
Mammalian Introns: When the Junk Generates Molecular Diversity



Abstract
:In eukaryotes, the process of making a messenger RNA (mRNA) involves the co-transcriptional excision of introns in the nucleus, whereas joined-exons are exported to the cytoplasm to be translated. As a consequence, introns are inherently non-protein-coding sequences in that they are transcribed but not translated (usually) into protein. Because, and by definition, introns always lie between 2 exons, they were called “Intervening” or INtrons. By opposition, exons are those sequences that are EXpressed and EXported to the cytoplasm.
1. Introduction
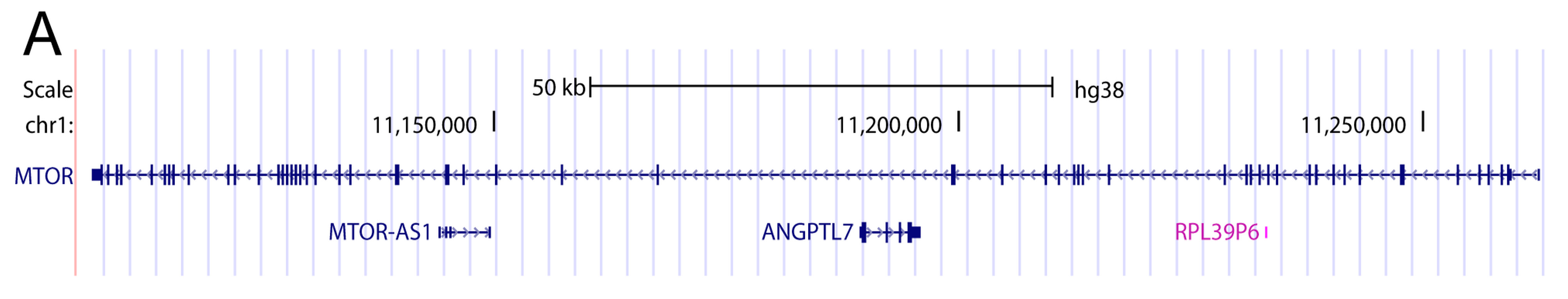
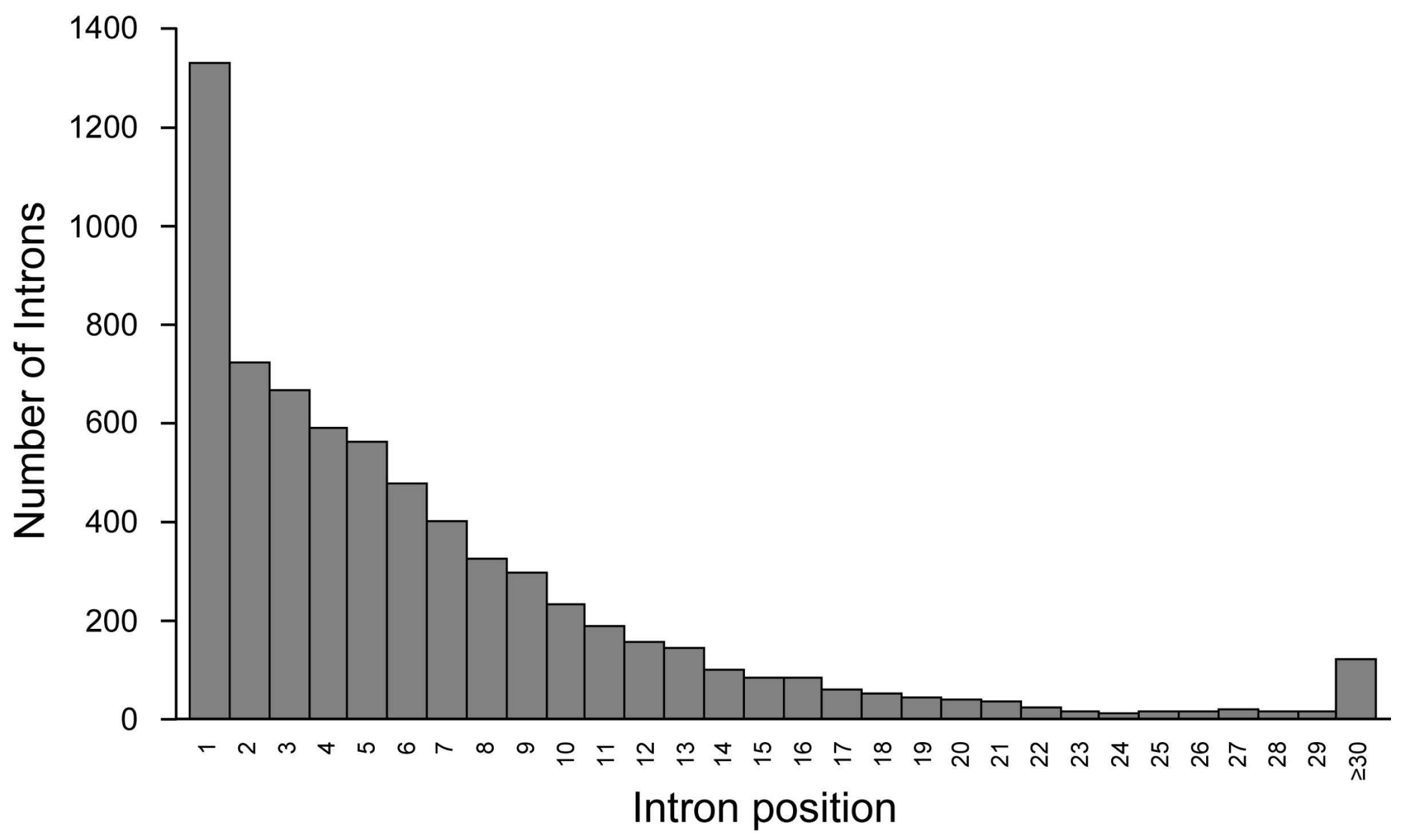
2. Introns Account for Half of the Genome—From a Single Nucleotide to One Megabase
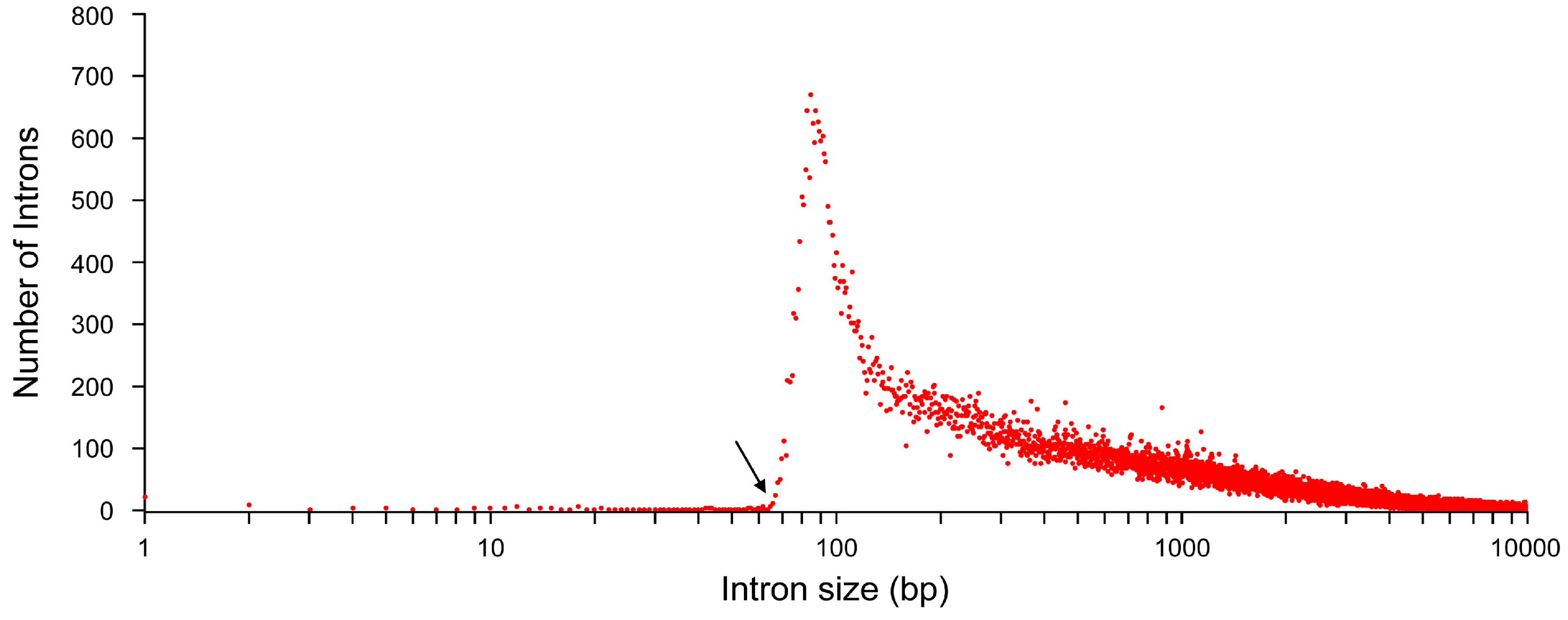

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
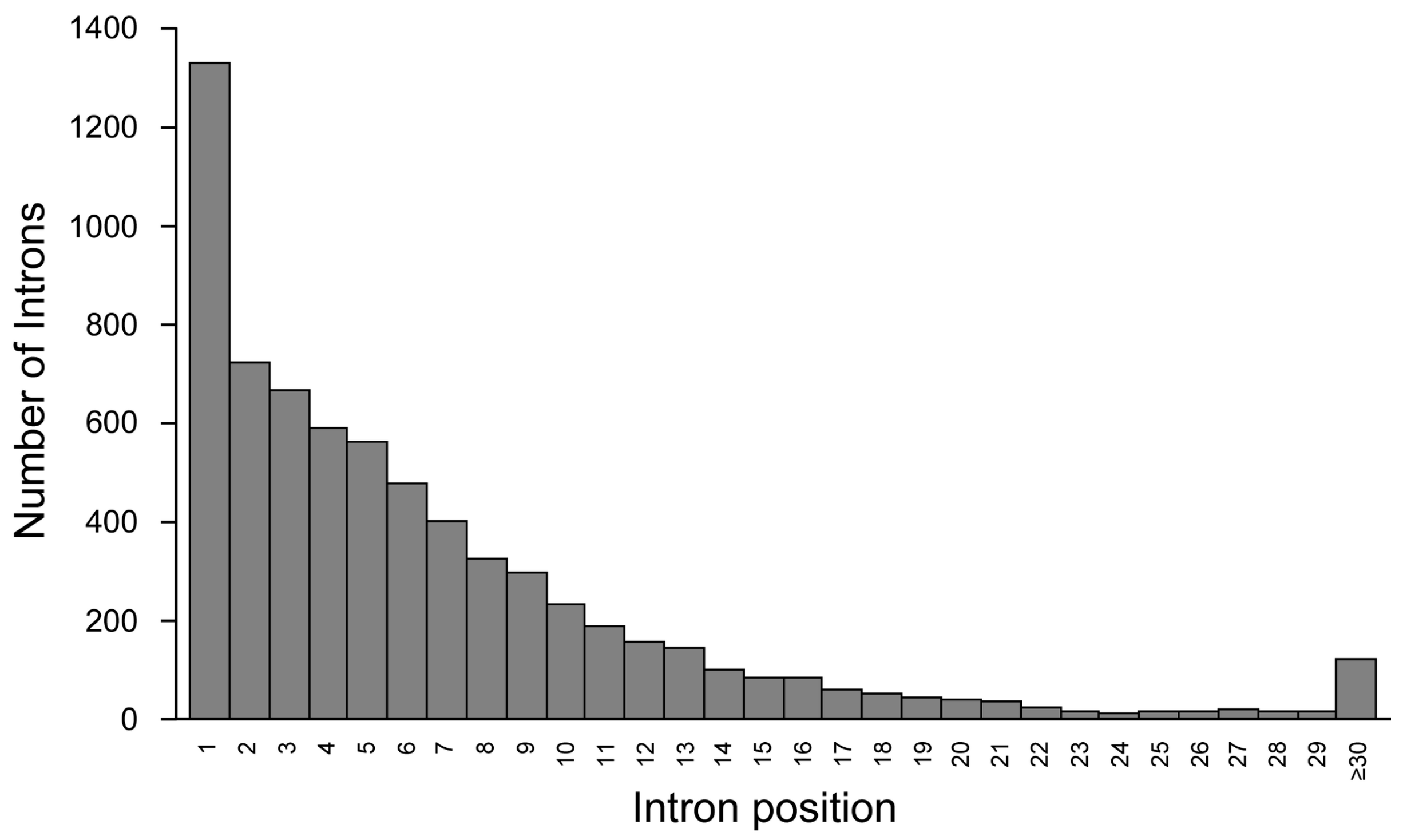{kind=link}
{kind=link}
{kind=link}
| Chr # | Total # Genes | Total # Exons | Total # Introns | Max # Exons/Gene | Chromosome Size (bp) | Avg # of Exons/Gene | Avg Length (bp) ± Std Dev | Total Length (bp) | Shortest (bp) | Longest (bp) | Genes/Millions | Intronless Genes | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exon | Intron | Exon | Intron | Exon | Intron | Gene | Exon | Intron | Gene | |||||||||
| 1 | 3592 | 31,744 | 28,152 | 138 | 248,956,422 | 8.8 | 313 ± 705 | 5283 ± 16,017 | 9,934,447 | 148,725,908 | 3 | 1 | 41 | 12,573 | 451,448 | 1,491,100 | 14.4 | 544 |
| 2 | 2208 | 24,805 | 22,597 | 363 | 242,193,529 | 11.2 | 299 ± 718 | 6574 ± 21,058 | 7,407,514 | 147,572,357 | 1 | 14 | 49 | 17,969 | 866,400 | 1,900,275 | 9.1 | 203 |
| 3 | 1916 | 18,292 | 16,376 | 118 | 198,295,559 | 9.5 | 317 ± 759 | 7669 ± 25,134 | 5,801,464 | 125,580,914 | 3 | 1 | 21 | 24,927 | 842,378 | 1,502,150 | 9.7 | 181 |
| 4 | 1234 | 10,880 | 9646 | 84 | 190,214,555 | 8.8 | 343 ± 740 | 8687 ± 26,112 | 3,736,564 | 83,794,301 | 8 | 1 | 44 | 9856 | 912,253 | 1,474,687 | 6.5 | 197 |
| 5 | 1496 | 13,193 | 11,697 | 90 | 181,538,259 | 8.8 | 342 ± 778 | 7678 ± 23,233 | 4,508,195 | 89,805,936 | 6 | 12 | 43 | 22,753 | 772,519 | 1,519,058 | 8.2 | 205 |
| 6 | 1793 | 16,106 | 14,313 | 146 | 170,805,979 | 9.0 | 326 ± 719 | 6876 ± 20,012 | 5,256,442 | 98,421,513 | 6 | 1 | 50 | 15,177 | 478,750 | 1,987,246 | 10.5 | 227 |
| 7 | 1629 | 15,280 | 13,651 | 108 | 159,345,973 | 9.4 | 315 ± 763 | 7770 ± 24,375 | 4,806,589 | 106,063,166 | 2 | 1 | 53 | 21,017 | 657,297 | 2,304,636 | 10.2 | 187 |
| 8 | 1207 | 10,041 | 8834 | 86 | 145,138,636 | 8.3 | 335 ± 779 | 8354 ± 26,109 | 3,363,013 | 73,802,469 | 5 | 12 | 23 | 15,980 | 955,098 | 2,059,454 | 8.3 | 160 |
| 9 | 1402 | 12,839 | 11,437 | 98 | 138,394,717 | 9.2 | 314 ± 714 | 6073 ± 16,910 | 4,029,327 | 69,453,705 | 3 | 5 | 54 | 10,345 | 344,501 | 2,298,478 | 10.1 | 200 |
| 10 | 1346 | 12,626 | 11,280 | 68 | 133,797,422 | 9.4 | 321 ± 723 | 7821 ± 23,764 | 4,058,525 | 88,222,345 | 5 | 67 | 50 | 11,090 | 482,575 | 1,783,674 | 10.1 | 131 |
| 11 | 2112 | 17,709 | 15,597 | 90 | 135,086,622 | 8.4 | 311 ± 959 | 5306 ± 20,073 | 5,501,031 | 82,757,607 | 2 | 1 | 50 | 91,671 | 811,152 | 1,468,409 | 15.6 | 377 |
| 12 | 1773 | 17,618 | 15,845 | 173 | 133,275,309 | 9.9 | 300 ± 686 | 5076 ± 15,654 | 5,281,875 | 80,428,519 | 9 | 5 | 50 | 14,194 | 403,400 | 1,249,864 | 13.3 | 169 |
| 13 | 715 | 5982 | 5267 | 83 | 114,364,328 | 8.4 | 339 ± 957 | 9101 ± 29,323 | 2,025,232 | 47,933,871 | 5 | 66 | 48 | 37,567 | 740,920 | 1,468,616 | 6.3 | 94 |
| 14 | 1171 | 9771 | 8600 | 116 | 107,043,718 | 8.3 | 319 ± 734 | 6212 ± 20,436 | 3,115,048 | 53,426,644 | 4 | 14 | 46 | 17,546 | 479,079 | 1,464,560 | 10.9 | 229 |
| 15 | 1188 | 11,480 | 10,292 | 104 | 101,991,189 | 9.7 | 306 ± 698 | 5672 ± 17,698 | 3,514,500 | 58,372,375 | 8 | 21 | 33 | 11,532 | 732,200 | 887,042 | 11.6 | 220 |
| 16 | 1468 | 13,212 | 11,744 | 63 | 90,338,345 | 9.0 | 285 ± 607 | 3892 ± 17,423 | 3,766,310 | 45,712,994 | 3 | 1 | 51 | 10,024 | 778,855 | 1,694,208 | 16.3 | 159 |
| 17 | 2069 | 19,482 | 17,413 | 85 | 83,257,441 | 9.4 | 289 ± 615 | 3531 ± 13,106 | 5,622,686 | 61,484,756 | 8 | 1 | 47 | 10,345 | 1,043,910 | 1,143,719 | 24.9 | 251 |
| 18 | 495 | 4568 | 4073 | 75 | 80,373,285 | 9.2 | 362 ± 850 | 10,552 ± 25,422 | 1,653,586 | 42,976,591 | 9 | 75 | 50 | 14,862 | 411,175 | 1,195,732 | 6.2 | 58 |
| 19 | 2449 | 18,534 | 16,085 | 106 | 58,617,616 | 7.6 | 297 ± 655 | 2382 ± 6158 | 5,507,290 | 38,316,474 | 6 | 2 | 47 | 21,693 | 255,789 | 301,152 | 41.8 | 358 |
| 20 | 983 | 8035 | 7052 | 80 | 64,444,167 | 8.2 | 312 ± 675 | 5515 ± 18,323 | 2,509,662 | 38,891,514 | 8 | 31 | 50 | 10,441 | 544,980 | 2,057,697 | 15.3 | 103 |
| 21 | 463 | 3745 | 3282 | 47 | 46,709,983 | 8.1 | 309 ± 728 | 6401 ± 18,061 | 1,157,367 | 21,006,837 | 9 | 9 | 60 | 13,351 | 323,564 | 834,698 | 9.9 | 95 |
| 22 | 797 | 7029 | 6232 | 55 | 50,818,468 | 8.8 | 312 ± 712 | 4321 ± 12,968 | 2,194,256 | 26,927,006 | 4 | 1 | 52 | 12,955 | 355,998 | 701,852 | 15.7 | 84 |
| X | 1152 | 8062 | 6910 | 84 | 156,040,895 | 7.0 | 350 ± 873 | 6898 ± 24,018 | 2,820,104 | 47,662,441 | 7 | 67 | 48 | 37,027 | 536,479 | 1,368,337 | 7.4 | 244 |
| Y | 198 | 1318 | 1120 | 46 | 57,227,415 | 6.7 | 259 ± 540 | 10,637 ± 33,468 | 341,789 | 14,179,080 | 22 | 67 | 64 | 8690 | 493,512 | 686,139 | 3.5 | 11 |
| Chr # | Total # Genes | Total # Exons | Total # Introns | Max # Exons/Gene | Chromosome Size (bp) | Avg # of Exons/Gene | Avg Length (bp) ± Std Dev | Total Length (bp) | Shortest (bp) | Longest (bp) | Genes/Millions | Intronless Genes | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exon | Intron | Exon | Intron | Exon | Intron | Gene | Exon | Intron | Gene | |||||||||
| 1 | 2729 | 28,554 | 25,825 | 138 | 248,956,422 | 10.5 | 307 ± 702 | 5136 ± 15,685 | 8,778,766 | 132,633,451 | 3 | 1 | 270 | 12,573 | 451,448 | 1,491,100 | 11.0 | 149 |
| 2 | 1653 | 22,244 | 20,591 | 363 | 242,193,529 | 13.5 | 294 ± 723 | 6283 ± 20,404 | 6,534,873 | 128,437,100 | 1 | 37 | 582 | 17,969 | 866,400 | 1,900,275 | 6.8 | 35 |
| 3 | 1448 | 16,336 | 14,888 | 118 | 198,295,559 | 11.3 | 312 ± 765 | 7535 ± 24,995 | 5,098,251 | 5,098,251 | 3 | 3 | 294 | 24,927 | 24,927 | 1,502,150 | 7.3 | 44 |
| 4 | 989 | 9991 | 9002 | 84 | 190,214,555 | 10.1 | 338 ± 729 | 8471 ± 25,955 | 3,372,765 | 76,252,775 | 8 | 1 | 576 | 9856 | 912,253 | 1,474,687 | 5.2 | 107 |
| 5 | 1103 | 11,709 | 10,606 | 90 | 181,538,259 | 10.6 | 337 ± 785 | 7240 ± 22,309 | 3,944,829 | 76,788,283 | 6 | 21 | 530 | 22,753 | 772,519 | 1,519,058 | 6.1 | 85 |
| 6 | 1432 | 14,660 | 13,228 | 146 | 170,805,979 | 10.2 | 317 ± 713 | 6665 ± 19,530 | 4,653,325 | 88,160,529 | 6 | 1 | 354 | 15,177 | 478,750 | 1,987,246 | 8.4 | 109 |
| 7 | 1189 | 13,187 | 11,998 | 108 | 159,345,973 | 11.1 | 306 ± 744 | 7897 ± 24,804 | 4,032,145 | 94,745,609 | 2 | 1 | 600 | 14,889 | 657,297 | 2,304,636 | 7.5 | 55 |
| 8 | 869 | 8779 | 7910 | 86 | 145,138,636 | 10.1 | 327 ± 768 | 7942 ± 24,970 | 2,871,568 | 62,820,216 | 5 | 12 | 663 | 15,980 | 955,098 | 2,059,454 | 6.0 | 26 |
| 9 | 1041 | 11,352 | 10,311 | 98 | 138,394,717 | 10.9 | 304 ± 703 | 5971 ± 16,684 | 3,453,046 | 61,566,944 | 3 | 5 | 411 | 10,345 | 344,501 | 2,298,478 | 7.5 | 73 |
| 10 | 978 | 11,096 | 10,118 | 68 | 133,797,422 | 11.3 | 310 ± 715 | 7926 ± 24,491 | 3,440,785 | 80,199,711 | 5 | 67 | 563 | 11,090 | 482,575 | 1,783,674 | 7.3 | 26 |
| 11 | 1703 | 16,196 | 14,493 | 90 | 135,086,622 | 9.5 | 301 ± 665 | 5136 ± 19,917 | 4,880,180 | 74,431,941 | 2 | 1 | 484 | 18,173 | 811,152 | 1,468,409 | 12.6 | 211 |
| 12 | 1407 | 16,186 | 14,779 | 173 | 133,275,309 | 11.5 | 294 ± 688 | 4939 ± 15,284 | 4,757,204 | 72,993,897 | 9 | 5 | 396 | 14,194 | 403,400 | 1,249,864 | 10.6 | 50 |
| 13 | 426 | 4920 | 4494 | 83 | 114,364,328 | 11.5 | 330 ± 864 | 8615 ± 29,149 | 1,624,165 | 38,717,496 | 5 | 66 | 1310 | 21,022 | 740,920 | 1,468,616 | 3.7 | 10 |
| 14 | 832 | 8745 | 7913 | 116 | 107,043,718 | 10.5 | 319 ± 741 | 6107 ± 20,447 | 2,786,125 | 48,327,558 | 4 | 14 | 465 | 17,546 | 479,079 | 1,464,560 | 7.8 | 38 |
| 15 | 769 | 9686 | 8917 | 104 | 101,991,189 | 12.6 | 299 ± 689 | 5508 ± 16,227 | 2,900,248 | 49,116,901 | 8 | 21 | 918 | 10,227 | 550,366 | 887,042 | 7.5 | 23 |
| 16 | 1117 | 11,738 | 10,621 | 63 | 90,338,345 | 10.5 | 281 ± 607 | 3831 ± 17,251 | 3,296,823 | 40,686,903 | 5 | 1 | 397 | 10,024 | 778,855 | 1,694,208 | 12.4 | 20 |
| 17 | 1619 | 17,644 | 16,025 | 85 | 83,257,441 | 10.9 | 280 ± 605 | 3427 ± 13,031 | 4,943,096 | 54,914,295 | 8 | 1 | 445 | 9719 | 1,043,910 | 1,143,719 | 19.4 | 68 |
| 18 | 364 | 4082 | 3718 | 75 | 80,373,285 | 11.2 | 360 ± 872 | 10,356 ± 24,257 | 1,469,520 | 38,504,335 | 9 | 75 | 906 | 14,862 | 411,175 | 1,195,732 | 4.5 | 17 |
| 19 | 1904 | 16,757 | 14,853 | 106 | 58,617,616 | 8.8 | 296 ± 659 | 2283 ± 5233 | 4,964,098 | 33,906,702 | 6 | 2 | 541 | 21,693 | 121,730 | 301,152 | 32.5 | 54 |
| 20 | 741 | 7114 | 6373 | 80 | 64,444,167 | 9.6 | 307 ± 669 | 5564 ± 18,912 | 2,180,506 | 35,459,411 | 11 | 66 | 666 | 10,441 | 544,980 | 2,057,697 | 11.5 | 23 |
| 21 | 311 | 3183 | 2872 | 47 | 46,709,983 | 10.2 | 292 ± 700 | 5831 ± 16,574 | 930,502 | 16,745,676 | 9 | 9 | 147 | 11,938 | 323,564 | 834,698 | 6.7 | 54 |
| 22 | 598 | 6190 | 5592 | 55 | 50,818,468 | 10.4 | 300 ± 702 | 4156 ± 11,646 | 1,857,403 | 23,242,726 | 8 | 1 | 686 | 12,955 | 322,908 | 701,852 | 11.8 | 14 |
| X | 854 | 7222 | 6368 | 84 | 156,040,895 | 8.5 | 344 ± 774 | 6428 ± 22,011 | 2,483,142 | 40,934,557 | 10 | 67 | 501 | 10,363 | 536,479 | 1,368,337 | 5.5 | 79 |
| Y | 101 | 849 | 748 | 46 | 57,227,415 | 8.4 | 267 ± 576 | 9483 ± 32,196 | 226,383 | 5,888,994 | 24 | 67 | 737 | 8690 | 493,512 | 686,139 | 1.8 | 9 |
| Chr # | Total # Genes | Total # Exons | Total # Introns | Max # Exons/Gene | Chromosome Size (bp) | Avg # of Exons/Gene | Avg length (bp) ± Std Dev | Total length (bp) | Shortest (bp) | Longest (bp) | Genes/Millions | Intronless Genes | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exon | Intron | Exon | Intron | Exon | Intron | Gene | Exon | Intron | Gene | |||||||||
| 1 | 863 | 3190 | 2327 | 46 | 248,956,422 | 3.7 | 362 ± 731 | 6916 ± 19,245 | 1,155,681 | 16,092,457 | 4 | 44 | 41 | 11,846 | 300,899 | 670,478 | 3.5 | 395 |
| 2 | 555 | 2561 | 2006 | 55 | 242,193,529 | 4.6 | 341 ± 668 | 9539 ± 26,653 | 872,641 | 19,135,257 | 3 | 14 | 49 | 11,633 | 415,325 | 1,126,123 | 2.3 | 168 |
| 3 | 468 | 1956 | 1488 | 45 | 198,295,559 | 4.2 | 360 ± 702 | 9009 ± 26,455 | 703,213 | 13,405,364 | 12 | 67 | 21 | 8244 | 427,004 | 581,065 | 2.4 | 137 |
| 4 | 245 | 889 | 644 | 36 | 190,214,555 | 3.6 | 409 ± 847 | 11,710 ± 28,055 | 363,799 | 7,541,526 | 12 | 60 | 44 | 9848 | 250,403 | 491,647 | 1.3 | 90 |
| 5 | 393 | 1484 | 1091 | 30 | 181,538,259 | 3,8 | 380 ± 722 | 11,932 ± 30,494 | 563,366 | 13,017,653 | 15 | 12 | 43 | 8875 | 340,222 | 932,203 | 2.2 | 120 |
| 6 | 361 | 1446 | 1085 | 36 | 170,805,979 | 4.0 | 417 ± 772 | 9457 ± 25,027 | 603,117 | 10,260,984 | 7 | 73 | 50 | 8695 | 326,934 | 621,277 | 2.1 | 118 |
| 7 | 440 | 2093 | 1653 | 48 | 159,345,973 | 4.8 | 370 ± 871 | 6847 ± 20,981 | 774,444 | 11,317,557 | 13 | 70 | 53 | 21,017 | 414,132 | 630,440 | 2.8 | 132 |
| 8 | 338 | 1262 | 924 | 29 | 145,138,636 | 3.7 | 389 ± 844 | 11,886 ± 34,163 | 491,445 | 10,982,253 | 15 | 71 | 23 | 12,722 | 499,303 | 541,308 | 2.3 | 134 |
| 9 | 361 | 1487 | 1126 | 45 | 138,394,717 | 4.1 | 388 ± 791 | 7004 ± 18,841 | 576,281 | 7,886,761 | 14 | 68 | 54 | 7835 | 308,685 | 310,090 | 2.6 | 127 |
| 10 | 368 | 1530 | 1162 | 28 | 133,797,422 | 4.2 | 404 ± 776 | 6904 ± 16,076 | 617,740 | 8,022,634 | 15 | 73 | 50 | 7617 | 212,605 | 337,030 | 2.8 | 105 |
| 11 | 409 | 1513 | 1104 | 29 | 135,086,622 | 3.7 | 410 ± 2455 | 7541 ± 21,908 | 620,851 | 8,325,666 | 5 | 44 | 50 | 91,671 | 295,436 | 663,821 | 3.0 | 166 |
| 12 | 366 | 1432 | 1066 | 27 | 133,275,309 | 3.9 | 366 ± 666 | 6974 ± 19,998 | 524,671 | 7,434,622 | 17 | 15 | 50 | 10,432 | 266,879 | 373,979 | 2.7 | 119 |
| 13 | 289 | 1062 | 773 | 26 | 114,364,328 | 3.7 | 378 ± 1306 | 11,923 ± 30,179 | 401,067 | 9,216,375 | 14 | 76 | 48 | 37,567 | 330,963 | 562,471 | 2.5 | 84 |
| 14 | 339 | 1026 | 687 | 46 | 107,043,718 | 3.0 | 321 ± 679 | 7422 ± 20,278 | 328,923 | 5,099,086 | 23 | 76 | 46 | 8430 | 289,502 | 437,743 | 3.2 | 191 |
| 15 | 419 | 1794 | 1375 | 35 | 101,991,189 | 4.3 | 342 ± 742 | 6731 ± 25,217 | 614,252 | 9,255,474 | 10 | 21 | 33 | 11,532 | 732,200 | 797,140 | 4.1 | 197 |
| 16 | 351 | 1474 | 1123 | 50 | 90,338,345 | 4.2 | 319 ± 601 | 4476 ± 18,977 | 469,487 | 5,026,091 | 3 | 47 | 51 | 7148 | 368,335 | 531,096 | 3.9 | 139 |
| 17 | 450 | 1838 | 1388 | 61 | 83,257,441 | 4.1 | 370 ± 699 | 4734 ± 13,885 | 679,590 | 6,570,461 | 15 | 70 | 47 | 10,345 | 220,687 | 325,488 | 5.4 | 183 |
| 18 | 131 | 486 | 355 | 22 | 80,373,285 | 3.7 | 379 ± 642 | 12,598 ± 35,371 | 184,066 | 4,472,256 | 26 | 89 | 50 | 4791 | 326,668 | 545,072 | 1.6 | 41 |
| 19 | 545 | 1777 | 1232 | 30 | 58,617,616 | 3.3 | 306 ± 611 | 3579 ± 12,785 | 543,192 | 4,409,772 | 11 | 62 | 47 | 11,194 | 255,789 | 292,306 | 9.3 | 304 |
| 20 | 242 | 921 | 679 | 43 | 64,444,167 | 3.8 | 357 ± 720 | 5055 ± 11,393 | 329,156 | 3,432,103 | 8 | 31 | 50 | 10,441 | 138,007 | 195,695 | 3.8 | 80 |
| 21 | 152 | 562 | 410 | 32 | 46,709,983 | 3.7 | 404 ± 869 | 10,393 ± 25,888 | 226,865 | 4,261,161 | 16 | 79 | 60 | 13,351 | 256,374 | 539,254 | 3.3 | 41 |
| 22 | 199 | 839 | 640 | 30 | 50,818,468 | 4.2 | 401 ± 775 | 5757 ± 21,234 | 336,853 | 3,684,280 | 4 | 9 | 52 | 8320 | 355,998 | 411,958 | 3.9 | 70 |
| X | 298 | 840 | 542 | 17 | 156,040,895 | 2.8 | 401 ± 1472 | 12,413 ± 40,403 | 336,962 | 6,727,884 | 7 | 78 | 48 | 37,027 | 405,107 | 1,033,350 | 1.9 | 165 |
| Y | 97 | 469 | 372 | 26 | 57,227,415 | 4.8 | 246 ± 467 | 16,221 ± 58,381 | 115,406 | 1,313,894 | 22 | 90 | 64 | 5836 | 353,508 | 320,464 | 1.7 | 2 |
3. Introns May Contain Independent Coding and Non-Coding Genes
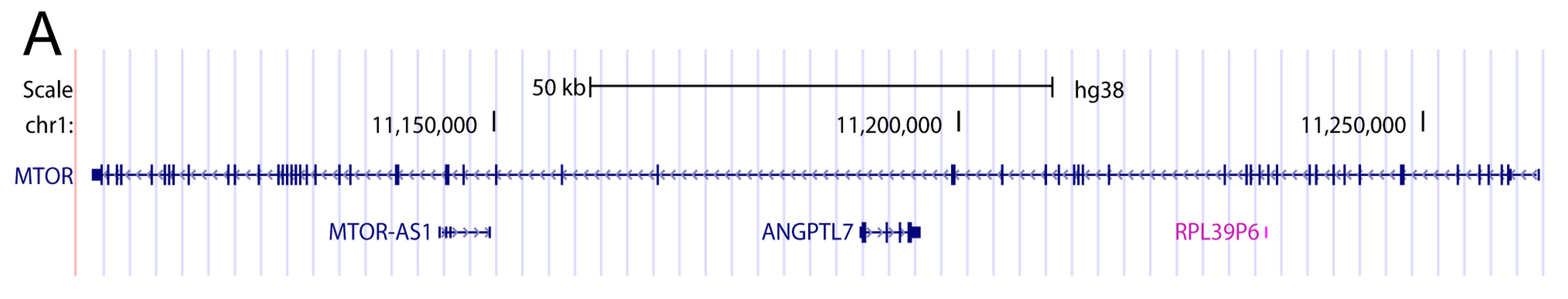

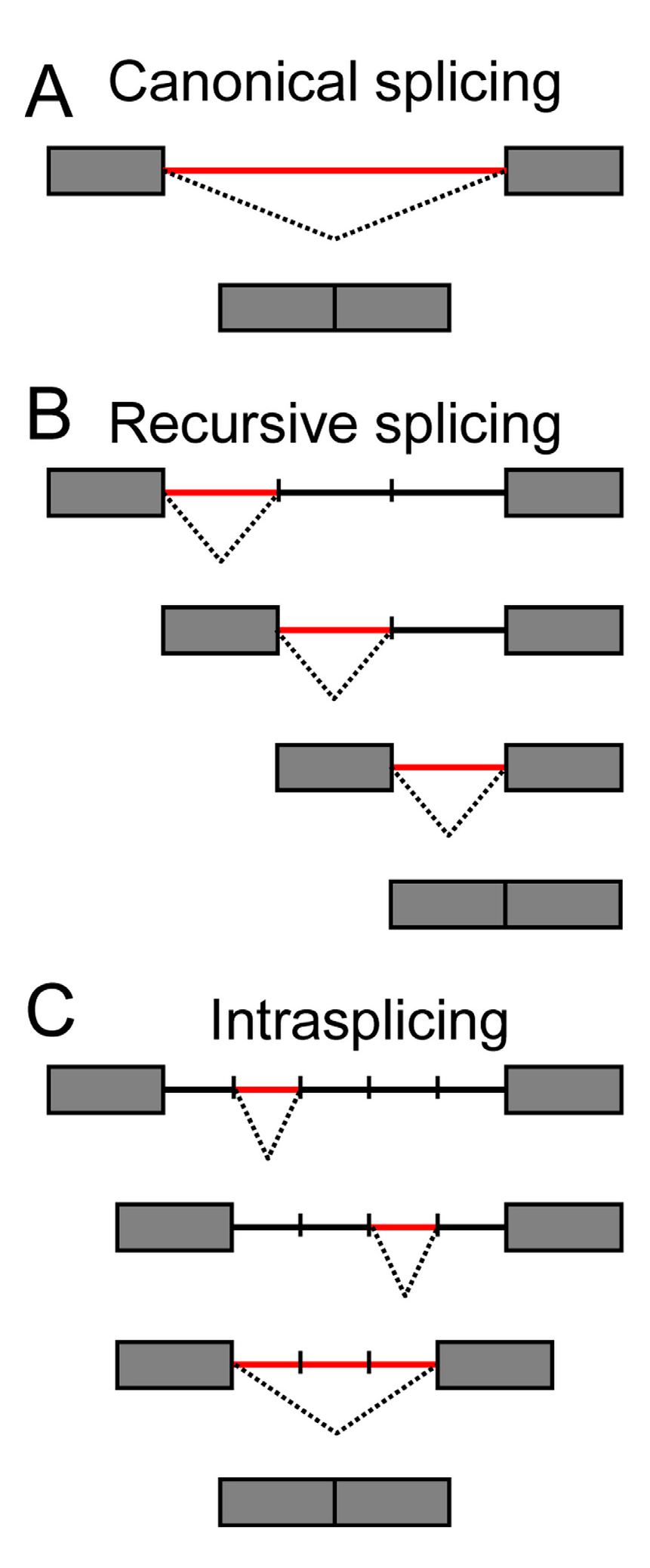
4. When Introns Are too Big to Be Spliced at Once—Intron Re-Splicing
5. Cytoplasmic Splicing—Adding to the Complexity of Transcriptional Regulation
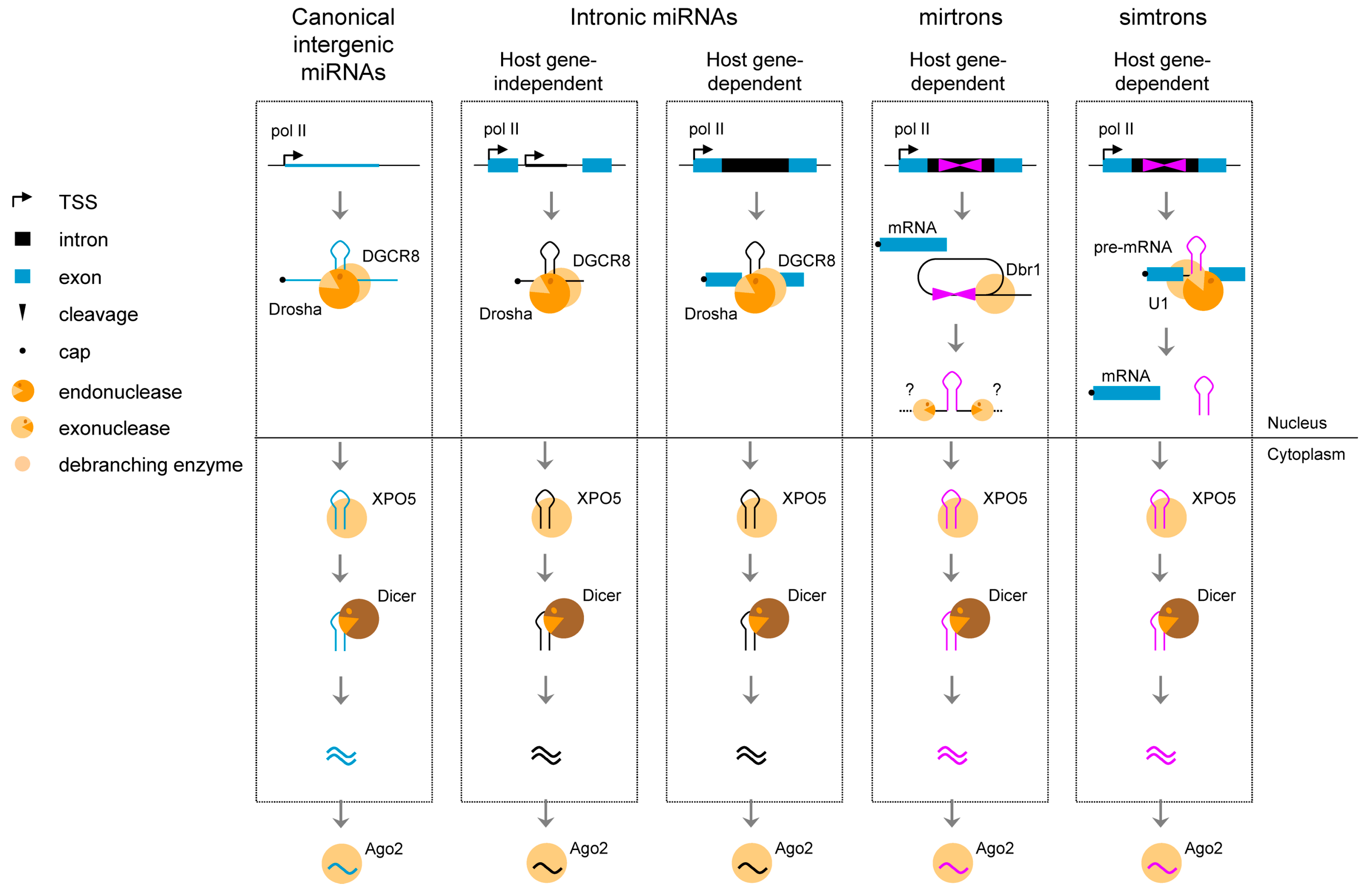
6. Splicing of Introns to Produce Small Non-Coding Transcripts
| Name | Genome Position | Host Intron | Host Gene | Genome Position | Gene Function | |
|---|---|---|---|---|---|---|
| snoRNA | ACA67 | chr21:33,749,496–33,749,631 | Intron 5 | URB Ribosome Biogenesis 1 homolog (URB1) | chr21:33,683,330–33,765,312 | Ribosome biogenesis |
| HBI-43 | chr20:17,943,353–17,943,589 | Intron 1 | Sorting Nexin 5 (SNX5) | chr20:17,922,244–17,949,490 | Member of the sorting nexin family, involved in intracellular trafficking | |
| SNORD119 | chr20:2,443,605–2,443,686 | Intron 2 | Small Nuclear Ribonucleoprotein Polypeptides B and B1 (SNRPB) | chr20:2,442,288–2,451,499 | Nuclear proteins that are found in common among U1, U2, U4/U6, and U5 small ribonucleoprotein particles (snRNPs) | |
| U101 | chr6:133136446–133136518 | Intron 3 | Ribosomal protein S12 (RPS12) | chr6:133,135,708–133,138,703 | Component of the ribosomal 40S subunit | |
| HBII-429 | chr6:133137941–133138016 | Intron 4 | Ribosomal protein S12 (RPS12) | chr6:133,135,708–133,138,703 | Component of the ribosomal 40S subunit | |
| ACA33 | chr6:133138358–133138490 | Intron 5 | Ribosomal protein S12 (RPS12) | chr6:133,135,708–133,138,703 | Component of the ribosomal 40S subunit | |
| ACA37 | chr18:51,748,654–51,748,782 | Intron 1 | Methyl-CpG Binding Domain protein 2 (MBD2) | chr18:51,677,971–51,751,158 | Repress transcription from methylated gene promoters | |
| miRNA | hsa-mir-643 | chr19:52,785,050–52,785,146 | Intron 1 | Zinc Finger protein 766 (ZNF766) | chr19:52,772,824–52,795,976 | Unknown |
| hsa-mir-220c | chr19:49,063,529–49,063,611 | Intron 1 | Sulfotransferase family, cytosolic, 2B, member 1 (SULT2B1) | chr19:49,055,429–49,102,684 | Catalyze the sulfate conjugation of many hormones, neurotransmitters, drugs, and xenobiotic compounds | |
| hsa-mir-3191 | chr19:47,730,201–47,730,276 | Intron 2 | BCL2 Binding Component 3 (BBC3) | chr19:47,724,079–47,736,023 | Member of the BCL-2 family of proteins, cooperates with direct activator proteins to induce mitochondrial outer membrane permeabilization and apoptosis | |
| hsa-mir-770 | chr14:101,318,727–101,318,824 | Intron 9 | Maternally Expressed 3 (non-protein coding) (MEG3) | chr14:101,292,445–-101,327,360 | Long ncRNA tumor suppressor. Interacts with the tumor suppressor p53, and regulates p53 target gene expression | |
| hsa-mir-1273d | chr1:10287776–10287861 | Intron 1 | Kinesin family member 1B (KIF1B) | chr1:10,270,764–10,441,661 | Transports mitochondria and synaptic vesicle precursors | |
| hsa-mir-3190 | chr19:47,730,199–47,730,278 | Intron 2 | BCL2 Binding Component 3 (BBC3) | chr19:47,724,079–47,736,023 | Member of the BCL-2 family of proteins, cooperates with direct activator proteins to induce mitochondrial outer membrane permeabilization and apoptosis | |
| hsa-mir-942 | chr1:117,637,265–117,637,350 | Intron 18 | Transcription Termination Factor, RNA polymerase II (TTF2) | chr1:117,602,949–117,645,491 | Member of the SWI2/SNF2 family of proteins | |
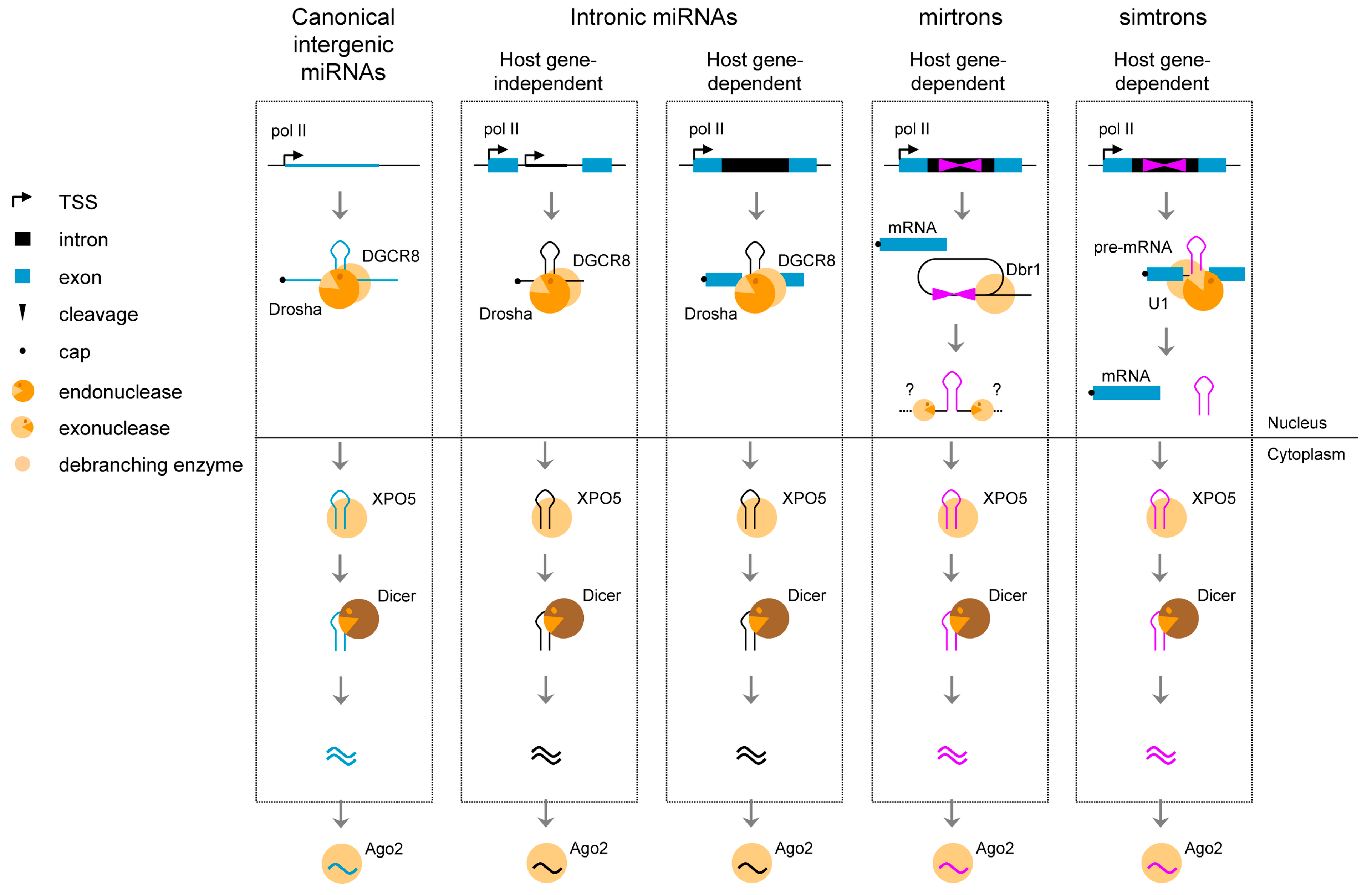
Splicing of introns may also lead to the production of long non-coding RNAs with a circular structure (ciRNA). In human cells, Zhang and colleagues [58] have recently identified hundreds of such RNAs, ranging from 200 nt to over 3000 nt in length, with no ORF, and suggested to be expressed in a cell-specific manner. The mechanism of biogenesis was described as follows [58]; the lariat structure of introns that contain a consensus 7 nt GU-rich pattern in 5' splice site and an 11 nt C-rich element near the branchpoint are digested from the 3' end to the branchpoint to preserve the loop portion of the lariat intron. In contrast to circular RNAs originating from circularized exons and acting as miRNA sponges in the cytoplasm [59,60], intronic ciRNAs seem to function as positive regulators of Pol II transcription and play a role in the efficient transcription of their host gene.
7. Introns Can Be Retained within Coding Segments—When Alternative Splicing of Introns Participates in Proteome Diversification
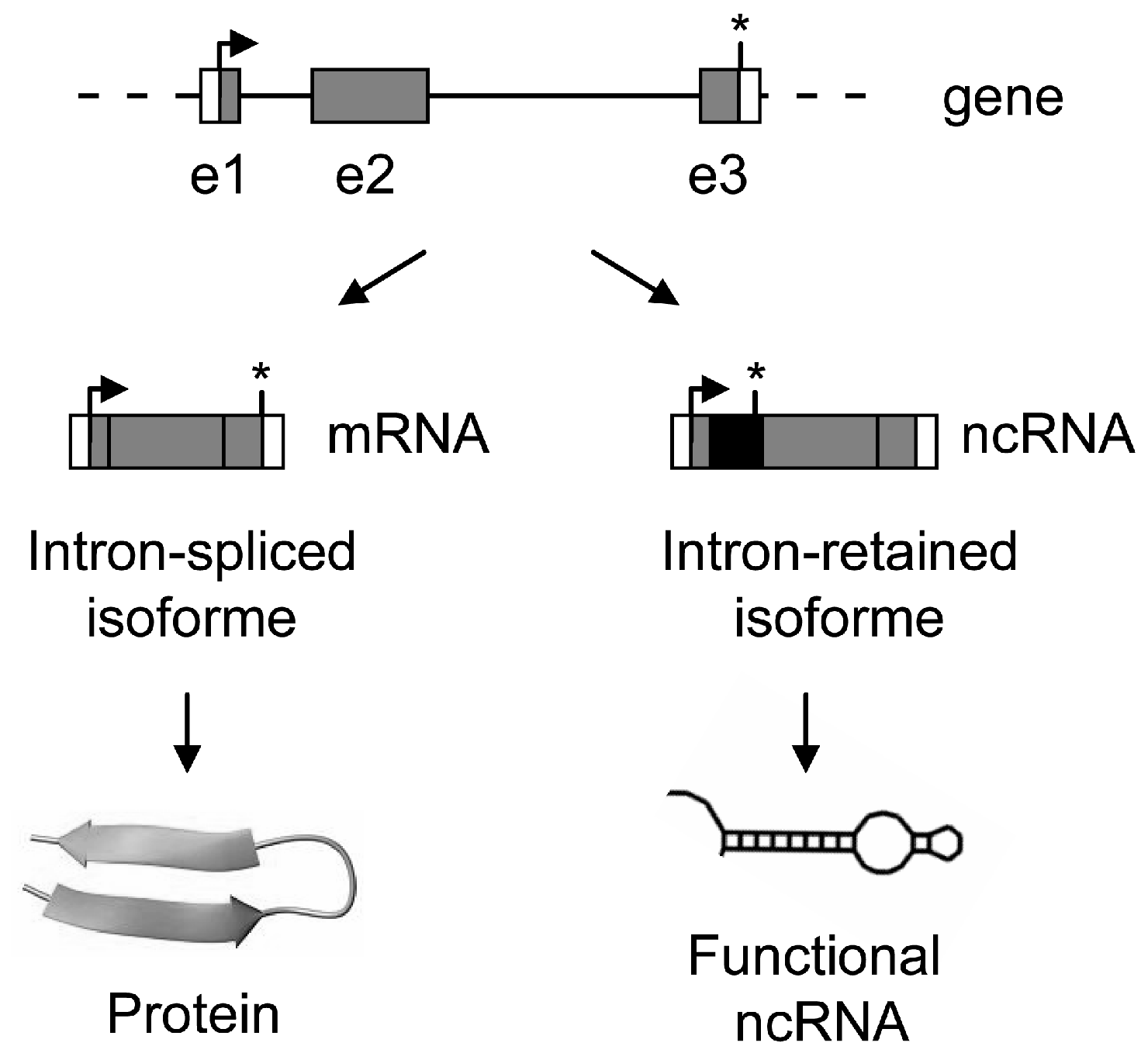
8. Introns as a Switch to Produce Coding or Non-Coding RNAs—When Alternative Splicing of Introns Generates Transcriptome Diversity
9. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rogozin, I.B.; Carmel, L.; Csuros, M.; Koonin, E.V. Origin and evolution of spliceosomal introns. Biol. Direct. 2012, 7, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Q. Statistical features of human exons and their flanking regions. Hum. Mol. Genet. 1998, 7, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Abelson, J.; Trotta, C.R.; Li, H. tRNA splicing. J. Biol. Chem. 1998, 273, 12685–12688. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R. Ribozymes, the first 20 years. Biochem. Soc. Trans. 2002, 30, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. The origin of introns and their role in eukaryogenesis: A compromise solution to the introns-early versus introns-late debate? Biol. Direct. 2006, 1, 1–22. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hare, M.P.; Palumbi, S.R. High intron sequence conservation across three mammalian orders suggests functional constraints. Mol. Biol. Evol. 2003, 20, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Sasaki-Haraguchi, N.; Shimada, M.K.; Taniguchi, I.; Ohno, M.; Mayeda, A. Mechanistic insights into human pre-mRNA splicing of human ultra-short introns: Potential unusual mechanism identifies G-rich introns. Biochem. Biophys. Res. Commun. 2012, 423, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Sakharkar, M.K.; Chow, V.T.; Ghosh, K.; Chaturvedi, I.; Lee, P.C.; Bagavathi, S.P.; Shapshak, P.; Subbiah, S.; Kangueane, P. Computational prediction of SEG (single exon gene) function in humans. Front Biosci. 2005, 10, 1382–1395. [Google Scholar] [CrossRef] [PubMed]
- Sakharkar, M.K.; Kangueane, P.; Petrov, D.A.; Kolaskar, A.S.; Subbiah, S. SEGE: A database on “intron less/single exonic” genes from eukaryotes. Bioinformatics 2002, 18, 1266–1267. [Google Scholar] [CrossRef] [PubMed]
- Sakharkar, M.K.; Kangueane, P. Genome SEGE: A database for “intronless” genes in eukaryotic genomes. BMC Bioinform. 2004, 5, 67. [Google Scholar] [CrossRef]
- Brosius, J. Genomes were forged by massive bombardments with retroelements and retrosequences. Genetica 1999, 107, 209–238. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A. An overview of nested genes in eukaryotic genomes. Eukaryot. Cell 2009, 8, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Chang, H.H. The evolution and functional significance of nested gene structures in Drosophila melanogaster. Genome Biol. Evol. 2013, 5, 1978–1985. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Ma, D.; Xu, M. Nested genes in the human genome. Genomics 2005, 86, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Makunin, I.V. Small regulatory RNAs in mammals. Hum. Mol. Genet. 2005, 14, R121–R132. [Google Scholar] [CrossRef] [PubMed]
- Penny, D.; Hoeppner, M.P.; Poole, A.M.; Jeffares, D.C. An overview of the introns-first theory. J. Mol. Evol. 2009, 69, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Westholm, J.O.; Lai, E.C. Mirtrons: microRNA biogenesis via splicing. Biochimie 2011, 93, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Monteys, A.M.; Spengler, R.M.; Wan, J.; Tecedor, L.; Lennox, K.A.; Xing, Y.; Davidson, B.L. Structure and activity of putative intronic miRNA promoters. RNA 2010, 16, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F.; Poling, L.L.; Wang, Z.; Liu, H.; Liu, X.S.; Roeder, R.G.; Zhang, X.; Song, J.S.; Fisher, D.E. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008, 22, 3172–3183. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, U.; Gueroussov, S.; Plocik, A.M.; Graveley, B.R.; Blencowe, B.J. Dynamic integration of splicing within gene regulatory pathways. Cell 2013, 152, 1252–1269. [Google Scholar] [CrossRef] [PubMed]
- Burnette, J.M.; Miyamoto-Sato, E.; Schaub, M.A.; Conklin, J.; Lopez, A.J. Subdivision of large introns in Drosophila by recursive splicing at nonexonic elements. Genetics 2005, 170, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Hatton, A.R.; Subramaniam, V.; Lopez, A.J. Generation of alternative Ultrabithorax isoforms and stepwise removal of a large intron by resplicing at exon-exon junctions. Mol. Cell 1998, 2, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.; Tamada, Y.; Bannai, H.; Nakai, K.; Miyano, S. Intrasplicing—Analysis of long intron sequences. Pac. Symp. Biocomput. 2003, 339–350. [Google Scholar]
- Suzuki, H.; Kameyama, T.; Ohe, K.; Tsukahara, T.; Mayeda, A. Nested introns in an intron: Evidence of multi-step splicing in a large intron of the human dystrophin pre-mRNA. FEBS Lett. 2013, 587, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Parra, M.K.; Gallagher, T.L.; Amacher, S.L.; Mohandas, N.; Conboy, J.G. Deep intron elements mediate nested splicing events at consecutive AG dinucleotides to regulate alternative 3' splice site choice in vertebrate 4.1 genes. Mol. Cell Biol. 2012, 32, 2044–2053. [Google Scholar] [CrossRef]
- Brogna, S.; Wen, J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nat. Struct. Mol. Biol. 2009, 16, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Jurica, M.S.; Moore, M.J. Pre-mRNA splicing: Awash in a sea of proteins. Mol. Cell 2003, 12, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Turunen, J.J.; Niemela, E.H.; Verma, B.; Frilander, M.J. The significant other: Splicing by the minor spliceosome. Wiley Interdiscip. Rev. RNA 2013, 4, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.; Durbin, R. A computational scan for U12-dependent introns in the human genome sequence. Nucleic Acids Res. 2001, 29, 4006–4013. [Google Scholar] [CrossRef] [PubMed]
- Konig, H.; Matter, N.; Bader, R.; Thiele, W.; Muller, F. Splicing segregation: The minor spliceosome acts outside the nucleus and controls cell proliferation. Cell 2007, 131, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Pessa, H.K.; Will, C.L.; Meng, X.; Schneider, C.; Watkins, N.J.; Perala, N.; Nymark, M.; Turunen, J.J.; Luhrmann, R.; Frilander, M.J. Minor spliceosome components are predominantly localized in the nucleus. Proc. Natl. Acad. Sci. USA 2008, 105, 8655–8660. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Bernabeu, C. Alternative splicing factor or splicing factor-2 plays a key role in intron retention of the endoglin gene during endothelial senescence. Aging Cell 2011, 10, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Caceres, J.F.; Misteli, T. Division of labor: Minor splicing in the cytoplasm. Cell 2007, 131, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Denis, M.M.; Tolley, N.D.; Bunting, M.; Schwertz, H.; Jiang, H.; Lindemann, S.; Yost, C.C.; Rubner, F.J.; Albertine, K.H.; Swoboda, K.J.; et al. Escaping the nuclear confines: Signal-dependent pre-mRNA splicing in anucleate platelets. Cell 2005, 122, 379–391. [Google Scholar]
- Racca, C.; Gardiol, A.; Eom, T.; Ule, J.; Triller, A.; Darnell, R.B. The neuronal splicing factor nova co-localizes with target RNAs in the dendrite. Front. Neural Circuits 2010, 4, 5. [Google Scholar] [PubMed]
- Glanzer, J.; Miyashiro, K.Y.; Sul, J.Y.; Barrett, L.; Belt, B.; Haydon, P.; Eberwine, J. RNA splicing capability of live neuronal dendrites. Proc. Natl. Acad. Sci. USA 2005, 102, 16859–16864. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H. Unconventional splicing of XBP-1 mRNA in the unfolded protein response. Antioxid. Redox Signal. 2007, 9, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- Back, S.H.; Lee, K.; Vink, E.; Kaufman, R.J. Cytoplasmic IRE1α-mediated XBP1 mRNA splicing in the absence of nuclear processing and endoplasmic reticulum stress. J. Biol. Chem. 2006, 281, 18691–18706. [Google Scholar] [CrossRef] [PubMed]
- Ruegsegger, U.; Leber, J.H.; Walter, P. Block of HAC1 mRNA translation by long-range base pairing is released by cytoplasmic splicing upon induction of the unfolded protein response. Cell 2001, 107, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.E.; Walter, P. Translational attenuation mediated by an mRNA intron. Curr. Biol. 1997, 7, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.H.; Sun, Y.M.; Chang, W.C.; Chiang-Hsieh, P.Y.; Lee, T.Y.; Tsai, W.C.; Horng, J.T.; Tsou, A.P.; Huang, H.D. Identifying transcriptional start sites of human microRNAs based on high-throughput sequencing data. Nucleic Acids Res. 2011, 39, 9345–9356. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, D.L.; Pandit, K.V.; Gordon, B.; Bhattacharjee, A.; Kaminski, N.; Benos, P.V. Features of mammalian microRNA promoters emerge from polymerase II chromatin immunoprecipitation data. PLoS One 2009, 4, e5279. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, P.; Palanichamy, J.K.; Singh, A.; Das, P.; Bhagat, M.; Kassab, M.A.; Sinha, S.; Chattopadhyay, P. Biogenesis of intronic miRNAs located in clusters by independent transcription and alternative splicing. RNA 2014, 20, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Kim, V.N. Processing of intronic microRNAs. EMBO J. 2007, 26, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.M.; Steitz, J.A. Classification of gas5 as a multi-small-nucleolar-RNA (snoRNA) host gene and a member of the 5'-terminal oligopyrimidine gene family reveals common features of snoRNA host genes. Mol. Cell Biol. 1998, 18, 6897–6909. [Google Scholar] [PubMed]
- Falaleeva, M.; Stamm, S. Processing of snoRNAs as a new source of regulatory non-coding RNAs: SnoRNA fragments form a new class of functional RNAs. Bioessays 2013, 35, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass Drosha processing. Nature 2007, 448, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Ladewig, E.; Okamura, K.; Flynt, A.S.; Westholm, J.O.; Lai, E.C. Discovery of hundreds of mirtrons in mouse and human small RNA data. Genome Res. 2012, 22, 1634–1645. [Google Scholar] [CrossRef] [PubMed]
- Janas, M.M.; Khaled, M.; Schubert, S.; Bernstein, J.G.; Golan, D.; Veguilla, R.A.; Fisher, D.E.; Shomron, N.; Levy, C.; Novina, C.D. Feed-Forward microprocessing and splicing activities at a microRNA-containing intron. PLoS Genet. 2011, 7, e1002330. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Reich, A.A.; Duelli, D.M.; Hastings, M.L. Biogenesis of mammalian microRNAs by a non-canonical processing pathway. Nucleic Acids Res. 2012, 40, 4626–4640. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lu, M.; Miao, J.; Li, T.; Wang, E.; Cui, Q. Cepred: Predicting the co-expression patterns of the human intronic microRNAs with their host genes. PLoS One 2009, 4, e4421. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Denli, A.M.; Gage, F.H. The enemy within: Intronic miR-26b represses its host gene, ctdsp2, to regulate neurogenesis. Genes Dev. 2012, 26, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Hinske, L.C.; Galante, P.A.; Kuo, W.P.; Ohno-Machado, L. A potential role for intragenic miRNAs on their hosts’ interactome. BMC Genomics 2010, 11, 533. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Reich, A.A.; Hastings, M.L. Drosha promotes splicing of a pre-microRNA-like alternative exon. PLoS Genet. 2014, 10, e1004312. [Google Scholar] [CrossRef] [PubMed]
- Granat-Tamir, L.; Shomron, N.; Sperling, J.; Sperling, R. Interplay between pre-mRNA splicing and microRNA biogenesis within the supraspliceosome. Nucleic Acids Res. 2014, 42, 4640–4651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.; Yang, L.; Chen, L.L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Agarwal, V.; Guo, H.; Bartel, D.P. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014, 15, 409. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Pheasant, M.; Mattick, J.S. Raising the estimate of functional human sequences. Genome Res. 2007, 17, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Ast, G. The alternative genome. Sci. Am. 2005, 292, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Sugnet, C.W.; Kent, W.J.; Ares, M., Jr.; Haussler, D. Transcriptome and genome conservation of alternative splicing events in humans and mice. Pac. Symp. Biocomput. 2004, 9, 66–77. [Google Scholar]
- De Lima Morais, D.A.; Harrison, P.M. Large-scale evidence for conservation of NMD candidature across mammals. PLoS One 2010, 5, e11695. [Google Scholar] [CrossRef] [PubMed]
- Buckley, P.T.; Lee, M.T.; Sul, J.Y.; Miyashiro, K.Y.; Bell, T.J.; Fisher, S.A.; Kim, J.; Eberwine, J. Cytoplasmic intron sequence-retaining transcripts can be dendritically targeted via ID element retrotransposons. Neuron 2011, 69, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Calvanese, V.; Mallya, M.; Campbell, R.D.; Aguado, B. Regulation of expression of two LY-6 family genes by intron retention and transcription induced chimerism. BMC Mol. Biol. 2008, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.; Ritchie, W.; Ebner, O.A.; Selbach, M.; Wong, J.W.; Huang, Y.; Gao, D.; Pinello, N.; Gonzalez, M.; Baidya, K.; et al. Orchestrated intron retention regulates normal granulocyte differentiation. Cell 2013, 154, 583–595. [Google Scholar]
- Braunschweig, U.; Barbosa-Morais, N.L.; Pan, Q.; Nachman, E.N.; Alipanahi, B.; Gonatopoulos-Pournatzis, T.; Frey, B.; Irimia, M.; Blencowe, B.J. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 2014, 24, 1774–1786. [Google Scholar] [CrossRef] [PubMed]
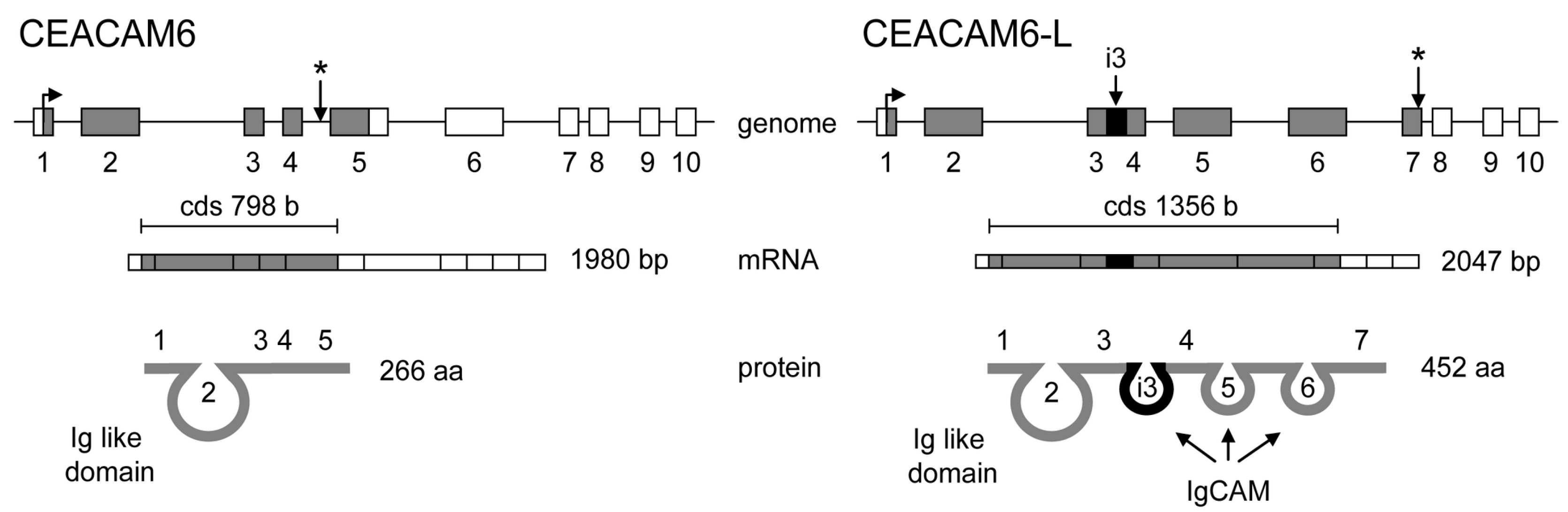
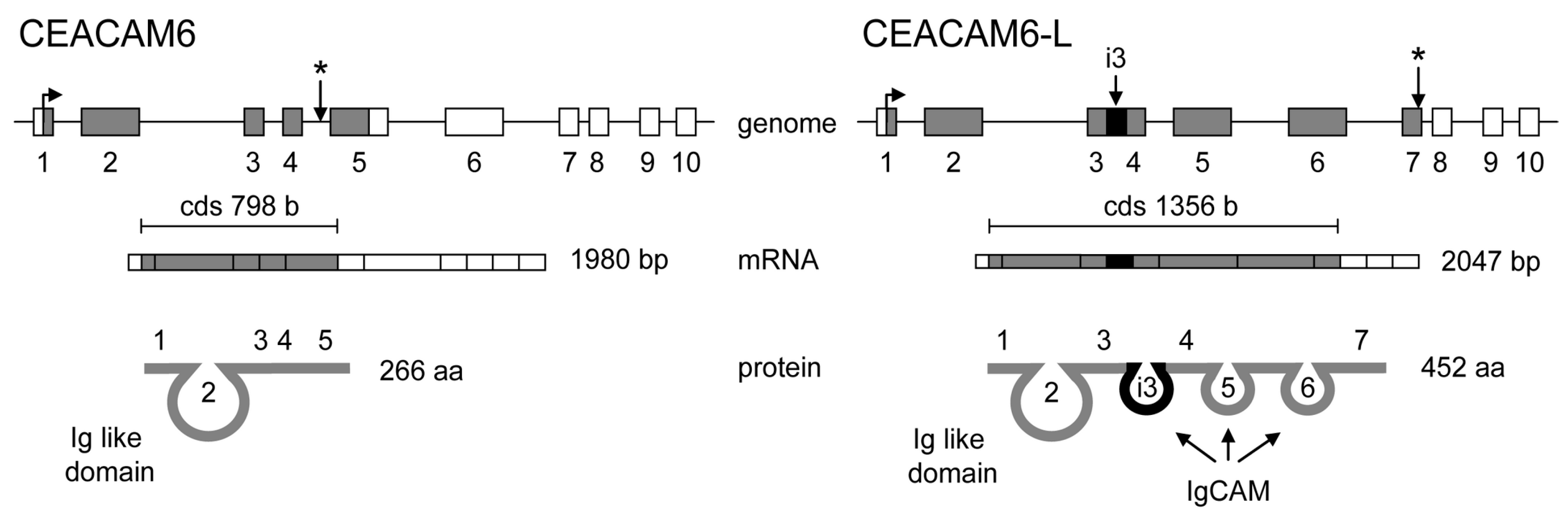
- Kurio, H.; Murayama, E.; Kaneko, T.; Shibata, Y.; Inai, T.; Iida, H. Intron retention generates a novel isoform of CEACAM6 that may act as an adhesion molecule in the ectoplasmic specialization structures between spermatids and sertoli cells in rat testis. Biol. Reprod. 2008, 79, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Buckley, P.T.; Khaladkar, M.; Kim, J.; Eberwine, J. Cytoplasmic intron retention, function, splicing, and the sentinel RNA hypothesis. Wiley Interdiscip. Rev. RNA 2014, 5, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Chooniedass-Kothari, S.; Emberley, E.; Hamedani, M.K.; Troup, S.; Wang, X.; Czosnek, A.; Hube, F.; Mutawe, M.; Watson, P.H.; Leygue, E. The steroid receptor RNA activator is the first functional RNA encoding a protein. FEBS Lett. 2004, 566, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.; Vincett, D.; Yan, Y.; Hamedani, M.K.; Myal, Y.; Leygue, E. Steroid receptor RNA activator bi-faceted genetic system: Heads or Tails? Biochimie 2011, 93, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Dinger, M.E.; Pang, K.C.; Mercer, T.R.; Mattick, J.S. Differentiating protein-coding and noncoding RNA: Challenges and ambiguities. PLoS Comput. Biol. 2008, 4, e1000176. [Google Scholar] [CrossRef] [PubMed]
- Dinger, M.E.; Gascoigne, D.K.; Mattick, J.S. The evolution of RNAs with multiple functions. Biochimie 2011, 93, 2013–2018. [Google Scholar] [CrossRef]
- Francastel, C.; Hube, F. Coding or non-coding: Need they be exclusive? Biochimie 2011, 93, 6–7. [Google Scholar] [CrossRef]
- Hube, F.; Guo, J.; Chooniedass-Kothari, S.; Cooper, C.; Hamedani, M.K.; Dibrov, A.A.; Blanchard, A.A.; Wang, X.; Deng, G.; Myal, Y.; et al. Alternative splicing of the first intron of the steroid receptor RNA activator (SRA) participates in the generation of coding and noncoding RNA isoforms in breast cancer cell lines. DNA Cell Biol. 2006, 25, 418–428. [Google Scholar]
- Hube, F.; Velasco, G.; Rollin, J.; Furling, D.; Francastel, C. Steroid receptor RNA activator protein binds to and counteracts SRA RNA-mediated activation of MyoD and muscle differentiation. Nucleic Acids Res. 2011, 39, 513–525. [Google Scholar] [CrossRef]
- Ulveling, D.; Francastel, C.; Hube, F. Identification of potentially new bifunctional RNA based on genome-wide data-mining of alternative splicing events. Biochimie 2011, 93, 2024–2027. [Google Scholar] [CrossRef] [PubMed]
- Ulveling, D.; Francastel, C.; Hube, F. When one is better than two: RNA with dual functions. Biochimie 2011, 93, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Lanz, R.B.; McKenna, N.J.; Onate, S.A.; Albrecht, U.; Wong, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 1999, 97, 17–27. [Google Scholar]
- Lanz, R.B.; Razani, B.; Goldberg, A.D.; O’Malley, B.W. Distinct RNA motifs are important for coactivation of steroid hormone receptors by steroid receptor RNA activator (SRA). Proc. Natl. Acad. Sci. USA 2002, 99, 16081–16086. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.; Guo, J.; Yan, Y.; Chooniedass-Kothari, S.; Hube, F.; Hamedani, M.K.; Murphy, L.C.; Myal, Y.; Leygue, E. Increasing the relative expression of endogenous non-coding Steroid Receptor RNA Activator (SRA) in human breast cancer cells using modified oligonucleotides. Nucleic Acids Res. 2009, 37, 4518–4531. [Google Scholar] [CrossRef] [PubMed]
- Emberley, E.; Huang, G.J.; Hamedani, M.K.; Czosnek, A.; Ali, D.; Grolla, A.; Lu, B.; Watson, P.H.; Murphy, L.C.; Leygue, E. Identification of new human coding steroid receptor RNA activator isoforms. Biochem. Biophys. Res. Commun. 2003, 301, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Walker, D.; Bernardo, A.; Brodbeck, J.; Balestra, M.E.; Huang, Y. Intron-3 retention/splicing controls neuronal expression of apolipoprotein E in the CNS. J. Neurosci. 2008, 28, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Ishida, E.; Matsumoto, S.; Shibusawa, N.; Okada, S.; Monden, T.; Satoh, T.; Yamada, M.; Mori, M. A liver X receptor (LXR)-β alternative splicing variant (LXRBSV) acts as an RNA co-activator of LXR-β. Biochem. Biophys. Res. Commun. 2009, 390, 1260–1265. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hubé, F.; Francastel, C. Mammalian Introns: When the Junk Generates Molecular Diversity. Int. J. Mol. Sci. 2015, 16, 4429-4452. https://doi.org/10.3390/ijms16034429
Hubé F, Francastel C. Mammalian Introns: When the Junk Generates Molecular Diversity. International Journal of Molecular Sciences. 2015; 16(3):4429-4452. https://doi.org/10.3390/ijms16034429
Chicago/Turabian StyleHubé, Florent, and Claire Francastel. 2015. "Mammalian Introns: When the Junk Generates Molecular Diversity" International Journal of Molecular Sciences 16, no. 3: 4429-4452. https://doi.org/10.3390/ijms16034429
APA StyleHubé, F., & Francastel, C. (2015). Mammalian Introns: When the Junk Generates Molecular Diversity. International Journal of Molecular Sciences, 16(3), 4429-4452. https://doi.org/10.3390/ijms16034429