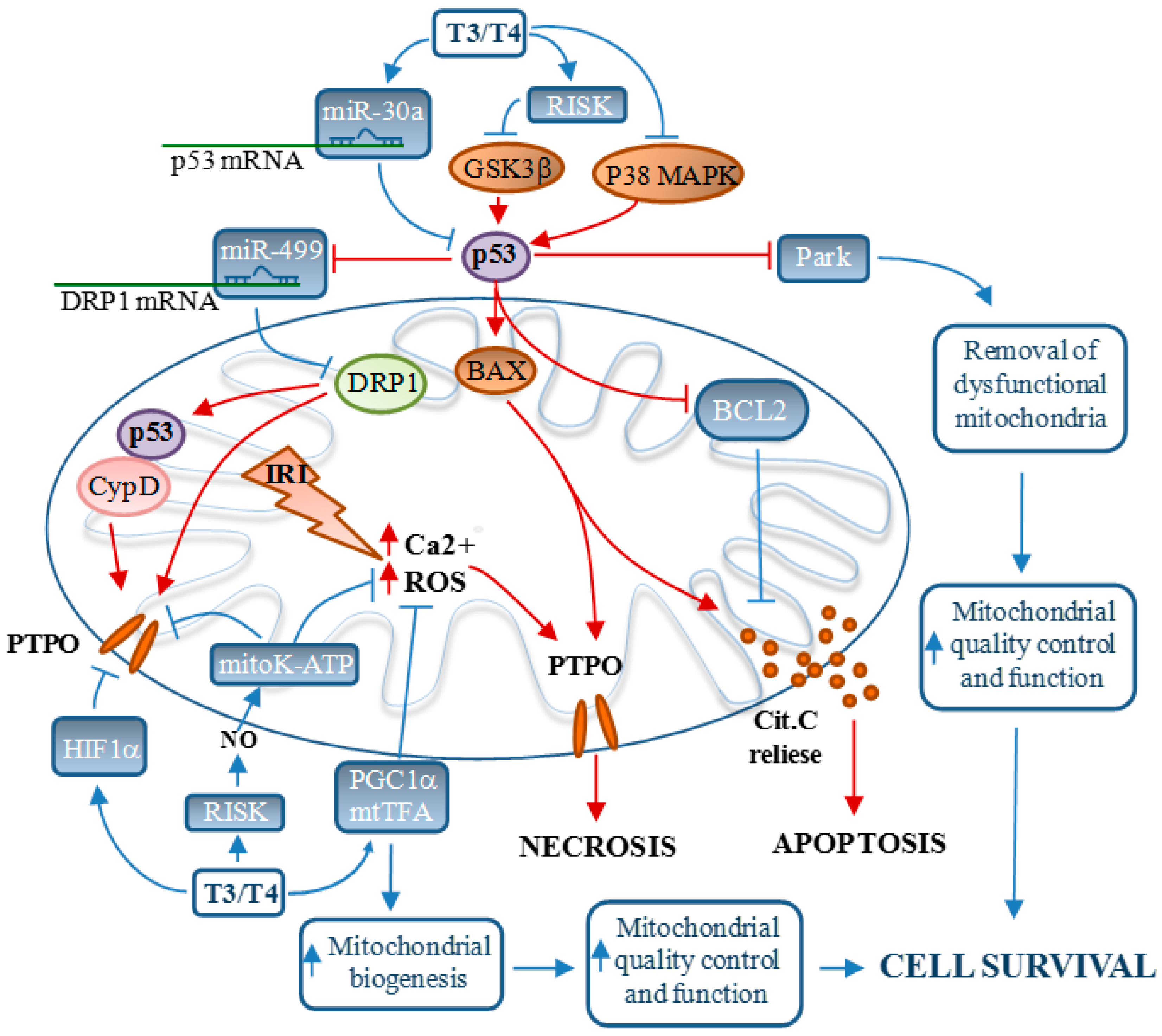
:
{kind=link}
{kind=link}
1. Introduction
2. Triggers of Mitochondrial-Dependent Cardiomyocyte Death in Ischemia/Reperfusion
2.1. Mitochondrial Dysfunction in Ischemia/Reperfusion
2.2. The p38 Mitogen-Activated Protein Kinase Intracellular Signaling
TH Inhibits p38MAPK under Stress Conditions
2.3. Tumor Suppressor Protein p53
2.3.1. p53 and Cardiomyocyte Death: Direct Action
2.3.2. p53 Regulation of Mitochondrial Morphology
2.3.3. p53 Effect on Mitophagy
3. Promoters of Mitochondria-Mediated Cardioprotection in Ischemia/Reperfusion
3.1. The Reperfusion Injury Salvage Pathway
Role of Thyroid Hormone in the Activation of Reperfusion Injury Salvage Pathway
3.2. Inhibition of p53 Signaling
Role of Thyroid Hormone in the Inhibition of p53 Signaling
3.3. Targeting Mitochondrial Oxidative Stress
Role of Thyroid Hormone in the Inhibition of Mitochondrial Oxidative Stress
3.4. Inhibition of Mitochondrial Permeability Transition Pore Opening
Role of TH in the Inhibition of Permeability Transition Pore Opening
3.5. Mitochondrial Biogenesis
Thyroid Hormone Is Key Regulator of Mitochondrial Biogenesis
3.6. MicroRNAs
Role of Thyroid Hormone in the Regulation of Cardioprotective miRNA
4. Closing Remarks and Conclusions
Acknowledgments
Abbreviations
| AMI | acute myocardial infarction |
| CypD | Cyclophilin D |
| DRP1 | dynamin-related protein |
| GSK3β | glycogen synthase kinase 3-β |
| HIF1α | Hypoxia inducible factor 1 α |
| IRI | ischemia/reperfusion injuries |
| Low T3 syndrome | Low-T3S |
| mitoK-ATP | mitochondrial ATP-dependent potassium channel |
| mtTFA | mitochondrial transcription factor A |
| Park | parkin |
| PGC1-α | peroxisome proliferator-activated receptor-γ coactivator-1α |
| PTPO | permeability transition pore opening |
| P38MAPK | P38 mitogen activated protein kinase |
| RISK | reperfusion injury salvage kinase |
Conflicts of Interest
References
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Kyle, D.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Whelan, R.S.; Kaplinskiy, V.; Kitsis, R.N. Cell death in the pathogenesis of heart disease mechanisms and significance. Annu. Rev. Physiol. 2010, 72, 19–44. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res. Cardiol. 2009, 104, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J.; Goldenthal, M.J. Mitochondrial centrality in heart failure. Heart Fail. Rev. 2008, 13, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signaling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Ovize, M.; Thibault, H.; Przyklenk, K. Myocardial conditioning: Opportunities for clinical translation. Circ. Res. 2013, 113, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, A.M.; Iervasi, G. Thyroid replacement therapy and heart failure. Circulation 2010, 122, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, A.; Chen, Y.; Gerdes, A.M.; Iervasi, G. Acute myocardial infarction and thyroid function: New pathophysiological and therapeutic perspectives. Ann. Med. 2012, 44, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Mourouzis, I.; Forini, F.; Pantos, C.; Iervasi, G. Thyroid hormone and cardiac disease: From basic concepts to clinical application. J. Thyroid Res. 2011, 2011. [Google Scholar] [CrossRef]
- Nicolini, G.; Pitto, L.; Kusmic, C.; Balzan, S.; Sabatino, L.; Iervasi, G.; Forini, F. New insights into mechanisms of cardioprotection mediated by thyroid hormones. J. Thyroid Res. 2013, 2013. [Google Scholar] [CrossRef]
- Hamilton, M.A.; Stevenson, L.W.; Luu, M.; Walden, J.A. Altered thyroid hormone metabolism in advanced heart failure. J. Am. Coll. Cardiol. 1990, 16, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Wiersinga, W.M.; Lie, K.I.; Touber, J.L. Thyroid hormones in acute myocardial infarction. Clin. Endocrinol. (Oxf.) 1981, 14, 367–374. [Google Scholar] [CrossRef]
- Friberg, L.; Drvota, V.; Bjelak, A.H.; Eggertsen, G.; Ahnve, S. Association between increased levels of reverse triiodothyronine and mortality after acute myocardial infarction. Am. J. Med. 2001, 111, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Iervasi, G.; Pingitore, A.; Landi, P.; Raciti, M.; Ripoli, A.; Scarlattini, M.; L’Abbate, A.; Donato, L. Low-T3 syndrome: A strong prognostic predictor of death in patients with heart disease. Circulation 2003, 107, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, A.; Galli, E.; Barison, A.; Iervasi, A.; Scarlattini, M.; Nucci, D.; L’abbate, A.; Mariotti, R.; Iervasi, G. Acute effects of triiodothyronine (T3) replacement therapy in patients with chronic heart failure and low-T3 syndrome: A randomized, placebo-controlled study. J. Clin. Endocrinol. Metab. 2008, 93, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Henderson, K.K.; Danzi, S.; Paul, J.T.; Leya, G.; Klein, I.; Samarel, A.M. Physiological replacement of t3 improves left ventricular function in an animal model of myocardial infarction-induced congestive heart failure. Circ. Heart Fail. 2009, 2, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Kobayashi, S.; Chen, J.; Redetzke, R.A.; Said, S.; Liang, Q.; Gerdes, A.M. Short term triiodo-Lthyronine treatment inhibits cardiac myocyte apoptosis in border area after myocardial infarction in rats. J. Mol. Cell. Cardiol. 2008, 44, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Pantos, C.; Mourouzis, I.; Markakis, K.; Tsagoulis, N.; Panagiotou, M.; Cokkinos, D.V. Long term thyroid hormone administration reshapes left ventricular chamber and improves cardiac function after myocardial infarction in rats. Basic Res. Cardiol. 2008, 103, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Wrutniak-Cabello, C.; Casas, F.; Cabello, G. Thyroid hormone action in mitochondria. J. Mol. Endocrinol. 2001, 26, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Goldenthal, M.J.; Ananthakrishnan, R.; Marín-García, J. Nuclear-mitochondrial cross-talk in cardiomyocyte T3 signaling: A time-course analysis. J. Mol. Cell. Cardiol. 2005, 39, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J. Thyroid hormone and myocardial mitochondrial biogenesis. Vascul. Pharmacol. 2010, 52, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Shintani-Ishida, K.; Inui, M.; Yoshida, K. Ischemia-reperfusion induces myocardial infarction through mitochondrial Ca2+ overload. J. Mol. Cell. Cardiol. 2012, 53, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Maundrell, K.; Martinou, J.C. Life without the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1398–1400. [Google Scholar] [CrossRef] [PubMed]
- Tompkinsa, A.J.; Burwellb, L.; Digernessc, S.B.; Zaragozac, C.; Holmanc, W.L.; Brookesa, P.S. Mitochondrial dysfunction in cardiac ischemia-reperfusion injury: ROS from complex I, without inhibition. BBA Mol. Basis Dis. 2006, 1762, 223–231. [Google Scholar] [CrossRef]
- Dorn, G.W., II. Apoptotic and non-apoptotic programmed cardiomyocyte death in ventricular remodeling. Cardiovasc. Res. 2009, 81, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.1.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012, 748, 145–169. [Google Scholar] [PubMed]
- Becker, L.B. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc. Res. 2004, 61, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Assaly, R.; d’Anglemont de Tassigny, A.; Paradis, S.; Jacquin, S.; Berdeaux, A.; Morin, D. Oxidative stress, mitochondrial permeability transition pore opening and cell death during hypoxia-reoxygenation in adult Cardiomyocytes. Eur. J. Pharmacol. 2012, 675, 6–14. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chen, T.; Szeto, H.; Nieves-Cintrón, M.; Kutyavin, V.; Santana, L.F.; Rabinovitch, P.S. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J. Am. Coll. Cardiol. 2011, 58, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P. The cardiac mitochondrion: Nexus of stress. Annu. Rev. Physiol. 2010, 72, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Brady, N.R.; Hamacher-Brady, A.; Gottlieb, R.A. Proapoptotic BCL-2 family members and mitochondrial dysfunction during ischemia/reperfusion injury, a study employing cardiac HL-1 cells and GFP biosensors. Biochim. Biophys. Acta 2006, 1757, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar]
- Garcia-Dorado, D.; Ruiz-Meana, M.; Inserte, J.; Rodriguez-Sinovas, A.; Piper, H.M. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc. Res. 2012, 94, 168–180. [Google Scholar] [CrossRef] [PubMed]
- McStay, G.P.; Clarke, S.J.; Halestrap, A.P. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem. J. 2002, 367, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.W.C.; Varanyuwatana, P.; Halestrap, A.P. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition J. Biol. Chem. 2008, 283, 26312–26323. [Google Scholar] [CrossRef]
- Kwong, J.; Davis, C.P.; Baines, M.A.; Sargent, J.; Karch, X.; Wang, X.; Huang, T.; Molkentin, J.D. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014, 21, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Wilson, S.; Belham, C.M.; Robinson, C.J.; Scott, P.H.; Gould, G.W.; Plevin, R. Stress-activated protein kinases: Activation, regulation and function. Cell Signal. 1997, 9, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.L.; Kumar, S.; Gao, F.; Louden, C.S.; Lopez, B.L.; Christopher, T.A.; Wang, C.; Lee, J.C.; Feuerstein, G.Z.; Yue, T.L. Inhibition of p38 mitogen-activated protein kinase decreases cardiomyocyte apoptosis and improves cardiac function after myocardial ischemia and reperfusion. Circulation 1999, 99, 1685–1689. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, S.; Sharma, A.K.; Singla, D.K.; Singal, P.K. p38 and ERK1/2 MAPKs mediate the interplay of TNF-α and IL-10 in regulating oxidative stress and cardiac myocyte apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3524–H3531. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.I.; Ebner, M.; Wallner, C.; Haller, M.; Khalid, S.; Schwelberger, H.; Koziel, K.; Enthammer, M.; Hermann, M.; Sickinger, S.; et al. A p38MAPK/MK2 signaling pathway leading to redox stress, cell death and ischemia/reperfusion injury. Cell Commun. Signal. 2014, 12. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, S.; Kovacs, A.; Wang, Y.; Muslin, A.J. Role of p38α MAPK in cardiac apoptosis and remodeling after myocardial infarction. J. Mol. Cell. Cardiol. 2005, 38, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Capano, M.; Crompton, M. Bax translocates to mitochondria of heart cells during simulated ischaemia: Involvement of AMP-activated and p38 mitogen-activated protein kinases. Biochem. J. 2006, 395, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.D.; de Nicola, G.F.; Marber, M.S. New therapeutic targets in cardiology: p38 α mitogen-activated protein kinase for ischemic heart disease. Circulation 2012, 126, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.A.; Bueno, O.F.; Lips, D.J.; Doevendans, P.A.; Jones, F.; Kimball, T.F.; Molkentin, J.D. Targeted inhibition of p38 mitogen-activated protein kinase antagonizes cardiac injury and cell death following ischemia-reperfusion in vivo. J. Biol. Chem. 2004, 279, 15524–15530. [Google Scholar] [CrossRef] [PubMed]
- Jeong, C.W.; Yoo, K.Y.; Lee, S.H.; Jeong, H.J.; Lee, C.S.; Kim, S. Curcumin protects against regional myocardial ischemia/reperfusion injury through activation of RISK/GSK-3β and inhibition of p38 MAPK and JNK. J. Cardiovasc. Pharmacol. Ther. 2012, 17, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Genovese, T.; Impellizzeri, D.; Ahmad, A.; Cornelius, C.; Campolo, M.; Cuzzocrea, S.; Esposito, E. Post ischaemic thyroid hormone treatment in a rat model of acute stroke. Brain Res. 2013, 1513, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Pantos, C.; Malliopoulou, V.; Mourouzis, I.; Karamanoli, E.; Tzeis, S.M.; Carageorgiou, H.; Varonos, D.; Cokkinos, D.V. Long-term thyroxine administration increases HSP70 mRNA expression and attenuates p38 MAP kinase activity in response to ischaemia. J. Endocrinol. 2001, 70, 207–215. [Google Scholar] [CrossRef]
- Pantos, C.; Mourouzis, I.; Saranteas, T.; Clavé, G.; Ligeret, H.; Noack-Fraissignes, P.; Renard, P.Y.; Massonneau, M.; Perimenis, P.; Spanou, D.; et al. Thyroid hormone improves postischaemic recovery of function while limiting apoptosis: A new therapeutic approach to support hemodynamics in the setting of ischaemia-reperfusion? Basic Res. Cardiol. 2009, 104, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Mourouzis, I.; Kostakou, E.; Galanopoulos, G.; Mantzouratou, P.; Pantos, C. Inhibition of thyroid hormone receptor α1 impairs post ischemic cardiac performance after myocardial infarction in mice. Mol. Cell. Biochem. 2013, 379, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H. p53, death star. Cell 2000, 103, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Reed, J. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Boluyt, M.O.; Hipolito, M.; Lundberg, M.S.; Zheng, J.S.; O’Neill, L.; Cirielli, C.; Lakatta, E.G.; Crow, M.T. p53 and the hypoxia-induced apoptosis of cultured neonatal rat cardiac myocytes. J. Clin. Investig. 1997, 99, 2635–2643. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010, 88, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Dieter, A.; Kubli, Å.B. Gustafsson mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [PubMed]
- Dimmer, K.S.; Scorrano, L. (De)constructing mitochondria: What for? Physiology 2006, 21, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Scorrano, L. Targeting cell death. Clin. Pharmacol. Ther. 2007, 82, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Zheng, M.; Cao, C.; Chen, C.; Tang, J.; Zhang, W.; Cheng, H.; Chen, K.H.; Xiao, R.P. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J. Biol. Chem. 2007, 282, 23354–23361. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2202. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Xu, L.; Yu, Y.; Zhu, W.; Andrews, D.W.; Yoon, Y.; Kuo, T.H. Regulation of Ca2+-induced permeability transition by Bcl-2 is antagonized by Drpl and hFis1. Mol. Cell Biochem. 2005, 272, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiao, J.; Li, Q.; Long, B.; Wang, K.; Liu, J.; Li, Y.; Li, P. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 2011, 17, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Sesaki, H.H.; Qi, X. Drp1 stabilizes p53 on the mitochondria to trigger necrosis under oxidative stress conditions in vitro and in vivo. Biochem. J. 2014, 461, 137–146. [Google Scholar] [PubMed]
- Whelan, R.S.; Konstantinidis, K.; Wei, A.C.; Chen, Y.; Reyna, D.E.; Jha, S.; Yang, Y.; Calvert, J.W.; Lindsten, T.; Thompson, C.B.; et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc. Natl. Acad. Sci. USA 2012, 109, 6566–6571. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, S.E.; Calvani, R.; Marzetti, E. The interplay between autophagy and mitochondrial dysfunction in oxidative stress-induced cardiac aging and pathology. J. Mol. Cell. Cardiol. 2014, 71, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W., II. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Hoshino, A.; Matoba, S.; Iwai-Kanai, E.; Nakamura, H.; Kimata, M.; Nakaoka, M.; Katamura, M.; Okawa, Y.; Ariyoshi, M.; Mita, Y.; et al. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J. Mol. Cell. Cardiol. 2012, 52, 1175–1184. [Google Scholar] [CrossRef]
- Cohen, P.; Frame, S. The renaissance of GSK3B. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Sussman, M.A.; Völkers, M.; Fischer, K.; Bailey, B.; Cottage, C.T.; Din, S.; Gude, N.; Avitabile, D.; Alvarez, R.; Sundararaman, B.; et al. Myocardial AKT: The omnipresent nexus. Physiol. Rev. 2011, 91, 1023–1070. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. New directions for protecting the heart against ischaemia–reperfusion injury: Targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc. Res. 2004, 61, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Linseman, D.A.; Butts, B.D.; Precht, T.A.; Phelps, R.A.; Le, S.S.; Laessig, T.A.; Bouchard, R.J.; Florez-McClure, M.L.; Heidenreich, K.A. Glycogen synthase kinase-3β phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J. Neurosci. 2004, 24, 9993–10002. [Google Scholar] [CrossRef] [PubMed]
- Juhaszova, M.; Zorov, D.B.; Kim, S.H.; Pepe, S.; Fu, Q.; Fishbein, K.W.; Ziman, B.D.; Wang, S.; Ytrehus, K.; Antos, C.L.; et al. Glycogen synthase kinase-3β mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Investig. 2004, 113, 1535–1549. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Paillard, M.; Thibault, H.; Derumeaux, G.; Ovize, M. Inhibition of GSK3β by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation 2008, 117, 2761–2768. [Google Scholar] [CrossRef] [PubMed]
- Watcharasit, P.; Bijur, G.N.; Song, L.; Zhu, J.; Chen, X.; Jope, R.S. Glycogen synthase kinase-3b(GSK3b) binds to and promotes the actions of p53. J. Biol. Chem. 2003, 278, 48872–48879. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Sciacovelli, M.; Chiara, F.; Pantic, B.; Brusilow, W.S.; Bernardi, P. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc. Natl. Acad. Sci. USA 2010, 107, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Phukan, S.; Babu, V.S.; Kannoji, A.; Hariharan, R.; Balaji, V.N. GSK3β: Role in therapeutic landscape and development of modulators. Br. J. Pharmacol. 2010, 160, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Tanno, M.; Kuno, A.; Ishikawa, S.; Miki, T.; Kouzu, H.; Yano, T.; Murase, H.; Tobisawa, T.; Ogasawara, M.; Horio, Y.; et al. Translocation of Glycogen Synthase Kinase-3β (GSK-3β), a trigger of permeability transition, is kinase activity dependent and mediated by interaction with voltage-dependent anion channel 2 (VDAC2). J. Biol. Chem. 2014, 289, 29285–29296. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.J.; Hsu, A.; Pfeiffer, A.W.; Nithipatikom, K. Roles of endothelial nitric oxide synthase (eNOS) and mitochondrial permeability transition pore (MPTP) in epoxyeicosatrienoic acid (EET)-induced cardioprotection against infarction in intact rat hearts. J. Mol. Cell. Cardiol. 2013, 59, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Sato, T.; Ohler, A.; O'Rourke, B.; Marbán, E. Activation of mitochondrial ATP-dependent potassium channels by nitric oxide. Circulation 2000, 101, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Rastaldo, R.; Mancardi, D.; Raimondo, S.; Cappello, S.; Gattullo, D.; Losano, G.; Pagliario, P. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res. Cardiol. 2006, 101, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Gucek, M.; Murphy, E. What can we learn about cardioprotection from the cardiac mitochondrial proteome? Cardiovasc. Res. 2010, 88, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Burwell, L.S.; Brookes, P.S. Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxid. Redox Signal. 2008, 10, 579–599. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hall, A.R.; Dongworth, R.K.; Kalkhoran, S.; Pyakurel, A.; Scorrano, L.; Hausenloy, D.J. Akt protects the heart against ischaemia/reperfusion injury by modulating mitochondrial morphology. Thromb. Haemost. 2015, 113, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Kenessey, A.; Ojamaa, K. Thyroid hormone stimulates protein synthesis in the cardiomyocyte by activating the Akt-mTOR and p70S6Kpathways. J. Biol. Chem. 2006, 28, 20666–20672. [Google Scholar] [CrossRef]
- Kuzman, J.A.; Gerdes, A.M.; Kobayashi, S.; Liang, Q. Thyroid hormone activates Akt and prevents serum starvation-induced cell death in neonatal rat cardiomyocytes. J. Mol. Cell. Cardiol. 2005, 39, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Kuzman, J.A.; Vogelsang, K.A.; Thomas, T.A.; Gerdes, A.M. L-Thyroxine activates Akt signaling in the heart. J. Mol. Cell. Cardiol. 2005, 39, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Mourouzis, I.; Mantzouratou, P.; Galanopoulos, G.; Kostakou, E.; Roukounakis, N.; Kokkinos, A.D.; Cokkinos, D.V.; Pantos, C. Dose-dependent effects of thyroid hormone on post-ischemic cardiac performance: Potential involvement of Akt and ERK signalings. Mol. Cell. Biochem. 2012, 363, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Kehat, I.; Davis, J.; Tiburcy, M.; Accornero, F.; Saba-El-Leil, M.K.; Maillet, M.; York, A.J.; Lorenz, J.N.; Zimmermann, W.H.; Meloche, S.; et al. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ. Res. 2010, 108, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Naito, A.T.; Okada, S.; Minamino, T.; Iwanaga, K.; Liu, M.L.; Sumida, T.; Nomura, S.; Sahara, N.; Mizoroki, T.; Takashima, A.; et al. Promotion of chip-mediated p53 degradation protects the heart from ischemic injury. Circ. Res. 2010, 106, 1692–1702. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Davis, P.J.; Tang, H.Y.; Mousa, S.A.; Luidens, M.K.; Hercbergs, A.H.; Davis, F.B. The pro-apoptotic action of stilbene-induced COX-2 in cancer cells: Convergence with the anti-apoptotic effect of thyroid hormone. Cell Cycle 2009, 8, 1877–1882. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Tang, H.Y.; Keating, T.; Wu, Y.H.; Shih, A.; Hammond, D.; Sun, M.; Hercbergs, A.; Davis, F.B.; Davis, P.J. Resveratrol is pro-apoptotic and thyroid hormone is anti-apoptotic in glioma cells: Both actions are integrin and ERK mediated. Carcinogenesis 2008, 29, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Yap, N.; Yu, C.L.; Cheng, S.Y. Modulation of thetrascriptional activity ofthyroid hormone receptor by the tumor suppressor p53. Proc. Natl. Acad. Sci. USA 1996, 93, 4273–4277. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.K.; Yu, C.l.; Yap, N.; Zhan, Q.; Hayashi, Y.; Seth, P. Tumor suppressor p53 is a negative regulator in thyroid hormone receptor signaling pathways. J. Biol. Chem. 1997, 272, 28989–28993. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Kusmic, C.; Nicolini, G.; Mariani, L.; Zucchi, R.; Matteucci, M.; Iervasi, G.; Pitto, L. Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/p53 axis. Endocrinology 2014, 155, 4581–4590. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Lionetti, V.; Ardehali, H.; Pucci, A.; Cecchetti, F.; Ghanefar, M.; Nicolini, G.; Ichikawa, Y.; Nannipieri, M.; Recchia, F.A.; et al. Early long-term L-T3 replacement rescues mitochondria and prevents ischemic cardiac remodelling in rats. J. Cell. Mol. Med. 2011, 15, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Dagenais, G.; Pogue, J.; Bosch, J.; Sleight, P. Vitamin E supplementation and cardiovascular events in high-risk patients. The heart outcomes prevention evaluation study investigators. N. Engl. J. Med. 2000, 342, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Hare, J.M.; Mangal, B.; Brown, J.; Fisher, C., Jr.; Freudenberger, R.; Colucci, W.S.; Mann, D.L.; Liu, P.; Givertz, M.M.; Schwarz, R.P.; OPT-CHF Investigators. Impact of oxypurinol in patients with symptomatic heart failure: Results of the OPT-CHF study. J. Am. Coll. Cardiol. 2008, 5, 2301–2309. [Google Scholar] [CrossRef]
- Brown, D.A.; Hale, S.L.; Baines, C.P.; del Rio, C.L.; Hamlin, R.L.; Yueyama, Y.; Kijtawornrat, A.; Yeh, S.T.; Frasier, C.R.; Stewart, L.M.; et al. Reduction of early reperfusion injury with the mitochondria-targeting peptide bendavia. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antiox. Redox Signal. 2008, 10, 601–619. [Google Scholar] [CrossRef]
- Zhao, K.; Zhao, G.M.; Wu, D.; Soong, Y.; Birk, A.V.; Schiller, P.W.; Szeto, H.H. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 2004, 279, 34682–34690. [Google Scholar] [CrossRef] [PubMed]
- De Castro, A.L.; Tavares, A.V.; Campos, C.; Fernandes, R.O.; Siqueira, R.; Conzatti, A.; Bicca, A.M.; Fernandes, T.R.; Sartório, C.L.; Schenkel, P.C.; et al. Cardioprotective effects of thyroid hormones ina rat model of myocardial infarction are associated with oxidative stress reduction. Mol. Cell. Endocrinol. 2014, 391, 22–29. [Google Scholar] [CrossRef]
- Di Lisa, F.; Menabò, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 2001, 276, 2571–2575. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.J.; McStay, G.P.; Halestrap, A.P. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J. Biol. Chem. 2002, 277, 34793–34799. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Chen, X.; Baines, C.P.; Klevitsky, R.; Zhang, X.; Zhang, H.; Jaleel, N.; Chua, B.H.; Hewett, T.E.; Robbins, J.; et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Investig. 2007, 117, 2431–2444. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Akao, M.; Matsumoto-Ida, M.; Makiyama, T.; Iguchi, M.; Takeda, T.; Shimizu, S.; Kita, T. The targeting of cyclophilin D by RNAi as a novel cardioprotective therapy: Evidence from two-photon imaging. Cardiovasc. Res. 2009, 83, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Elrod, J.W.; Wong, R.; Mishra, S.; Vagnozzi, R.J.; Sakthievel, B.; Goonasekera, S.A.; Karch, J.; Gabel, S.; Farber, J.; Force, T.; et al. Cyclophilin D controls mitochondrial pore-dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Investig. 2010, 120, 3680–3687. [Google Scholar] [CrossRef] [PubMed]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–448. [Google Scholar] [CrossRef] [PubMed]
- Fuglesteg, B.N.; Suleman, N.; Tiron, C.; Kanhema, T.; Lacerda, L.; Andreasen, T.V.; Sack, M.N.; Janassen, A.K.; Mjos, O.D.; Opie, L.H.; et al. Signal transducer and activator of transcription 3 is involved in the cardioprotective signaling pathway activated by insulin therapy at reperfusion. Bas. Res. Cardiol. 2008, 103, 444–453. [Google Scholar] [CrossRef]
- Lacerda, L.; Somers, S.; Opie, L.H.; Lecour, S. Ischemic postconditioning protect against reperfusion injury via SAFE pathway. Cardiovasc. Res. 2009, 84, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Hilfiker-Kleiner, D.; Heusch, G.; Schulz, R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 2010, 105, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Hataishi, R.; Rodrigues, A.C.; Neilan, T.G.; Morgan, J.G.; Buys, E.; Shiva, S.; Tambouret, R.; Jassal, D.S.; Raher, M.J.; Furutani, E.; et al. Inhaled nitric oxide decreases infarction size and improves left ventricular function in a murine model of myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H379–H384. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, Y.; Fernandez, B.O.; Garcia-Saura, M.F.; Petersen, B.; Ichinose, F.; Bloch, K.D.; Feelisch, M.; Zapol, W.M. Brief periods of nitric oxide inhalation protect against myocardial ischemia-reperfusion injury. Anesthesiology 2008, 109, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, Y.; Pokreisz, P.; Vermeersch, P.; Marsboom, G.; Swinnen, M.; Verbeken, E.; Santos, J.; Pellens, M.; Gillijns, H.; et al. Nitric oxide inhalation improves microvascular flow and decreases infarction size after myocardial ischemia and reperfusion. J. Am. Coll. Cardiol. 2007, 50, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Liem, D.A.; Vondriska, T.M.; Honda, H.M.; Korge, P.; Pantaleon, D.M.; Qiao, X.; Wang, Y.; Weiss, J.N.; Ping, P. Nitric oxide donors protect murine myocardium against infarction via modulation of mitochondrial permeability transition. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1290–H1295. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.G.; Hausenloy, D.J. Hypoxia-inducible factor as a therapeutic target for cardioprotection. Pharmacol. Ther. 2012, 136, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.G.; Lee, W.H.; Theodorou, L.; Kodo, K.; Lim, S.Y.; Shukla, D.H.; Briston, T.; Kiriakidis, S.; Ashcroft, M.; Davidson, S.M.; et al. HIF-1 reduces ischaemia-reperfusion injury in the heart by targeting the mitochondrial permeability transition pore. Cardiovasc. Res. 2014, 104, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Benard, G.; Bellance, N.; Jose, C.; Melser, S.; Nouette-Gaulain, K.; Rossignol, R. Multi-site control and regulation of mitochondrial energy production. Biochim. Biophys. Acta 2010, 1797, 698–709. [Google Scholar] [CrossRef] [PubMed]
- McLeod, C.J.; Pagel, I.; Sack, M.N. The mitochondrial biogenesis regulatory program adaptation to ischemia-A putative target for therapeutic intervention. Trends Cardiovasc. Med. 2005, 15, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Hock, M.B.; Kralli, A. Transcriptional control of mitochondrial biogenesis and function. Annu. Rev. Physiol. 2009, 71, 177–203. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1α. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V.; Joubert, F. Bioenergetics of the failing heart. Biochim. Biophys. Acta 2011, 1813, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Garnier, A.; Fortin, D.; Deloménie, C.; Momken, I.; Veksler, V.; Ventura-Clapier, R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J. Physiol. 2003, 551, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Reusch, J.E.; McCune, S.A.; Leinwand, L.A.; Luckey, S.W.; Konhilas, J.P. Restoration of CREB function is linked to completion and stabilization of adaptive cardiac hypertrophy in response to exercise. Am. J. Physiol. 2007, 293, H246–H259. [Google Scholar]
- Lehman, J.J.; Kelly, D.P. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin. Exp. Pharmacol. Physiol. 2002, 29, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPARγ coactivator 1α. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef] [PubMed]
- Sihag, S.; Li, A.Y.; Cresci, S.; Sucharov, C.C.; Lehman, J.J. PGC-1α and ERRα target gene down-regulation is a signature of the failing human heart. J. Mol. Cell. Cardiol. 2009, 46, 201–212. [Google Scholar] [CrossRef]
- Ahuja, P.; Zhao, P.; Angelis, E.; Ruan, H.; Korge, P.; Olson, A.; Wang, Y.; Jin, E.S.; Jeffrey, F.M.; Portman, M.; et al. Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J. Clin. Investig. 2010, 120, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zhang, H.; Liu, P.; Wang, H.; Liu, J.; Gao, C.; Liu, Y.; Lian, K.; Yang, L.; Sun, L.; et al. Impaired mitochondrial biogenesis due to dysfunctional adiponectin-AMPK-PGC-1α signaling contributing to increased vulnerability in diabetic heart. Basic Res. Cardiol. 2013, 108. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhao, M.; Yu, X.J.; Wang, H.; He, X.; Liu, J.K.; Zang, W.J. Cardioprotection by acetylcholine: A novel mechanism via mitochondrial biogenesis and function involving the PGC-1α pathway. J. Cell. Physiol. 2013, 228, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Lin, J.; Krauss, S.; Tarr, P.T.; Yang, R.; Newgard, C.B.; Spiegelman, B.M. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1α and 1β (PGC-1α and PGC-1β) in muscle cells. J. Biol. Chem. 2003, 278, 26597–26603. [Google Scholar] [CrossRef] [PubMed]
- Kajander, O.A.; Karhunen, P.J.; Jacobs, H.T. The relationship between somatic mtDNA rearrangements, human heart disease and aging. Hum. Mol. Genet. 2002, 11, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Naya, F.J.; Black, B.L.; Wu, H.; Bassel-Duby, R.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat. Med. 2002, 8, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Lebrecht, D.; Setzer, B.; Ketelsen, U.P.; Haberstroh, J.; Walker, U.A. Timedependent and tissue-specific accumulation of mtDNA and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation 2003, 108, 2423–2429. [Google Scholar] [CrossRef] [PubMed]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Utsumi, H.; Kang, D.; Hattori, N.; Uchida, K.; Arimura, K.; Egashira, K.; Takeshita, A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ. Res. 1999, 85, 357–363. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Wilhelmsson, H.; Hansson, A.; Thoren, P.; Duffy, J.; Rustin, P.; Larsson, N.G. Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proc. Natl. Acad. Sci. USA 2000, 97, 3467–3472. [Google Scholar]
- Wang, J.; Wilhelmsson, H.; Graff, C.; Li, H.; Oldfors, A.; Rustin, P.; Bruning, J.C.; Kahn, C.R.; Clayton, D.A.; Barsh, G.S.; et al. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat. Genet. 1999, 21, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, M.; Matsusaka, H.; Kang, D.; Matsushima, S.; Ide, T.; Kubota, T.; Fujiwara, T.; Hamasaki, N.; Takeshita, A.; Sunagawa, K.; et al. Overexpression of mitochondrial transcription factor a ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation 2005, 112, 683–669. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Sirtuins in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Guarente, L. Sirtuins at a glance. J. Cell Sci. 2011, 124, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Vigilanza, P.; Baldelli, S.; Pagliei, B.; Rotilio, G.; Ciriolo, M.R. Peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) and sirtuin 1 (SIRT1) reside in mitochondria. Possible direct function ion mitochondrila biogenesis. J. Biol. Chem. 2010, 285, 21590–21599. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [PubMed]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.P.; Zhai, P.; Yamamoto, T.; Maejima, Y.; Matsushima, S.; Hariharan, N.; Shao, D.; Takagi, H.; Oka, S.; Sadoshima, J. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 2010, 122, 2170–2182. [Google Scholar] [CrossRef] [PubMed]
- Orallo, F.; Alvarez, E.; Camina, M.; Leiro, J.M.; Gomez, E.; Fernandez, P. The possible implication of trans-Resveratrol in the cardioprotective effects of long-term moderate wine consumption. Mol. Pharmacol. 2002, 61, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Biala, A.; Tauriainen, E.; Siltanen, A.; Shi, J.; Merasto, S.; Louhelainen, M.; Martonen, E.; Finckenberg, P.; Muller, D.N.; Mervaala, E. Resveratrol induces mitochondrial biogenesis and ameliorates Ang II-induced cardiac remodeling in transgenic rats harboring human renin and angiotensinogen genes. Blood Press. 2010, 19, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, J.M.; Iwen, K.A. Coordination of mitochondrial biogenesis by thyroid hormone. Mol. Cell. Endocrinol. 2011, 342, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; Bari, A.; di Stefano, L.; Cardone, A.; della Ragione, F.; D’Esposito, M.; di Meo, S. Involvement of PGC-1, NRF-1, and NRF-2 in metabolic response by rat liver to hormonal and environmental signals. Mol. Cell. Endocrinol. 2009, 305, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Wulf, A.; Harneit, A.; Kröger, M.; Kebenko, M.; Wetzel, M.G.; Weitzel, J.M. T3-mediated expression of PGC-1α via a far upstream located thyroid hormone response element. Mol. Cell. Endocrinol. 2008, 287, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, C.L.; Guilbeau-Frugier, C.; Sicard, P.; Lairez, O.; Ordener, C.; Duparc, T.; de Paulis, D.; Couderc, B.; Spreux-Varoquaux, O.; Tortosa, F.; et al. p53-PGC-1α pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A up-regulation: Role in chronic left ventricular dysfunction in mice. Antioxid. Redox Signal. 2013, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Ingwall, J.S. Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 2009, 81, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Ardehali, H.; Sabbah, H.N.; Burke, M.A.; Sarma, S.; Liu, P.P.; Cleland, J.G.; Maggioni, A.; Fonarow, G.C.; Abel, E.D.; Campia, U.; et al. Targeting myocardial substrate metabolism in heart failure: Potential for new therapies. Eur. J. Heart Fail. 2012, 14, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Wrutniak-Cabello, C.; Carazo, A.; Casas, F.; Cabello, G. Triiodothyronine mitochondrial receptors: Import and molecular mechanisms. J. Soc. Biol. 2008, 202, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Saelim, N.; Holstein, D.; Chocron, E.S.; Camacho, P.; Lechleiter, J.D. Inhibition of apoptotic potency by ligand stimulated thyroid hormone receptors located in mitochondria. Apoptosis 2007, 12, 1781–1794. [Google Scholar] [CrossRef] [PubMed]
- Saelim, N.; John, L.M.; Wu, J.; Park, J.S.; Bai, Y.; Camacho, P.; Lechleiter, J.D. Nontranscriptional modulation of intracellular Ca2+ signaling by ligand stimulated thyroid hormone receptor. J. Cell Biol. 2004, 167, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Barringhaus, K.G.; Zamore, P.D. MicroRNAs: Regulating a change of heart. Circulation 2009, 119, 2217–2224. [Google Scholar] [CrossRef] [PubMed]
- Divakaran, V.; Mann, D.L. The emerging role of microRNAs in cardiac remodeling and heart failure. Circ. Res. 2008, 103, 1072–1083. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Galuppo, P.; Wolf, C.; Fiedler, J.; Kneitz, S.; van Laake, L.W.; Doevendans, P.A.; Mummery, C.L.; Borlak, J.; Haverich, A.; et al. MicroRNAs in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation 2007, 116, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Marshall, W.; Olson, E. Toward microRNA-based therapeutics for heart disease the sense in antisense. Circ. Res. 2008, 103, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive micrornas that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; diMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Khanna, S.; Hussain, S.R.; Biswas, S.; Azad, A.; Rink, C.; Gnyawali, S.; Shilo, S.; Nuovo, G.J.; Sen, C.K. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc. Res. 2009, 82, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.P.; Wu, J.; Wang, X.; Sartor, M.A.; Qian, J.; Jones, K.; Nicolaou, P.; Pritchard, T.J.; Fan, G.C. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation 2009, 119, 2357–2366. [Google Scholar] [CrossRef] [PubMed]
- Sripada, L.; Tomar, D.; Singh, R. Mitochondria: One of the destinations of miRNAs. Mitochondrion 2012, 12, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Perez-Polo, J.R.; Qian, J.; Birnbaum, Y. The role of microRNA in modulating myocardial ischemia-reperfusion injury. Physiol. Genomics 2011, 43, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Aurora, A.B.; Mahmoud, A.I.; Luo, X.; Johnson, B.A.; van Rooij, E.; Matsuzaki, S.; Humphries, K.M.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; et al. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. J. Clin. Investig. 2012, 122, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Ren, X.P.; Chen, J.; Liu, H.; Yang, J.; Medvedovic, M.; Hu, Z.; Fan, G.C. MicroRNA494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion-induced cardiac injury. Circulation 2010, 122, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Donath, S.; Li, Y.; Qin, D.; Prabhakar, B.; Li, P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010, 6, e1000795. [Google Scholar] [CrossRef] [PubMed]
- Duisters, R.F.; Tijsen, A.J.; Schroen, B.; Leenders, J.J.; Lentink, V.; van der Made, I.; Herias, V.; van Leeuwen, R.E.; Schellings, M.W.; Barenbrug, P.; et al. miR-133 and miR-30 regulate connective tissue growth factor: Implications for a role of microRNAs in myocardial matrix remodeling. Circ. Res. 2009, 104, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Gambacciani, C.; Kusmic, C.; Chiavacci, E.; Meghini, F.; Rizzo, M.; Mariani, L.; Pitto, L. miR-29a and miR-30c negatively regulate DNMT3a in cardiac ischemic tissues: Implications for cardiac remodelling. microRNA Diagn. Ther. 2013, 2013, 34–44. [Google Scholar]
- Pantos, C.; Mourouzis, I.; Cokkinos, D.V. Thyroid hormone and cardiac repair/regeneration: From Prometheus myth to reality? Can. J. Physiol. Pharmacol. 2012, 90, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Braunwald, E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 1990, 81, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, A.; Eriksson, S.V.; Hall, C.; Kahan, T.; Swedberg, K. Early neurohormonal effects of trandolapril in patients with left ventricular dysfunction and a recent acute myocardial infarction: A double-blind, randomized, placebo-controlled multicentre study. Eur. J. Heart Fail. 2001, 3, 69–78. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forini, F.; Nicolini, G.; Iervasi, G. Mitochondria as Key Targets of Cardioprotection in Cardiac Ischemic Disease: Role of Thyroid Hormone Triiodothyronine. Int. J. Mol. Sci. 2015, 16, 6312-6336. https://doi.org/10.3390/ijms16036312
Forini F, Nicolini G, Iervasi G. Mitochondria as Key Targets of Cardioprotection in Cardiac Ischemic Disease: Role of Thyroid Hormone Triiodothyronine. International Journal of Molecular Sciences. 2015; 16(3):6312-6336. https://doi.org/10.3390/ijms16036312
Chicago/Turabian StyleForini, Francesca, Giuseppina Nicolini, and Giorgio Iervasi. 2015. "Mitochondria as Key Targets of Cardioprotection in Cardiac Ischemic Disease: Role of Thyroid Hormone Triiodothyronine" International Journal of Molecular Sciences 16, no. 3: 6312-6336. https://doi.org/10.3390/ijms16036312
APA StyleForini, F., Nicolini, G., & Iervasi, G. (2015). Mitochondria as Key Targets of Cardioprotection in Cardiac Ischemic Disease: Role of Thyroid Hormone Triiodothyronine. International Journal of Molecular Sciences, 16(3), 6312-6336. https://doi.org/10.3390/ijms16036312