
Intravenous Single-Dose Toxicity of Redaporfin-Based Photodynamic Therapy in Rodents

 , ,
, , 
Abstract
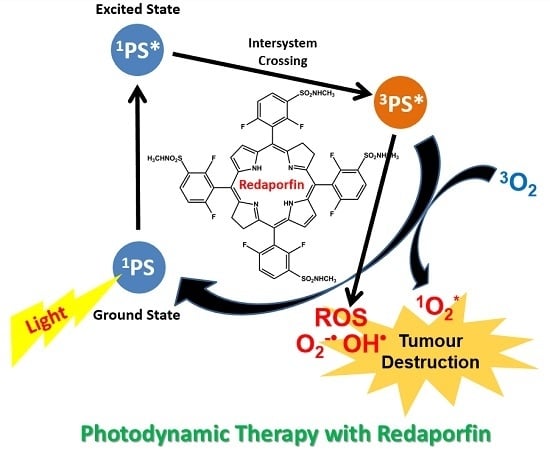
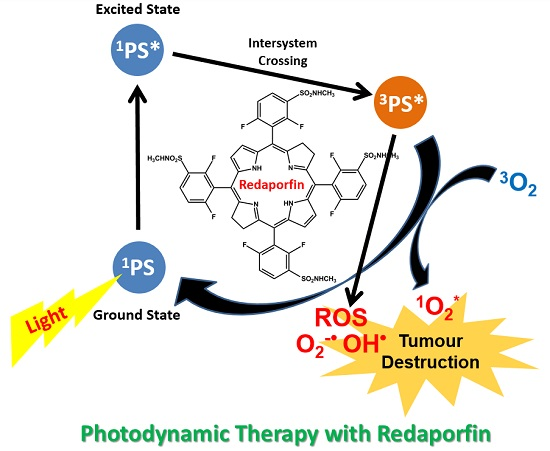
:
1. Introduction
2. Results
2.1. Dose Escalation Study
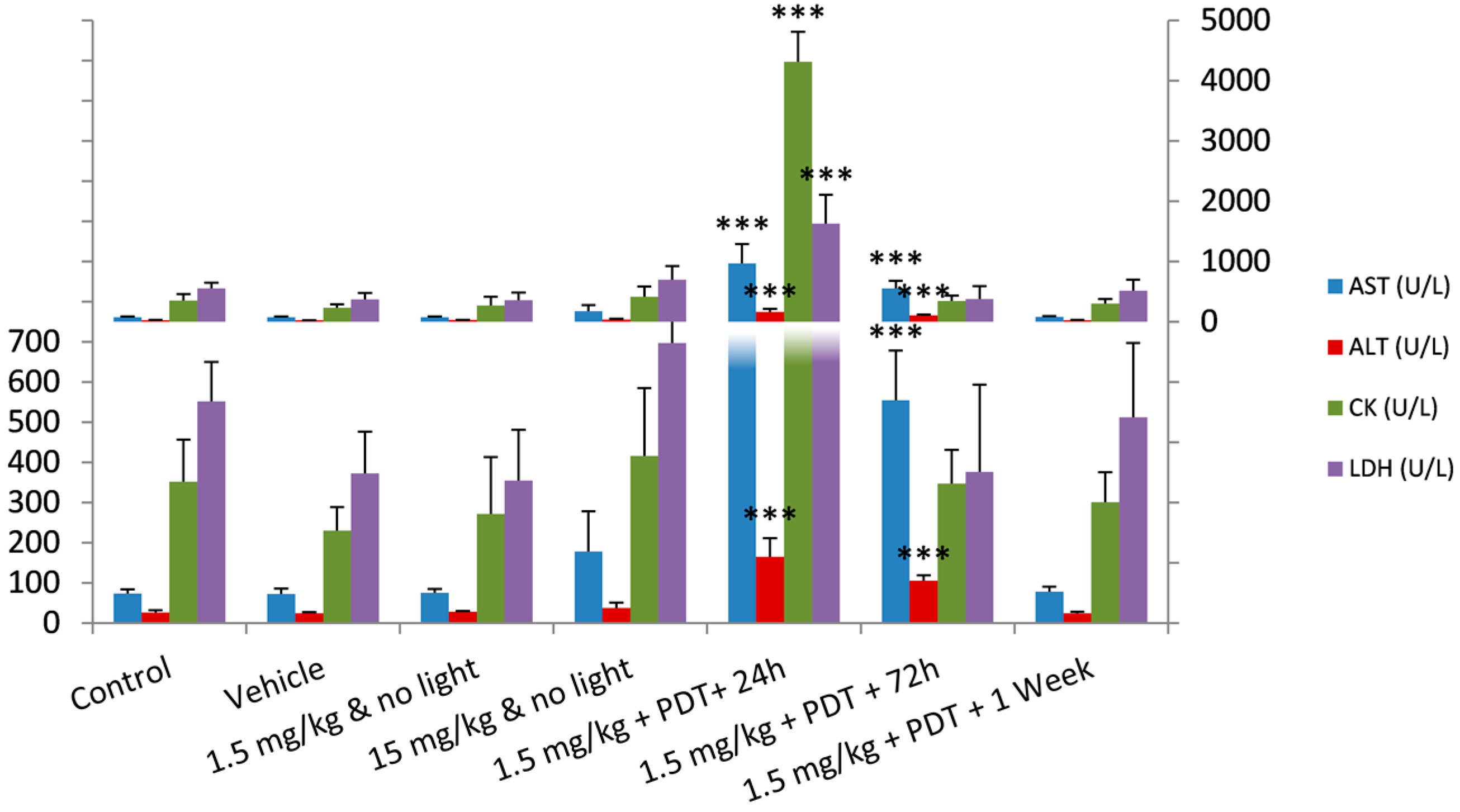
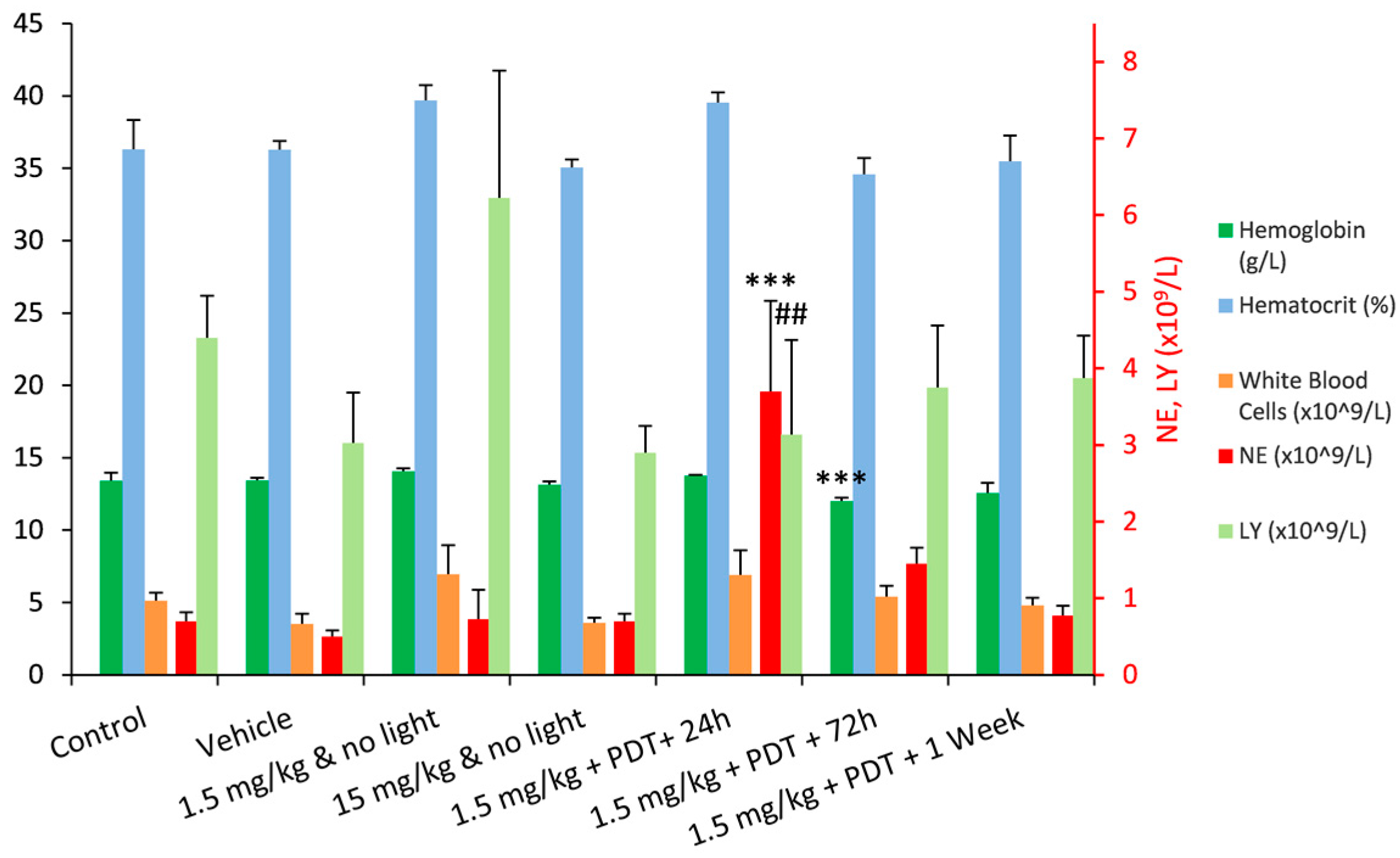
2.2. Safety Toxicology Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Redaporfin (mg/kg) | Days After Administration | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 4 | 7 | 11 | 14 | 16 | 21 | 28 | 35 | 46 | ||
| G0 | 0 | 21.9 ± 0.6 | - | 21.9 ± 0.8 | - | - | 22.2 ± 1.0 | - | 22.1 ± 1.2 | 22.4 ± 0.8 | - |
| G1 | 15 | 22.0 ± 0.7 | - | 21.8 ± 0.6 | - | - | 22.4 ± 0.6 | - | 22.9 ± 0.5 | 23.1 ± 0.6 | - |
| G2 | 21 | 22.0 ± 1.0 | - | 21.7 ± 0.8 | - | - | 22.1 ± 0.9 | - | 22.3 ± 1.0 | 22.1 ± 1.1 | - |
| G3 | 26.3 | 21.4 ± 0.5 | - | 20.8 ± 0.7 | - | - | 21.0 ± 0.7 | - | 21.9 ± 1.1 | 21.7 ± 0.4 | - |
| G4 | 30 | 22.8 ± 0.9 | - | 22.5 ± 0.9 | - | - | 22.6 ± 1.0 | - | 22.3 ± 0.9 | 23.4 ± 1.0 | - |
| G5 | 37.5 | 21.9 ± 1.2 | - | 21.5 ± 1.0 | - | - | 21.7 ± 1.0 | - | 21.8 ± 1.3 | 22.2 ± 1.0 | - |
| G6 | 75 | 24.4 ± 1.7 | 23.9 ± 1.6 | 24.2 ± 1.7 | 24.2 ± 1.6 | 24.2 ± 1.6 | - | 24.5 ± 1.8 | 25.5 ± 1.6 | - | 25.3 ± 1.7 |
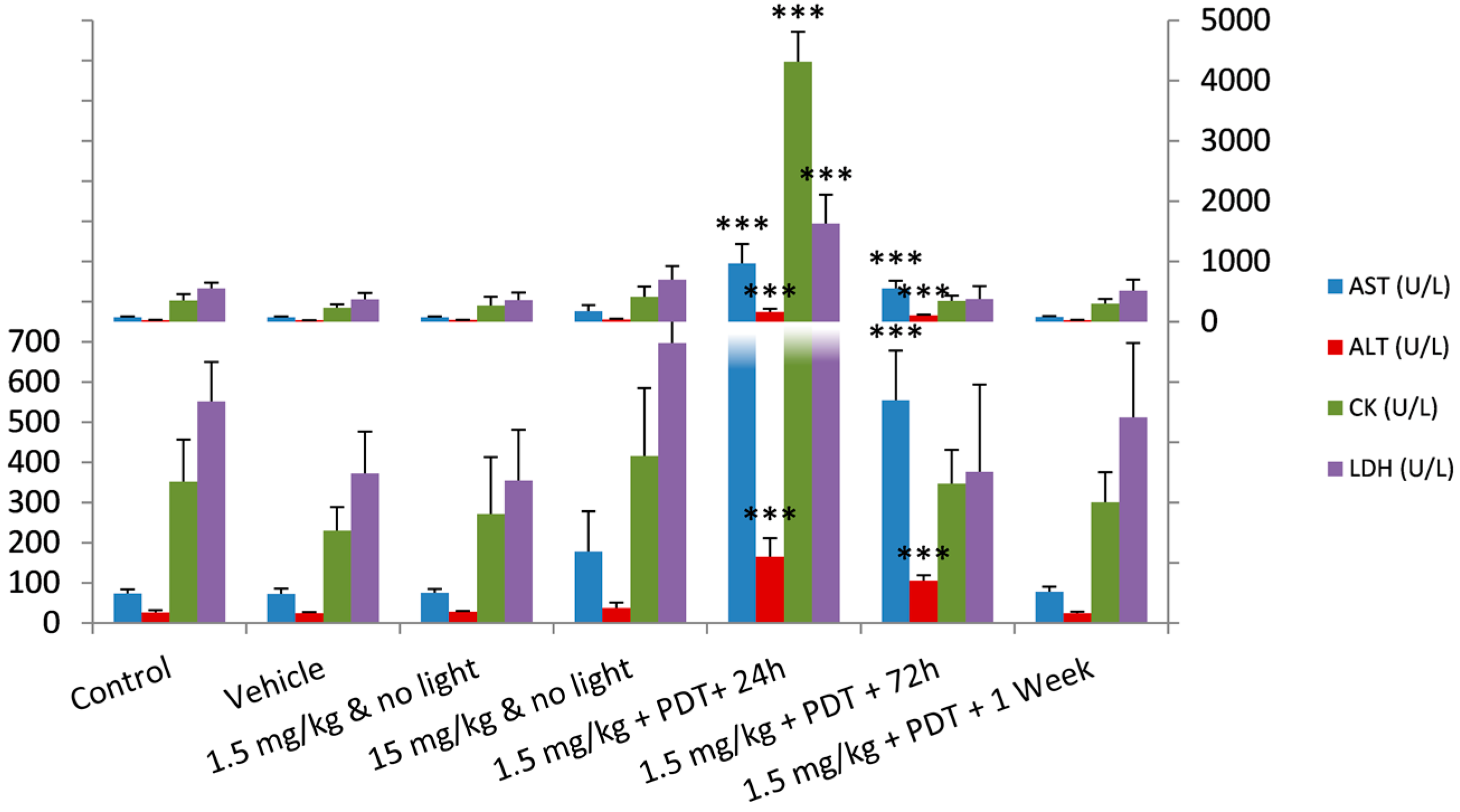

2.3. Photodynamic Efficacy of Redaporfin–PDT
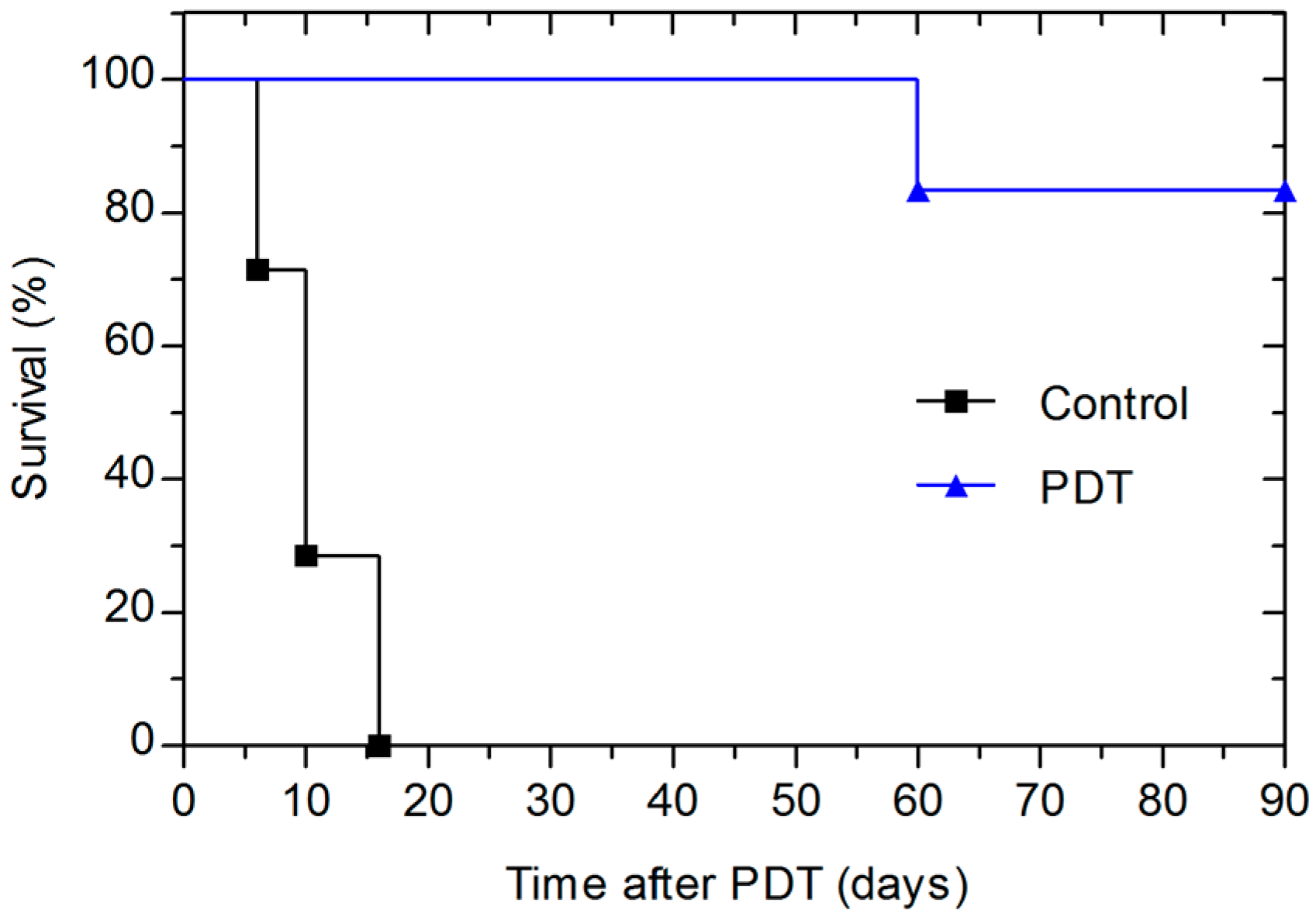
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Animals
4.3. Dose Escalation Study
4.3.1. Redaporfin Formulations
4.3.2. Intravenous Administration
| Test Group | N | Redaporfin (mg/kg) | Redaporfin (mg/mL) |
|---|---|---|---|
| G0 (Control) | 5 | 0.0 | 0.00 |
| G1 | 5 | 15.0 | 1.50 |
| G2 | 5 | 21.0 | 2.10 |
| G3 | 5 | 26.3 | 2.63 |
| G4 | 5 | 30.0 | 3.00 |
| G5 | 5 | 37.5 | 3.75 |
| G6 | 4 | 75.0 | 7.50 |
4.3.3. Mice Follow-up
4.4. Safety Toxicology Study
4.4.1. Redaporfin Formulation
4.4.2. Intravenous Injection, PDT and Blood Collection
4.4.3. Blood Analysis
4.5. PDT Treatment
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
Appendix A
| Haematology | Control | Vehicle | 1.5 mg/kg | 15 mg/kg | 1.5 mg/kg PDT + 24 h | 1.5 mg/kg PDT + 72 h | 1.5 mg/kg PDT + 1 week |
|---|---|---|---|---|---|---|---|
| RBC (×1012/L) | 6.5 ± 0.3 | 6.5 ± 0.3 | 7.2 ± 0.3 | 6.4 ± 0.2 | 6.7 ± 0.3 | 6.1 ± 0.2 | 6.2 ± 0.4 |
| Retic (%) | 1.3 ± 0.1 | 1.4 ± 0.1 | 1.3 ± 0.1 | 1.4 ± 0.1 | 1.2 ± 0.1 | 1.3 ± 0.1 | NP |
| Hg (g/L) | 13.4 ± 0.5 | 13.5 ± 0.2 | 14.1 ± 0.2 | 13.1 ± 0.2 | 13.8 ± 0.1 | 12.0 ± 0.2 | 12.6 ± 0.7 |
| HCT (%) | 36.3 ± 2.0 | 36.3 ± 0.6 | 39.7 ± 1.0 | 35.1 ± 0.6 | 39.5 ± 0.7 | 34.6 ± 1.1 | 35.5 ± 1.8 |
| MCV (fL) | 56.1 ± 1.7 | 55.1 ± 1.8 | 55.2 ± 1.3 | 55.0 ± 1.3 | 59.5 ± 2.0 | 56.9 ± 1.8 | 57.1 ± 1.0 |
| MCH (pg) | 20.8 ± 0.8 | 20.6 ± 1.1 | 19.6 ± 0.9 | 20.6 ± 0.7 | 20.7 ± 1.0 | 19.8 ± 0.5 | 20.2 ± 0.3 |
| MCHC (g/L) | 37.1 ± 0.8 | 37.3 ± 0.8 | 35.5 ± 0.8 | 37.4 ± 0.5 | 34.9 ± 0.5 | 34.8 ± 0.6 | 35.4 ± 0.6 |
| RDW (%) | 12.0 ± 0.4 | 12.3 ± 1.1 | 12.3 ± 1.3 | 12.7 ± 1.0 | 11.2 ± 0.6 | 11.9 ± 0.9 | 13.0 ± 1.3 |
| PLT (x109/L) | 740 ± 62 | 629 ± 90 | 801 ± 56 | 654 ± 83 | 615 ± 167 | 610 ± 34 | NM |
| MPV (fL) | 5.8 ± 0.2 | 5.6 ± 0.3 | 5.4 ± 0.1 | 5.7 ± 0.2 | 6.1 ± 0.4 | 5.7 ± 0.2 | 5.7 ± 0.2 |
| PCT (%) | 42.7 ± 2.4 | 35.0 ± 3.4 | 43.4 ± 2.4 | 37.4 ± 6.1 | 37.4 ± 8.6 | 35.0 ± 1.0 | 67.3 ± 9.4 |
| PDW (%) | 17.0 ± 0.2 | 16.7 ± 0.6 | 16.9 ± 0.8 | 16.8 ± 0.5 | 17.2 ± 0.4 | 16.7 ± 0.3 | 16.6 ± 0.3 |
| WBC (×109/L) | 5.1 ± 0.6 | 3.5 ± 0.7 | 7.0 ± 2.0 | 3.6 ± 0.4 | 6.9 ± 1.7 | 5.4 ± 0.8 | 4.8 ± 0.5 |
| NE (%) | 16.0 ± 6.4 | 14.0 ± 3.5 | 11.0 ± 5.0 | 20.7 ± 9.3 | 53.3 ± 10.2 | 28.3 ± 7.8 | NP |
| NE (×109/L) | 0.8 ± 0.2 | 0.5 ± 0.1 | 0.8 ± 0.5 | 0.8 ± 0.4 | 3.7 ± 1.0 | 1.5 ± 0.4 | NP |
| LY (%) | 82.0 ± 6.3 | 84.3 ± 3.4 | 85.3 ± 5.0 | 76.0 ± 9.5 | 41.3 ± 10.0 | 66.0 ± 7.5 | NP |
| LY (×109/L) | 4.2 ± 0.7 | 3.0 ± 0.6 | 5.9 ± 1.6 | 2.7 ± 0.2 | 2.9 ± 1.2 | 3.6 ± 0.7 | NP |
| MO (%) | 1.8 ± 1.5 | 1.8 ± 2.1 | 3.0 ± 0.8 | 3.3 ± 1.5 | 5.3 ± 0.6 | 5.0 ± 2.2 | NP |
| MO (×109/L) | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.2 ± 0.1 | 0.1 ± 0.1 | 0.4 ± 0.1 | 0.3 ± 0.1 | NP |
| EO (%) | 0.3 ± 0.5 | 0.0 ± 0.0 | 0.8 ± 1.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.8 ± 1.0 | NP |
| EO (×109/L) | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.1 ± 0.1 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.1 | NP |
| BA (%) | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | NP |
| BA (×109/L) | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | NP |
| Biochemistry | Control | Vehicle | 1.5 mg/kg | 15 mg/kg | 1.5 mg/kg PDT + 24 h | 1.5 mg/kg PDT + 72 h | 1.5 mg/kg PDT + 1 week |
|---|---|---|---|---|---|---|---|
| GLU (mg/dL) | 229 ± 54 | 218 ± 17 | 217 ± 46 | 222 ± 9 | 174 ± 47 | 171 ± 21 | 191 ± 34 |
| Urea (mg/dL) | 42 ± 3 | 33 ± 2 | 33 ± 4 | 32 ± 5 | 30 ± 2 | 40 ± 8 | 44 ± 2 |
| CHOL (mg/dL) | 52 ± 7 | 43 ± 7 | 51 ± 4 | 41 ± 7 | 68 ± 12 | 54 ± 4 | 55 ± 8 |
| TG (mg/mL) | 114 ± 45 | 84 ± 9 | 120 ± 62 | 66 ± 20 | 61 ± 16 | 58 ± 14 | 117 ± 53 |
| AST (U/L) | 73 ± 11 | 73 ± 13 | 76 ± 10 | 178 ± 100 | 968 ± 317 | 555 ± 123 | 78 ± 12 |
| ALT (U/L) | 27 ± 5 | 24 ± 3 | 29 ± 2 | 38 ± 13 | 165 ± 47 | 106 ± 13 | 25 ± 3 |
| CRE (mg/dl) | 0.47 ± 0.02 | 0.44 ± 0.03 | 0.45 ± 0.01 | 0.44 ± 0.05 | 0.48 ± 0.07 | 0.43 ± 0.02 | 0.49 ± 0.03 |
| γ-GT (U/L) | NM | NM | NM | NM | NM | NM | NM |
| ALP (U/L) | 88 ± 15 | 54 ± 6 | 93 ± 37 | 90 ± 12 | 97 ± 38 | 56 ± 13 | 60 ± 8 |
| BIL (mg/dL) | 0 ± 0 | 0.1 ± 0.0 | 0 ± 0 | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.1 ± 0.0 |
| TP (g/L) | 5.6 ± 0.2 | 5.2 ± 0.3 | 5.3 ± 0.2 | 5.6 ± 0.3 | 4.7 ± 0.3 | 5.3 ± 0.1 | 5.6 ± 0.1 |
| CK (U/L) | 352 ± 105 | 230 ± 58 | 272 ± 142 | 416 ± 169 | 4314 ± 496 | 347 ± 84 | 301 ± 74 |
| BUN (mg/dL) | 20 ± 1 | 15.3 ± 0.8 | 15 ± 2 | 15.1 ± 2.2 | 14.2 ± 1.0 | 18.6 ± 3.7 | 20.5 ± 0.7 |
| LDH (U/L) | 552 ± 97 | 372 ± 104 | 355 ± 127 | 698 ± 227 | 1625 ± 483 | 376 ± 217 | 513 ± 185 |
References
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: an update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, J.M.; Arnaut, L.G. Photodynamic therapy (PDT) of cancer: From local to systemic treatment. Photochem. Photobiol. Sci. 2015, 14, 1765–1780. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part three—Photosensitizer pharmacokinetics, biodistribution, tumor localization and modes of tumor destruction. Photodiag. Photodyn. Ther. 2005, 2, 91–106. [Google Scholar] [CrossRef]
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 2006, 6, 535–545. [Google Scholar] [CrossRef] [PubMed]
- ICH, Guideline S9—Nonclinical Evaluation for Anticancer Pharmaceuticals. Available online: http://122.3.253.116/attachments/article/99526/S9%20Step%204.pdf. URL (accessed on 29 October 2015).
- Chevalier, S.; Cury, F.L.; Scarlata, E.; El-Zayat, E.; Hamel, L.; Rocha, J.; Zouanat, F.Z.; Moussa, S.; Scherz, A.; Elhilali, M.; et al. Endoscopic Vascular Targeted Photodynamic Therapy with the Photosensitizer WST11 for Benign Prostatic Hyperplasia in the Preclinical Dog Model. J. Urol. 2013, 190, 1946–1953. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Li, C.; Wang, Z.; Zhang, J.; Ye, X.; Gao, W.; Wang, A.; Jin, H.; Wei, J. A safety study of a novel photosensitizer, sinoporphyrin sodium, for photodynamic therapy in Beagle dogs. Photochem. Photobiol. Sci. 2015, 14, 815–832. [Google Scholar] [CrossRef] [PubMed]
- Arnaut, L.G.; Pereira, M.M.; Dabrowski, J.M.; Silva, E.F.; Schaberle, F.A.; Abreu, A.R.; Rocha, L.B.; Barsan, M.M.; Urbanska, K.; Stochel, G.; et al. Photodynamic therapy efficacy enhanced by dynamics: The role of charge transfer and photostability in the selection of photosensitizers. Chemistry 2014, 20, 5346–5357. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, R.; Rocha, L.B.; Dabrowski, J.M.; Arnaut, L.G. Modulation of biodistribution, pharmacokinetics, and photosensitivity with the delivery vehicle of a bacteriochlorin photosensitizer for photodynamic therapy. ChemMedChem 2014, 9, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Rocha, L.B.; Gomes-da-Silva, L.C.; Dąbrowski, J.M.; Arnaut, L.G. Elimination of primary tumours and control of metastasis with rationally designed bacteriochlorin photodynamic therapy regimens. Eur. J. Cancer 2015, 51, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Ten Tije, A.J.; Verweij, J.; Loos, W.J.; Sparreboom, A. Pharmacological effects of formulation vehicles: Implications for cancer chemotherapy. Clin. Pharmacokinet. 2003, 42, 665–685. [Google Scholar] [CrossRef] [PubMed]
- Woodburn, K.; Chang, C.K.; Lee, S.; Henderson, B.; Kessel, D. Biodistribution and PDT efficacy of a ketochlorin photosensitizer as a function of the delivery vehicle. Photochem. Photobiol. 1994, 60, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Muggia, F.M.; Alving, C.R. Complement activation by Cremophor EL as a possible contributor to hypersensitivity to paclitaxel: an in vitro study. J. Natl. Cancer Inst. 1998, 90, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor EL: The drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer 2001, 37, 1590–1598. [Google Scholar] [CrossRef]
- Boehm, D.K.; Maksymiuk, A.W. Paclitaxel premedication regimens. J. Natl. Cancer Inst. 1996, 88, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Commission, E. Community register of orphan medicinal products. Available online: http://ec.europa.eu/health/documents/community-register/html/o1470.htm (accessed on 11 November 2015).
- Photodynamic Therapy With LUZ11 in Advanced Head and Neck Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02070432?term=photodynamic+therapy&recr=Open&rank=37 (accessed on 9 October 2015).
- Zhang, Z.; Jin, H.; Bao, J.; Fang, F.; Wei, J.; Wang, A. Intravenous repeated-dose toxicity study of ZnPcS2P2-based-photodynamic therapy in Wistar rats. Photochem. Photobiol. Sci. 2006, 5, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Onodera, H.; Ueda, M.; Imai, T.; Hirose, M. A 13-week subchronic toxicity study of dietary administered morin in F344 rats. Food Chem. Toxicol. 2006, 44, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Green, T.; Dow, J.; Foster, J. Increased formic acid excretion and the development of kidney toxicity in rats following chronic dosing with trichloroethanol, a major metabolite of trichloroethylene. Toxicology 2003, 191, 109–119. [Google Scholar] [CrossRef]
- Charles River Laboratories. Baseline hematology and clinical chemistry values for charles river wistar rats. Available online: http://www.criver.com/files/pdfs/rms/wistar-rats/rm_rm_r_hematology_crl_wi_br_sex_age.aspx (accessed on 9 October 2015).
- Brancaccio, P.; Lippi, G.; Maffulli, N. Biochemical markers of muscular damage. Clin. Chem. Lab. Med. 2010, 48, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Mroz, P.; Castano, A.P.; Hamblin, M.R. Stimulation of dendritic cells enhances immune response after photodynamic therapy. Biophotonics Immune Responses 2009, 7178, 717803. [Google Scholar]
- Mroz, P.; Yaroslavsky, A.; Kharkwal, G.B.; Hamblin, M.R. Cell death pathways in photodynamic therapy of Cancer. Cancers 2011, 3, 2516–2539. [Google Scholar] [CrossRef] [PubMed]
- Krzykawska-Serda, M.; Dabrowski, J.M.; Arnaut, L.G.; Szczygiel, M.; Urbanska, K.; Stochel, G.; Elas, M. The role of strong hypoxia in tumors after treatment in the outcome of bacteriochlorin-based photodynamic therapy. Free Radic. Biol. Med. 2014, 73, 239–251. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency (EMA). European public assessment report (EPAR) for Foscan—Scientific Discussion. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000318/WC500024394.pdf (accessed on 22 October 2015).
- Freireich, E.J.; Gehan, E.A.; Rall, D.P.; Schmidt, L.H.; Skipper, H.E. Quantitative comparison of toxicity of anticancer agents in mouse, rat, hamster, dog, monkey, and man. Cancer Chemother. Rep. 1966, 50, 219–244. [Google Scholar] [PubMed]
- Schreckenberger, M.; Amberg, R.; Scheurich, A.; Lochmann, M.; Tichy, W.; Klega, A.; Siessmeier, T.; Grunder, G.; Buchholz, H.-G.; Landvogt, C.; et al. Acute alcohol effects on neuronal and attentional processing: Striatal reward system and inhibitory sensory interactions under acute ethanol challenge. Neuropsychopharmacology 2004, 29, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhao, L. Developing early formulations: Practice and perspective. Int. J. Pharm. 2007, 341, 1–19. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha, L.B.; Schaberle, F.; Dąbrowski, J.M.; Simões, S.; Arnaut, L.G. Intravenous Single-Dose Toxicity of Redaporfin-Based Photodynamic Therapy in Rodents. Int. J. Mol. Sci. 2015, 16, 29236-29249. https://doi.org/10.3390/ijms161226162
Rocha LB, Schaberle F, Dąbrowski JM, Simões S, Arnaut LG. Intravenous Single-Dose Toxicity of Redaporfin-Based Photodynamic Therapy in Rodents. International Journal of Molecular Sciences. 2015; 16(12):29236-29249. https://doi.org/10.3390/ijms161226162
Chicago/Turabian StyleRocha, Luis B., Fábio Schaberle, Janusz M. Dąbrowski, Sérgio Simões, and Luis G. Arnaut. 2015. "Intravenous Single-Dose Toxicity of Redaporfin-Based Photodynamic Therapy in Rodents" International Journal of Molecular Sciences 16, no. 12: 29236-29249. https://doi.org/10.3390/ijms161226162
APA StyleRocha, L. B., Schaberle, F., Dąbrowski, J. M., Simões, S., & Arnaut, L. G. (2015). Intravenous Single-Dose Toxicity of Redaporfin-Based Photodynamic Therapy in Rodents. International Journal of Molecular Sciences, 16(12), 29236-29249. https://doi.org/10.3390/ijms161226162