
Diversity and Inheritance of Intergenic Spacer Sequences of 45S Ribosomal DNA among Accessions of Brassica oleracea L. var. capitata


 ,
,
Abstract
:
1. Introduction
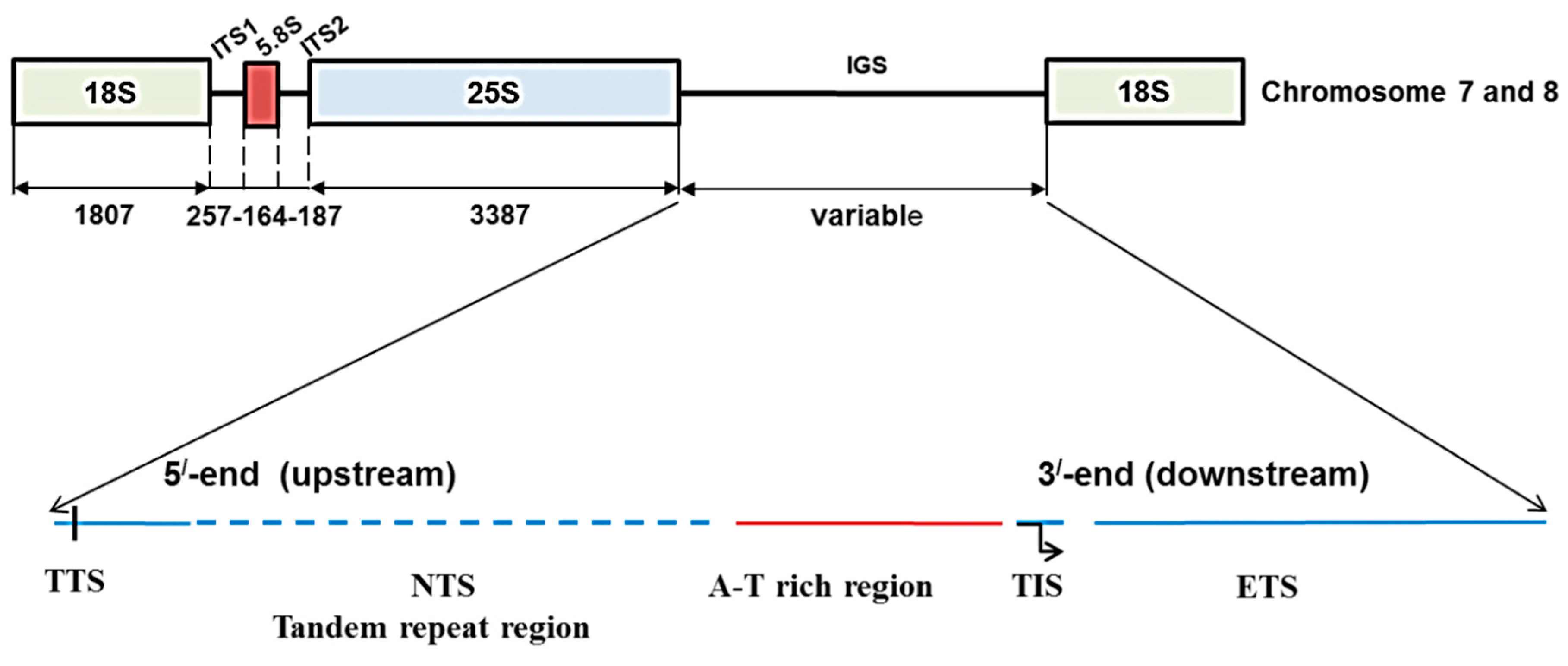
2. Results
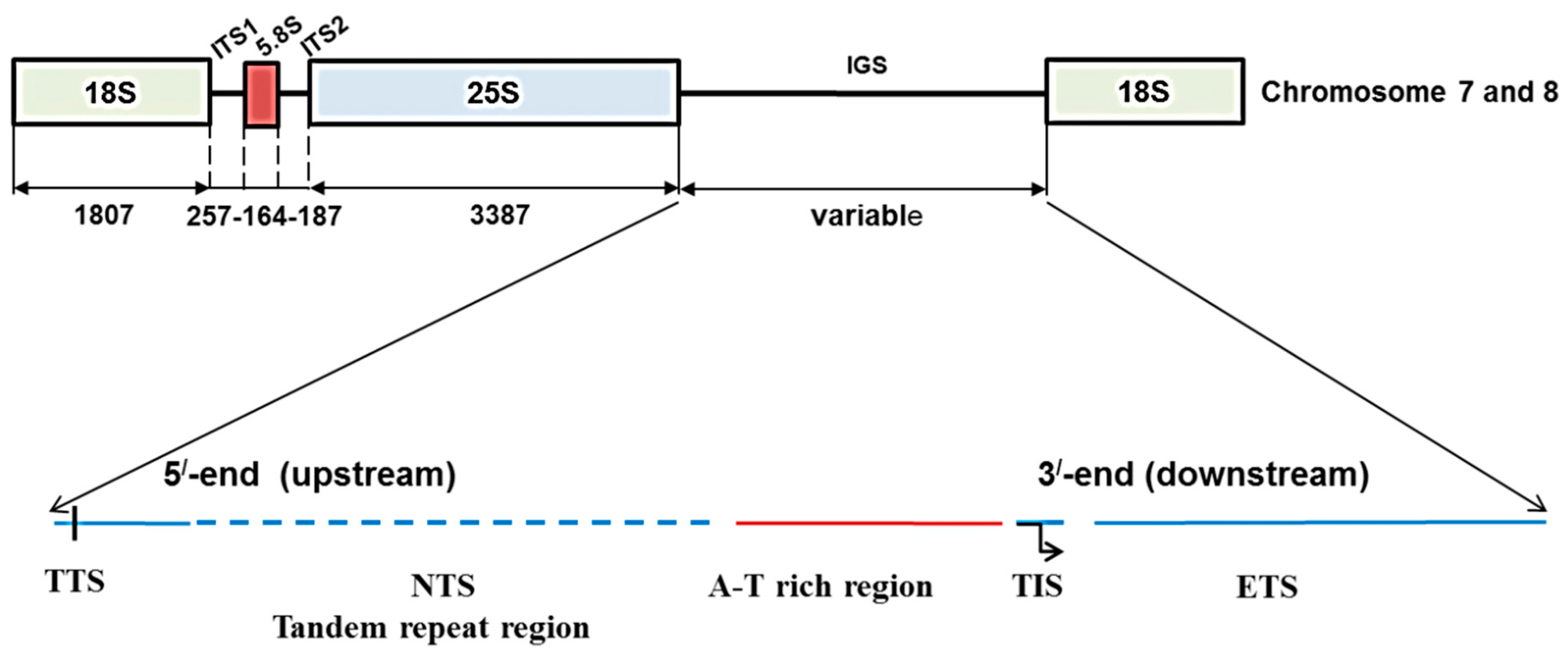
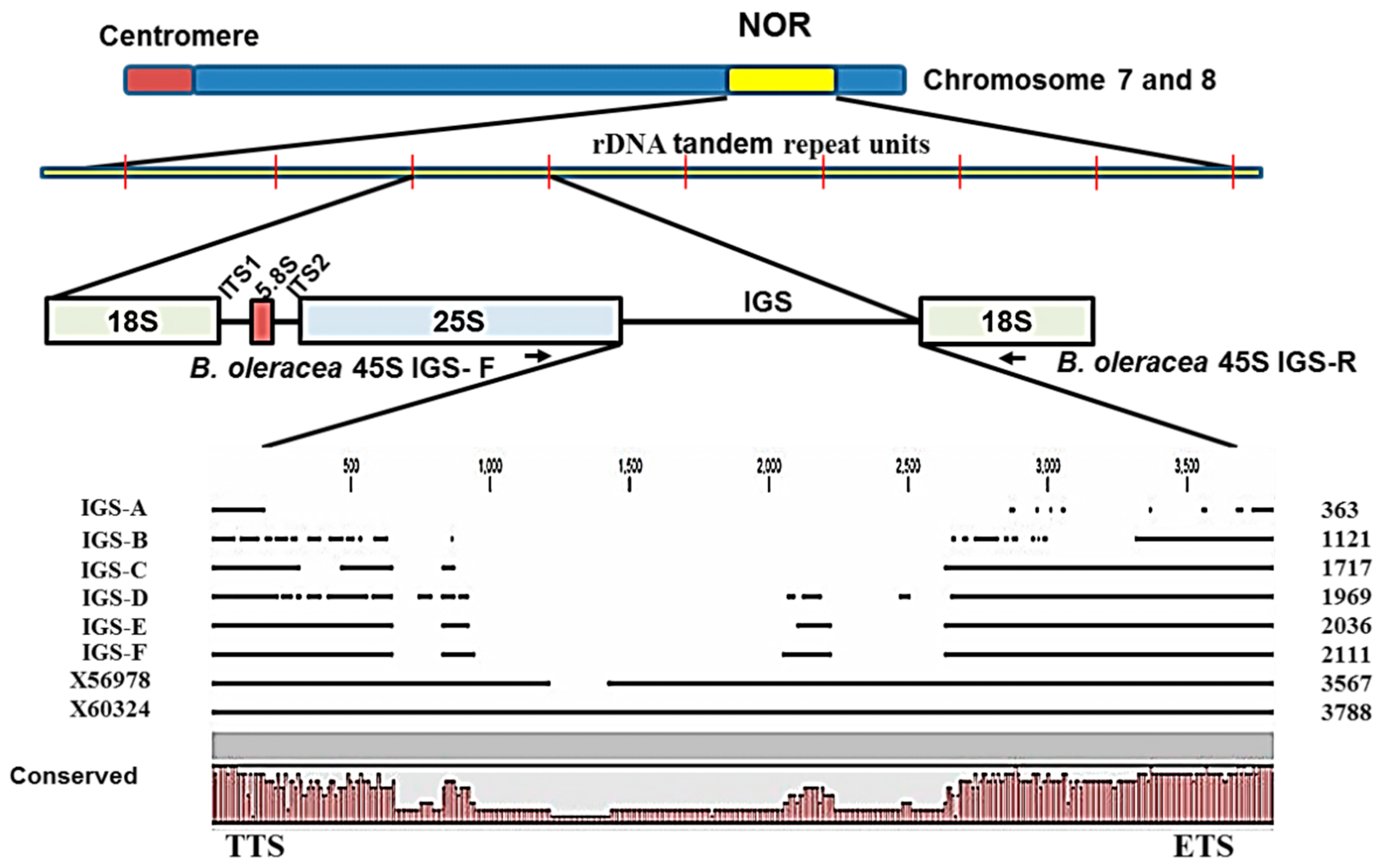
2.1. Identification of 45S Ribosomal DNA
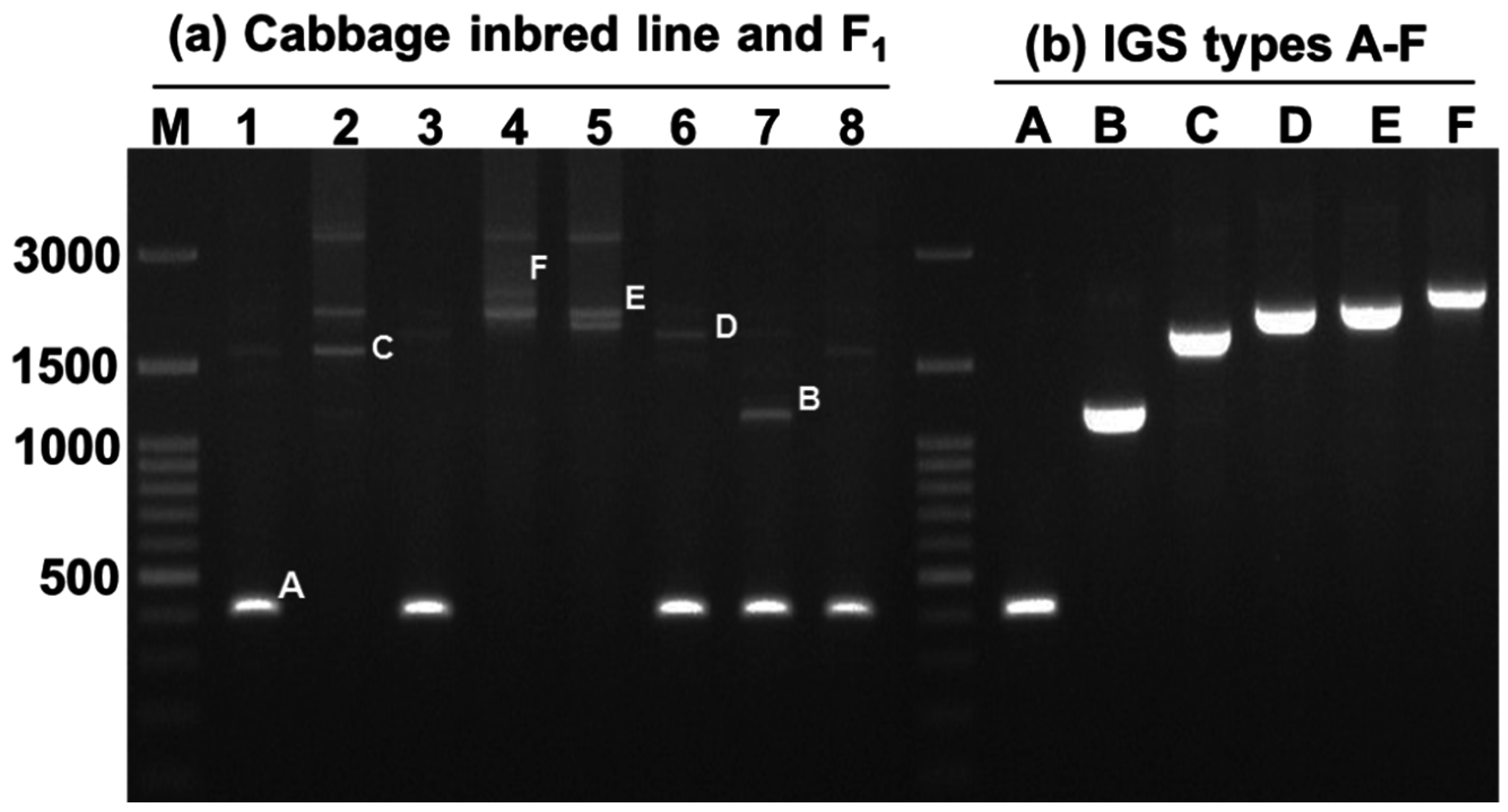
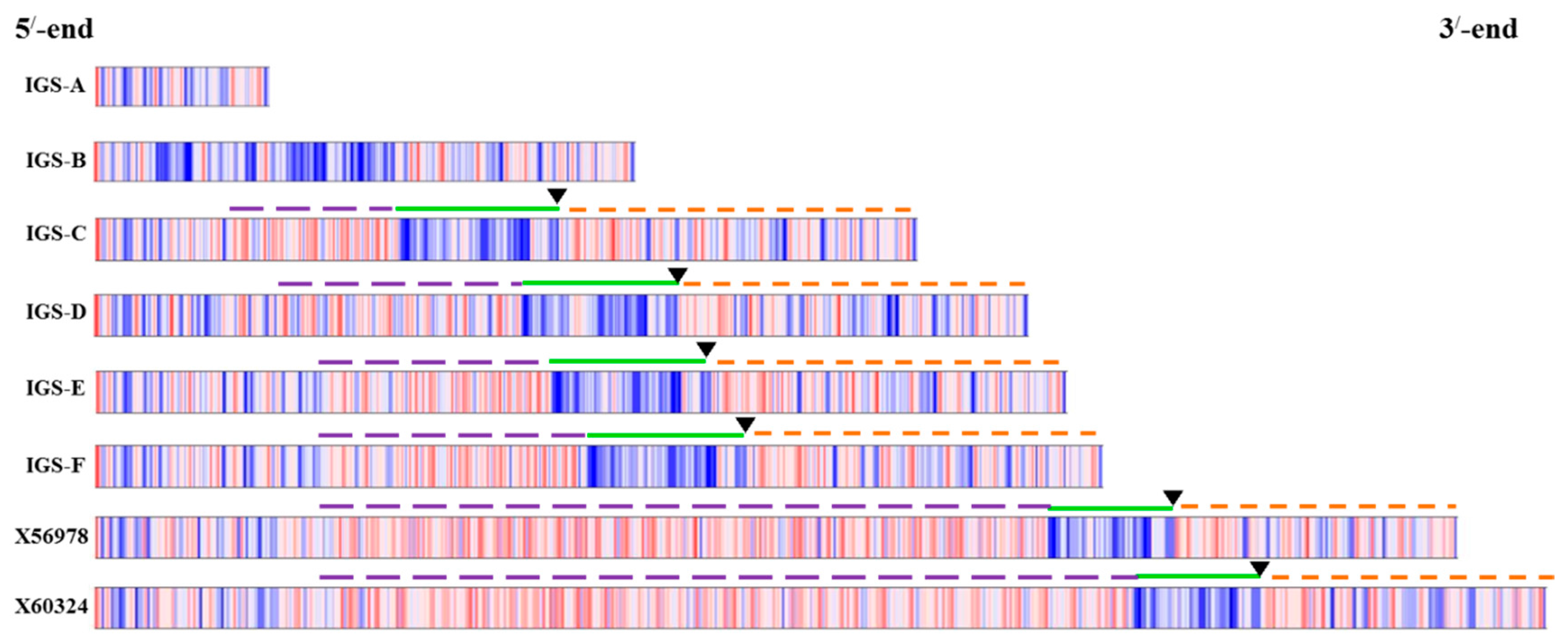
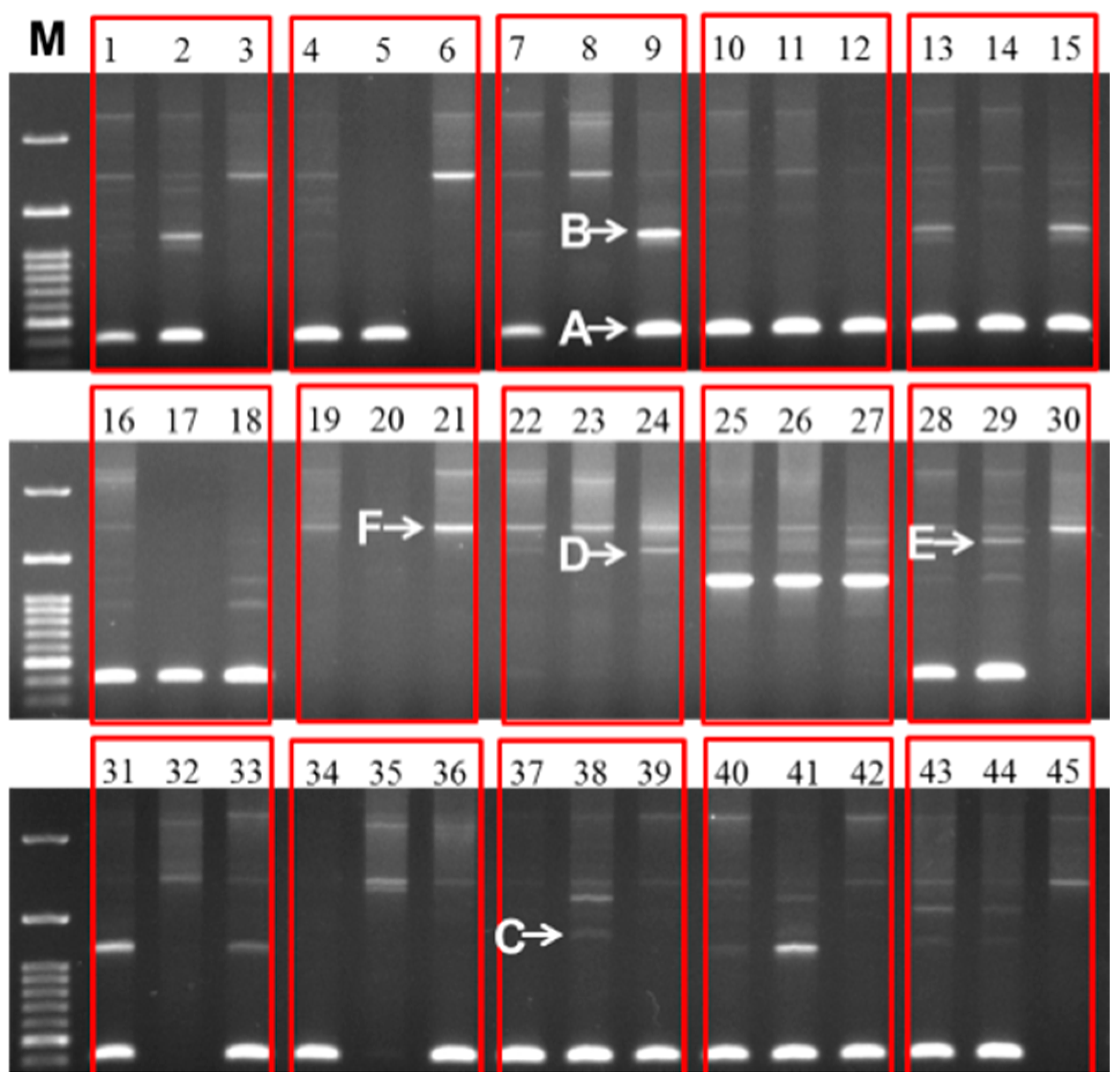
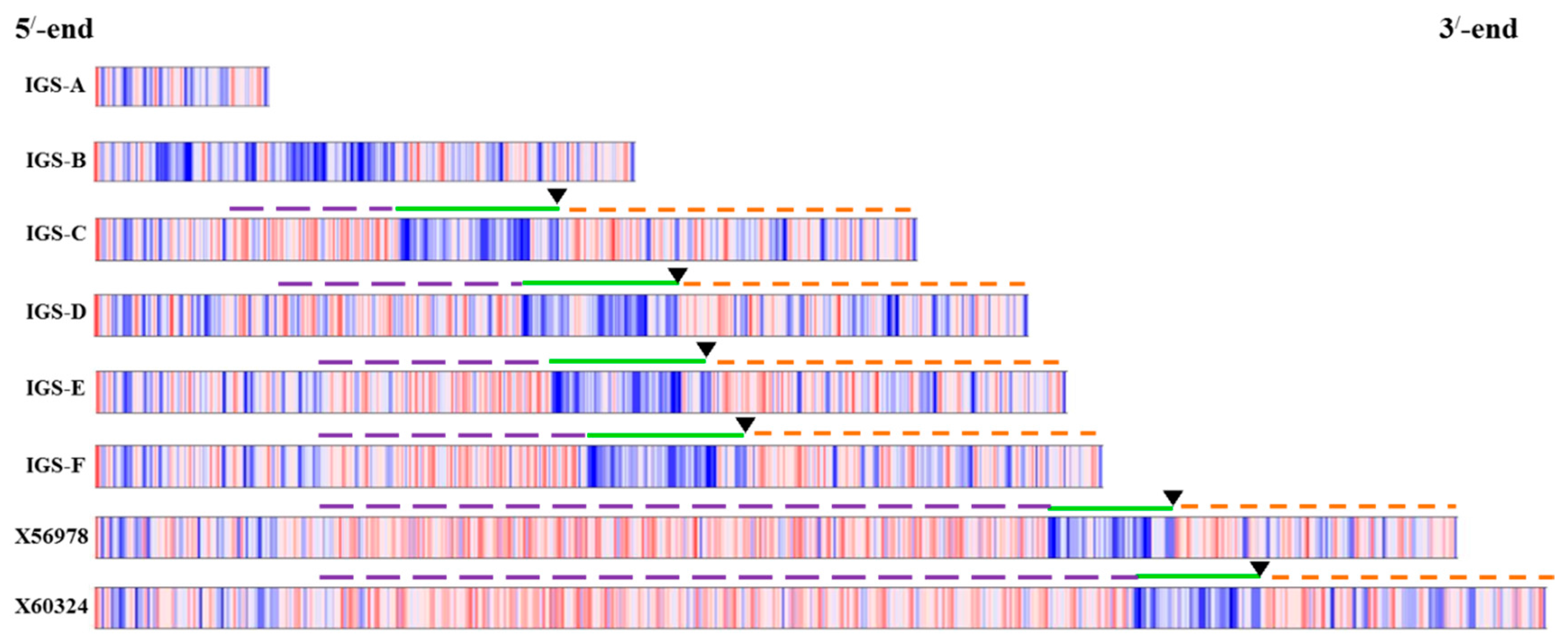
2.2. Variation in 45S-IGS
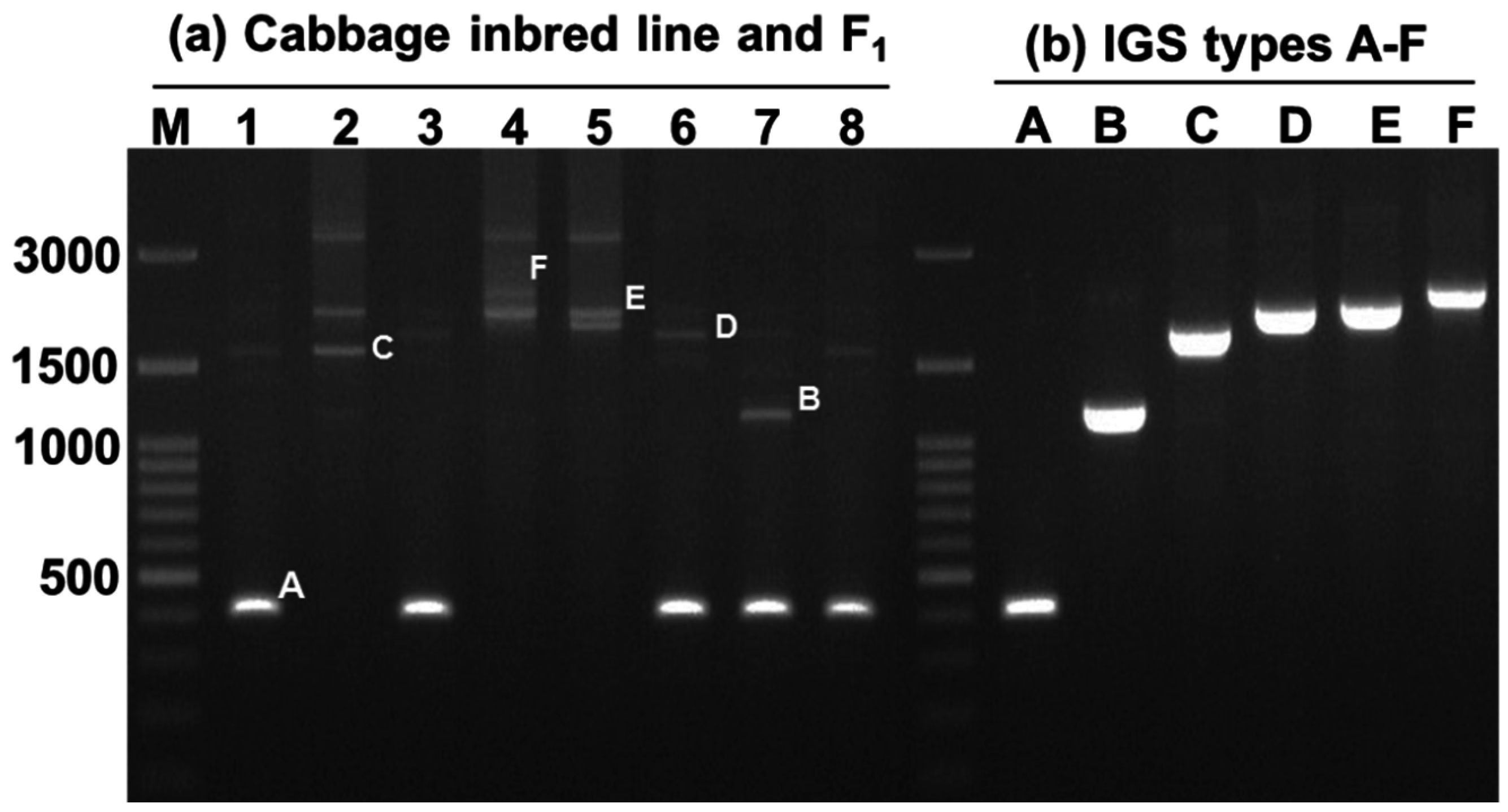
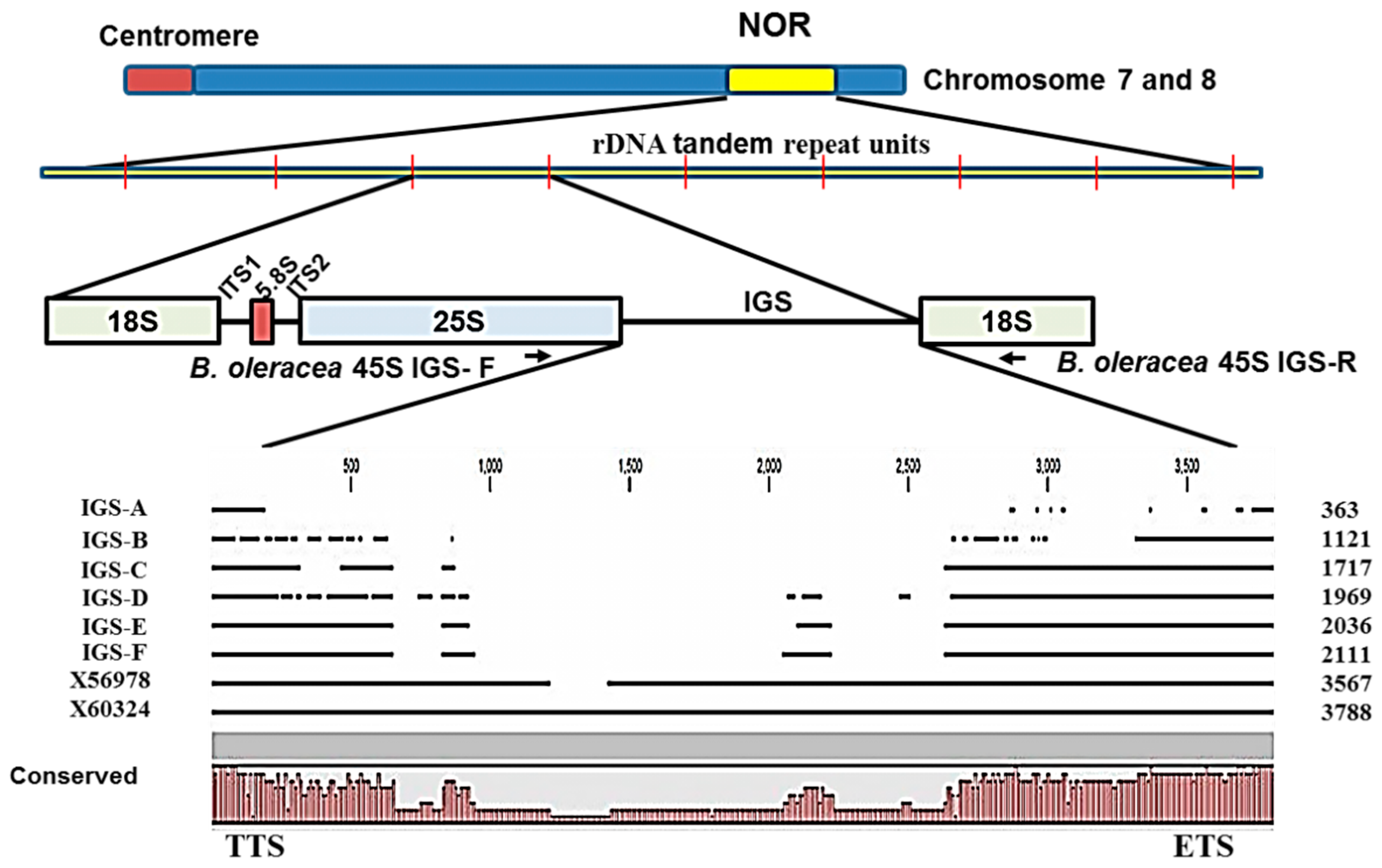

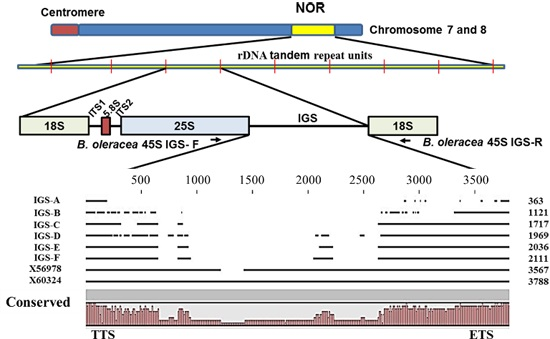{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IGS Type | Description | Query Cover (%) | E Value | Identity | Accession | GC Content (%) |
|---|---|---|---|---|---|---|
| IGS_A | B.oleracea 25S & 18S rRNA genes | 73 | 7 × 10−85 | 98% | X60324.1 | 44.08 |
| IGS_B | B.oleracea 25S & 18S rRNA genes | 44 | 0.0 | 98% | X60324.1 | 39.52 |
| IGS_C | B.oleracea 25S & 18S rRNA genes | 100 | 0.0 | 96% | X60324.1 | 46.53 |
| IGS_D | B.oleracea 25S & 18S rRNA genes | 85 | 0.0 | 87% | X60324.1 | 44.59 |
| IGS_E | B.oleracea 25S & 18S rRNA genes | 100 | 0.0 | 94% | X60324.1 | 46.37 |
| IGS_F | B.oleracea 25S & 18S rRNA genes | 100 | 0.0 | 95% | X60324.1 | 46.80 |
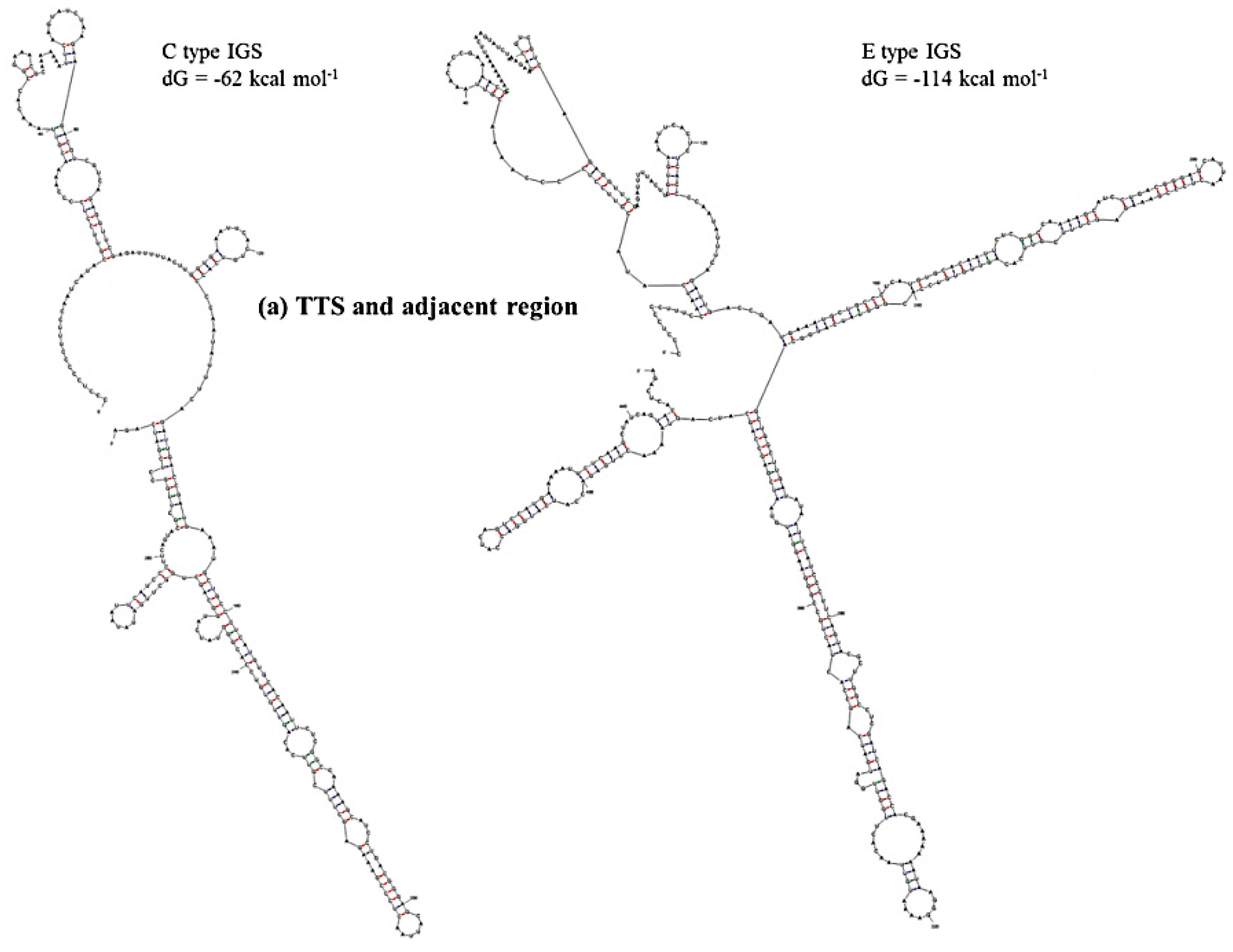
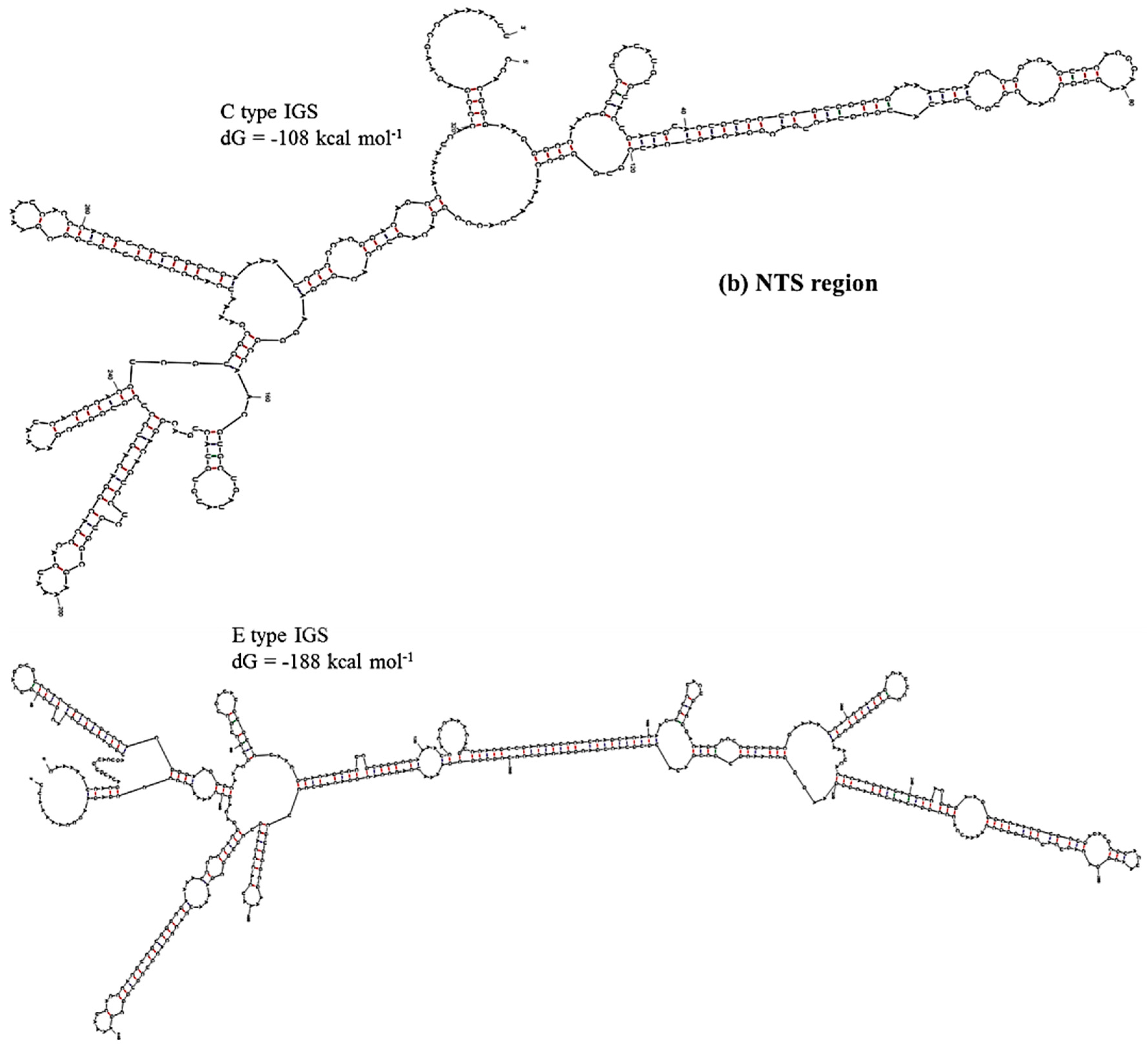
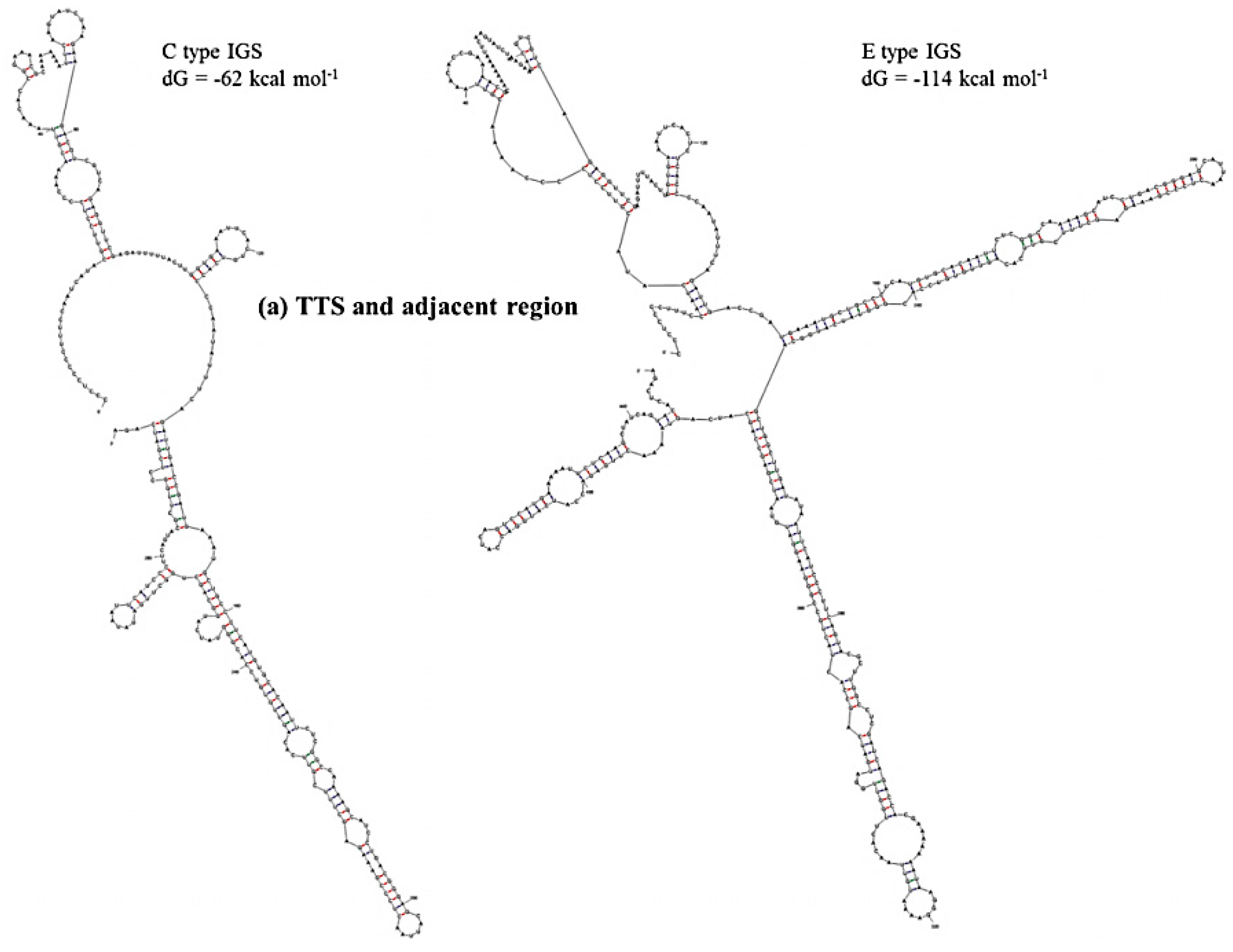
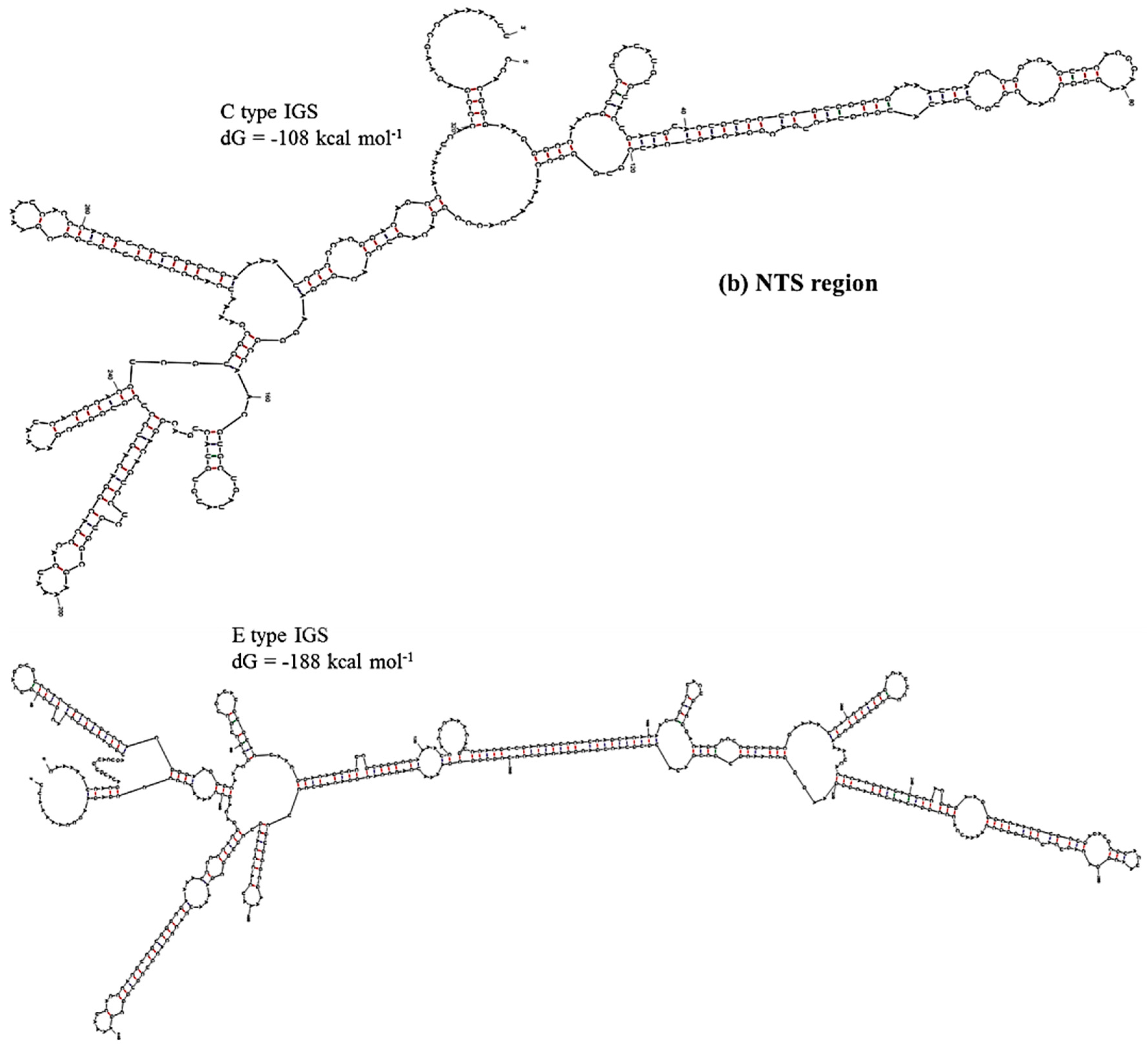
2.3. Functional Elements and Domains
| IGS Type | TTS Sequence and Position | Repeat Position | Consensus Size of Tandem Repeats (bp) | Percent Match | AT-rich Position (AT%) | TIS Sequence and Position | ETS Size (GC %) |
|---|---|---|---|---|---|---|---|
| IGS_A | CCCTCCCCCTTTCT (1–14) | None | None | None | None | ||
| IGS_B | CCCTCCCCCTTTCC (1–14) | None | None | None | None | ||
| IGS_C | CCCTCCCCCTTTCC (1–14) | 304–640 | 12–72 | 85–96 | 634–962 (69.91) | TATATAAGGGG (963–973) | 744 (49.19) |
| IGS_D | CCCTCCCCCATTCC (1–14) | 395–905 | 19–58 | 85–97 | 899–1220 (68.94) | TATATAAAGGG (1221–1231) | 738 (46.34) |
| IGS_E | CCCTCCCCTTTCC (1–13) | 454–959 | 12–72 | 84–96 | 953–1281 (69.60) | TATATAAGGGG (1282–1292) | 744 (49.19) |
| IGS_F | CCCTCCCCCTTTCC (1–14) | 455–1034 | 12–74 | 84–96 | 1028–1356 (69.60) | TATATAAGGGG (1357–1367) | 744 (49.19) |
| X56978 | CCCTCCCCCTTTCT (1–14) | 456–2495 | 12–454 | 68–96 | 2489–2817 (70.21) | TATATAAGGGG (2818–2828) | 739 (49.80) |
| X60324 | CCCTCCCCCTTTCT (1–14) | 457–2711 | 12–250 | 67–97 | 2705–3033 (70.21) | TATATAAGGGG (3034–3044) | 744 (49.33) |
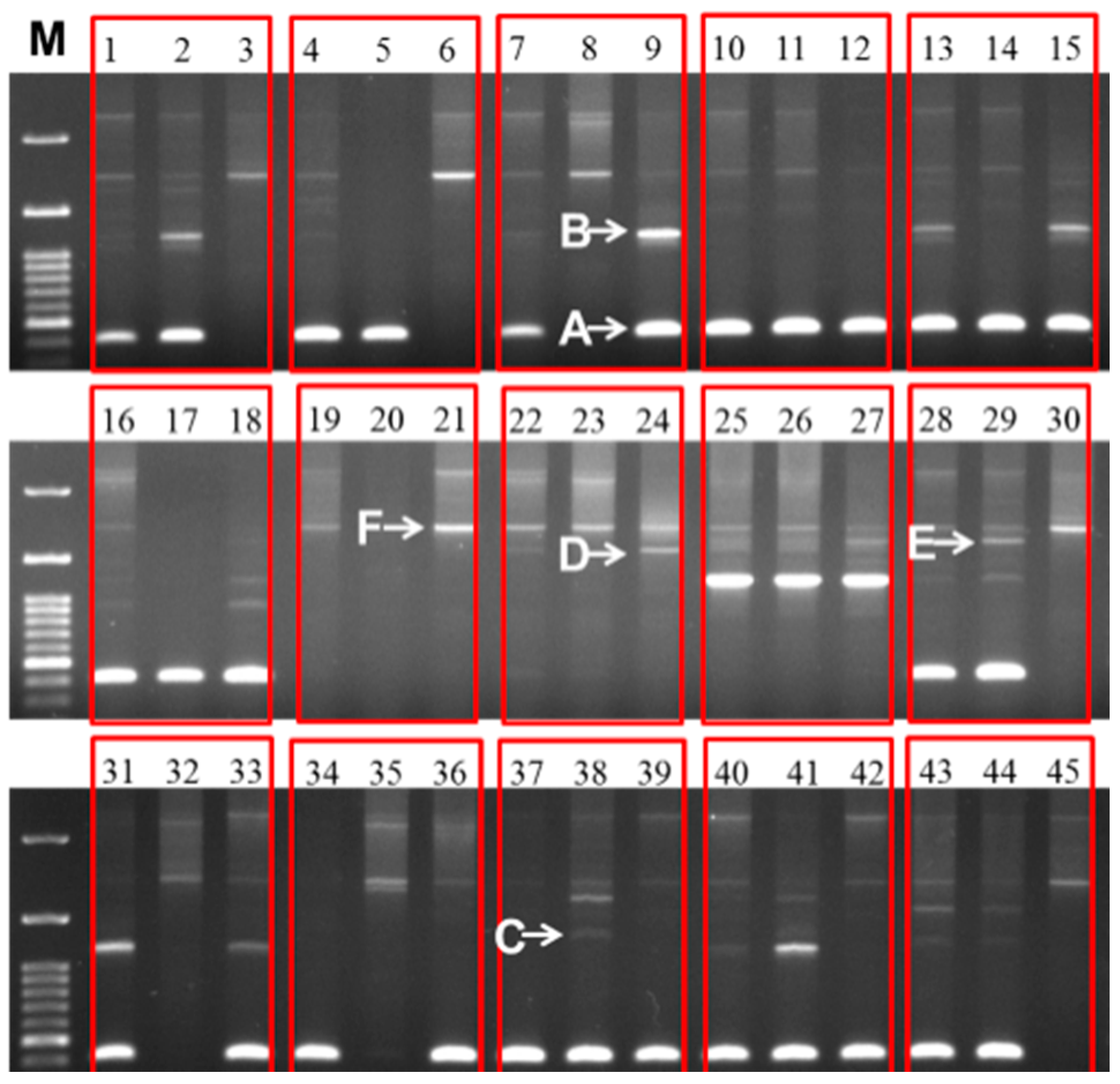
2.4. Variable IGS Inheritance
3. Discussion
4. Materials and Methods
4.1. Plant Materials and DNA Extraction
4.2. Whole-Genome Sequencing
4.3. PCR Amplification Cloning and Sequencing
| (A) Inbred Lines for NGS | (B) F1 Combination or Parents | (C) Lines and F1s to Test 45S IGS Variation | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sl. no. | Line | Sl. no. | Lines or cross combinations | Type | Sl. No. | Lines or cross combinations | Type | Sl. No. | Lines or cross combinations | Type | Sl. No. | Lines or cross combinations | Type |
| 1 | Asiaseed-14 | 1 | 8352 × 636 | F1 | 1 | 1464 × 621 | F1 | 16 | 26S × NC1 | F1 | 31 | 337S × 94 | F1 |
| 2 | Asiaseed-17 | 2 | 2418 × CT5-51 | F1 | 2 | 1464 | F | 17 | 26S | F | 32 | 337S | F |
| 3 | Asiaseed-28 | 3 | 842 × 621 | F1 | 3 | 621 | M | 18 | NC1 | M | 33 | 94 | M |
| 4 | Asiaseed-31 | 4 | 94 | Inbred line | 4 | 8352 × 636 | F1 | 19 | 337 × P15-41 | F1 | 34 | IB14 × DWB | F1 |
| 5 | IB14 | Inbred line | 5 | 8352 | F | 20 | 337 | F | 35 | IB14 | F | ||
| 6 | 842 | Inbred line | 6 | 636 | M | 21 | P15-41 | M | 36 | DWB | M | ||
| 7 | 496B × 2409 | F1 | 7 | 225 × 95 | F1 | 22 | 2418 × CT5-51 | F1 | 37 | 842 × 2409 | F1 | ||
| 8 | 9051 × PKT-41 | F1 | 8 | 225 | F | 23 | 2418 | F | 38 | 842 | F | ||
| 9 | 94 | M | 24 | CT5-51 | M | 39 | 2409 | M | |||||
| 10 | 9051 × 3074 | F1 | 25 | 632 × 755 | F1 | 40 | 496B × 2409 | F1 | |||||
| 11 | 9051 | F | 26 | 632 | F | 41 | 496B | F | |||||
| 12 | 3074 | M | 27 | 755 | M | 42 | 2409 | M | |||||
| 13 | 2409 × 8S8-7 | F1 | 28 | 842 × 621 | F1 | 43 | 9051 × PKT-41 | F1 | |||||
| 14 | 2409 | F | 29 | 842 | F | 44 | 9051 | F | |||||
| 15 | 8S8-7 | M | 30 | 621 | M | 45 | PKT-41 | M | |||||
| 45S IGS | TTS + 3’ETS Region, Region 1 | NTS Region, Region 2 | AT-rich Region, Region 3 | 5’ETS Region, Region 4 |
|---|---|---|---|---|
| C type | −62 | −108 | −48 | −273 |
| D type | −67 | −171 | −46 | −239 |
| E type | −114 | −188 | −58 | −273 |
| F type | −113 | −209 | −58 | −273 |
| X56978 | −110 | Not obtained | −54 | −277 |
| X60324 | −107 | Not obtained | −54 | −276 |
| Name | Forward and Revers Primers | Product Size | Comment |
|---|---|---|---|
| Bo 5S IGS | F: ACTAGGATGGGTGACCTCCCG; | 484 | 5S-IGS product size |
| R: CGCTTAACTGCGGAGTTCTGA | |||
| Bo ITS1 and 2 | F: CGCGAGAAGTCCACTAAACC; | 823 | Identify sequence differences |
| R: ACGTCCGATTTTCAAGCTGG | |||
| Bo 45S IGS | F: ATTCAGCCCTTTGTCGCTAA; | variable | 45S-IGS product size |
| R: ATGACTACTGGCAGGATCAACCAG | |||
| IGS inter-D | ATCGTGTAGGACCGAGAAGT | D type intergenic | |
| IGS inter-CEF | GGTAGGCACAAAAGGAATGC | C, E and F type intergenic |
4.4. Sequence Analysis
5. Conclusions
| KT377443 | 45S intergenic spacer (A type) | PCR-primer amplified |
| KT377444 | 45S intergenic spacer (B type) | |
| KT377445 | 45S intergenic spacer (C type) | |
| KT377446 | 45S intergenic spacer (D type) | |
| KT377447 | 45S intergenic spacer (E type) | |
| KT377448 | 45S intergenic spacer (F type) | |
| KT377450 | 5S intergenic spacer | |
| KT377452 | Internal transcribed spacer 1 | |
| KT377454 | Internal transcribed spacer 2 |
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Moss, T.; Stefanovsky, V.Y. Promotion and regulation of ribosomal transcription in eukaryotes by RNA polymerase 1. Prog. Nucl. Acid Res. Mol. Biol. 1995, 50, 25–66. [Google Scholar]
- Paule, M.R.; Lofquist, A.K. Organization and expression of eukaryotic ribosomal RNA genes. In Ribosomal RNA Structure, Evolution, Processing and Function in Protein Biosynthesis; Zimmermann, R.A., Dahlberg, A.E., Eds.; CRC Press: New York, NY, USA, 1996; pp. 395–419. [Google Scholar]
- Appels, R.; Honeycutt, R.L. rDNA: evolution over a billion years. In DNA Systematics; Dutta, S.K., Ed.; CRC Press: Boca Raton, FL, USA, 1986; Volume 2, pp. 81–135. [Google Scholar]
- Kahl, G. Architecture of Eukaryotic Genes; VCH Verlagsgesellschaft: Weinheim, Germany, 1988. [Google Scholar]
- Poczai, P.; Hyvonen, J. Nuclear ribosomal spacer regions in plant phylogenetics: Problems and prospects. Mol. Biol. Rep. 2010, 37, 1897–1912. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, G.D.; Dalton, M.W.; Fallon, A.M. Is higher-order structure conserved in eukaryotic ribosomal DNA intergenic spacers? J. Mol. Evol. 1992, 35, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Reeder, R.H. Regulatory elements of the generic ribosomal gene. Curr. Opin. Cell Biol. 1989, 1, 466–474. [Google Scholar] [CrossRef]
- Rogers, S.O.; Bendich, A.J. Ribosomal RNA genes in plants: Variability in copy number and in the intergenic spacer. Plant Mol. Biol. 1987, 9, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Gerstner, J.; Schiebel, K.; von Waldburg, G.; Hemleben, V. Complex organization of the length heterogeneous 50 external spacer of mung bean (Vigna radiata) ribosomal DNA. Genome 1988, 30, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Hemleben, V.; Ganal, M.; Gerstner, J.; Schiebel, K.; Torres, R.A. Organization and length heterogeneity of plant ribosomal RNA genes. In The Architecture of Eukaryotic Genes; Kahl, G., Ed.; VHC: Weinheim, Germany, 1988; pp. 371–383. [Google Scholar]
- Jorgansen, R.A.; Cluster, P.D. Modes and tempos in the evolution of nuclear ribosomal DNA: New characters for evolutionary studies and new markers for genetic and population studies. Ann. Mo. Bot. Gard. 1988, 75, 1238–1247. [Google Scholar] [CrossRef]
- Schaal, B.A.; Learn, G.H.J. Ribosomal DNA variation with and among plant populations. Ann. Mo. Bot. Gard. 1988, 75, 1207–1216. [Google Scholar] [CrossRef]
- Weider, L.J.; Elser, J.J.; Crease, T.J.; Mateos, M.; Cotner, J.B. The functional significance of ribosomal (r) DNA variation: Impacts on the evolutionary ecology of organisms. Annu. Rev. Ecol. Evol. Syst. 2005, 36, 219–242. [Google Scholar] [CrossRef]
- Kim, K.J.; Mabry, T.J. Phylogenetic and evolutionary implications of nuclear ribosomal DNA variation in dwarf dandelions (Krigia, Lactuceae, Asteraceae). Plant Syst. Evol. 1991, 177, 53–69. [Google Scholar] [CrossRef]
- Yakura, K.; Kato, A.; Tanifuji, S. Length heterogeneity in the large spacer of Vicia faba rDNA is due to the differing number of a 325 bp repetitive sequence element. Mol. Gen. Genet. 1984, 193, 400–405. [Google Scholar] [CrossRef]
- Toloczyki, C.; Feix, G. Occurrence of nine homologous repeat units in the external spacer of a nuclear maize rRNA gene unit. Nucleic Acids Res. 1986, 14, 4969–4986. [Google Scholar] [CrossRef] [PubMed]
- Lakshmikumaran, M.; Negi, M.S. Structural analysis of two length variants of the rDNA intergenic spacer from Eruca sativa. Plant Mol. Biol. 1994, 24, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Negi, M.S.; Lakshmikumaran, M. Structural analysis of the rDNA intergenic spacer of Brassica nigra: Evolutionary divergence of the spacers of the three diploid Brassica species. J. Mol. Evol. 1996, 43, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.; Markow, T.A. Ribosomal intergenic spacer (IGS) length variation across the Drosophilinae (Diptera: Drosophilidae). BMC Evol. Biol. 2005, 5, 46. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maggini, F.; Tucci, G.; Demartis, A.; Gelati, M.T.; Avanzi, S. Ribosomal RNA genes of Phaseolus coccineus. I. Plant Mol. Biol. 1992, 18, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Sardana, R.; O’Dell, M.; Flavell, R.B. Correlation between the size of the intergenic regulatory region, the status of cytosine methylation of rRNA genes and nucleolar expression in wheat. Mol. Gen. Genet. 1993, 236, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Hemleben, V.; Zentgraf, U. Structural organization and regulation of transcription by RNA polymerase I of plant nuclear ribosomal RNA genes. In Results and Problems in Cell Differentiation 20. Plant Promoters and Transcription Factors; Nover, L., Ed.; Springer: Berlin, Germany, 1994; pp. 3–24. [Google Scholar]
- Da Rocha, P.S.C.F.; Bertrand, H. Structure and comparative analysis of the rDNA intergenic spacer of Brassica rapa: Implications for the function and evolution of the Cruciferae spacer. Eur. J. Biochem. 1995, 229, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Polanco, C.; Ruiz, M.L.; Pérez De La Vega, M. A comparative study of the structure of the rDNA intergenic spacer of Lens culinaris Medik., and other legume species. Genome 2000, 43, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Schiebel, K.; von Waldburg, G.; Gerstner, J.; Hemleben, V. Termination of transcription of ribosomal RNA genes of mung bean occurs within a 175 bp repetitive element of the spacer region. Mol. Gen. Genet. 1989, 218, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Warwick, S.I.; Francis, A.; Al-Shehbaz, I.A. Brassicaceae: Species checklist and database on CD-Rom. Plant Syst. Evol. 2006, 259, 249–258. [Google Scholar] [CrossRef]
- Johnston, J.S.; Pepper, A.E.; Hall, A.E.; Chen, Z.J.; Hodnett, G.; Drabek, J.; Price, H.J. Evolution of genome size in Brassicaceae. Ann. Bot. 2005, 95, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Parkin, A.C.; Koh, H.; Tang, S.J.; Robinson, S.; Kagale, W.E. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 2014, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Yi, G.E.; Robin, A.H.K.; Yang, K.W.; Park, J.I.; Kang, J.G. Identification and Expression Analysis of Glucosinolate Biosynthetic Genes and Estimation of Glucosinolate Contents in Edible Organs of Brassica oleracea subspecies. Molecules 2015, 20, 13089–13111. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.I.; Smith, A.G. The complete nucleotide sequence of the intergenic spacer region of an rDNA operon from Brassica oleracea and its comparison with other crucifers. Plant Mol. Biol. 1991, 16, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Tremousaygue, D.; Laudie, M.; Grellet, F.; Delseny, M. The Brassica oleracea rDNA spacer revisited. Plant Mol. Biol. 1992, 18, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Gruendler, P.; Unfried, I.; Pascher, K.; Schweizer, D. rDNA intergenic region from Arabidopsis thaliana Structural analysis, intraspecific variation and functional implications. J. Mol. Biol. 1991, 221, 1209–1222. [Google Scholar] [CrossRef]
- Doelling, J.H.; Gaudino, R.J.; Pikaard, C.S. Functional analysis of Arabidopsis thaliana rRNA gene and spacer promoters in vivo and by transient expression. Proc. Natl. Acad. Sci. USA 1993, 90, 7528–7532. [Google Scholar] [CrossRef] [PubMed]
- Doelling, J.H.; Pikaard, C.S. The minimal ribosomal RNA gene promoter of Arabidopsis thaliana includes a critical element at the transcription initiation site. Plant J. 1995, 8, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, A.; Gupta, V.; Lakshmikumaran, M.; Shivanna, K.R.; Prakash, S. Production of Eruca- Brassica hybrids by embryo rescue. Plant Breed. 1990, 104, 281–289. [Google Scholar] [CrossRef]
- Copenhaver, G.P.; Pikaard, C.S. RFLP and physical mapping with an rDNA-specific endonuclease reveals that nucleolus organizer regions of Arabidopsis thaliana adjoin the telomeres on chromosomes 2 and 4. Plant J. 1996, 9, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Abou-Ellail, M.; Cooke, R.; Sáez-Vásquez, J. Variations in a team: Major and minor variants of Arabidopsis thaliana rDNA genes. Nucleus 2011, 2, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Zentgraf, U.; Ganal, M.; Hemleben, V. Length heterogeneity of the rRNA precursor in cucumber (Cucumis sativus). Plant Mol. Biol. 1990, 15, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Bauer, N.; Horvat, T.; Birus, I.; Vicic, V.; Zoldos, V. Nucleotide sequence, structural organization and length heterogeneity of ribosomal DNA intergenic spacer in Quercus petraea (Matt.) Liebl. and Q. robur L. Mol. Genet. Gen. 2009, 281, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, C.; Crease, T. Evolution of the nuclear ribosomal DNA intergenic spacer in four species of the Daphnia pulex complex. BMC Genet. 2011, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Borisjuk, N.V.; Davidjuk, Y.M.; Kostishin, S.S.; Miroshnichenco, G.P.; Velasco, R. Structural analysis of rDNA in the genus Nicotiana. Plant Mol. Biol. 1997, 35, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Komarova, N.Y.; Grabe, T.; Huigen, D.J.; Hemleben, V.; Volkov, R.A. Organization, differential expression and methylation of rDNA in artificial Solanum allopolyploids. Plant Mol. Biol. 2004, 56, 439–463. [Google Scholar] [CrossRef] [PubMed]
- Delcasso-Tremousaygue, D.; Grellet, F.; Panabieres, F.; Ananiev, E.D.; Delseny, M. Structural and transcriptional characterization of the external spacer of a ribosomal RNA nuclear gene from a higher plant. Eur. J. Biochem. 1988, 172, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Bena, G.; Jubier, M.F.; Olivieri, I.; Lejeune, B. Ribosomal external and internal transcribed spacers: Combined use in the phylogenetic analysis of Medicago (Leguminosae). J. Mol. Evol. 1998, 46, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Cordesse, F.; Cooke, R.; Tremousaygue, D.; Grellet, F.; Delseny, M. Fine structure and evolution of the rDNA intergenic spacer in rice and other cereals. J. Mol. Evol. 1993, 36, 369–379. [Google Scholar] [CrossRef] [PubMed]
- McMullen, M.D.; Hunter, B.; Philips, R.L.; Rubenstein, I. The structure of the maize ribosomal DNA spacer region. Nucleic Acids Res. 1986, 14, 4953–4969. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.F.; Harberd, N.P.; Jarvis, M.G.; Flavell, R.B. Structure and evolution of the intergenic region of a ribosomal DNA repeat unit of wheat. J. Mol. Biol. 1988, 201, 1–17. [Google Scholar] [CrossRef]
- Taira, T.; Kato, A.; Tanifuji, S. Difference between two major size classes of carrot rDNA repeating units is due to reiteration of sequences of about 460 bp in the large spacer. Mol. Gen. Genet. 1988, 213, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Borisjuk, N.; Hemleben, V. Nucleotide sequence of the potato rDNA intergenic spacer. Plant Mol. Biol. 1993, 21, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Grellet, F.; Delcasso-Tremousaygue, D.; Delseny, M. Isolation and characterization of an unusual repeated sequence from the ribosomal intergenic spacer of the crucifer Sisymbrium irio. Plant Mol. Biol. 1989, 12, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Galián, J.A.; Rosato, M.; Rosselló, J.A. Incomplete sequence homogenization in 45S rDNA multigene families: intermixed IGS heterogeneity within the single NOR locus of the polyploid species Medicago arborea (Fabaceae). Ann. Bot. 2014, 114, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Siegel, A. The Cucurbita maxima ribosomal DNA intergenic spacer has a complex structure. Gene 1989, 80, 239–248. [Google Scholar] [CrossRef]
- Suzuki, A.; Tanifuji, S.; Komeda, Y.; Kato, A. Structural and functional characterization of the intergenic spacer region of the rDNA in Daucus carota. Plant Cell Physiol. 1996, 37, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Nickrent, D.L.; Patrick, J.A. The nuclear ribosomal DNA intergenic spacers of wild and cultivated soybean have low variation and cryptic subrepeats. Genome 1998, 41, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Achtziger, M.; Schultz, J.; Dandekar, T.; Müller, T. Homology modeling revealed more than 20,000 rRNA internal transcribed spacer 2 (ITS2) secondary structures. RNA 2005, 11, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Capesius, I. Sequence of the 5S rRNA gene from Sinapis alba. Plant Mol. Biol. 1991, 17, 169–170. [Google Scholar] [CrossRef] [PubMed]
- Capesius, I. Nucleotide sequence of a 25S rRNA gene from mustard (Sinapis alba). Plant Mol. Biol. 1991, 16, 1093–1094. [Google Scholar] [CrossRef] [PubMed]
- Inácio, V.; Rocheta, M.; Morais-Cecílio, L. Molecular Organization of the 25S–18S rDNA IGS of Fagus sylvatica and Quercus suber: A Comparative Analysis. PLoS ONE 2014, 9, 98678. [Google Scholar] [CrossRef] [PubMed]
- Loots, G.G.; Ovcharenko, I.; Pachter, L.; Dubchak, I.; Rubin, E.M. rVista for comparative sequence-based discovery of functional transcription factor binding sites. Genome Res. 2002, 12, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.A.; Madden, T.L. Blast 2 sequences—A new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 1999, 174, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, K.; Robin, A.H.K.; Yi, G.-E.; Lee, J.; Chung, M.-Y.; Yang, T.-J.; Nou, I.-S. Diversity and Inheritance of Intergenic Spacer Sequences of 45S Ribosomal DNA among Accessions of Brassica oleracea L. var. capitata. Int. J. Mol. Sci. 2015, 16, 28783-28799. https://doi.org/10.3390/ijms161226125
Yang K, Robin AHK, Yi G-E, Lee J, Chung M-Y, Yang T-J, Nou I-S. Diversity and Inheritance of Intergenic Spacer Sequences of 45S Ribosomal DNA among Accessions of Brassica oleracea L. var. capitata. International Journal of Molecular Sciences. 2015; 16(12):28783-28799. https://doi.org/10.3390/ijms161226125
Chicago/Turabian StyleYang, Kiwoung, Arif Hasan Khan Robin, Go-Eun Yi, Jonghoon Lee, Mi-Young Chung, Tae-Jin Yang, and Ill-Sup Nou. 2015. "Diversity and Inheritance of Intergenic Spacer Sequences of 45S Ribosomal DNA among Accessions of Brassica oleracea L. var. capitata" International Journal of Molecular Sciences 16, no. 12: 28783-28799. https://doi.org/10.3390/ijms161226125
APA StyleYang, K., Robin, A. H. K., Yi, G.-E., Lee, J., Chung, M.-Y., Yang, T.-J., & Nou, I.-S. (2015). Diversity and Inheritance of Intergenic Spacer Sequences of 45S Ribosomal DNA among Accessions of Brassica oleracea L. var. capitata. International Journal of Molecular Sciences, 16(12), 28783-28799. https://doi.org/10.3390/ijms161226125