
Reduction of Oxidative Stress Attenuates Lipoapoptosis Exacerbated by Hypoxia in Human Hepatocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
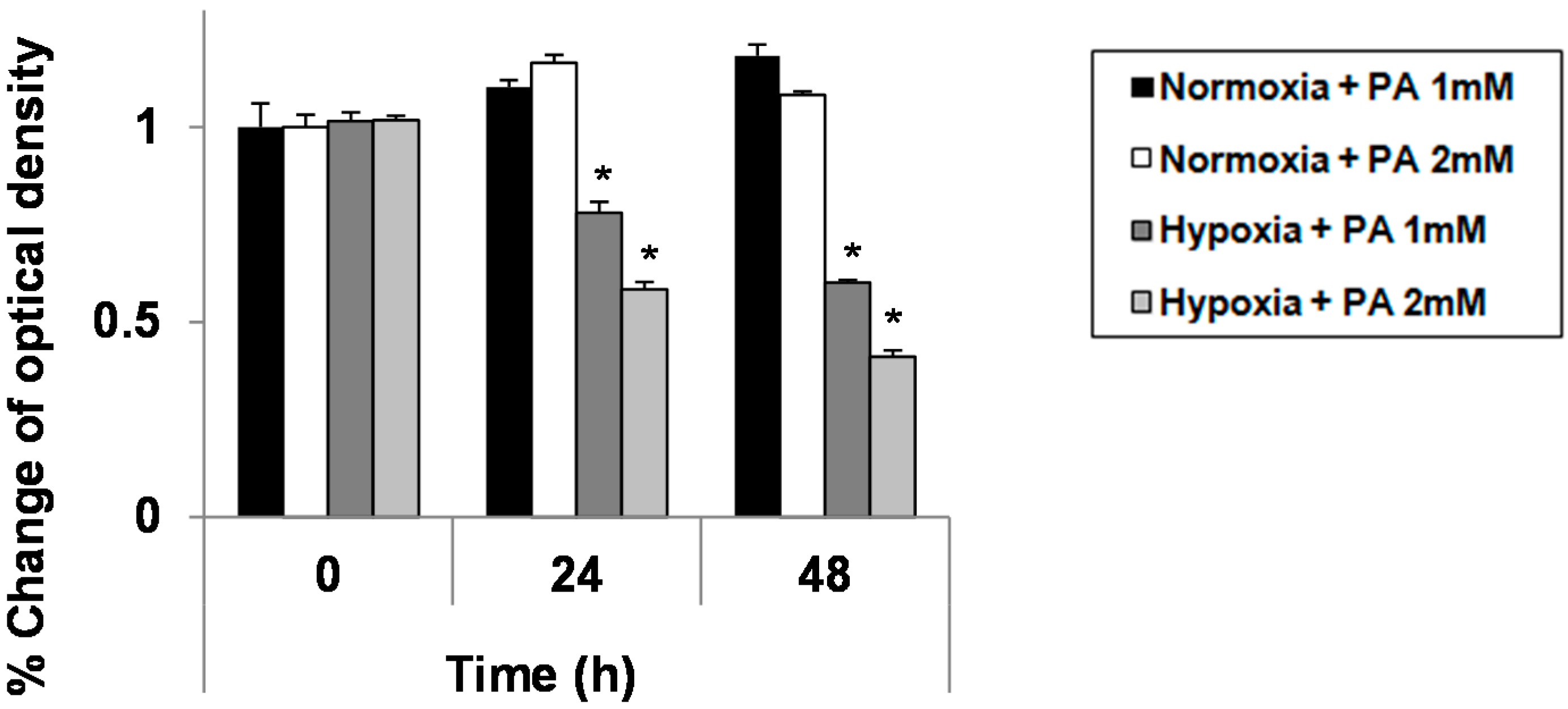
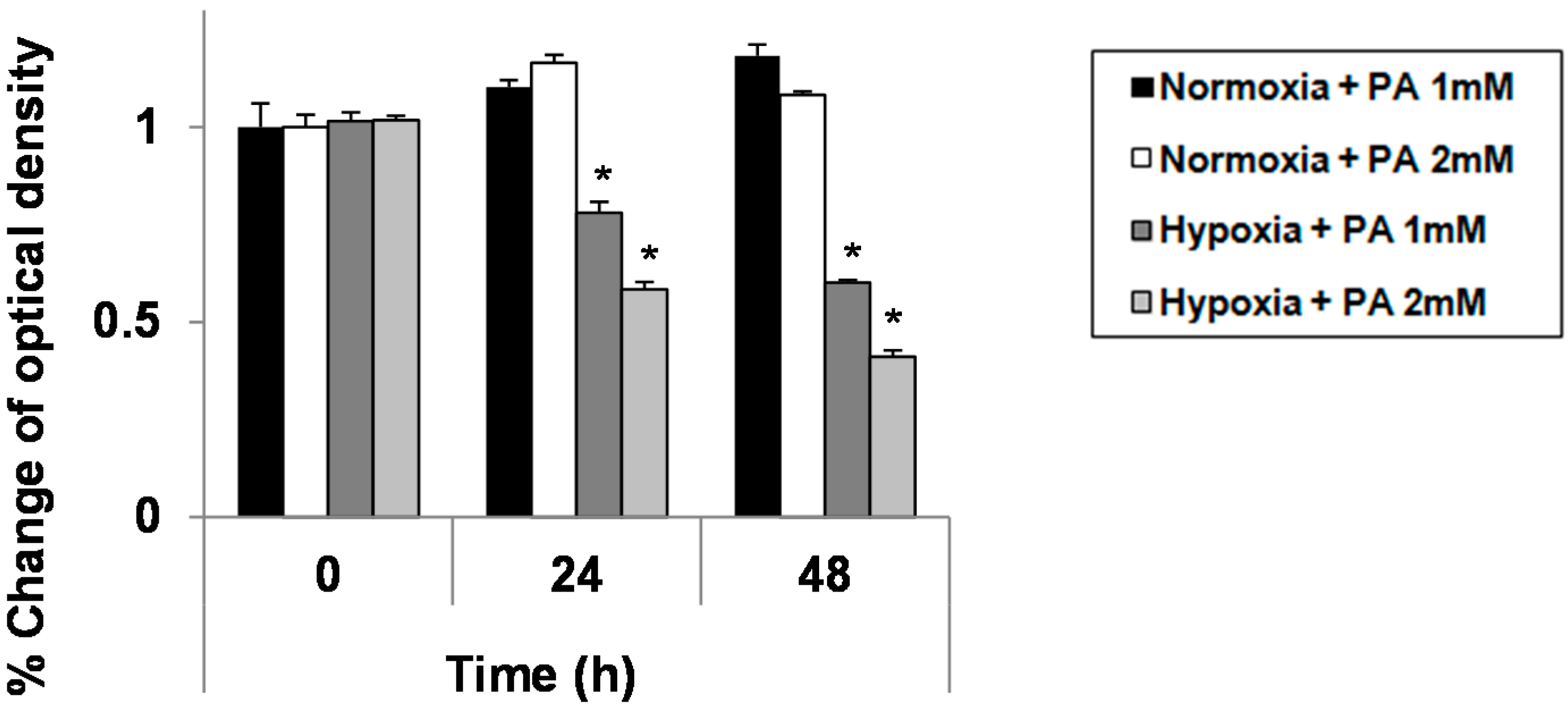
2.1. Hypoxia Exacerbates Growth Suppression in Hepatocytes Treated with Palmitic Acid (PA)
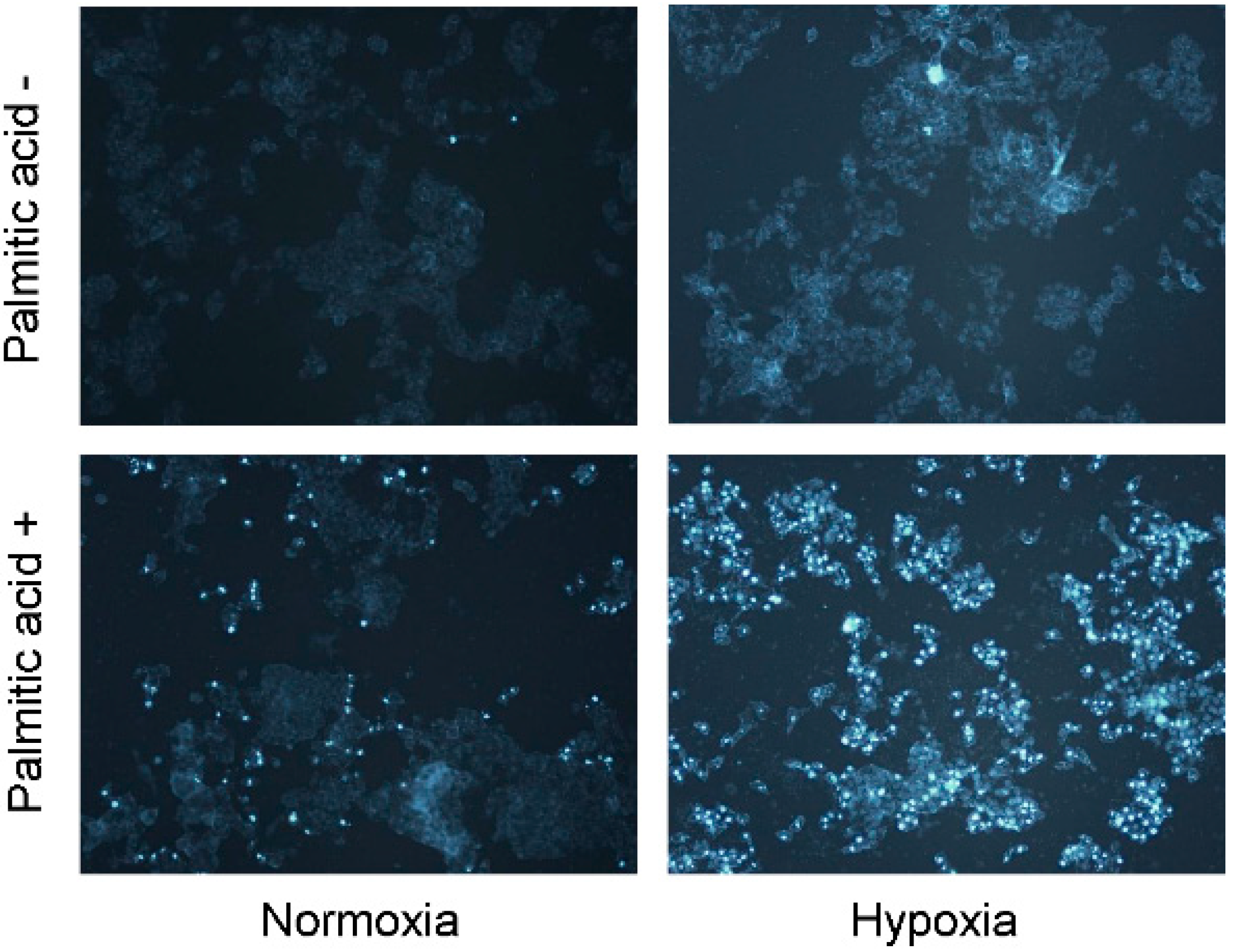
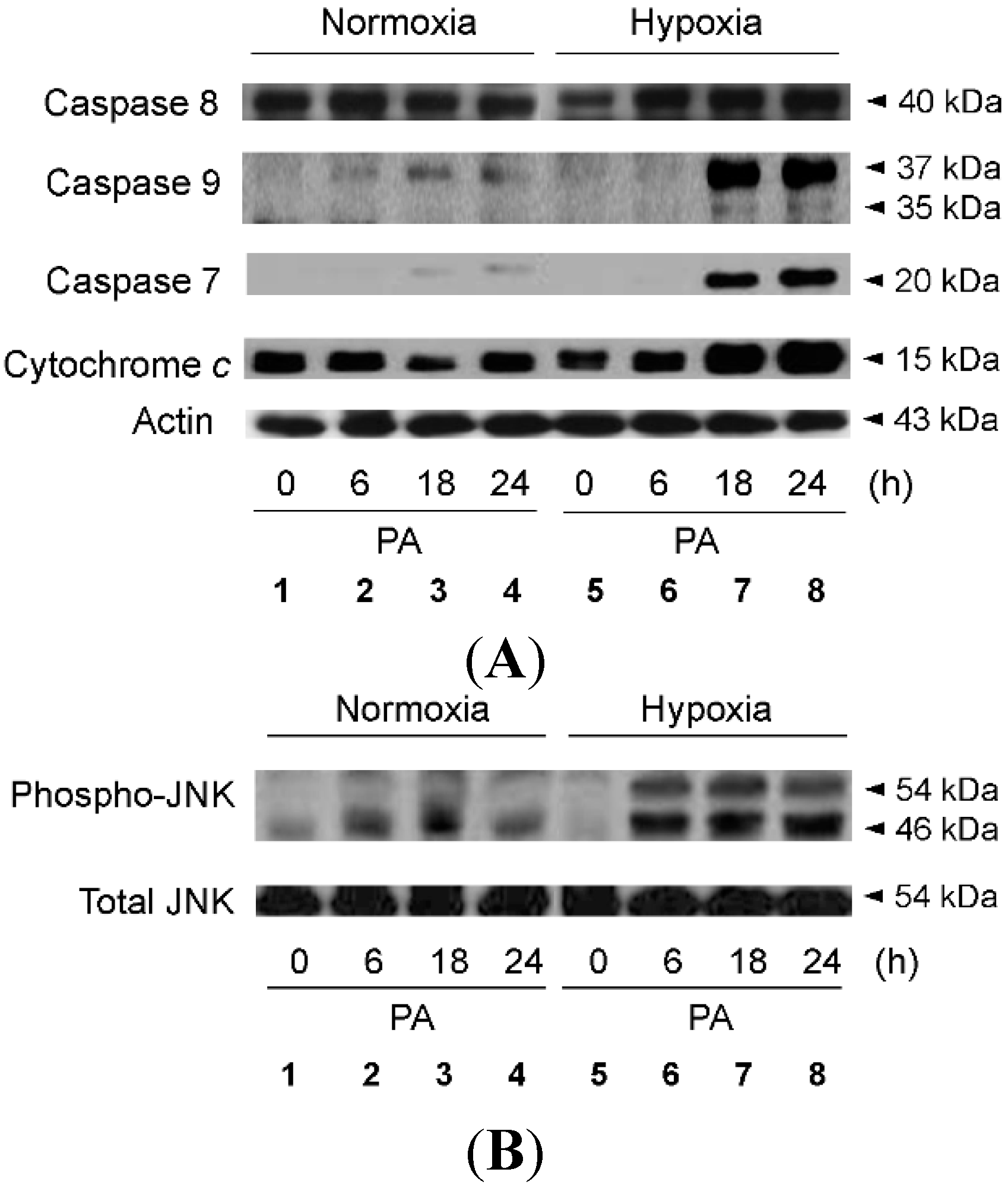
2.2. Hypoxia Exacerbates Lipoapoptosis Occurring via Mitochondrial Pathways
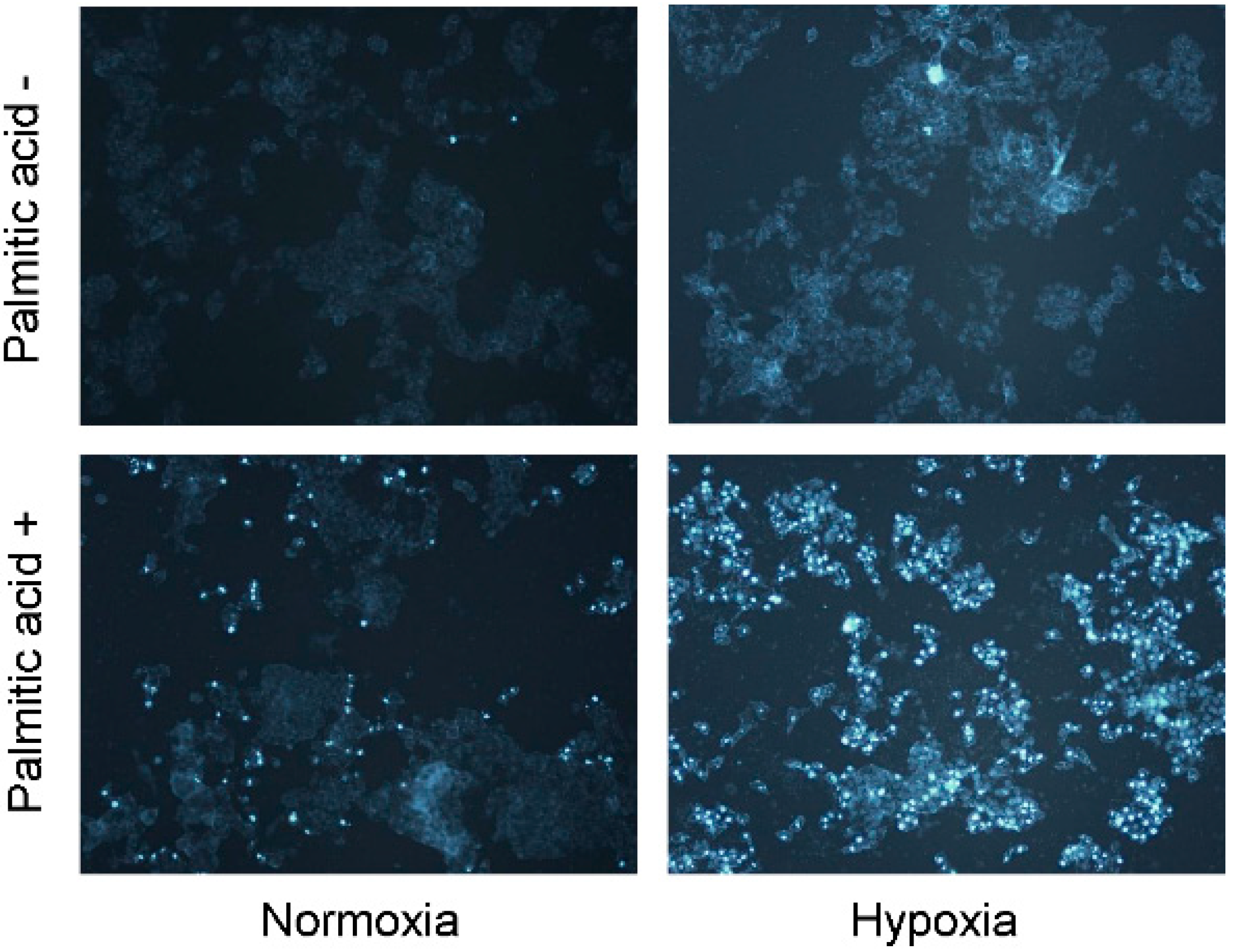
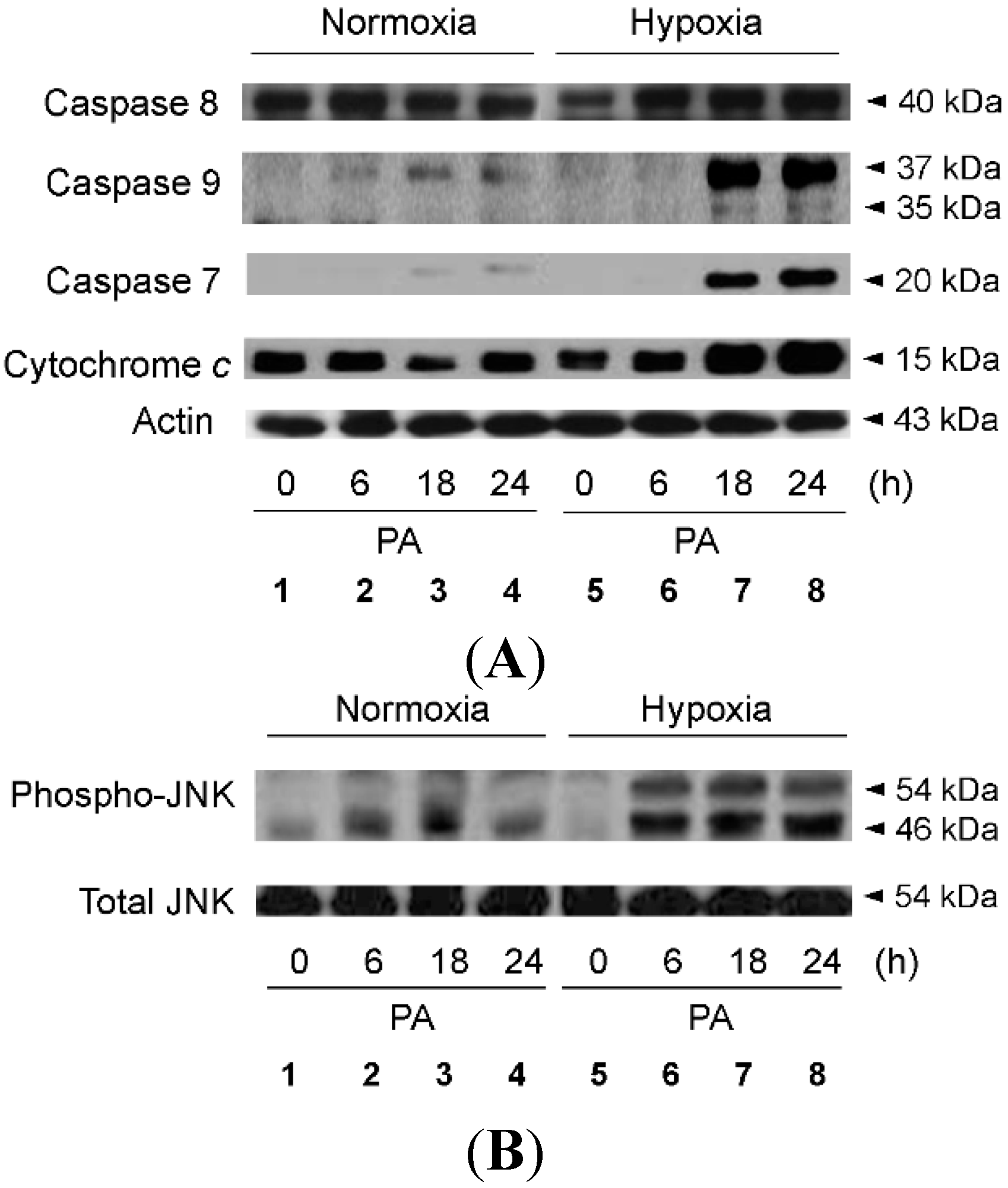
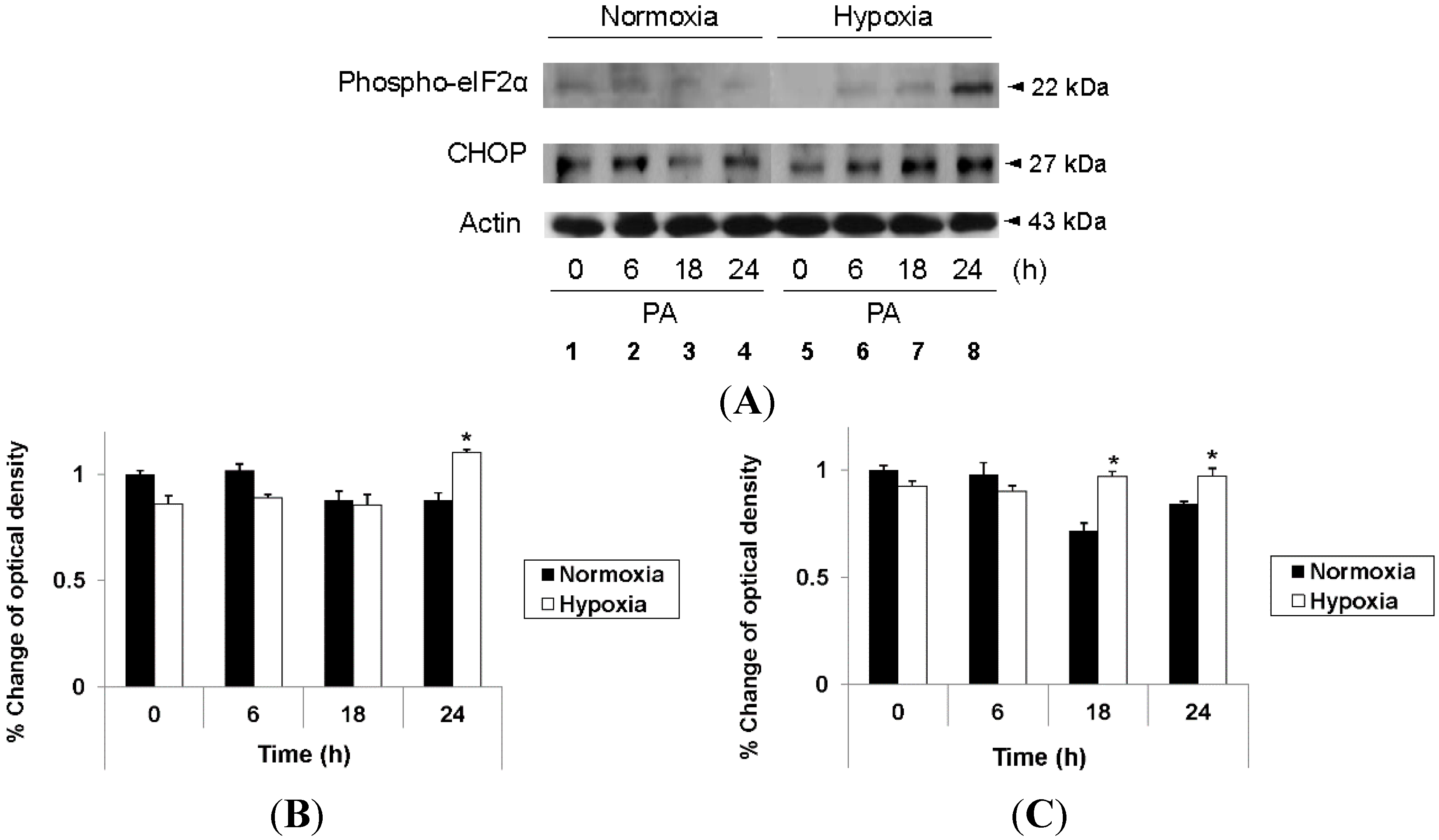
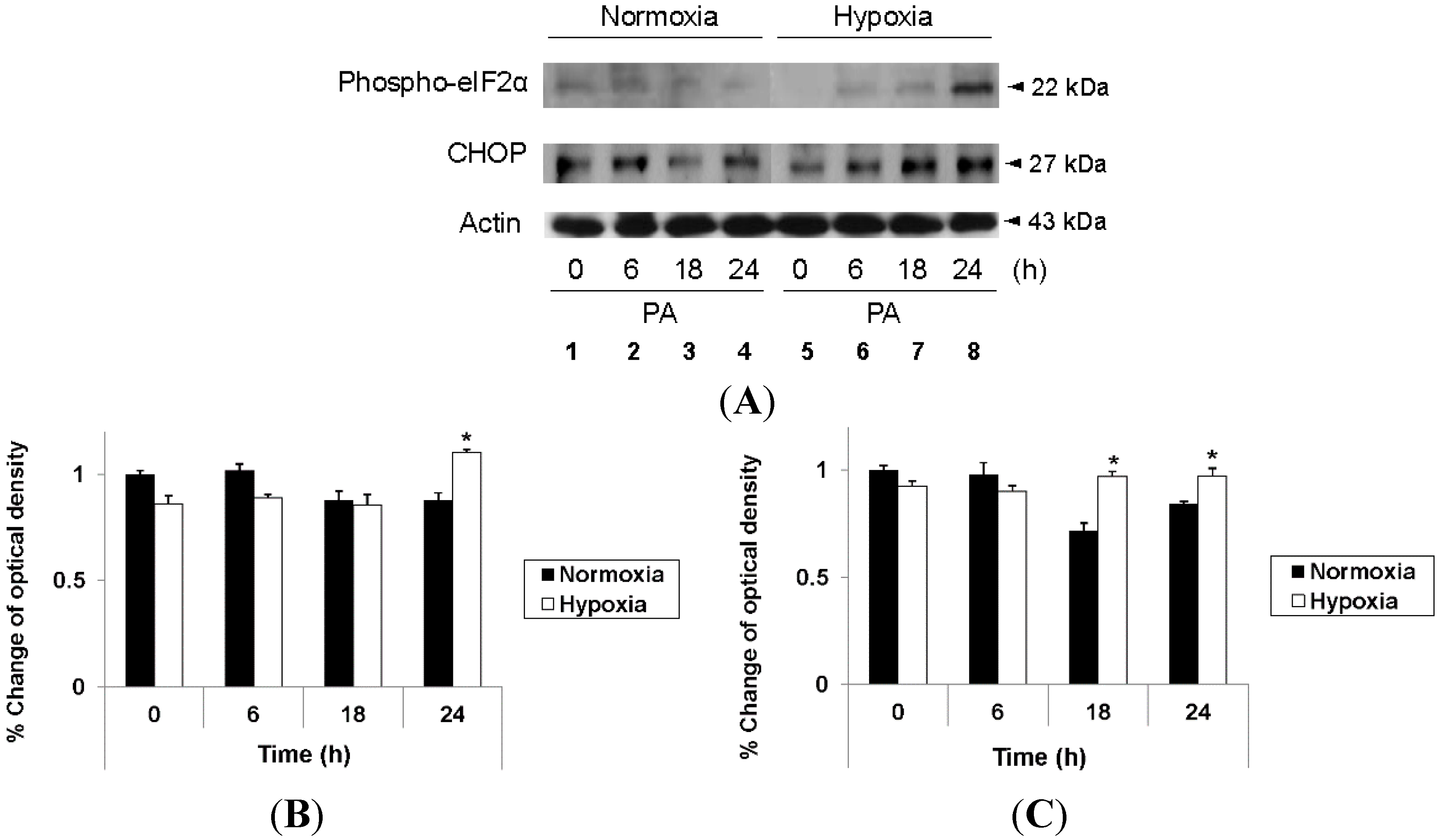
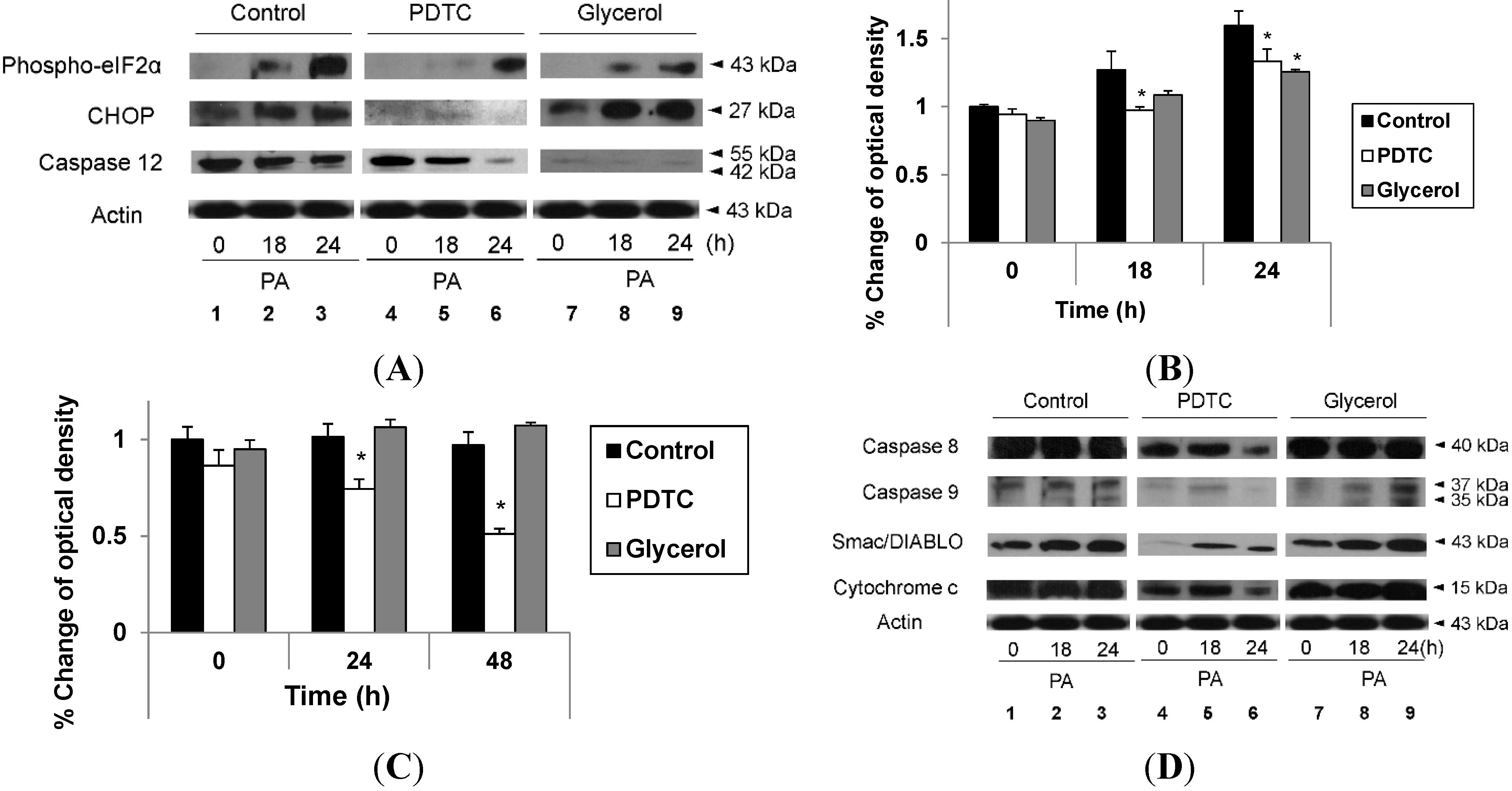
2.3. Hypoxia Exacerbates Endoplasmic Reticulum Stress Leading to Activation of Mitochondrial Apoptotic Pathways
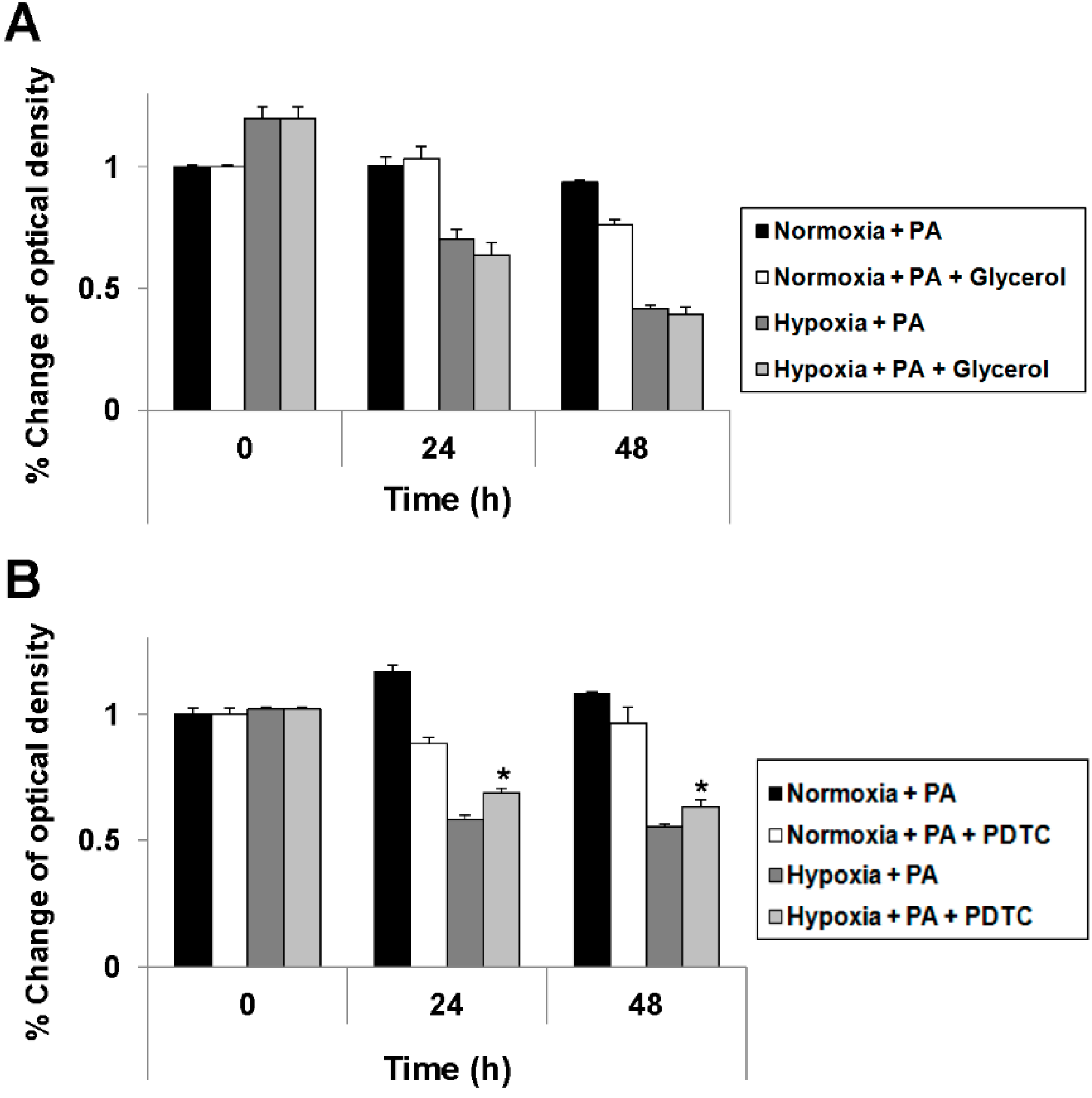
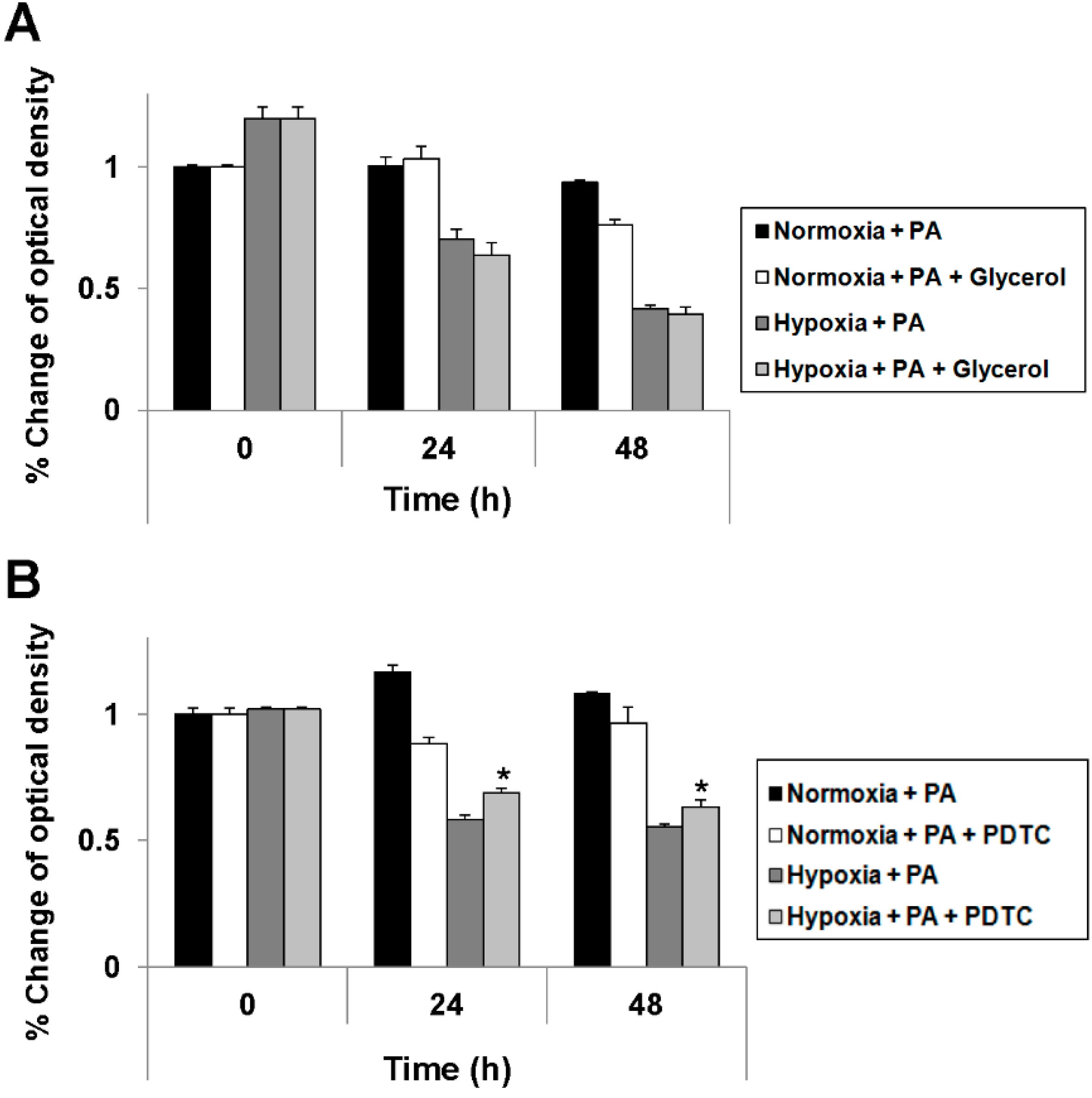
2.4. Reduction of Oxidative Stress Attenuates Exacerbation of Lipoapoptosis by Hypoxia
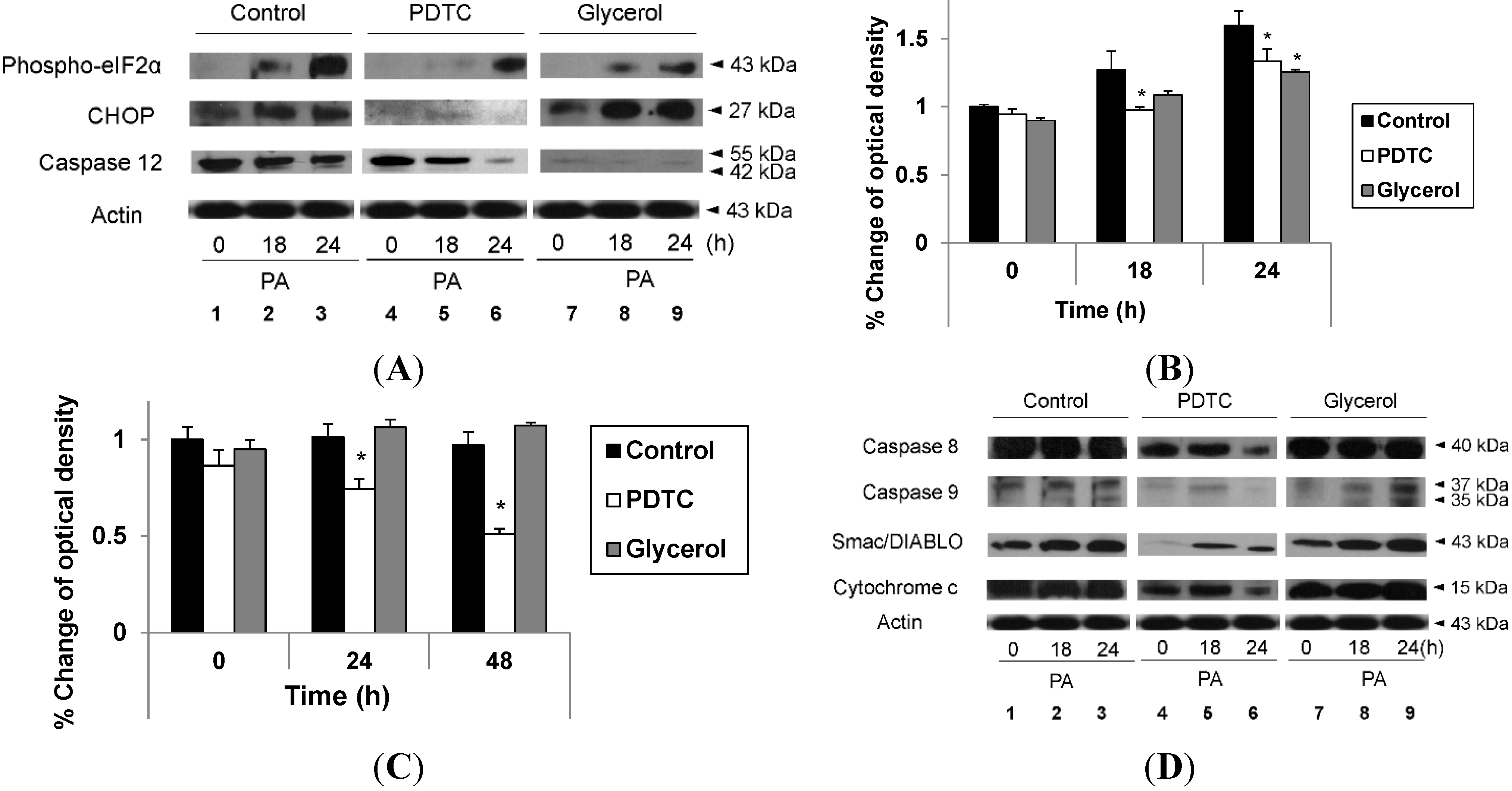
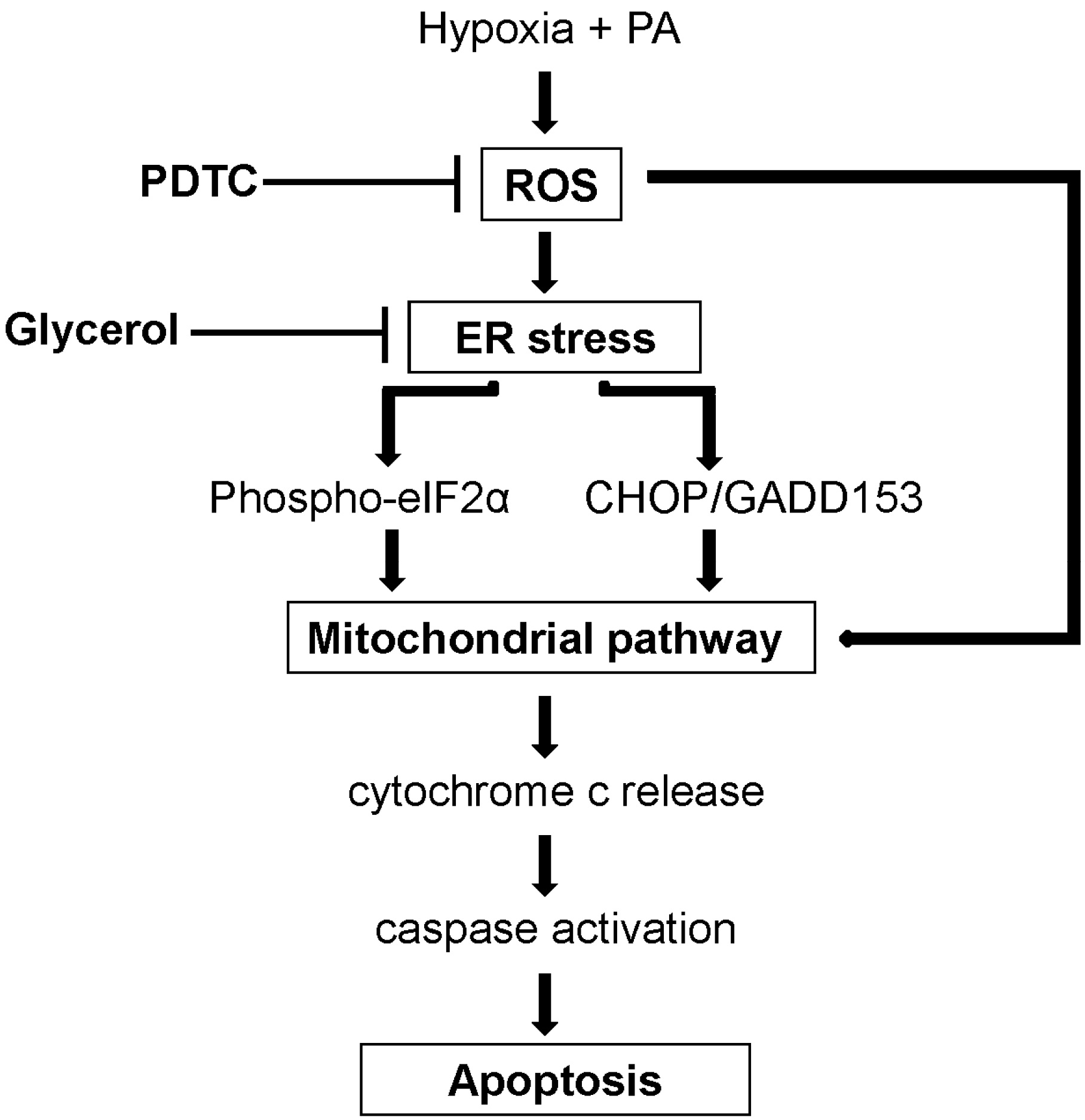
3. Discussion
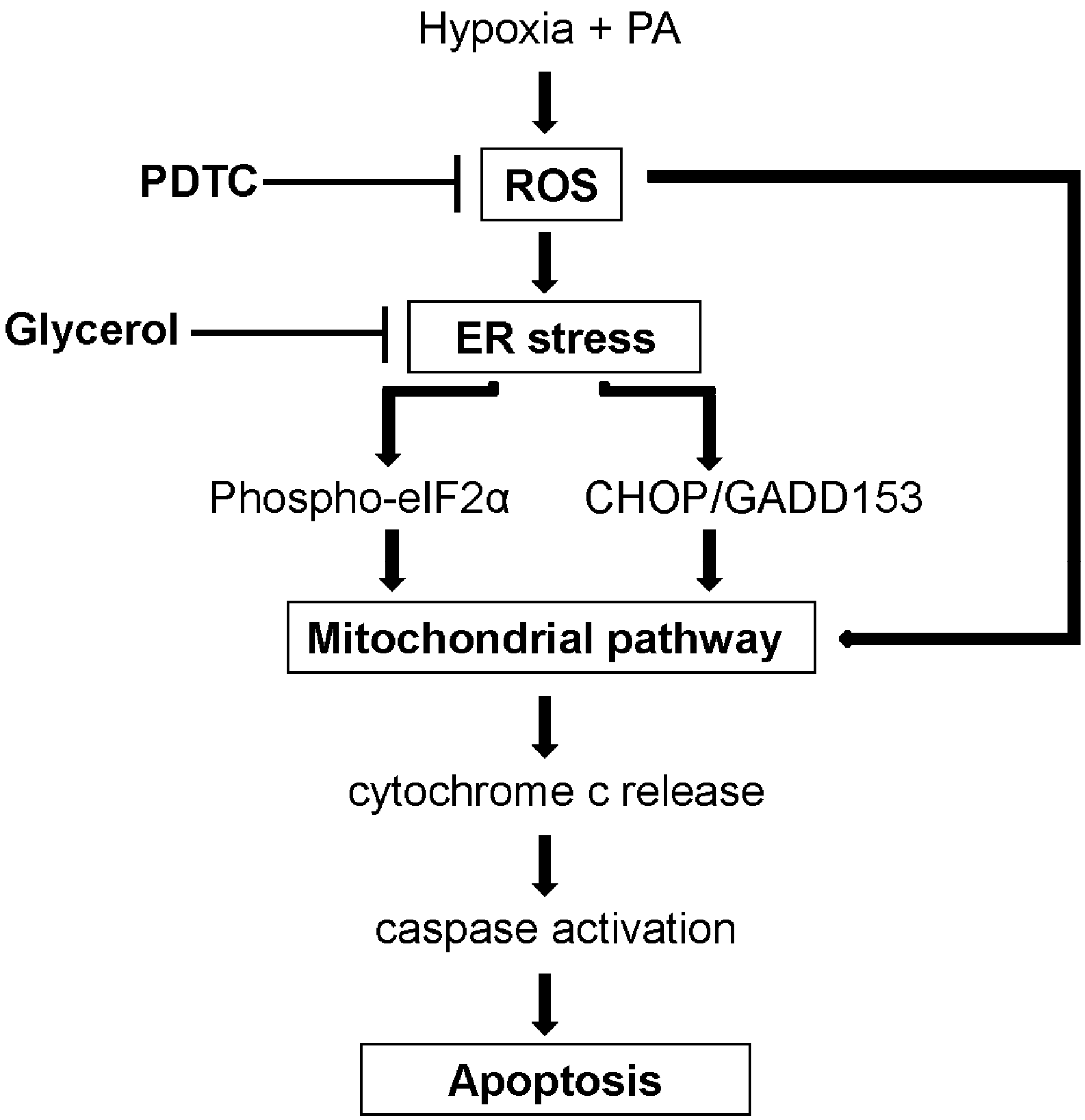
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Cell Proliferation Analysis
4.3. Reagents
4.4. Apoptosis Analysis
4.5. Immunoblot Analysis
4.6. Data Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [PubMed]
- Duvnjak, M.; Lerotic, I.; Barsic, N.; Tomasic, V.; Virovic Jukic, L.; Velagic, V. Pathogenesis and management issues for non-alcoholic fatty liver diseases in the management of hepatocelluar carcinoma. World J. Gastroenterol. 2007, 13, 4539–4950. [Google Scholar] [PubMed]
- Lazo, M.; Clark, J.M. The epidemiology of nonalcoholic fatty liver disease: A global perspective. Semin. Liver Dis. 2008, 28, 339–350. [Google Scholar] [CrossRef]
- Malhi, H.; Gores, G.J. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 360–369. [Google Scholar] [CrossRef]
- Takayama, F.; Egashira, T.; Kawasaki, H.; Mankura, M.; Nakamoto, K.; Okada, S.; Mori, A. A novel animal model of nonalcoholic steatohepatitis (NASH): Hypoxemia enhances the development of NASH. J. Clin. Biochem. Nutr. 2009, 45, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Tanne, F.; Gagnadoux, F.; Chazouilleres, O.; Fleury, B.; Wendum, D.; Lasnier, E.; Lebeau, B.; Poupon, R.; Serfaty, L. Chronic liver injury during obstructive sleep apnea. Hepatology 2005, 41, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Zamora-Valdes, D.; Mendez-Sanchez, N. Experimental evidence of obstructive sleep apnea syndrome as a second hit accomplice in nonalcoholic steatohepatitis pathogenesis. Ann. Hepatol. 2007, 6, 281–283. [Google Scholar] [PubMed]
- Savransky, V.; Nanayakkara, A.; Vivero, A.; Li, J.; Bevans, S.; Smith, P.L.; Torbnson, M.S.; Polotsky, V.Y. Chronic intermittent hypoxia predisposes to liver injury. Hepatology 2007, 45, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Day, C.; James, O. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufmann, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Araki, E.; Mori, M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic β-cells. Apoptosis 2002, 7, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [PubMed]
- Wang, X.Z.; Lawson, B.; Brewer, J.W.; Zinszner, H.; Sanjay, A.; Mi, L.J.; Boorstein, R.; Kreibic, G.; Hendershot, L.M.; Ron, D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol. Cell. Biol. 1996, 16, 4273–4280. [Google Scholar] [PubMed]
- Gotoh, T.; Oyadomari, S.; Mori, K.; Mori, M. Nitric oxide-induced apoptosis in RAW 264.7 macrophages is mediated by endoplasmic reticulum stress pathway involvingATF6 and CHOP. J. Biol. Chem. 2002, 277, 12343–12350. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.S.; Reddy, J.K. Peroxisomal β-oxidation and steatohepatitis. Semin. Liver Dis. 2001, 21, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Cande, C.; Vahsen, N.; Metivier, D.; Tourriere, H.; Chebli, K.; Garrido, C.; Tazi, J.; Kroemer, G. Regulation of cytoplasmic stress granules by apoptosis-inducing factor. J. Cell Sci. 2004, 117, 4461–4468. [Google Scholar] [CrossRef] [PubMed]
- Pharm, T.N.; Marion, M.; Denizeau, F.; Jumarie, C. Cadmium-induced apoptosis in rat hepatocytes does not necessarily involve caspase-dependent pathways. Toxicol. In Vitro 2006, 20, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.R.; Yu, C.J.; Chen, W.C.; Lee, H.S.; Chang, H.M.; Lee, Y.C.; Chien, C.T.; Chen, C.F. Green tea extract supplement reduces d-galactosamine-induced acute liver injury by inhibition of apoptotic and proinflammatory signaling. J. Biomed. Sci. 2009, 16, 35. [Google Scholar] [CrossRef] [PubMed]
- Jian, B.; Hsiceh, C.H.; Chen, J.; Choudhry, M.; Bland, K.; Chaudry, I.; Raju, R. Activation of endoplasmic reticulum stress response following trauma-hemorrhage. Biochim. Biophys. Acta 2008, 1782, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Kanki, K.; Kawamura, T.; Watanabe, Y. Control of ER stress by a chemical chaperone counteracts apoptotic signals in IFN-γ-treated murine hepatocytes. Apoptosis 2009, 14, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Ballestri, S.; Marchesini, G.; Angulo, P.; Loria, P. Nonalcoholic fatty liver disease: A precursor of the metabolic syndrome. Dig. Liver Dis. 2014. [Google Scholar] [CrossRef]
- Argo, C.K.; Northup, P.G.; Al-Osaimi, A.M.; Caldwell, S.H. Systematic review of risk factors for fibrosis progression in non-alcoholic steatohepatitis. J. Hepatol. 2009, 51, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Nugent, C.; Afendy, A.; Bai, C.; Bhatia, P.; Afendy, M.; Fang, Y.; Elariny, H.; Goodman, Z.; Younossi, Z.M. Apnoeic-hypopnoeic episodes during obstructive sleep apnoea are associated with histological nonalcoholic steatohepatitis. Liver Int. 2008, 28, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Pollock, R.; Uhanova, J.; Kryger, M.; Hawkins, K.; Minuk, G.Y. Symptoms of obstructive sleep apnea in patients with nonalcoholic fatty liver disease. Dig. Dis. Sci. 2005, 50, 2338–2343. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Olivetti, C.; Rosina, F.; Carbone, G.; Gambino, R. Association of obstructive sleep apnoea with the presence and severity of non-alcoholic fatty liver disease. A systematic review and meta-analysis. Obes. Rev. 2013, 14, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Cutrera, R.; Liccardo, D.; Pavone, M.; Devito, R.; Giorgio, V.; Verrillo, E.; Baviera, G.; Musso, G. Obstructive sleep apnea syndrome affects liver histology and inflammatory cell activation in pediatric nonalcoholic fatty liver disease, regardless of obesity/insulin resistance. Am. J. Respir. Crit. Care Med. 2014, 189, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Arnardottir, E.S.; Mackiewicz, M.; Gislason, T.; Teff, K.L.; Pack, A.I. Molecular signatures of obstructive sleep apnea in adults: A review and perspective. Sleep 2009, 32, 447–470. [Google Scholar] [PubMed]
- Nath, B.; Levin, I.; Csak, T.; Petrasek, J.; Mueller, C.; Kodys, K.; Catalano, D.; Mandrekar, P.; Szabo, G. Hepatocyte-specific hypoxia-inducible factor-1α is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology 2011, 53, 1526–1537. [Google Scholar] [CrossRef] [PubMed]
- Yoo, W.; Noh, K.H.; Ahn, J.H.; Yu, J.H.; Seo, J.A.; Kim, S.G.; Choi, K.M.; Baik, S.H.; Choi, D.S.; Kim, T.W.; et al. HIF-1α expression as a protective strategy of HepG2 cells against fatty acid-induced toxicity. J. Cell. Biochem. 2014, 115, 1147–1158. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, S.Y.; Yu, S.J.; Lee, J.-H.; Kim, H.Y.; Kim, Y.J. Reduction of Oxidative Stress Attenuates Lipoapoptosis Exacerbated by Hypoxia in Human Hepatocytes. Int. J. Mol. Sci. 2015, 16, 3323-3334. https://doi.org/10.3390/ijms16023323
Hwang SY, Yu SJ, Lee J-H, Kim HY, Kim YJ. Reduction of Oxidative Stress Attenuates Lipoapoptosis Exacerbated by Hypoxia in Human Hepatocytes. International Journal of Molecular Sciences. 2015; 16(2):3323-3334. https://doi.org/10.3390/ijms16023323
Chicago/Turabian StyleHwang, Sang Youn, Su Jong Yu, Jeong-Hoon Lee, Hwi Young Kim, and Yoon Jun Kim. 2015. "Reduction of Oxidative Stress Attenuates Lipoapoptosis Exacerbated by Hypoxia in Human Hepatocytes" International Journal of Molecular Sciences 16, no. 2: 3323-3334. https://doi.org/10.3390/ijms16023323
APA StyleHwang, S. Y., Yu, S. J., Lee, J.-H., Kim, H. Y., & Kim, Y. J. (2015). Reduction of Oxidative Stress Attenuates Lipoapoptosis Exacerbated by Hypoxia in Human Hepatocytes. International Journal of Molecular Sciences, 16(2), 3323-3334. https://doi.org/10.3390/ijms16023323