
Autophagy as a Regulatory Component of Erythropoiesis

Abstract
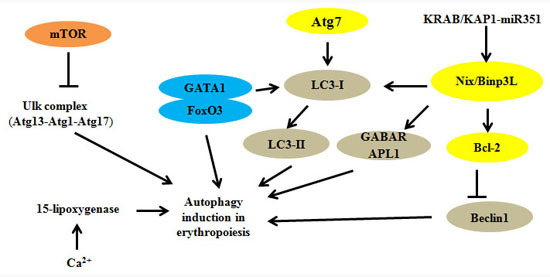
:
1. Introduction
2. Autophagy Regulators and Erythroid Maturation

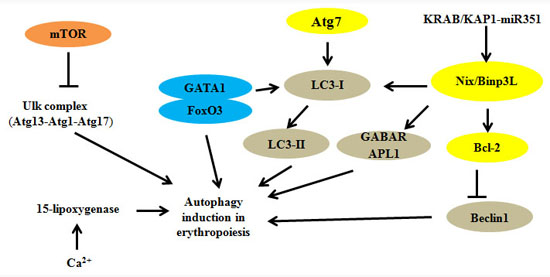{kind=link}
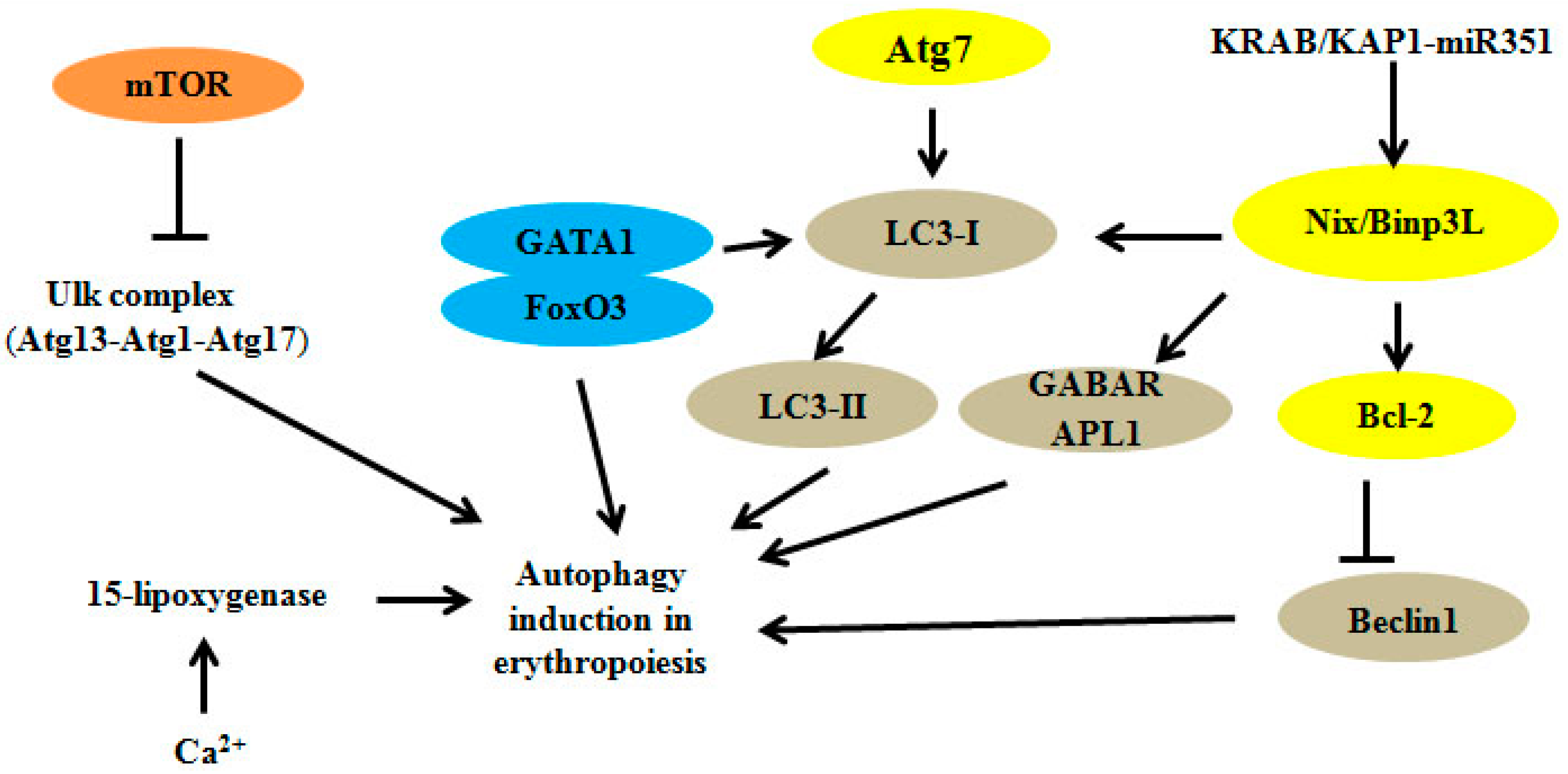{kind=link}
| Modulators | Interactions with Other Molecules or Targets | Functions | References |
|---|---|---|---|
| Atg1/Ulk1 | Atg13, Hsp90-Cdc37 | Regulation of mitochondrial and ribosomal clearance | [12,13,14] |
| Atg4 | - | Fusion of autophagosomes with lysosomes | [15] |
| Atg7 | Atg5 | Regulation of mitochondrial removal | [16,17,18,19,20,21] |
| Nix/Bnip3L | LC3, Atg8, miRNA | Modulation of mitochondrial clearance and autophagosome formation | [22,23,24,25,26,27] |
| GATA1 | FoxO3, LC3-I | Direct regulation of autophagy genes | [28,29,30,31,32,33] |
| KRAB/KAP1-miRNA | Nix/Bnip3L, Ulk1 | Participation in cascade controlling mitophagy | [34] |
| FIP200 | Ulk1, Atg13 | Essential autophagy gene in hematopoietic cells | [35,36] |
| Ca2+ and 15-lipoxygenase | - | Ca2+ promotes binding of 15-lipoxygenase to modulate the clearance of mitochondria | [37,38,39] |
2.1. Autophagy-Related Gene (Atg) Family
2.2. Uncoordinated 51-Like Autophagy Activating Kinase 1 (Ulk1)
2.3. Autophagy-Related 4 (Atg4)
2.4. Autophagy-Related 7 (Atg7)
2.5. Bcl-2 Family: Bcl-2/Adenovirus E1B 19 kDa Interacting Protein 3-Like (Nix/Binp3L)
2.6. Transcription Factors and KAP1
2.7. Other Modulators: FIP200, Ca2+ and 15-Lipoxygenase
3. Autophagy and β-Thalassemia
4. Perspectives
Acknowledgments
Author Contributions
Abbreviation
| 17AAG | 17-allylamino-17-demethoxygeldanamycin |
| AMPK | AMP-activated protein kinase |
| Atg | autophagy-related |
| Bcl-2 | B-cell lymphoma 2 |
| BFU-E | burst-forming-unit erythroid |
| CCCP | carbonyl cyanide 3-chlorophenylhydrazone |
| Cdc37 | cell division cycle 37 |
| CFU-E | colony-forming-unit erythroid |
| CNS | central nervous system |
| FIP200 | focal adhesion kinase family-interacting protein of 200-kDa |
| FoxO3 | forkhead box O3 |
| GABARAP | gamma aminobutyric acid A receptor-associated protein |
| GATA1 | globin transcription factor |
| HEL | human erythroleukemia |
| HSCs | hematopoietic stem cells |
| Hsp90 | 90 kDa heat shock protein |
| KRAB | Krueppel-associated box |
| KAP-1 | KRAB-associated protein 1 |
| KRIP-1 | KRAB-interacting protein 1 |
| LC3 | light chain 3 |
| LSK | Lin−Sca1+c-Kit+ |
| MCV | mean cell volume |
| MCH | mean corpuscular hemoglobin |
| MDS | myelodysplasia syndrome |
| MEL | mouse erythroleukemia |
| MER | minimal essential region |
| mTOR | mammalian target of rapamycin |
| Nix/Binp3L | Bcl-2/adenovirus E1B 19 kDa interacting protein 3-like |
| PE | phosphatidylethanolamine |
| RBCs | red blood cells |
| ROS | reactive oxygen species |
| RDW | relative distribution width |
| TRIM28 | tripartite motif containing 28 |
| TIF1β | transcription intermediary factor 1 beta |
| Ulk1 | uncoordinated 51-like autophagy activating kinase 1 |
Conflicts of Interest
References
- Stephenson, J.R.; Axelrad, A.A.; McLeod, D.L.; Shreeve, M.M. Induction of colonies of hemoglobin-synthesizing cells by erythropoietin in vitro. Proc. Natl. Acad. Sci. USA 1971, 68, 1542–1546. [Google Scholar] [CrossRef] [PubMed]
- Géminard, C.; de Gassart, A.; Vidal, M. Reticulocyte maturation: Mitoptosis and exosome release. Biocell 2002, 26, 205–215. [Google Scholar] [PubMed]
- Ashford, T.P.; Porter, K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 1962, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Deter, R.L.; DeDuve, C. Influence of glucagon, an inducer of cellular autophagy on some physical properties of rat liver lysosomes. J. Cell Biol. 1967, 33, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Ichimura, Y. Selective autophagy regulates various cellular functions. Genes Cells 2010, 10, 923–933. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, I.E. A century of mitochondrial research: Achievements and perspectives. Mitochondrion 2001, 1, 3–31. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria in health and disease: Perspectives on a new mitochondrial biology. Mol. Asp. Med. 2004, 25, 365–451. [Google Scholar] [CrossRef]
- Zhang, J.; Ney, P.A. Reticulocyte mitophagy: Monitoring mitochondrial clearance in a mammalian model. Autophagy 2010, 6, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Colombo, M.I. Multivesicular bodies and autophagy in erythrocyte maturation. Autophagy 2006, 2, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y.; Kir, S.; Tooze, S.A. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007, 282, 25464–25474. [Google Scholar] [CrossRef] [PubMed]
- Kundu, M.; Lindsten, T.; Yang, C.Y.; Wu, J.M.; Zhao, F.P.; Zhang, J.; Selak, M.A.; Ney, P.A.; Thompson, C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008, 112, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.H.; Dorsey, F.C.; Joshi, A.; Hennessy-Walters, K.M.; Rose, K.L.; McCastlain, K.; Zhang, J.; Iyengar, R.; Jung, C.H.; Suen, D.F.; et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol. Cell 2011, 43, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Betin, V.M.; Singleton, B.K.; Parsons, S.F.; Anstee, D.J.; Lane, J.D. Autophagy facilitates organelle clearance during differentiation of human erythroblasts: Evidence for a role for Atg4 paralogs during autophagosome maturation. Autophagy 2013, 9, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.E.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Randall, M.S.; Loyd, M.R.; Dorsey, F.C.; Kundu, M.; Cleveland, J.L.; Ney, P.A. Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation. Blood 2009, 114, 157–164. [Google Scholar] [PubMed]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Ferguson, D.J.; Edelmann, M.; Kessler, B.; Morten, K.J.; Komatsu, M.; Simon, A.K. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Simon, A.K. Nonredundant role of Atg7 in mitochondrial clearance during erythroid development. Autophagy 2010, 6, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Li-Harms, X.; Milasta, S.; Lynch, J.; Wright, C.; Joshi, A.; Iyengar, R.; Neale, G.; Wang, X.; Wang, Y.D.; Prolla, T.A.; et al. Mito-protective autophagy is impaired in erythroid cells of aged mtDNA-mutator mice. Blood 2015, 125, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Schwarten, M.; Mohrluder, J.; Ma, P.; Stoldt, M.; Thielmann, Y.; Stangler, T.; Hersch, N.; Hoffmann, B.; Merkel, R.; Willbold, D.; et al. Nix directly binds to GABARAP: A possible crosstalk between apoptosisand autophagy. Autophagy 2009, 5, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Nix induces mitochondrial autophagy in reticulocytes. Autophagy 2008, 4, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Sandoval, H.; Wang, J. Selective mitochondrial autophagy during erythroid maturation. Autophagy 2008, 4, 926–928. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.J.; Watts, J.A.; Vakoc, C.R.; Yao, Y.; Wang, H.; Hardison, R.C.; Blobel, G.A.; Chodosh, L.A.; Weiss, M.J. Gobal regulation of erythroid gene expression by transcription factor GATA-1. Blood 2004, 104, 3136–3147. [Google Scholar] [CrossRef] [PubMed]
- Layon, M.E.; Layon, C.J.; West, R.J.; Lowrey, C.H. Expression of GATA-1 in a non-hematopoietic cell line induces β-globin locus control region chromatin structure remode ling and an erythroid pattern of gene expression. J. Mol. Biol. 2007, 366, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.A.; Sanalkumar, R.; O’Geen, H.; Linnemann, A.K.; Chang, C.J.; Bouhassira, E.E.; Farnham, P.J.; Keles, S.; Bresnick, E.H. Autophagy driven by a master regulator of hematopoiesis. Mol. Cell. Biol. 2012, 32, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Spitali, P.; Grumati, P.; Hiller, M.; Chrisam, M.; Aartsma-Rus, A.; Bonaldo, P. Autophagy is impaired in the tibialis anterior of dystrophin null mice. PLoS Curr. 2013, 5, 1–9. [Google Scholar]
- Bakker, W.J.; van Dijk, T.B.; Parren-van Amelsvoort, M.; Kolbus, A.; Yamamoto, K.; Steinlein, P.; Verhaak, R.G.; Mak, T.W.; Beug, H.; Löwenberg, B.; et al. Differential regulation of FoxO3a target genes in erythropoiesis. Mol. Cell. Biol. 2007, 27, 3839–3854. [Google Scholar] [CrossRef] [PubMed]
- McIver, S.C.; Kang, Y.A.; DeVilbiss, A.W.; O’Driscoll, C.A.; Ouellette, J.N.; Pope, N.J.; Camprecios, G.; Chang, C.J.; Yang, D.; Bouhassira, E.E.; et al. The exosome complex establishes a barricade to erythroid maturation. Blood 2014, 124, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Barde, I.; Rauwel, B.; Marin-Florez, R.M.; Corsinotti, A.; Laurenti, E.; Verp, S.; Offner, S.; Marquis, J.; Kapopoulou, A.; Vanicek, J.; et al. A KRAB/KAP1-miRNA cascade regulates erythropoiesis through stage-specific control of mitophagy. Science 2013, 340, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lee, J.Y.; Wei, H.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, J.L. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; Belkner, J.; Wiesner, R. Subcellular distribution of lipoxygenase products in rabbit reticulocyte membranes. Eur. J. Biochem. 1990, 191, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Vijayvergiya, C.; DeAngelis, D.; Walther, M.; Kühn, H.; Duvoisin, R.M.; Smith, D.H.; Wiedmann, M. High-level expression of rabbit 15-lipoxygenase induces collapse of the mitochondrial pH gradient in cell culture. Biochemistry 2004, 43, 15296–15302. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.; Doherty, F.J. Calcium promotes membrane association of reticulocyte 15-lipoxygenase. Biochem. J. 1994, 298, 377–383. [Google Scholar] [PubMed]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy: Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Walls, K.C.; Ghosh, A.P.; Franklin, A.V.; Klocke, B.J.; Ballestas, M.; Shacka, J.J.; Zhang, J.; Roth, K.A. Lysosome dysfunction triggers Atg7-dependent neural apoptosis. J. Biol. Chem. 2010, 285, 10497–10507. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.M.; Malstrom, S.; Subramanian, T.; Venkatesh, L.K.; Schaeper, U.; Elangovan, B.; D’Sa-Eipper, C.; Chinnadurai, G. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell 1994, 79, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, M.; Fujiwara, T.; Takahashi, E.; Minaguchi, T.; Eguchi, Y.; Tsujimoto, Y.; Suzumori, K.; Nakamura, Y. Isolation, mapping, and functional analysis of a novel human cDNA (BNIP3L) encoding a protein homologous to human NIP3. Genes Chromosomes Cancer 1998, 21, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Loyd, M.R.; Randall, M.S.; Waddell, M.B.; Kriwacki, R.W.; Ney, P.A. A short linear motif in BNIP3L (Nix) mediates mitochondrial clearance in reticulocytes. Autophagy 2012, 8, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Aerbajinai, W.; Giattina, M.; Lee, Y.T.; Raffeld, M.; Miller, J.L. The proapoptotic factor Nix is coexpressed with Bcl-xL during terminal erythroid differentiation. Blood 2003, 102, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.S.; Mortensen, M.; Simon, A.K. Autophagy in the pathogenesis of myelodysplastic syndromeand acute myeloid leukemia. Cell Cycle 2011, 10, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Rhyasen, G.; Bolanos, L.; Rasch, C.; Varney, M.; Wunderlich, M.; Goyama, S.; Jansen, G.; Cloos, J.; Rigolino, C.; et al. Cytotoxic effects of bortezomib in myelodysplastic syndrome/acute myeloidleukemia depend on autophagy-mediated lysosomal degradation of TRAF6 and repression of PSMA1. Blood 2012, 120, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Lithanatudom, P.; Wannatung, T.; Leecharoenkiat, A.; Svasti, S.; Fucharoen, S.; Smith, D.R. Enhanced activation of autophagy in β-thalassemia/Hb E erythroblasts during erythropoiesis. Ann. Hematol. 2011, 90, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Tra, T.; Gong, L.; Kao, L.P.; Li, X.L.; Grandela, C.; Devenish, R.J.; Wolvetang, E.; Prescott, M. Autophagy in human embryonic stem cells. PLoS One 2011, 6, e27485. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Wu, K.; Xiao, X.; Liao, J.; Hu, Q.; Chen, H.; Liu, J.; An, X. Autophagy as a Regulatory Component of Erythropoiesis. Int. J. Mol. Sci. 2015, 16, 4083-4094. https://doi.org/10.3390/ijms16024083
Zhang J, Wu K, Xiao X, Liao J, Hu Q, Chen H, Liu J, An X. Autophagy as a Regulatory Component of Erythropoiesis. International Journal of Molecular Sciences. 2015; 16(2):4083-4094. https://doi.org/10.3390/ijms16024083
Chicago/Turabian StyleZhang, Jieying, Kunlu Wu, Xiaojuan Xiao, Jiling Liao, Qikang Hu, Huiyong Chen, Jing Liu, and Xiuli An. 2015. "Autophagy as a Regulatory Component of Erythropoiesis" International Journal of Molecular Sciences 16, no. 2: 4083-4094. https://doi.org/10.3390/ijms16024083
APA StyleZhang, J., Wu, K., Xiao, X., Liao, J., Hu, Q., Chen, H., Liu, J., & An, X. (2015). Autophagy as a Regulatory Component of Erythropoiesis. International Journal of Molecular Sciences, 16(2), 4083-4094. https://doi.org/10.3390/ijms16024083