
Regulatory Effects of Caffeic Acid Phenethyl Ester on Neuroinflammation in Microglial Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Caffeic Acid Phenethyl Ester (CAPE) Attenuates Microglial Activation Dose Dependently without Obvious Cytotoxicity
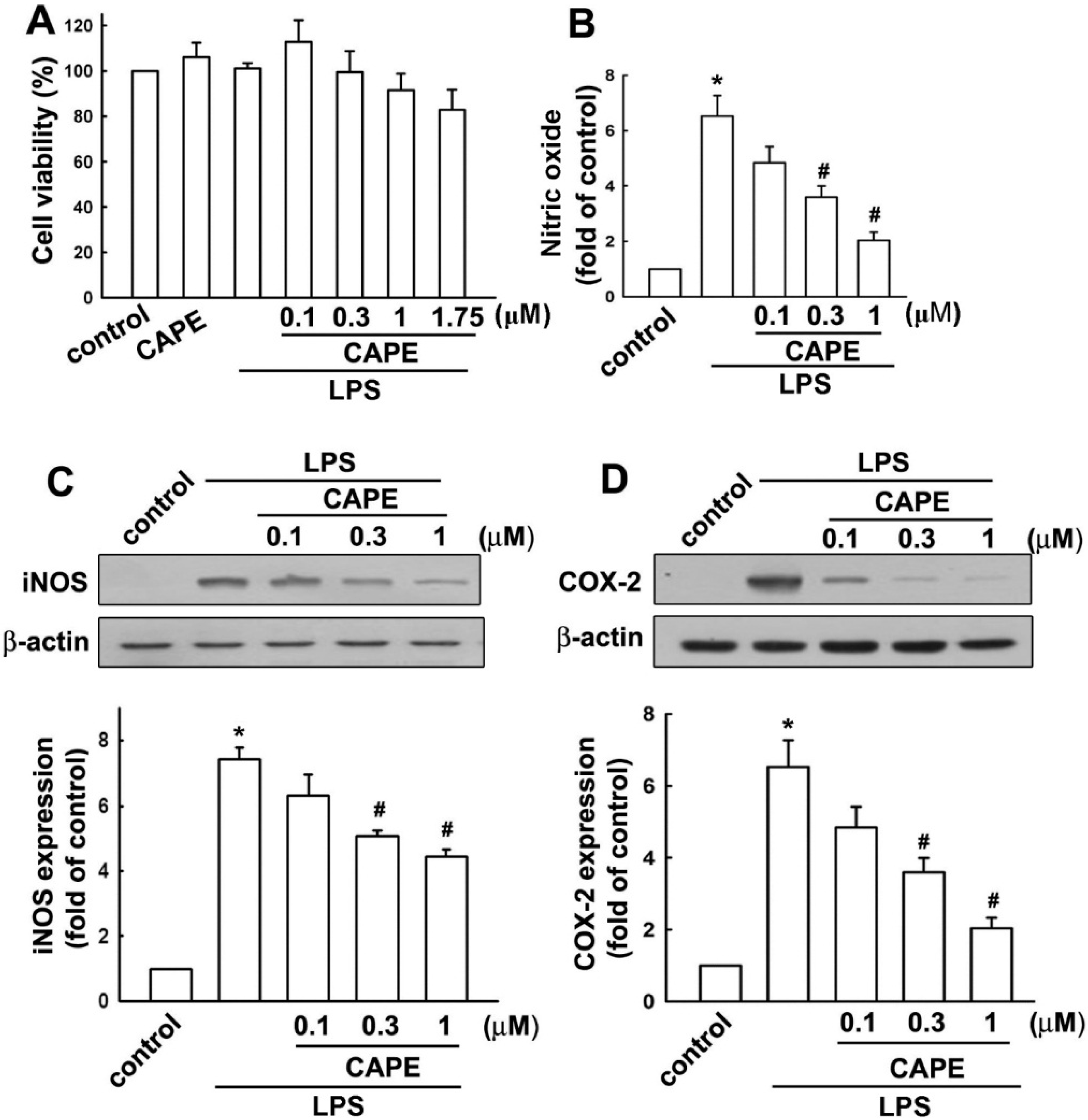
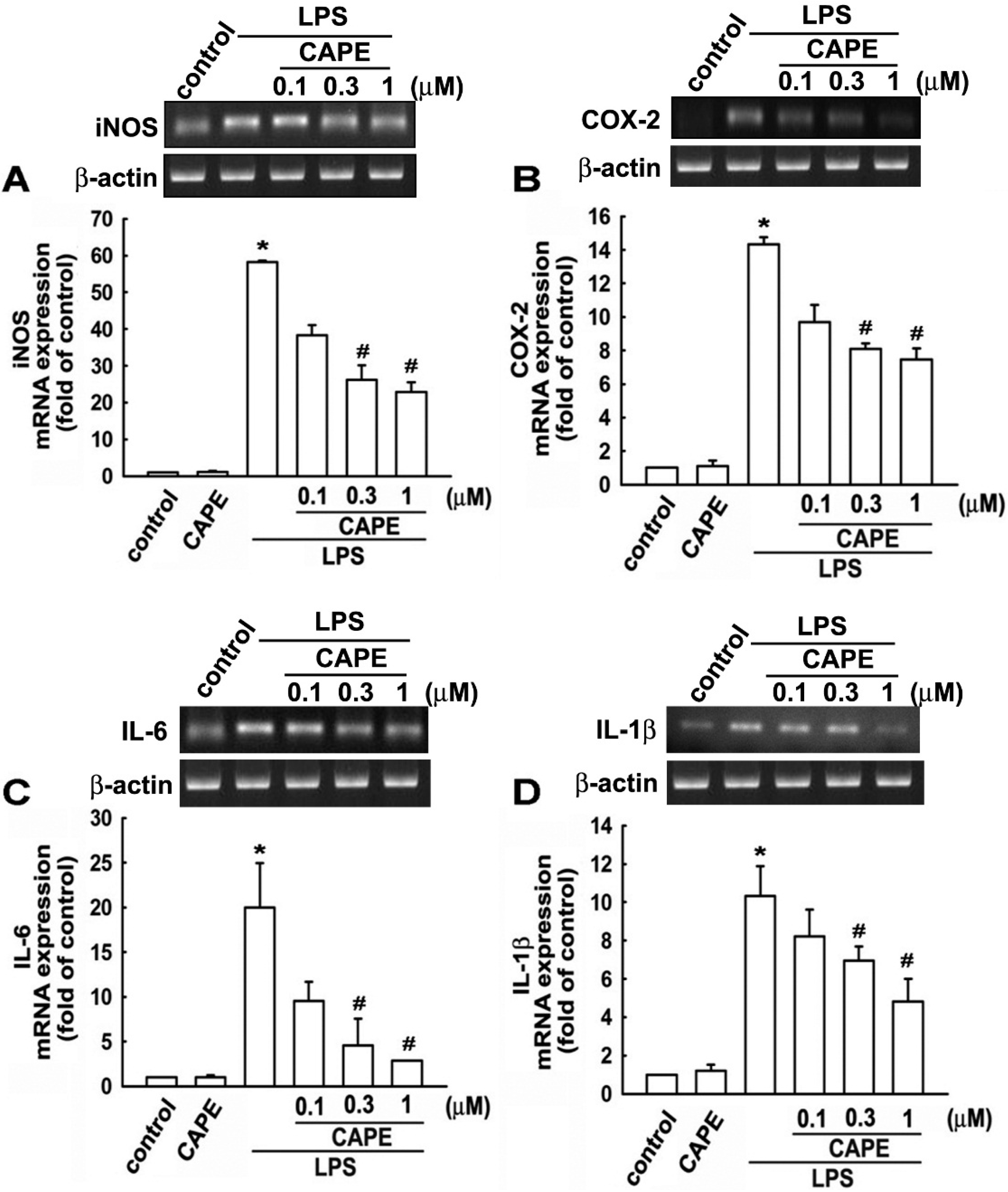
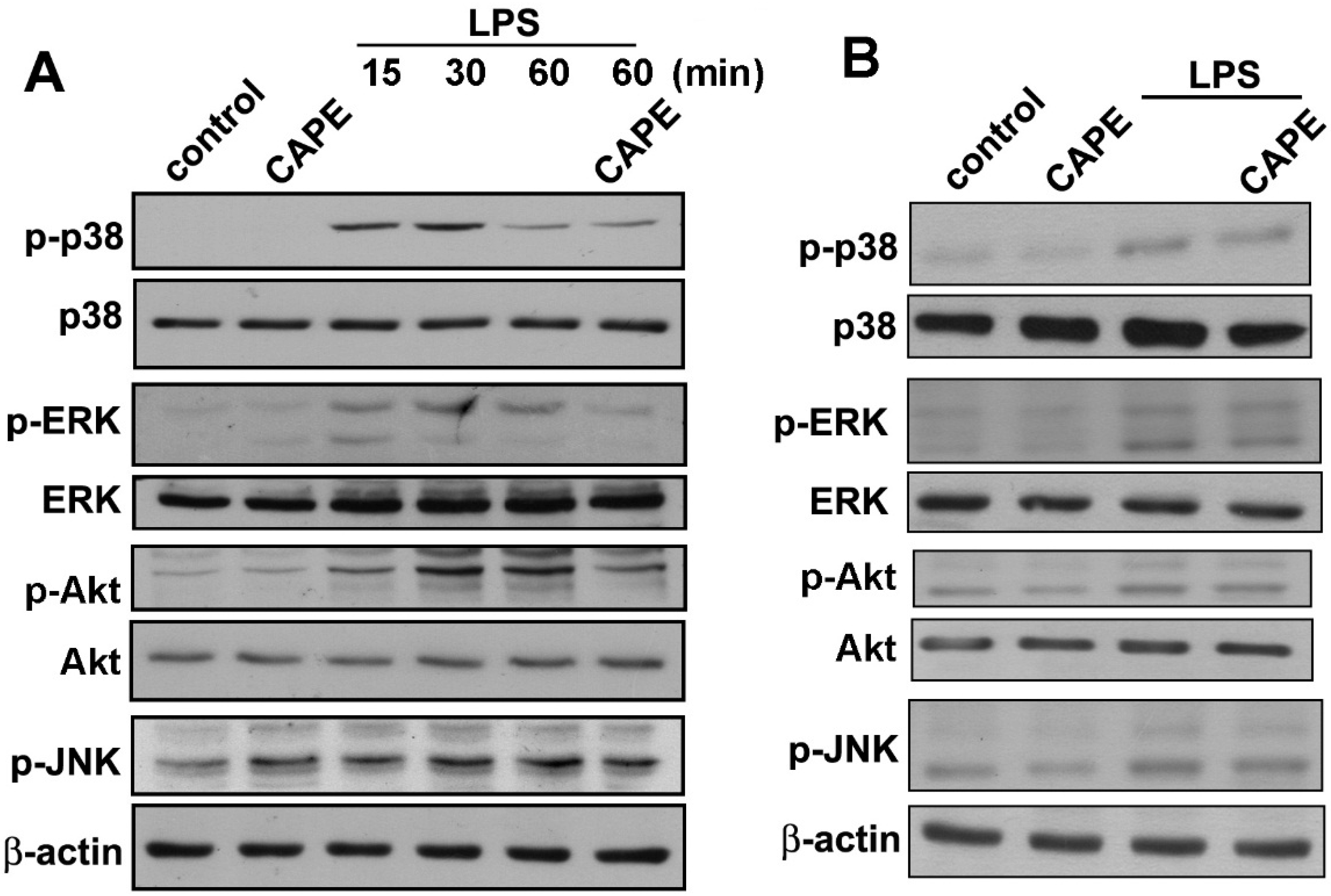
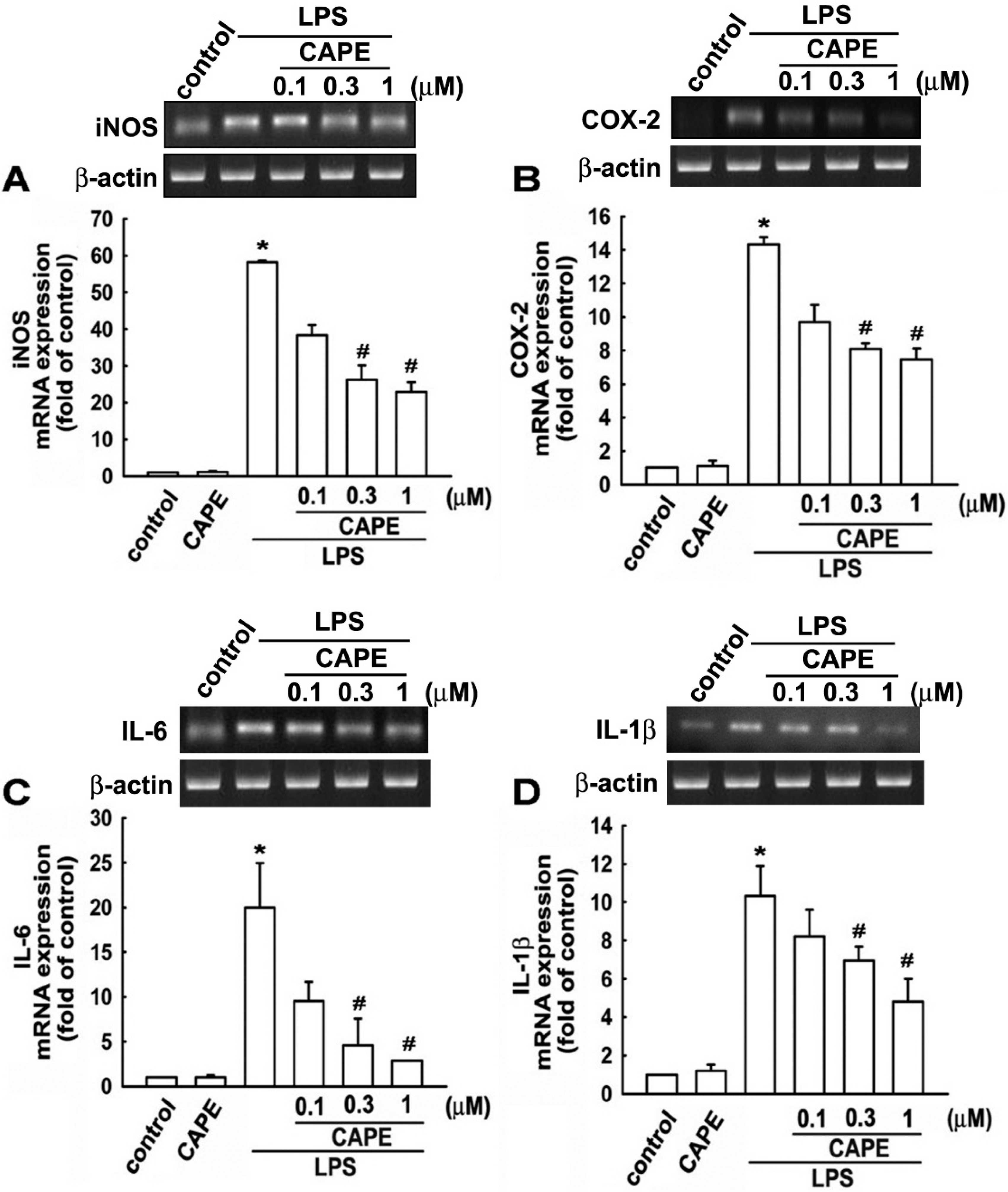
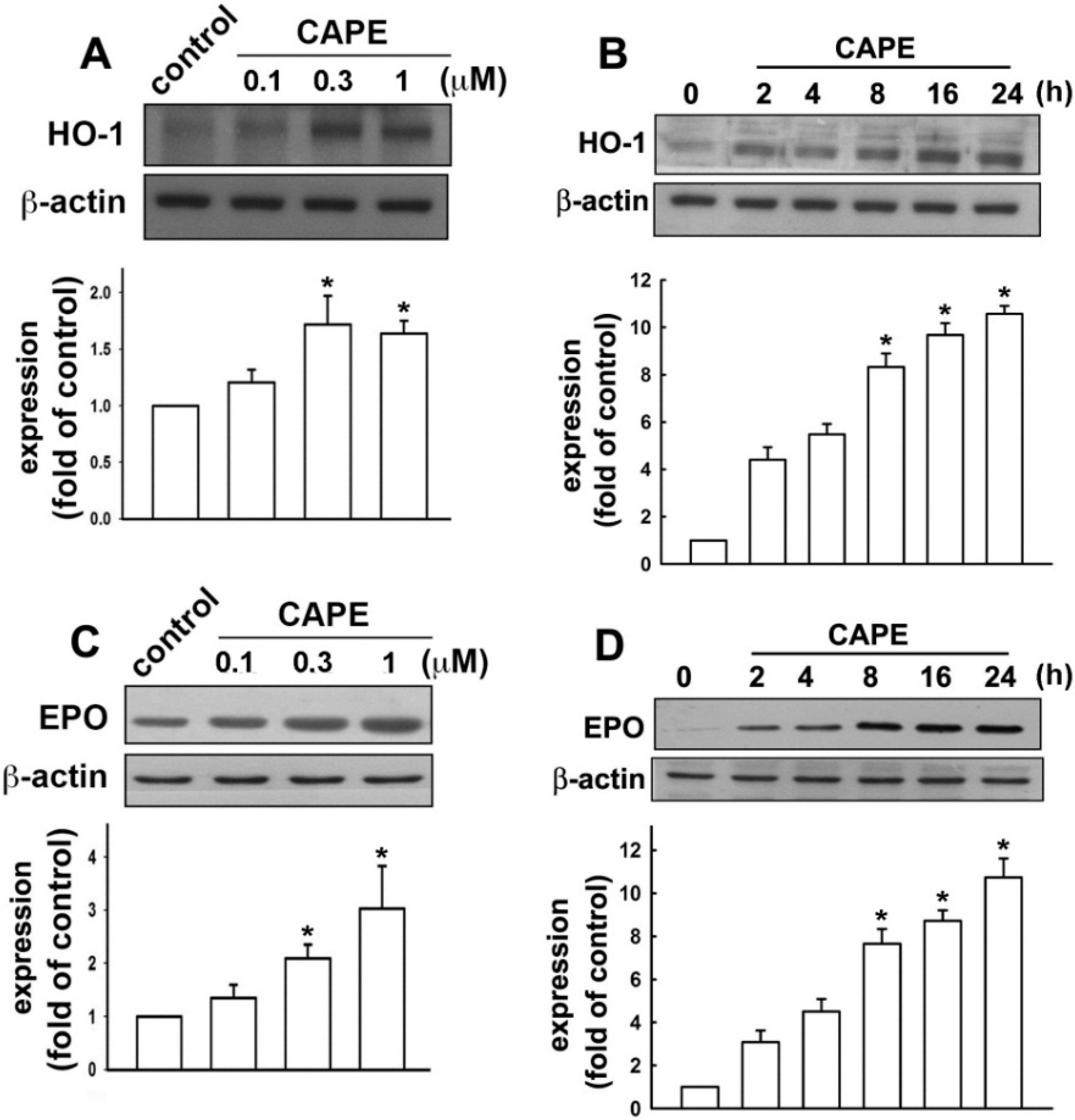
2.2. CAPE Induces Anti-Inflammatory and Protective Molecule Expressions in Microglial Cells
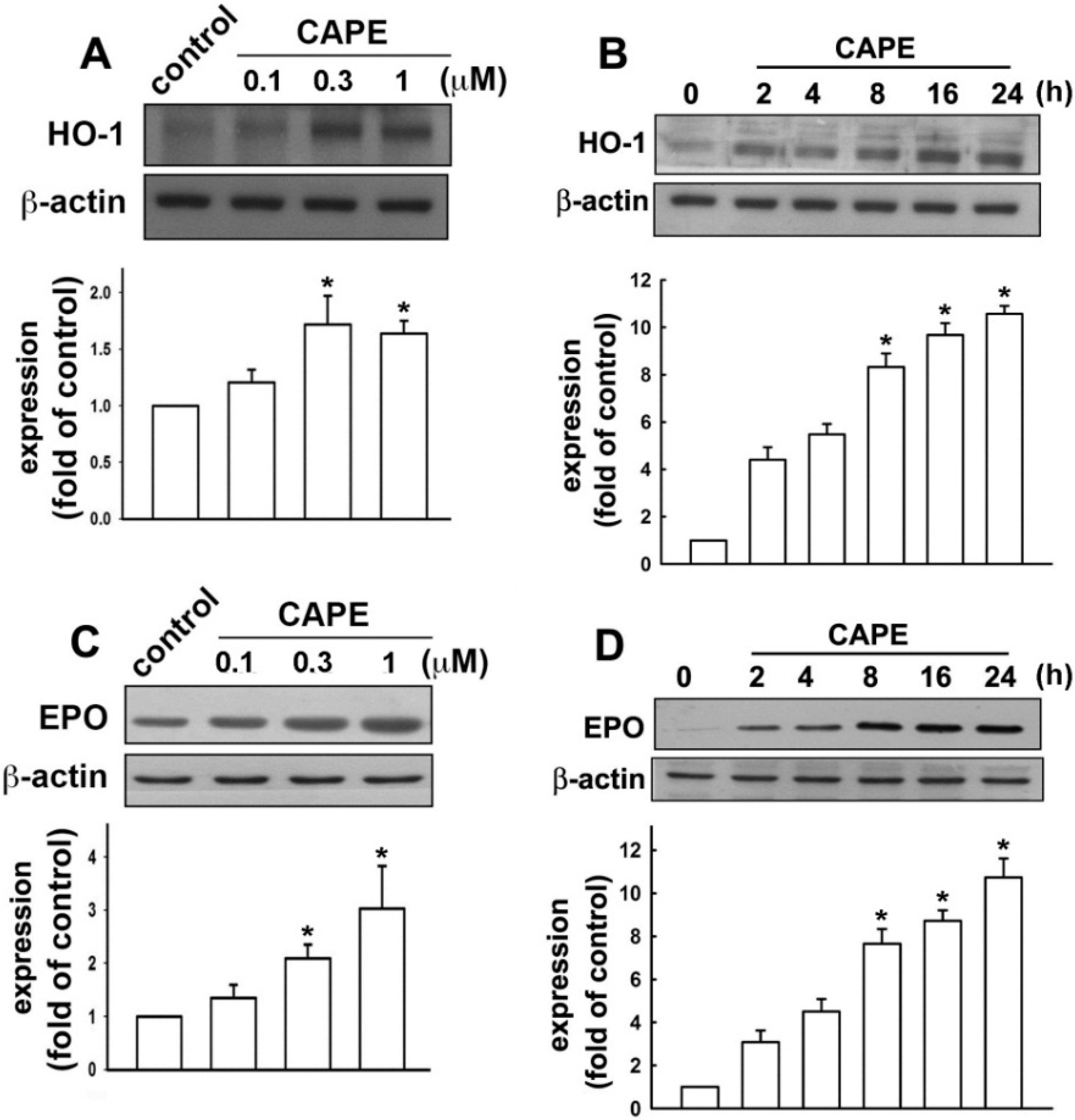
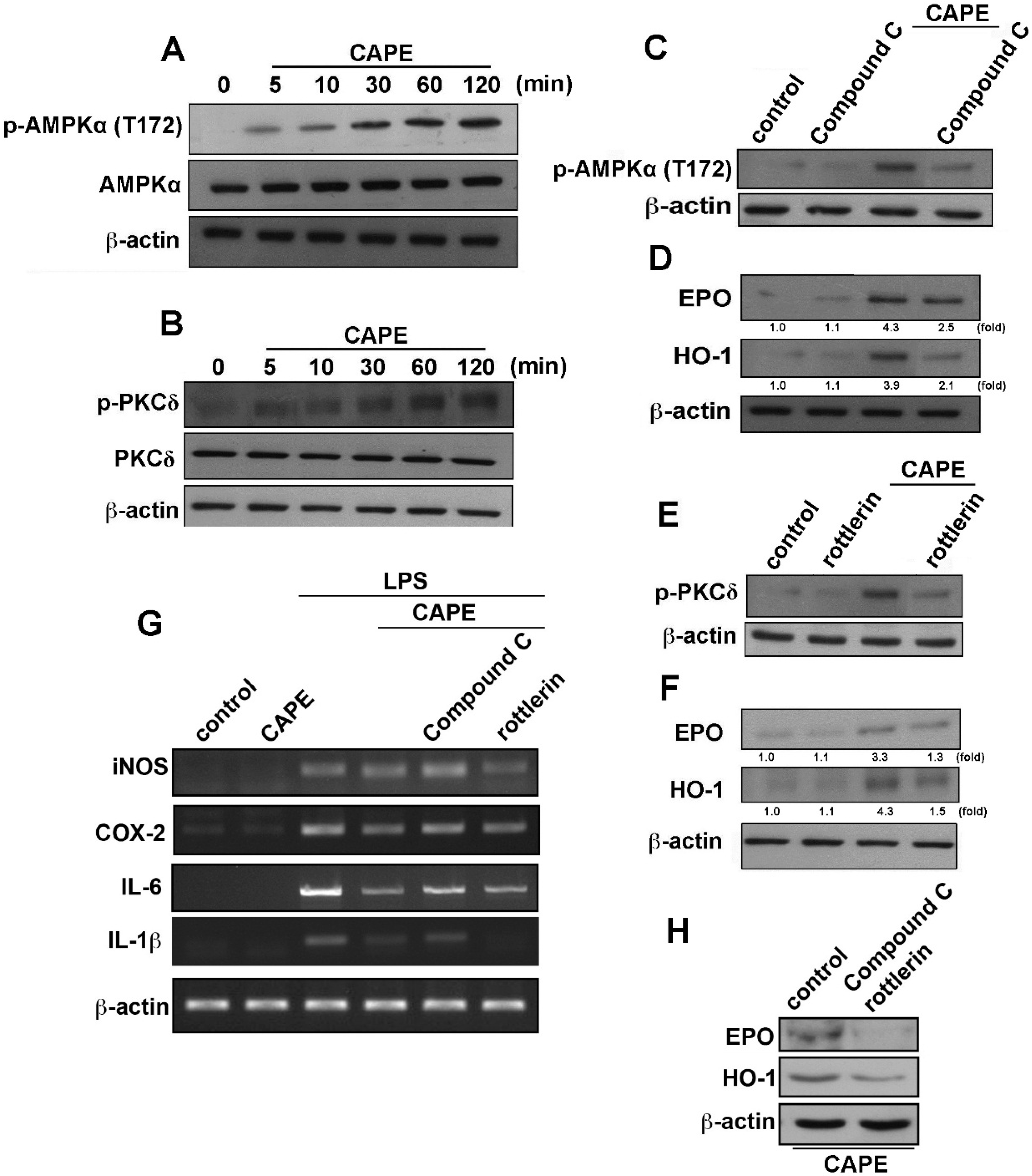
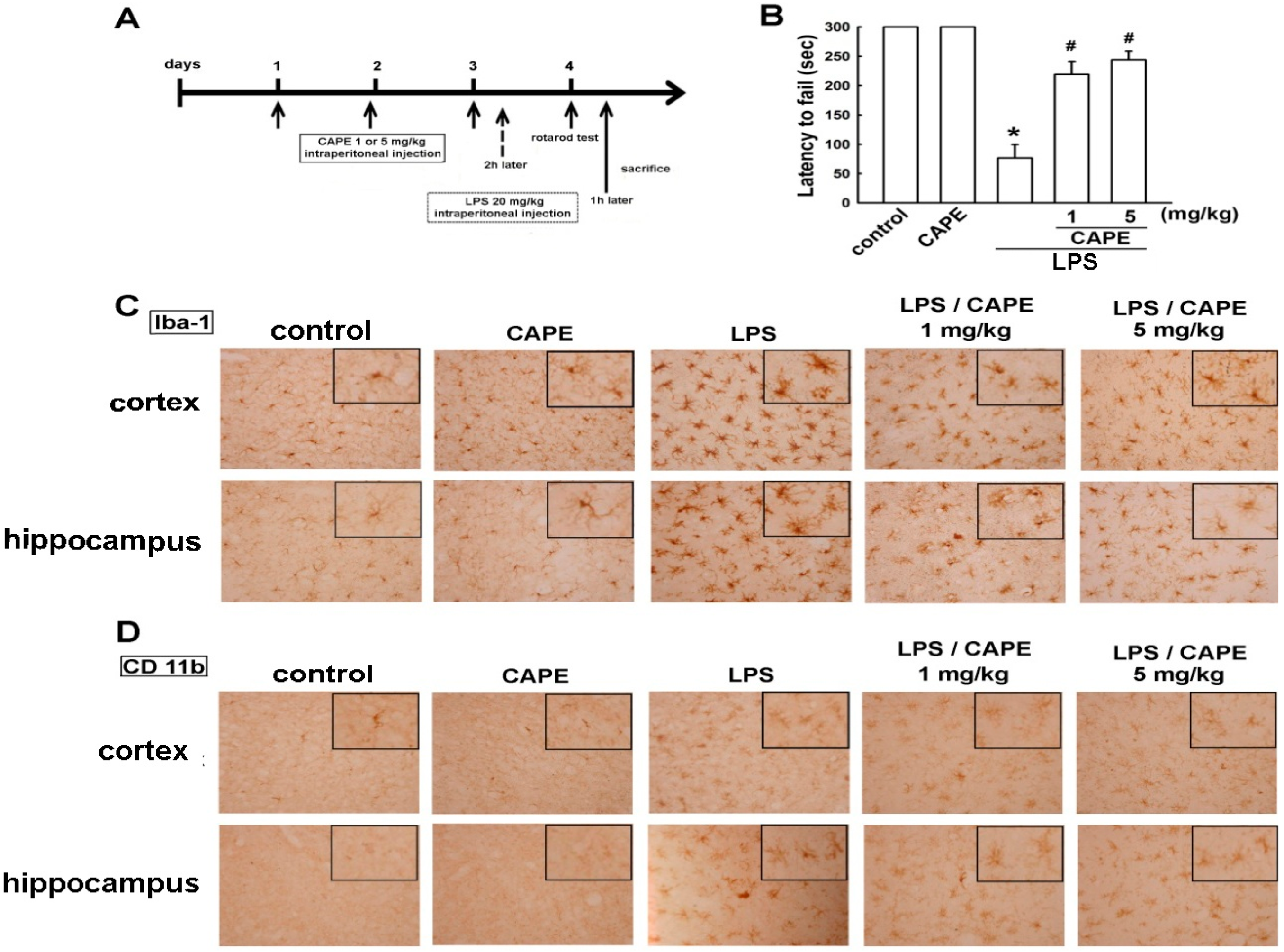
2.3. CAPE Ameliorates LPS-Induced Microglial Activation and Motor Incoordination
3. Discussion
4. Experimental Section
4.1. Reagents and Antibodies
4.2. Cell Culture and Animal Experiments
4.3. MTT Assay
4.4. Western Blot Analysis
4.5. Nitrite Oxide Assay
4.6. Reverse Transcriptase (RT)-PCR and Quantitative Real-Time PCR (qPCR)
4.7. Immunohistochemistry
4.8. Rotarod Analysis
4.9. Statistical Analyses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Lewis, C.A.; Zhang, R.; Rossi, F.M. The role of microglia in human disease: Therapeutic tool or target? Acta Neuropathol. 2014, 128, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Mol. Cell Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed]
- Maa, M.C.; Leu, T.H. Activation of toll-like receptors induces macrophage migration via the iNOS/Src/FAK pathway. Biomedicine 2011, 1, 11–15. [Google Scholar] [CrossRef]
- Weiner, H.L. The challenge of multiple sclerosis: How do we cure a chronic heterogeneous disease? Ann. Neurol. 2009, 65, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Politis, M.; Pavese, N.; Tai, Y.F.; Kiferle, L.; Mason, S.L.; Brooks, D.J.; Tabrizi, S.J.; Barker, R.A.; Piccini, P. Microglial activation in regions related to cognitive function predicts disease onset in Huntington’s disease: A multimodal imaging study. Hum. Brain Mapp. 2011, 32, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Wilms, H.; Zecca, L.; Rosenstiel, P.; Sievers, J.; Deuschl, G.; Lucius, R. Inflammation in Parkinson’s diseases and other neurodegenerative diseases: Cause and therapeutic implications. Curr. Pharm. Des. 2007, 13, 1925–1928. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Chen, J.; Crow, J.P.; Ye, Y.Z. Reactions of nitric oxide, superoxide and peroxynitrite with superoxide dismutase in neurodegeneration. Prog. Brain Res. 1994, 103, 371–380. [Google Scholar] [PubMed]
- Chao, C.C.; Hu, S.; Peterson, P.K. Modulation of human microglial cell superoxide production by cytokines. J. Leukoc. Biol. 1995, 58, 65–70. [Google Scholar] [PubMed]
- Wang, J.Y.; Shum, A.Y.; Ho, Y.J.; Wang, J.Y. Oxidative neurotoxicity in rat cerebral cortex neurons:Synergistic effects of H2O2 and NO on apoptosis involving activation of p38 mitogen-activated protein kinase and caspase-3. J. Neurosci. Res. 2003, 72, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wu, L.; Wang, Q.; Hand, T.; Bilak, M.; McCullough, L.; Andreasson, K. Function of COX-2 and prostaglandins in neurological disease. J. Mol. Neurosci. 2007, 33, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.L.; Lu, D.Y.; Liou, H.C.; Fu, W.M. A forward loop between glioma and microglia: Glioma-derived extracellular matrix-activated microglia secrete IL-18 to enhance the migration of glioma cells. J. Cell. Physiol. 2012, 227, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.R.; Chang, P.C.; Yeh, W.L.; Lee, C.H.; Tsai, C.F.; Lin, C.; Lin, H.Y.; Liu, Y.S.; Wu, C.Y.; Ko, P.Y.; et al. Anti-neuroinflammatory effects of the calcium channel blocker nicardipine on microglial cells: Implications for neuroprotection. PLoS One 2014, 9, e91167. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Takagi, T.; Higashimura, Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch. Biochem. Biophys. 2014, 564C, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Huang, B.R.; Yeh, W.L.; Lee, C.H.; Huang, S.S.; Lai, C.H.; Lin, H.; Lu, D.Y. Antineuroinflammatory effects of lycopene via activation of adenosine monophosphate-activated protein kinase-α1/heme oxygenase-1 pathways. Neurobiol. Aging 2014, 35, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.Y.; Huang, B.R.; Yeh, W.L.; Lin, H.Y.; Huang, S.S.; Liu, Y.S.; Kuo, Y.H. Anti-neuroinflammatory effect of a novel caffeamide derivative, KS370G, in microglial cells. Mol. Neurobiol. 2013, 48, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.R.; Tsai, C.F.; Lin, H.Y.; Tseng, W.P.; Huang, S.S.; Wu, C.R.; Lin, C.; Yeh, W.L.; Lu, D.Y. Interaction of inflammatory and anti-inflammatory responses in microglia by Staphylococcus aureus-derived lipoteichoic acid. Toxicol. Appl. Pharmacol. 2013, 269, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Yeh, W.L.; Huang, B.R.; Lin, C.; Lai, C.H.; Lin, H.; Lu, D.Y. Desipramine protects neuronal cell death and induces heme oxygenase-1 expression in MES23.5 dopaminergic neurons. PLoS One 2012, 7, e50138. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.Y.; Chen, J.H.; Tan, T.W.; Huang, C.Y.; Yeh, W.L.; Hsu, H.C. Resistin protects against 6-hydroxydopamine-induced cell death in dopaminergic-like MES23.5 cells. J. Cell. Physiol. 2013, 228, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Lee, S.S.; Chen, S.C.; Ho, F.M.; Lin, W.W. Oregonin inhibits lipopolysaccharide-induced iNOS gene transcription and up-regulates HO-1 expression in macrophages and microglia. Br. J. Pharmacol. 2005, 146, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chen, M.; Wang, M.; Wang, M.; Zhang, T.; Park, J.; Zhu, Y.; Guo, C.; Jia, Y.; Li, Y.; et al. Neuroprotection by acetyl-11-keto-β-boswellic acid, in ischemic brain injury involves the Nrf2/HO-1 defense pathway. Sci. Rep. 2014. [Google Scholar] [CrossRef]
- Vergadi, E.; Chang, M.S.; Lee, C.; Liang, O.D.; Liu, X.; Fernandez-Gonzalez, A.; Mitsialis, S.A.; Kourembanas, S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 2011, 123, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Palis, J. Primitive and definitive erythropoiesis in mammals. Front. Physiol. 2014, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Alural, B.; Duran, G.A.; Tufekci, K.U.; Allmer, J.; Onkal, Z.; Tunali, D.; Genc, K.; Genc, S. EPO mediates neurotrophic, neuroprotective, anti-oxidant, and anti-apoptotic effects via down-regulation of miR-451 and miR-885-5p in SH-SY5Y neuron-like cells. Front. Immunol. 2014, 5, 475. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Ding, H.; Tang, Y.; Dong, Q. Erythropoietin protects against hemorrhagic blood-brain barrier disruption through the effects of aquaporin-4. Lab. Investig. 2014, 94, 1042–1053. [Google Scholar] [CrossRef] [PubMed]
- Genc, K.; Egrilmez, M.Y.; Genc, S. Erythropoietin induces nuclear translocation of Nrf2 and heme oxygenase-1 expression in SH-SY5Y cells. Cell Biochem. Funct. 2010, 28, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Villa, P.; Bigini, P.; Mennini, T.; Agnello, D.; Laragione, T.; Cagnotto, A.; Viviani, B.; Marinovich, M.; Cerami, A.; Coleman, T.R.; et al. Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J. Exp. Med. 2003, 198, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Wang, Y.L.; Lo, W.T.; Wu, C.C.; Hsieh, C.W.; Huang, C.F.; Lan, Y.H.; Wang, C.C.; Chang, D.M.; Sytwu, H.K. Erythropoietin enhances endogenous haem oxygenase-1 and represses immune responses to ameliorate experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chong, Z.Z.; Maiese, K. Microglial integrity is maintained by erythropoietin through integration of Akt and its substrates of glycogen synthase kinase-3β, β-catenin, and nuclear factor-κB. Curr. Neurovasc. Res. 2006, 3, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Zang, M.; Zuccollo, A.; Hou, X.; Nagata, D.; Walsh, K.; Herscovitz, H.; Brecher, P.; Ruderman, N.B.; Cohen, R.A. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J. Biol. Chem. 2004, 279, 47898–47905. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Mounier, R.; Theret, M.; Arnold, L.; Cuvellier, S.; Bultot, L.; Goransson, O.; Sanz, N.; Ferry, A.; Sakamoto, K.; Foretz, M.; et al. AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 2013, 18, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.Y.; Tang, C.H.; Chen, Y.H.; Wei, I.H. Berberine suppresses neuroinflammatory responses through AMP-activated protein kinase activation in BV-2 microglia. J. Cell. Biochem. 2010, 110, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Kurauchi, Y.; Hisatsune, A.; Isohama, Y.; Mishima, S.; Katsuki, H. Caffeic acid phenethyl ester protects nigral dopaminergic neurons via dual mechanisms involving haem oxygenase-1 and brain-derived neurotrophic factor. Br. J. Pharmacol. 2012, 166, 1151–1168. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Guo, Y.; Ma, X.; Zhu, R.; Tian, C.; Zhang, Z.; Wang, X.; Ma, Z.; Liu, J. Design, synthesis and pharmacological evaluation of (E)-3,4-dihydroxy styryl sulfonamides derivatives as multifunctional neuroprotective agents against oxidative and inflammatory injury. Bioorg. Med. Chem. 2013, 21, 5589–5597. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.Y.; Tsai, C.F.; Chang, S.W.; Chiang, I.P.; Huang, S.M.; Lin, H.Y.; Yeh, W.L.; Lu, D.Y. Glial cell line-derived neurotrophic factor induces cell migration in human oral squamous cell carcinoma. Oral Oncol. 2013, 49, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.T.; Kwon, D.Y.; Yoon, S.H. AMP-activated protein kinase: A potential target for the diseases prevention by natural occurring polyphenols. New Biotechnol. 2009, 26, 17–22. [Google Scholar] [CrossRef]
- Guo, W.; Kong, E.; Meydani, M. Dietary polyphenols, inflammation, and cancer. Nutr. Cancer 2009, 61, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Meydani, M.; Hasan, S.T. Dietary polyphenols and obesity. Nutrients 2010, 2, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Kaur, C.; Wu, C.Y.; Lu, J.; Ling, E.A. Nuclear factor-κβ regulates Notch signaling in production of proinflammatory cytokines and nitric oxide in murine BV-2 microglial cells. Neuroscience 2011, 192, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Trocha, M.; Merwid-Lad, A.; Szuba, A.; Chlebda, E.; Piesniewska, M.; Sozanski, T.; Szelag, A. Effect of simvastatin on nitric oxide synthases (eNOS, iNOS) and arginine and its derivatives (ADMA, SDMA) in ischemia/reperfusion injury in rat liver. Pharmacol. Rep. 2010, 62, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, G.; Karim, S.; Akram, M.R.; Khan, S.A.; Azhar, S.; Mumtaz, A.; Bin Asad, M.H. Caffeic acid phenethyl ester and therapeutic potentials. BioMed Res. Int. 2014, 2014, 145342. [Google Scholar] [PubMed]
- Guney, M.; Oral, B.; Karahan, N.; Mungan, T. Protective effect of caffeic acid phenethyl ester (CAPE) on fluoride-induced oxidative stress and apoptosis in rat endometrium. Environ. Toxicol. Pharmacol. 2007, 24, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, L.H.; Maillet, J.; LeBlanc, L.M.; Jean-Francois, J.; Touaibia, M.; Flamand, N.; Surette, M.E. Caffeic acid phenethyl ester and its amide analogue are potent inhibitors of leukotriene biosynthesis in human polymorphonuclear leukocytes. PLoS One 2012, 7, e31833. [Google Scholar] [CrossRef] [PubMed]
- Fontanilla, C.V.; Ma, Z.; Wei, X.; Klotsche, J.; Zhao, L.; Wisniowski, P.; Dodel, R.C.; Farlow, M.R.; Oertel, W.H.; Du, Y. Caffeic acid phenethyl ester prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurodegeneration. Neuroscience 2011, 188, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Pati, S.; Redell, J. B.; Zhang, M.; Moore, A.N.; Dash, P.K. Caffeic acid phenethyl ester protects blood-brain barrier integrity and reduces contusion volume in rodent models of traumatic brain injury. J. Neurotrauma 2012, 29, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Barros Silva, R.; Santos, N.A.; Martins, N.M.; Ferreira, D.A.; Barbosa, F., Jr.; Oliveira Souza, V.C.; Kinoshita, A.; Baffa, O.; Del-Bel, E.; Santos, A.C. Caffeic acid phenethyl ester protects against the dopaminergic neuronal loss induced by 6-hydroxydopamine in rats. Neuroscience 2013, 233, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.H.; Lin, C.; Lin, H.Y.; Liu, Y.S.; Wu, C.Y.; Tsai, C.F.; Chang, P.C.; Yeh, W.L.; Lu, D.Y. Naringenin suppresses neuroinflammatory responses through inducing suppressor of cytokine signaling 3 expression. Mol. Neurobiol. 2015. [Google Scholar] [CrossRef]
- Chuang, J.Y.; Chang, P.C.; Shen, Y.C.; Lin, C.; Tsai, C.F.; Chen, J.H.; Yeh, W.L.; Wu, L.H.; Lin, H.Y.; Liu, Y.S.; et al. Regulatory effects of fisetin on microglial activation. Molecules 2014, 19, 8820–8839. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Forman, L.W.; Williams, R.M.; Faller, D.V. Protein kinase Cδ inactivation inhibits the proliferation and survival of cancer stem cells in culture and in vivo. BMC Cancer 2014, 14, 90. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.C.; Jeon, W.K.; Hong, H.Y.; Jeon, K.B.; Hahn, J.H.; Kim, Y.M.; Numazawa, S.; Yosida, T.; Park, E.H.; Lim, C.J. The anti-inflammatory activity of Phellinus linteus (Berk. & M.A. Curt.) is mediated through the PKCδ/Nrf2/ARE signaling to up-regulation of heme oxygenase-1. J. Ethnopharmacol. 2007, 113, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Bair, A.M.; Thippegowda, P.B.; Freichel, M.; Cheng, N.; Ye, R.D.; Vogel, S.M.; Yu, Y.; Flockerzi, V.; Malik, A.B.; Tiruppathi, C. Ca2+ entry via TRPC channels is necessary for thrombin-induced NF-κB activation in endothelial cells through AMP-activated protein kinase and protein kinase Cδ. J. Biol. Chem. 2009, 284, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.S.; Zheng, W.H.; Bastianetto, S.; Chabot, J.G.; Quirion, R. Neuroprotective effects of resveratrol against β-amyloid-induced neurotoxicity in rat hippocampal neurons: Involvement of protein kinase C. Br. J. Pharmacol. 2004, 141, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Yang, H.; Jeong, S.I.; Jin, Y.H.; Park, C.S.; Park, Y.S. Induction of heme oxygenase-1 inhibits cell death in crotonaldehyde-stimulated HepG2 cells via the PKCδ-p38-Nrf2 pathway. PLoS One 2012, 7, e41676. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.Y.; Leung, Y.M.; Su, K.P. Interferon-α induces nitric oxide synthase expression and haem oxygenase-1 down-regulation in microglia: Implications of cellular mechanism of IFN-α-induced depression. Int. J. Neuropsychopharmacol. 2013, 16, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Wang, X.W.; Du, Y.J. Protective effects of erythropoietin on myocardial infarction in rats: The role of AMP-activated protein kinase signaling pathway. Am. J. Med. Sci. 2011, 342, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.Y.; Chang, C.S.; Yeh, W.L.; Tang, C.H.; Cheung, C.W.; Leung, Y.M.; Liu, J.F.; Wong, K.L. The novel phloroglucinol derivative BFP induces apoptosis of glioma cancer through reactive oxygen species and endoplasmic reticulum stress pathways. Phytomedicine 2012, 19, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.R.; Lu, D.Y.; Lin, H.Y.; Yeh, W.L. Mesenchymal stem cell-induced doxorubicin resistance in triple negative breast cancer. BioMed Res. Int. 2014, 2014, 532161. [Google Scholar] [PubMed]
- Yeh, W.L.; Lin, H.Y.; Wu, H.M.; Chen, D.R. Combination treatment of tamoxifen with risperidone in breast cancer. PLoS One 2014, 9, e98805. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Chen, T.S.; Chiu, C.M.; Chang, L.K.; Liao, K.F.; Tan, H.M.; Yeh, W.L.; Chang, G.R.; Wang, M.Y.; et al. GDNF increases cell motility in human colon cancer through VEGF-VEGFR1 interaction. Endocr. Relat. Cancer 2014, 21, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Hockly, E.; Woodman, B.; Mahal, A.; Lewis, C.M.; Bates, G. Standardization and statistical approaches to therapeutic trials in the R6/2 mouse. Brain Res. Bull. 2003, 61, 469–479. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, C.-F.; Kuo, Y.-H.; Yeh, W.-L.; Wu, C.Y.-J.; Lin, H.-Y.; Lai, S.-W.; Liu, Y.-S.; Wu, L.-H.; Lu, J.-K.; Lu, D.-Y. Regulatory Effects of Caffeic Acid Phenethyl Ester on Neuroinflammation in Microglial Cells. Int. J. Mol. Sci. 2015, 16, 5572-5589. https://doi.org/10.3390/ijms16035572
Tsai C-F, Kuo Y-H, Yeh W-L, Wu CY-J, Lin H-Y, Lai S-W, Liu Y-S, Wu L-H, Lu J-K, Lu D-Y. Regulatory Effects of Caffeic Acid Phenethyl Ester on Neuroinflammation in Microglial Cells. International Journal of Molecular Sciences. 2015; 16(3):5572-5589. https://doi.org/10.3390/ijms16035572
Chicago/Turabian StyleTsai, Cheng-Fang, Yueh-Hsiung Kuo, Wei-Lan Yeh, Caren Yu-Ju Wu, Hsiao-Yun Lin, Sheng-Wei Lai, Yu-Shu Liu, Ling-Hsuan Wu, Jheng-Kun Lu, and Dah-Yuu Lu. 2015. "Regulatory Effects of Caffeic Acid Phenethyl Ester on Neuroinflammation in Microglial Cells" International Journal of Molecular Sciences 16, no. 3: 5572-5589. https://doi.org/10.3390/ijms16035572
APA StyleTsai, C.-F., Kuo, Y.-H., Yeh, W.-L., Wu, C. Y.-J., Lin, H.-Y., Lai, S.-W., Liu, Y.-S., Wu, L.-H., Lu, J.-K., & Lu, D.-Y. (2015). Regulatory Effects of Caffeic Acid Phenethyl Ester on Neuroinflammation in Microglial Cells. International Journal of Molecular Sciences, 16(3), 5572-5589. https://doi.org/10.3390/ijms16035572