
De Novo Sequencing and Analysis of the Safflower Transcriptome to Discover Putative Genes Associated with Safflor Yellow in Carthamus tinctorius L.

Abstract
:1. Introduction
2. Results
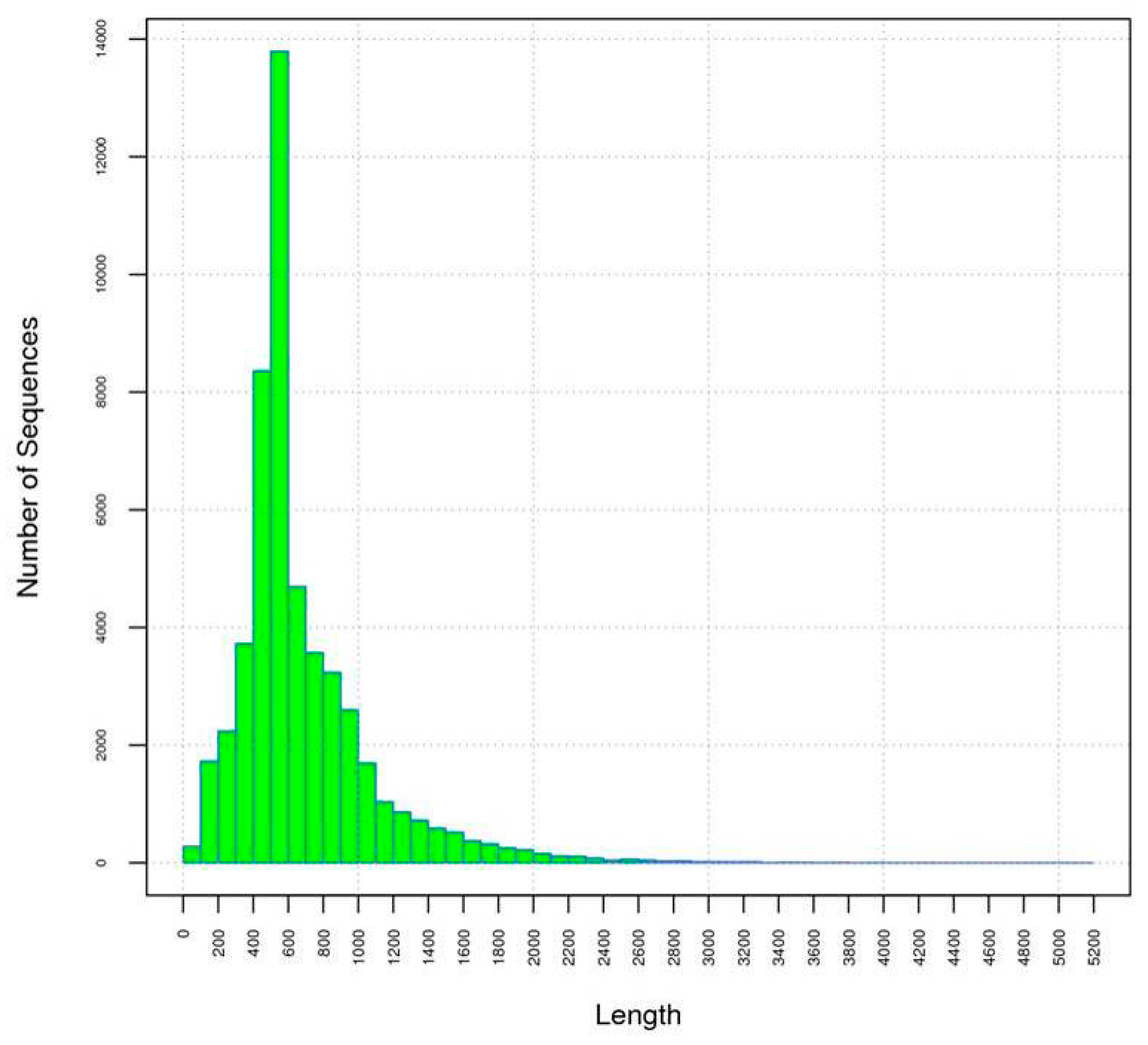
2.1. Preparation, Sequencing and de Novo Assembly of the Flower Transcriptome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
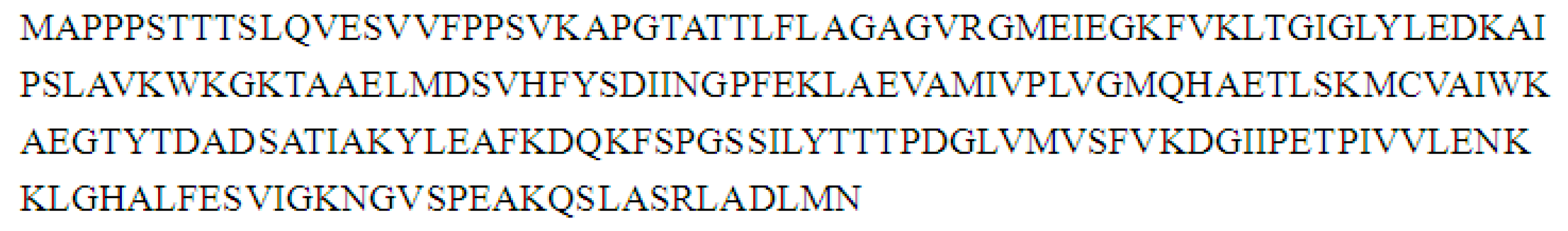{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Early | Full | |
|---|---|---|
| Raw reads | 583,440 | 567,884 |
| Low quality | 50 | 67 |
| Short reads (<50 bp) | 796 | 928 |
| Contamination sequences | 2785 | 2694 |
| Vector sequences | 2145 | 1265 |
| Clean reads | 577,664 | 562,930 |
| Average length | 427 bp | 436 bp |
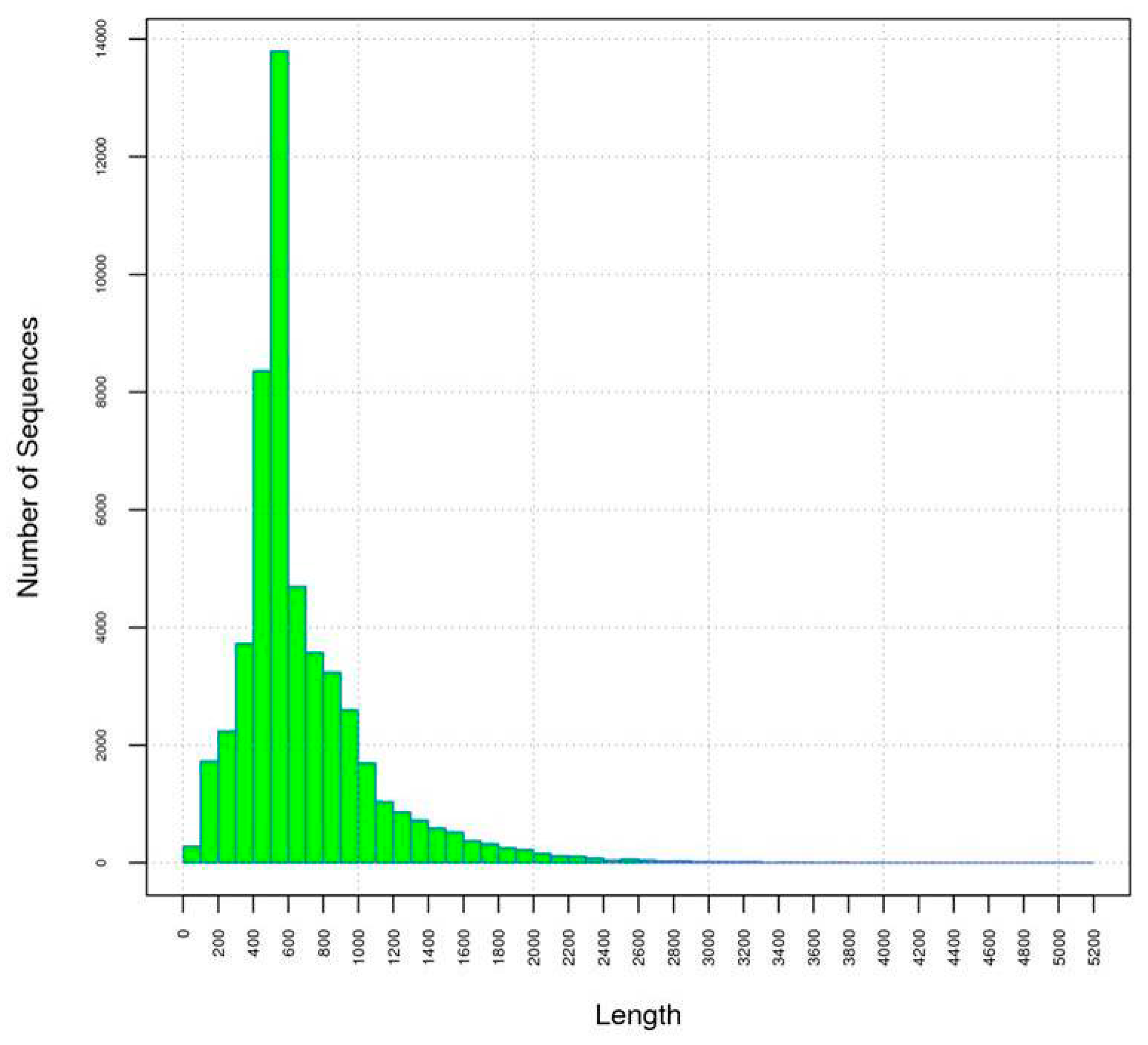
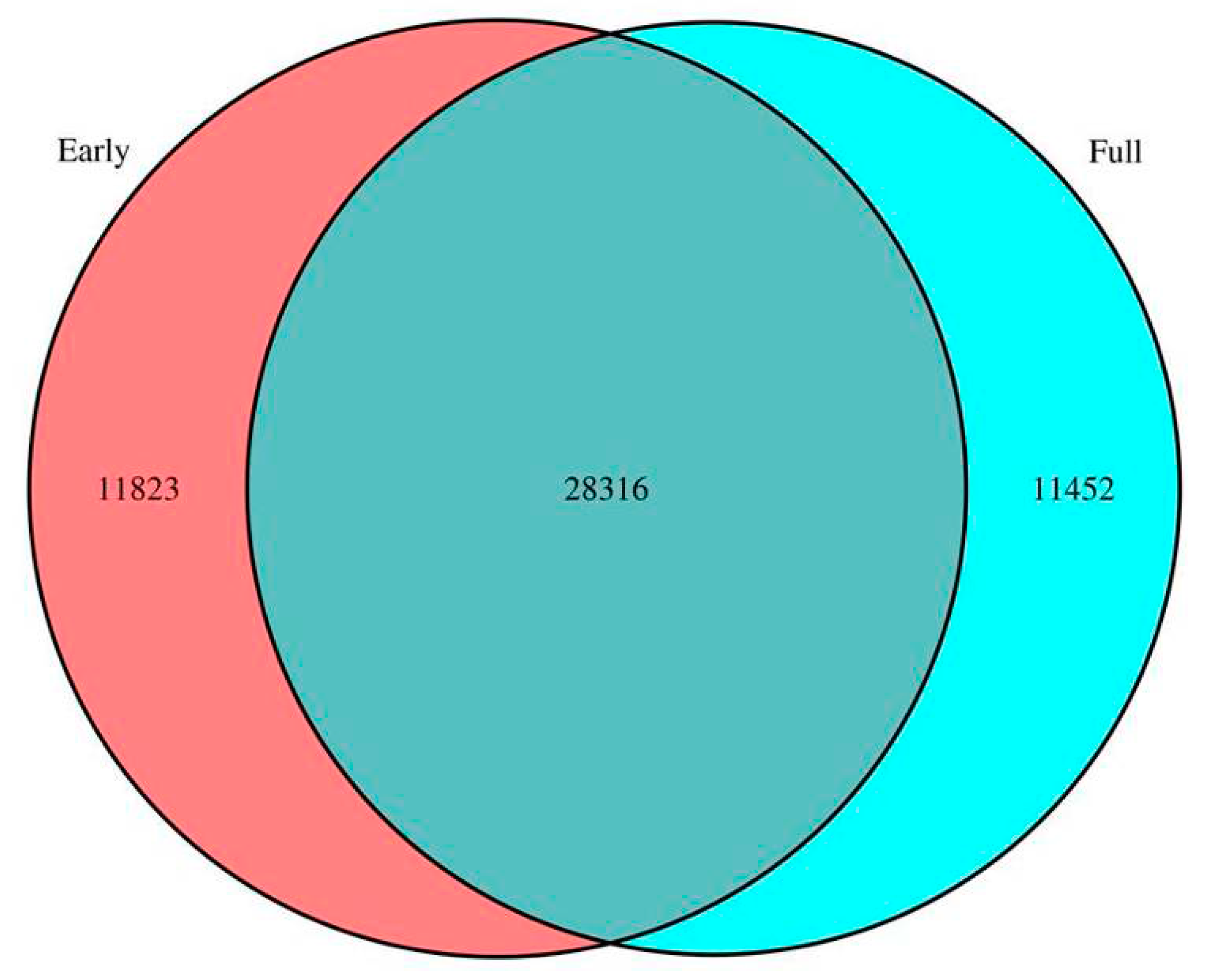
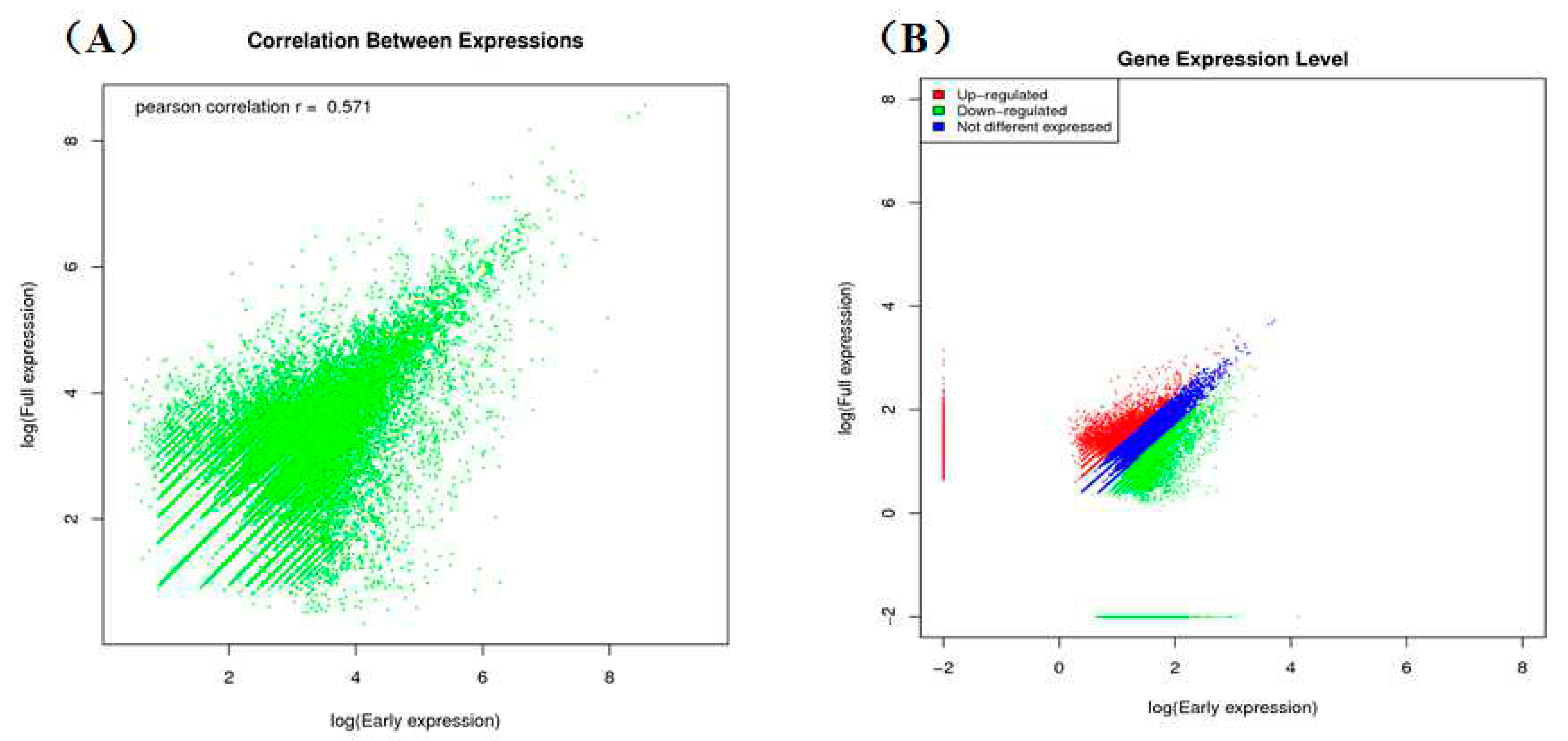
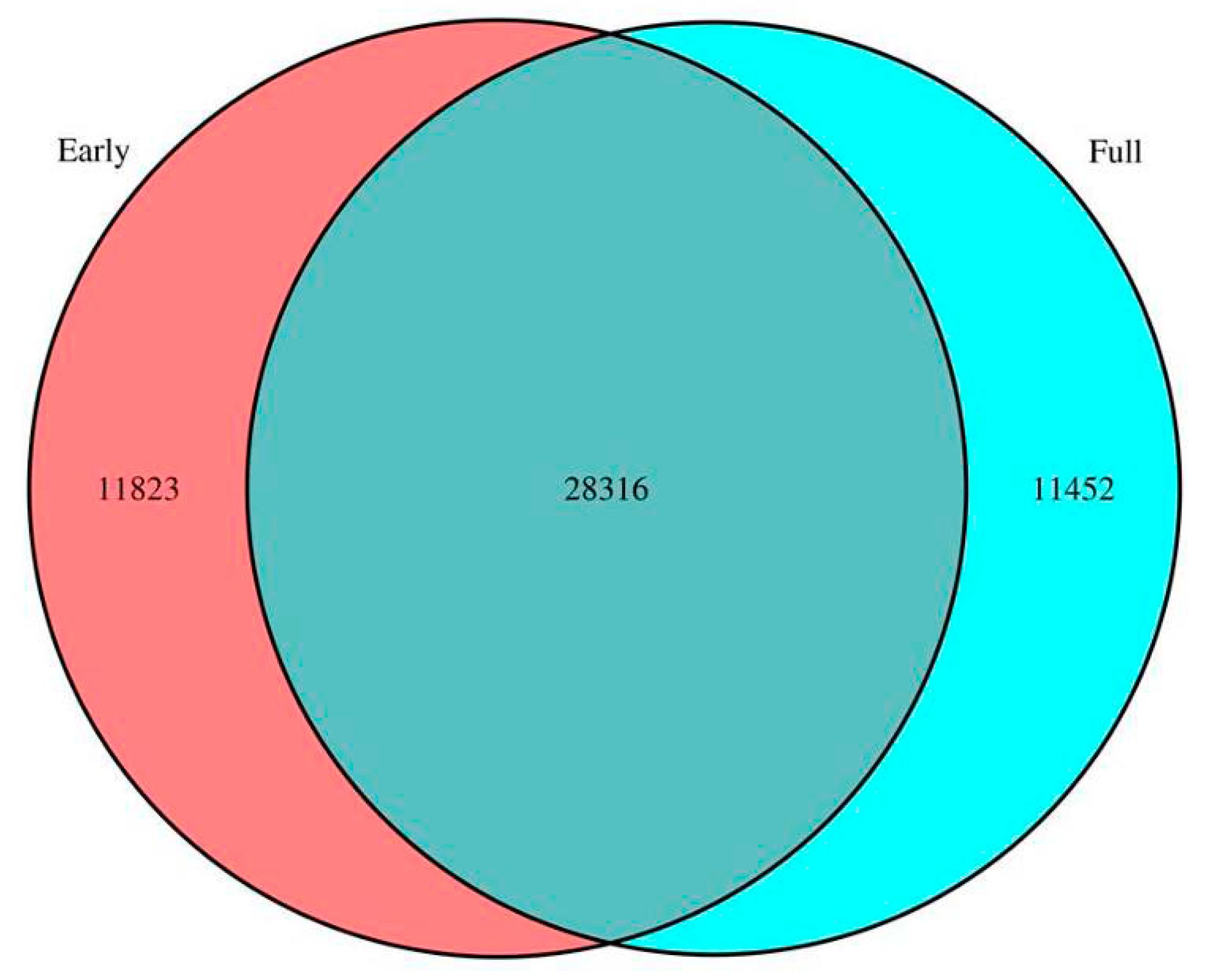
2.2. Comparison of Unigenes between Early and Full Flowering Stages
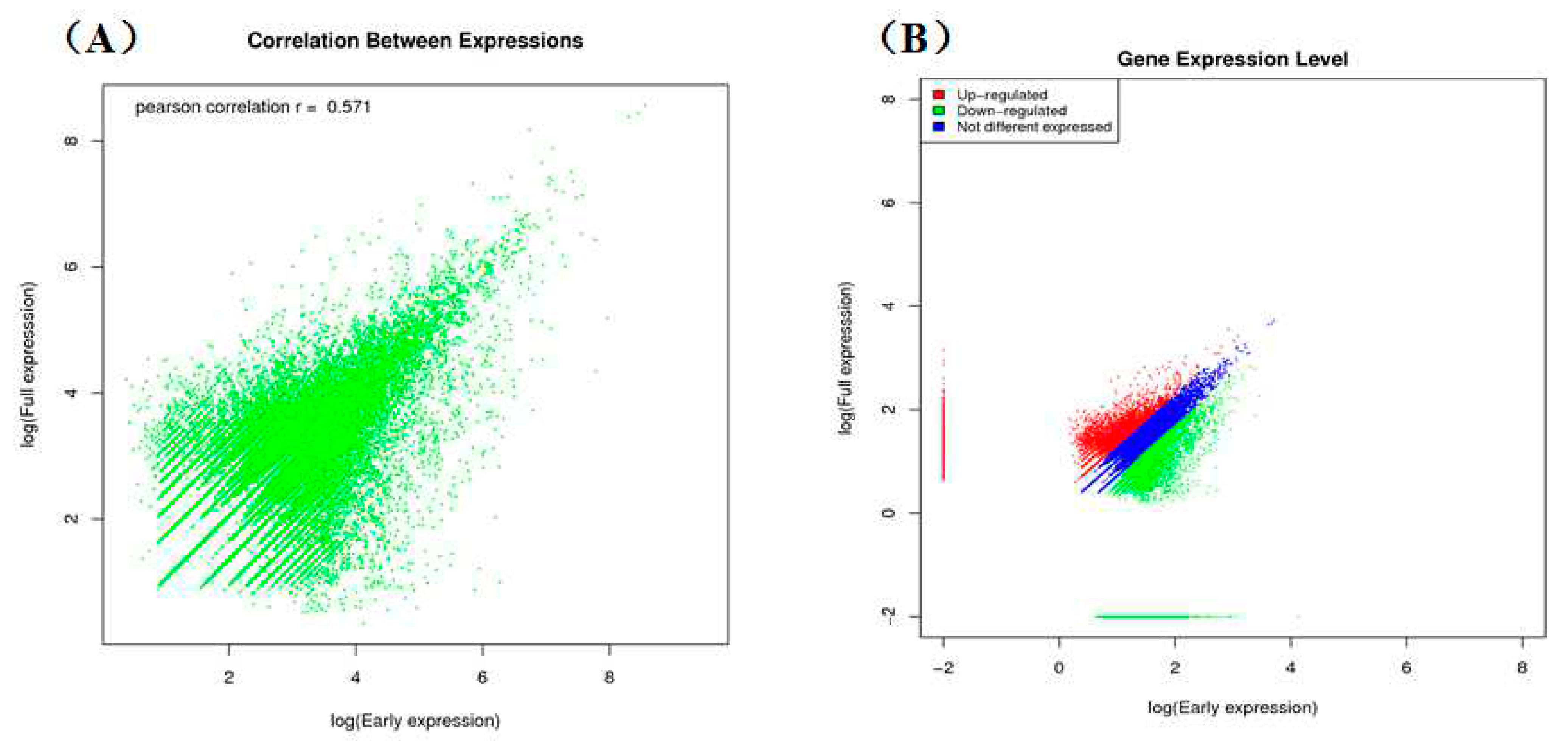
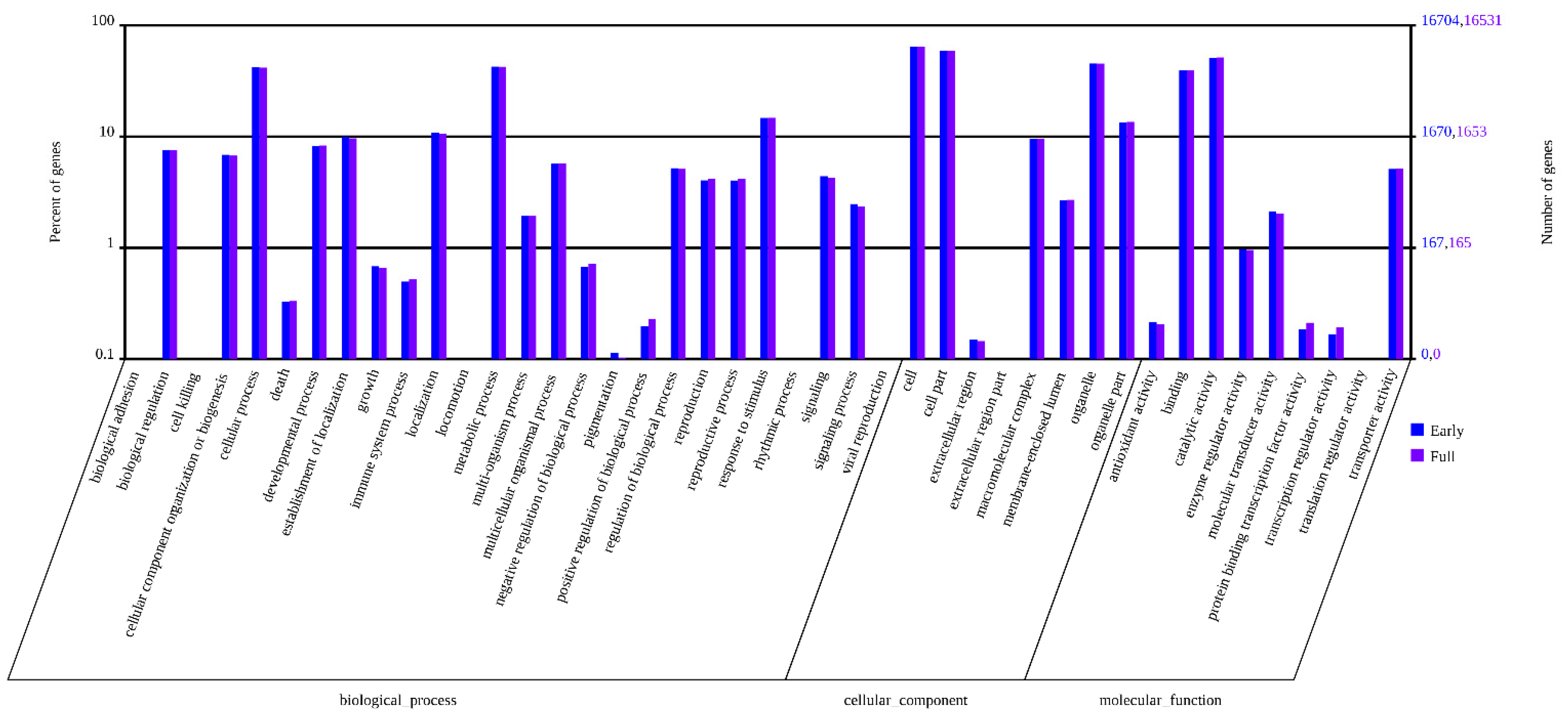
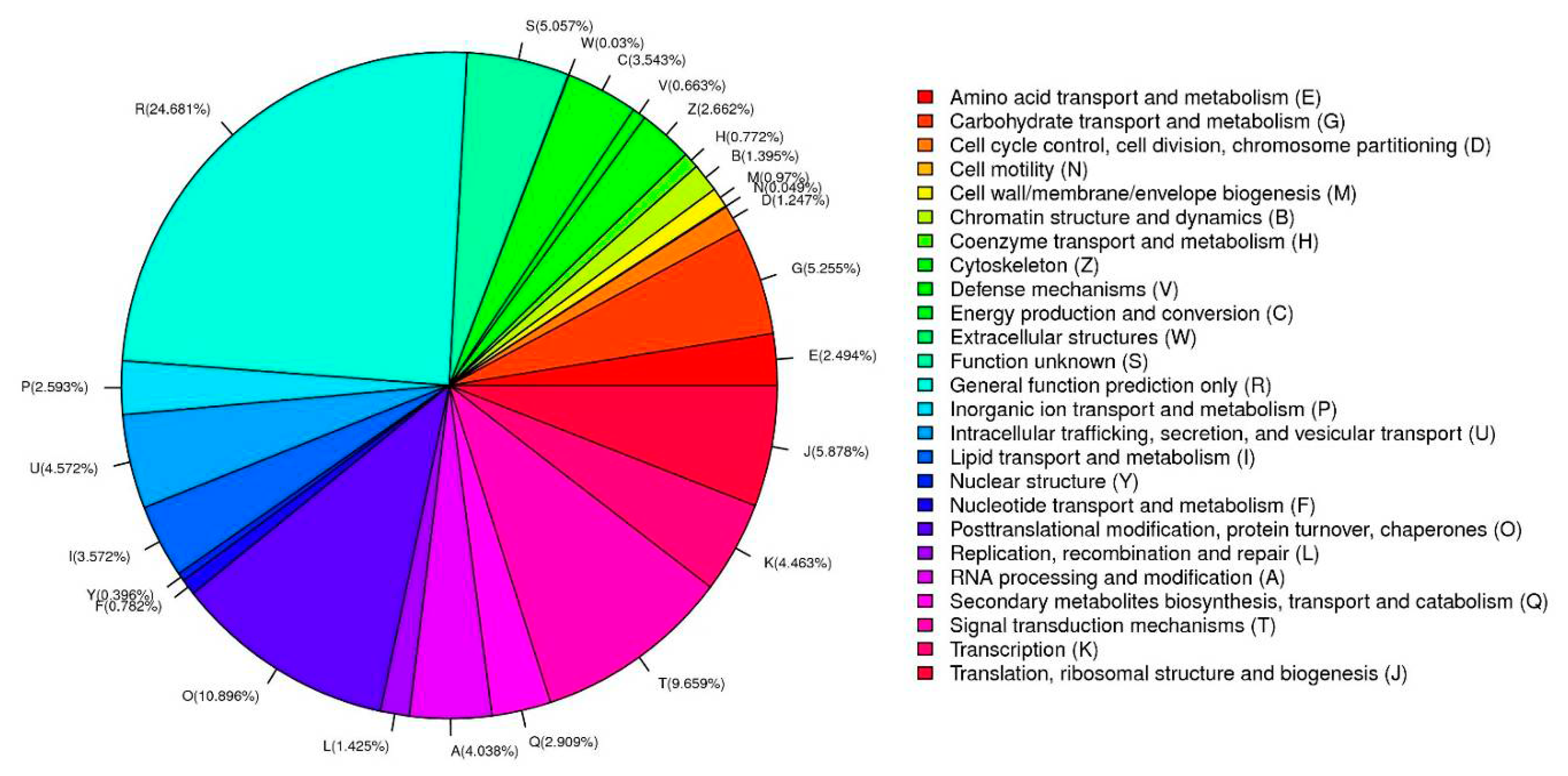
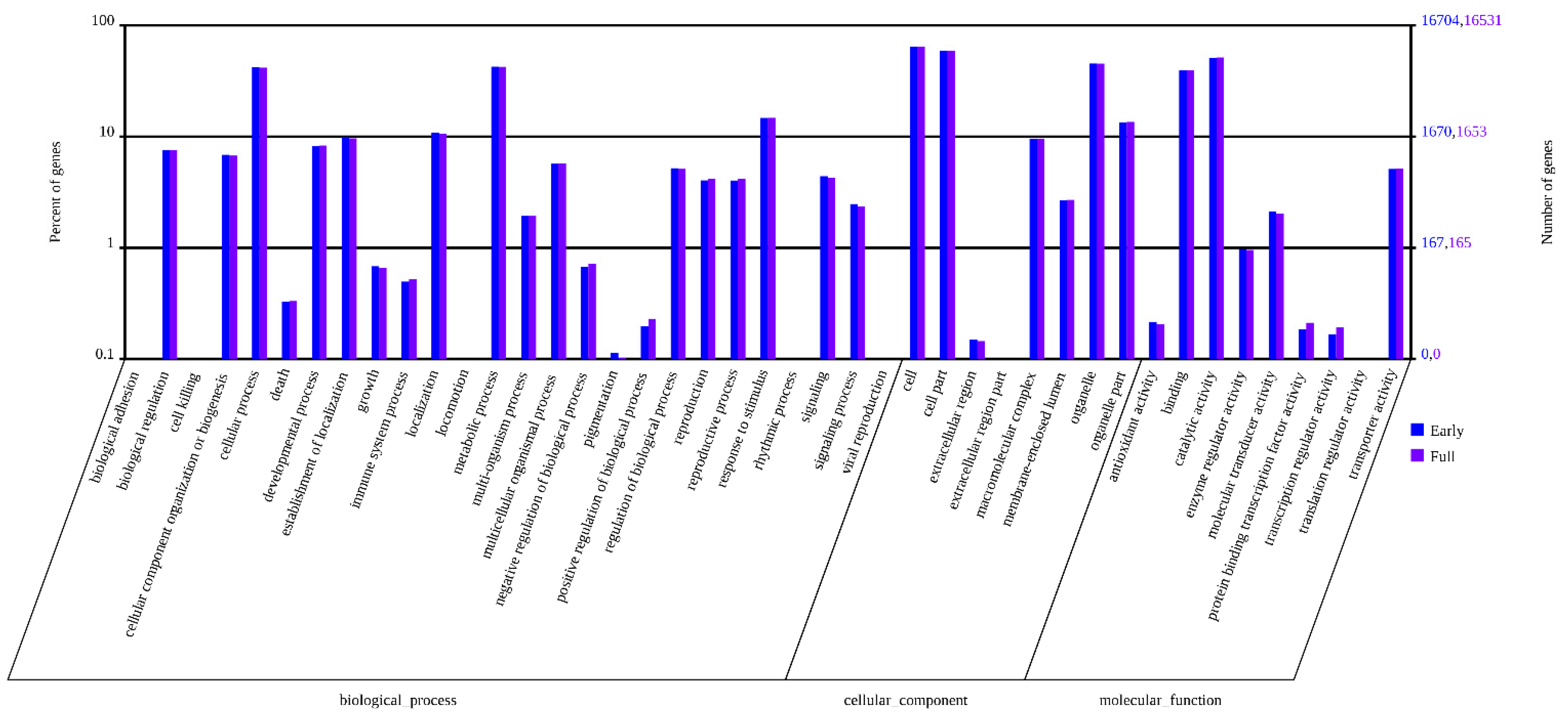
2.3. Classification of Gene Ontology (GO) and Functional Annotation

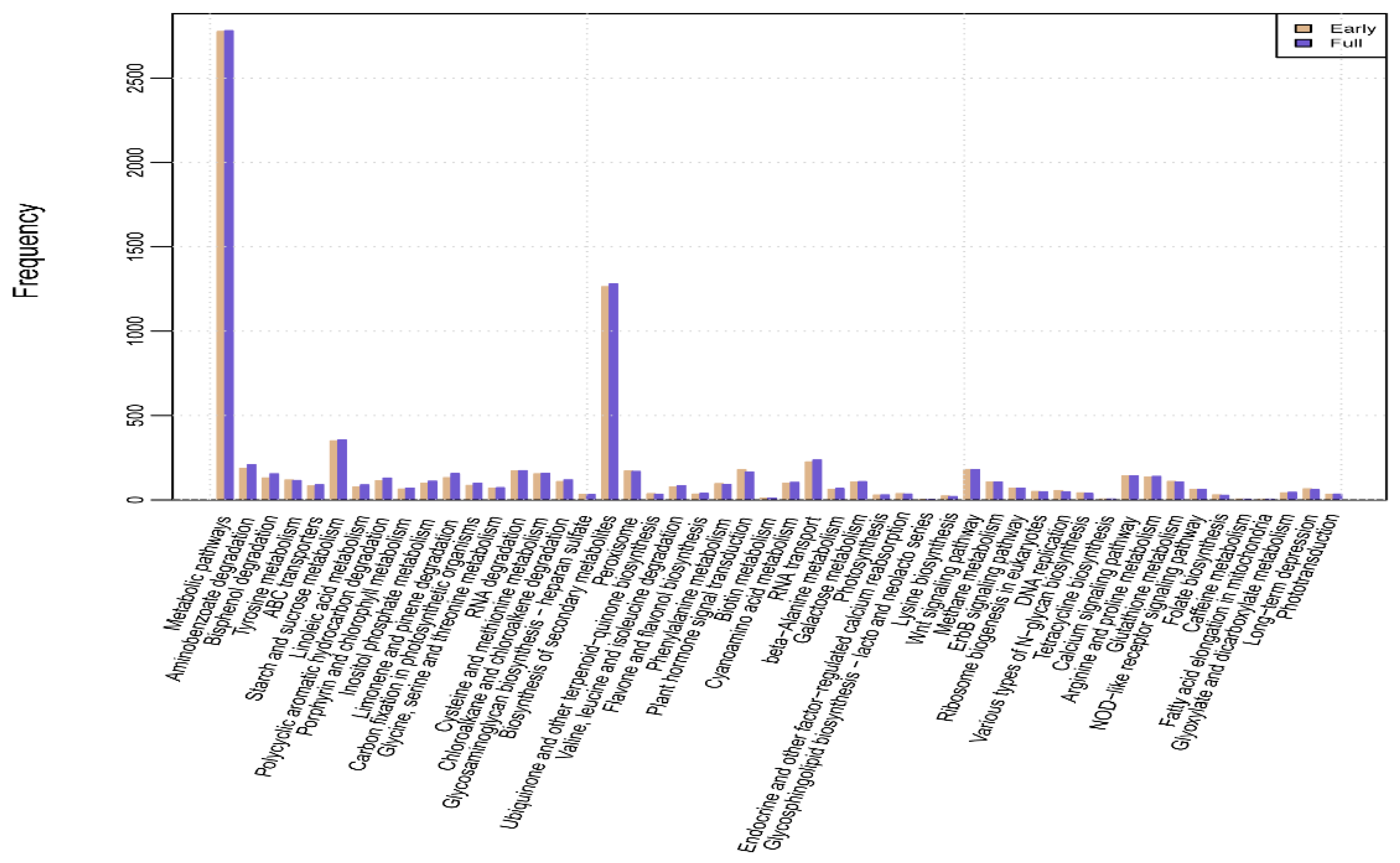
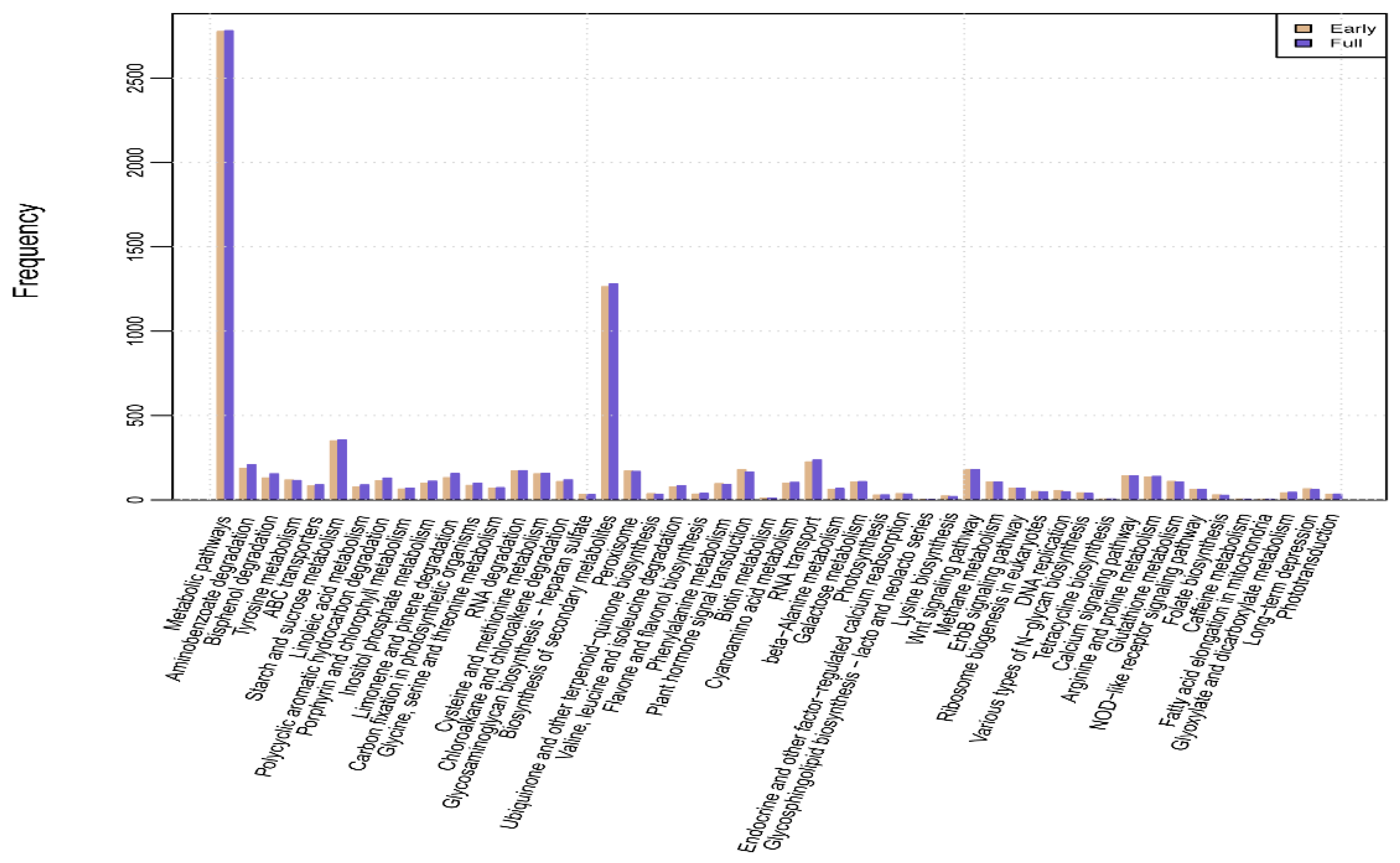
2.4. Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Mapping and Analysis of DEGs
| KEGG Pathway | Early | Full | p Value | Q Value | Pathway ID |
|---|---|---|---|---|---|
| up-regulated | |||||
| Bisphenol degradation | 84 | 165 | 3.04 × 10−8 | 8.50 × 10−6 | ko00363 |
| Polycyclic aromatic hydrocarbon degradation | 73 | 141 | 9.94 × 10−8 | 1.39 × 10−5 | ko00624 |
| Limonene and pinene degradation | 83 | 171 | 5.17 × 10−7 | 4.83 × 10−5 | ko00903 |
| Aminobenzoate degradation | 102 | 224 | 1.32 × 10−6 | 9.23 × 10−5 | ko00627 |
| Stilbenoid, diarylheptanoid and gingerol biosynthesis | 69 | 142 | 4.35 × 10−6 | 2.44 × 10−4 | ko00945 |
| Valine, leucine and isoleucine degradation | 44 | 97 | 0.00139643 | 0.0446 | ko00280 |
| Dioxin degradation | 7 | 8 | 0.00143353 | 0.0446 | ko00621 |
| Phenylpropanoid biosynthesis | 102 | 262 | 0.00205028 | 0.0574 | ko00940 |
| alpha-Linolenic acid metabolism | 48 | 110 | 0.00236695 | 0.0602 | ko00592 |
| Cyanoamino acid metabolism | 51 | 120 | 0.00345069 | 0.0805 | ko00460 |
| Fatty acid metabolism | 49 | 118 | 0.00698693 | 0.15 | ko00071 |
| Benzoxazinoid biosynthesis | 19 | 38 | 0.00912142 | 0.182 | ko00402 |
| Histidine metabolism | 34 | 80 | 0.01505098 | 0.262 | ko00340 |
| Glycosylphosphatidylinositol(GPI)-anchor biosynthesis | 39 | 95 | 0.01832198 | 0.262 | ko00563 |
| Flavone and flavonol biosynthesis | 20 | 43 | 0.01950833 | 0.262 | ko00944 |
| down-regulated | |||||
| Plant-pathogen interaction | 232 | 650 | 4.52 × 10−8 | 1.26 × 10−5 | ko04626 |
| Phagosome | 642 | 2069 | 1.41 × 10−7 | 1.30 × 10−5 | ko04145 |
| Focal adhesion | 607 | 1971 | 1.02 × 10−6 | 3.66 × 10−5 | ko04510 |
| Adherens junction | 587 | 1923 | 5.39 × 10−6 | 9.99 × 10−5 | ko04520 |
| Tight junction | 609 | 2015 | 1.33 × 10−5 | 2.32 × 10−4 | ko04530 |
| Monoterpenoid biosynthesis | 13 | 20 | 0.00031602 | 4.62 × 10−3 | ko00902 |
| Apoptosis | 47 | 117 | 0.00071701 | 9.97 × 10−3 | ko04210 |
| Biosynthesis of unsaturated fatty acids | 74 | 210 | 0.0025018 | 0.0316 | ko01040 |
| Gap junction | 45 | 120 | 0.00452067 | 0.0524 | ko04540 |
| SNARE interactions in vesicular transport | 36 | 96 | 0.01032285 | 0.115 | ko04130 |
| Calcium signaling pathway | 57 | 171 | 0.02407534 | 0.209 | ko04020 |
| Toll-like receptor signaling pathway | 40 | 118 | 0.04043678 | 0.331 | ko04620 |
| Glycerophospholipid metabolism | 51 | 157 | 0.04906638 | 0.39 | ko00564 |
| Sphingolipid metabolism | 29 | 83 | 0.05075266 | 0.392 | ko00600 |
| Cell cycle | 64 | 203 | 0.05448446 | 0.1 | ko04110 |
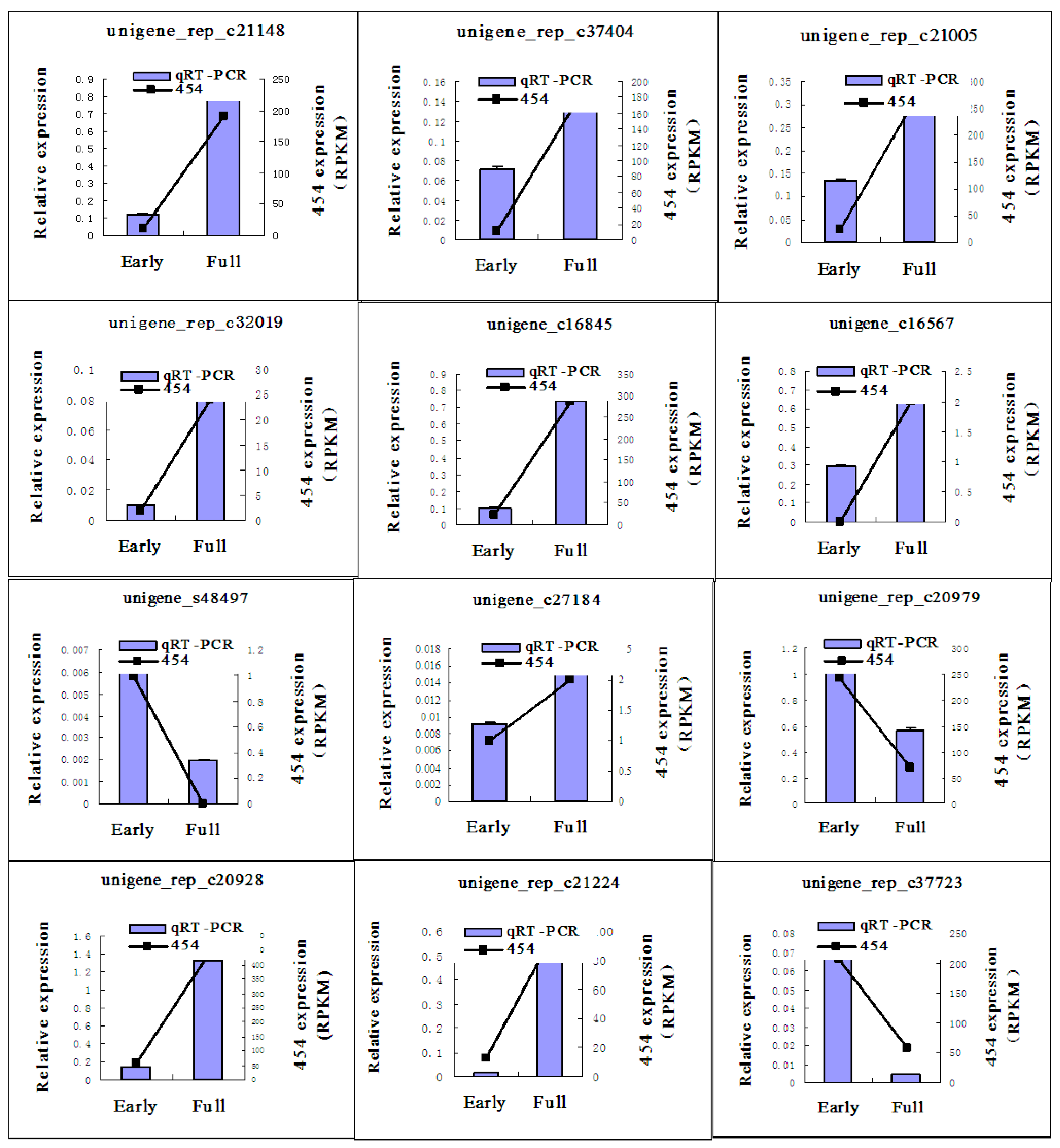
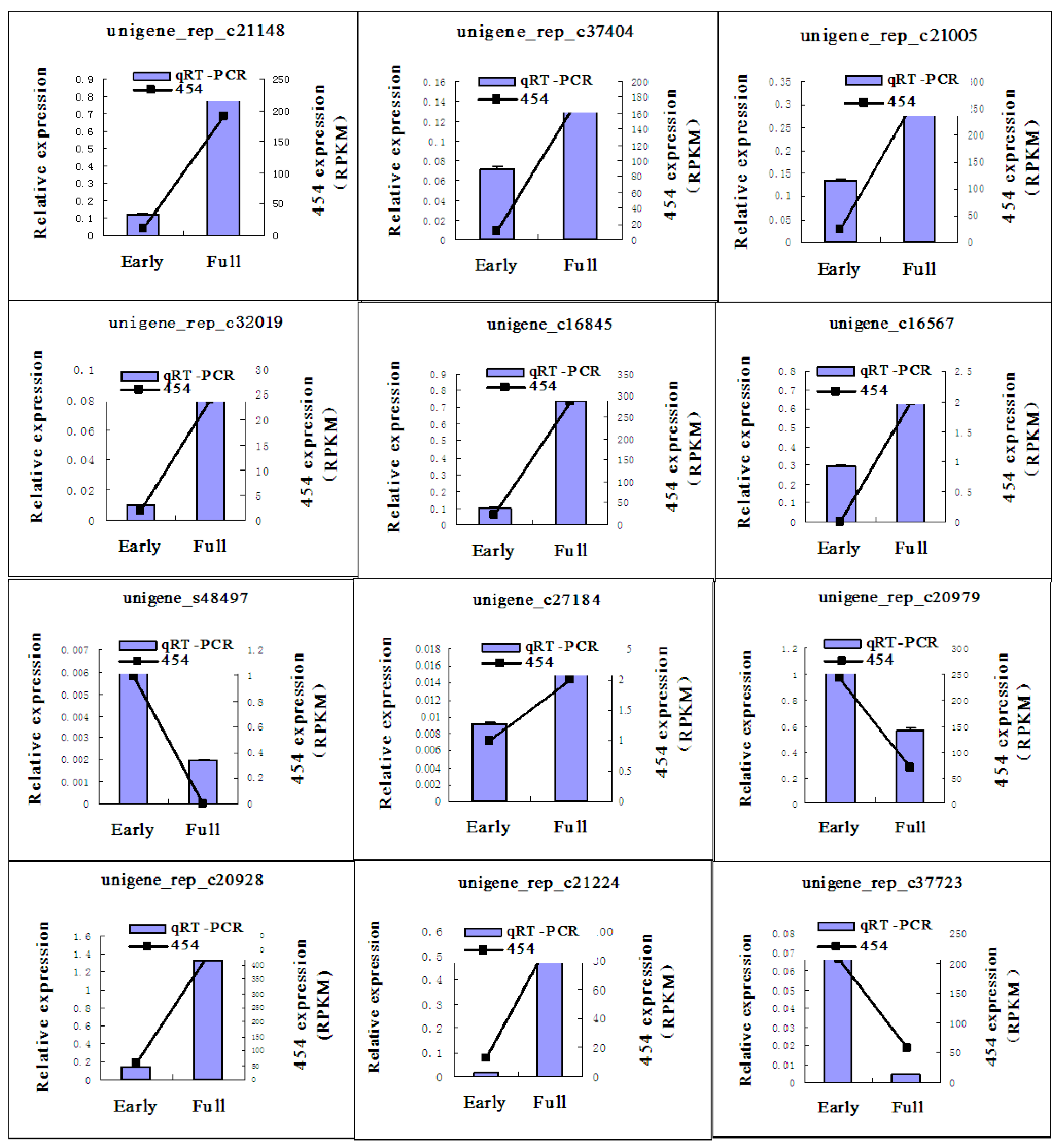
2.5. Quantitative Real Time-PCR (qRT-PCR) Validation of 454 Pyrosequencing
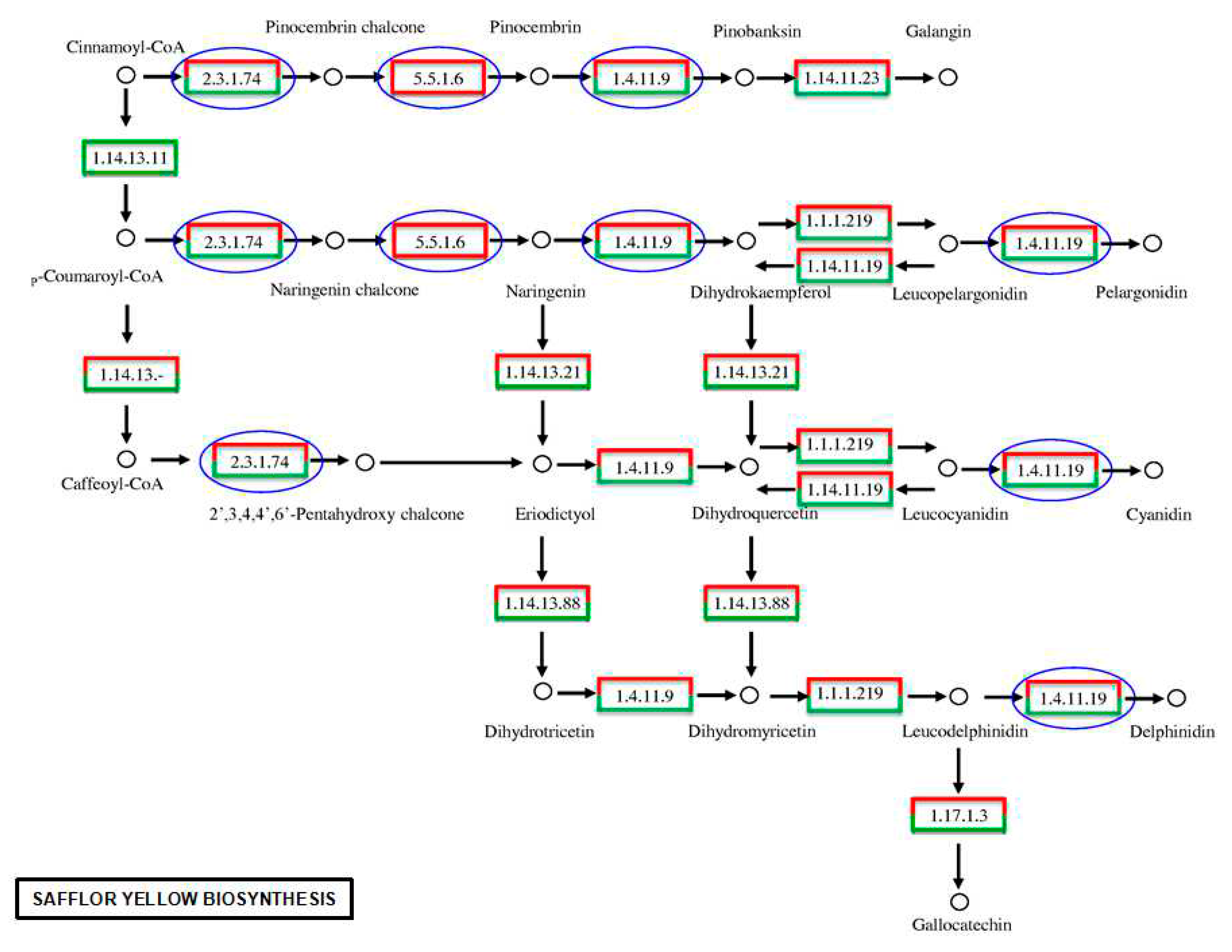
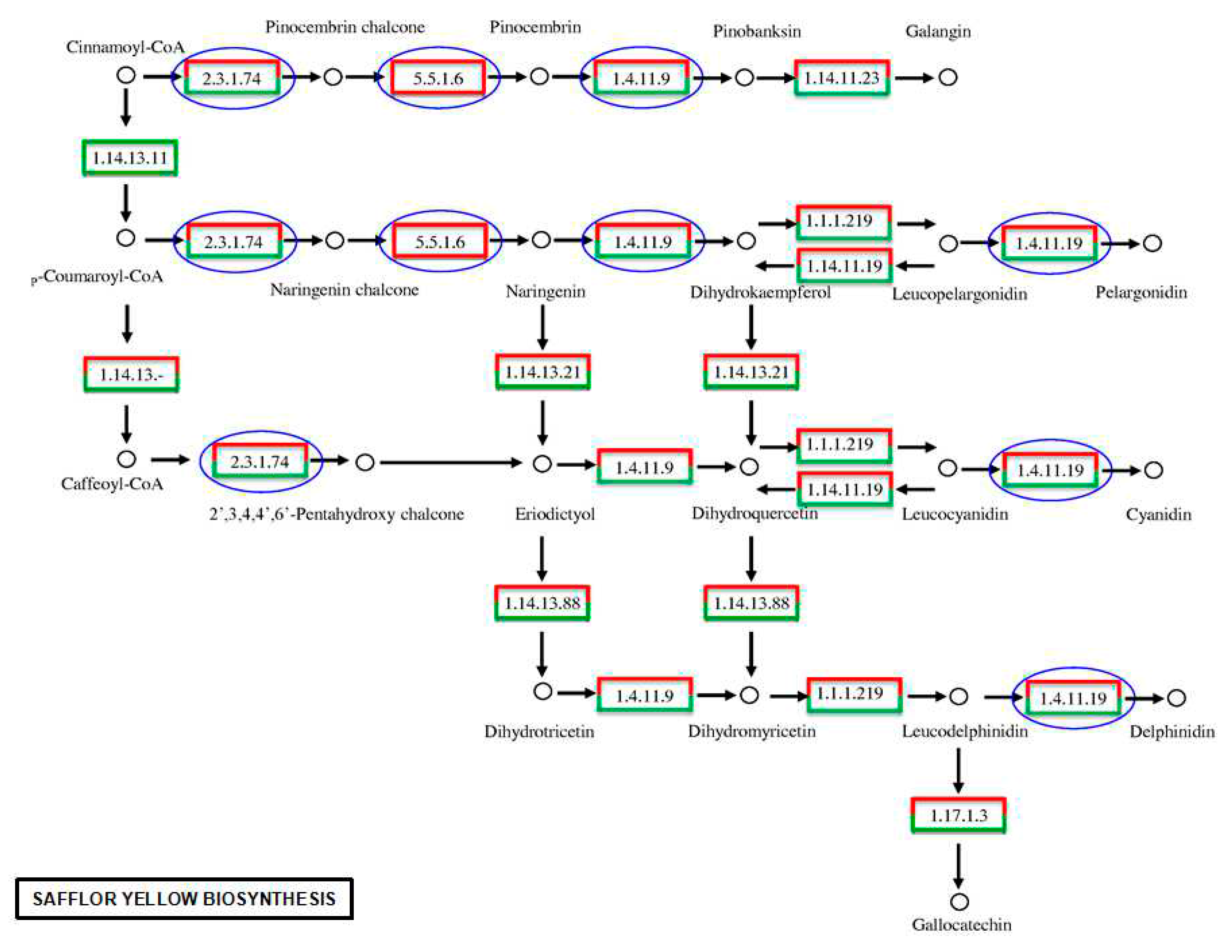
2.6. Analyses of the Flavonoid Biosynthesis Pathway and Putative Genes in the Transcriptome
| Gene | Code | unigene | DEG |
|---|---|---|---|
| chalcone synthase | EC 2.3.1.74 | unigene_c16567 (2) unigene_rep_c22629 (1.07111231014049) | Up-regulated |
| unigene_rep_c21289 (-3.00570328691034) unigene_c9855 (-2) unigene_rep_c33061 (-2) unigene_rep_c32553 (-2) unigene_rep_c30381 (-2) unigene_c13467 (-1.5809643864392) unigene_rep_c31854 (-1.29812149952523) | Down-regulated | ||
| chalcone isomerase | EC 5.5.1.6 | unigene_c27184 (1.07111231014049) | Up-regulated |
| anthocyanidin synthase | EC 1.14.11.19 | unigene_rep_c32431 (3.39304040502786) unigene_c5812 (2.8784672321981) unigene_rep_c20891 (2.13404356586695) unigene_c20381 (2) unigene_s42103 (2) unigene_c2096 (1.91910921669544) unigene_rep_c32424 (1.65607481086165) unigene_c9873 (1.65607481086165) | Up-regulated |
| unigene_rep_c35725 (-2) unigene_s48497 (-2) unigene_rep_c33425 (-2) unigene_c20561 (-1.06639121360944) | Down-regulated |
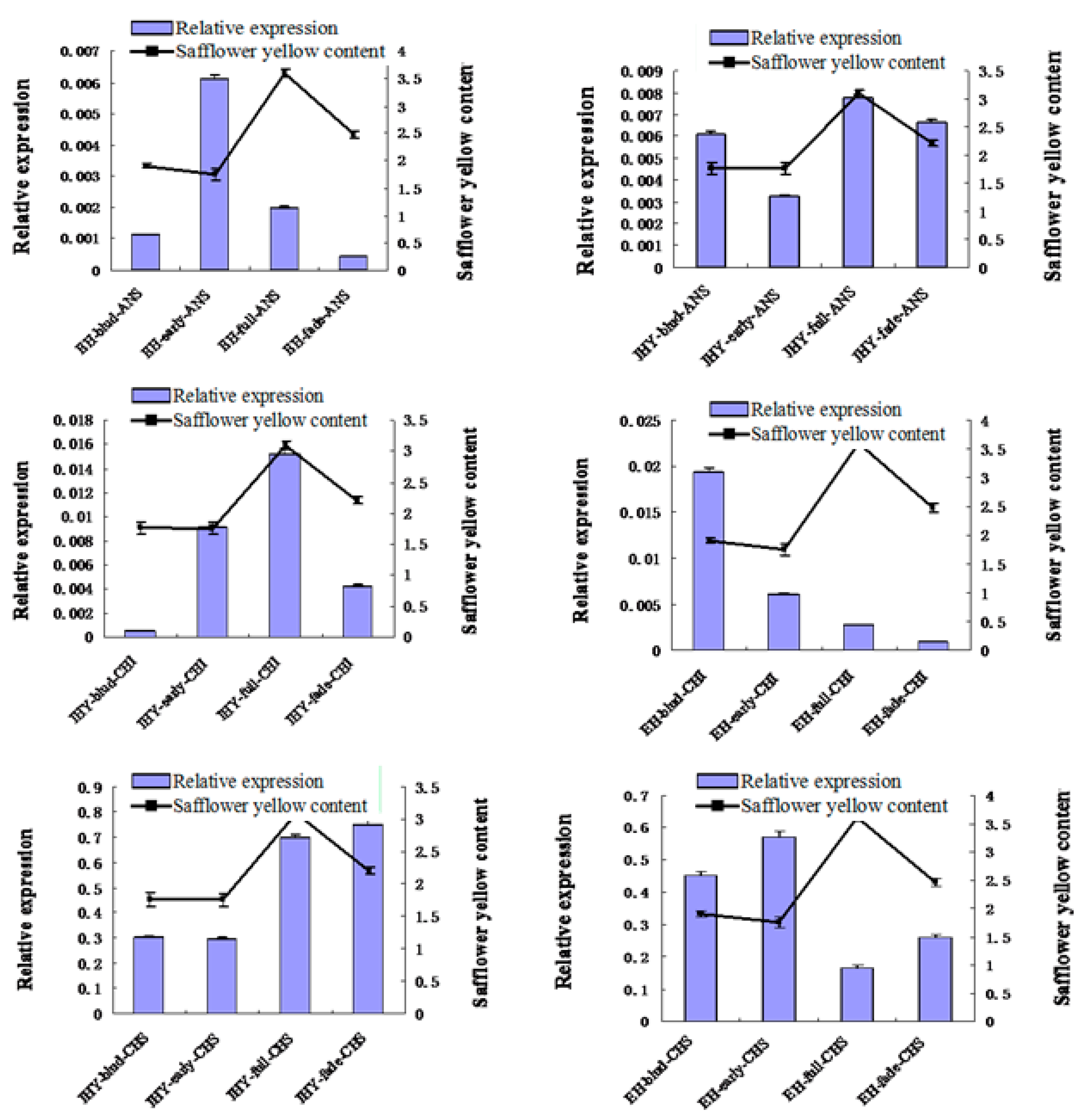
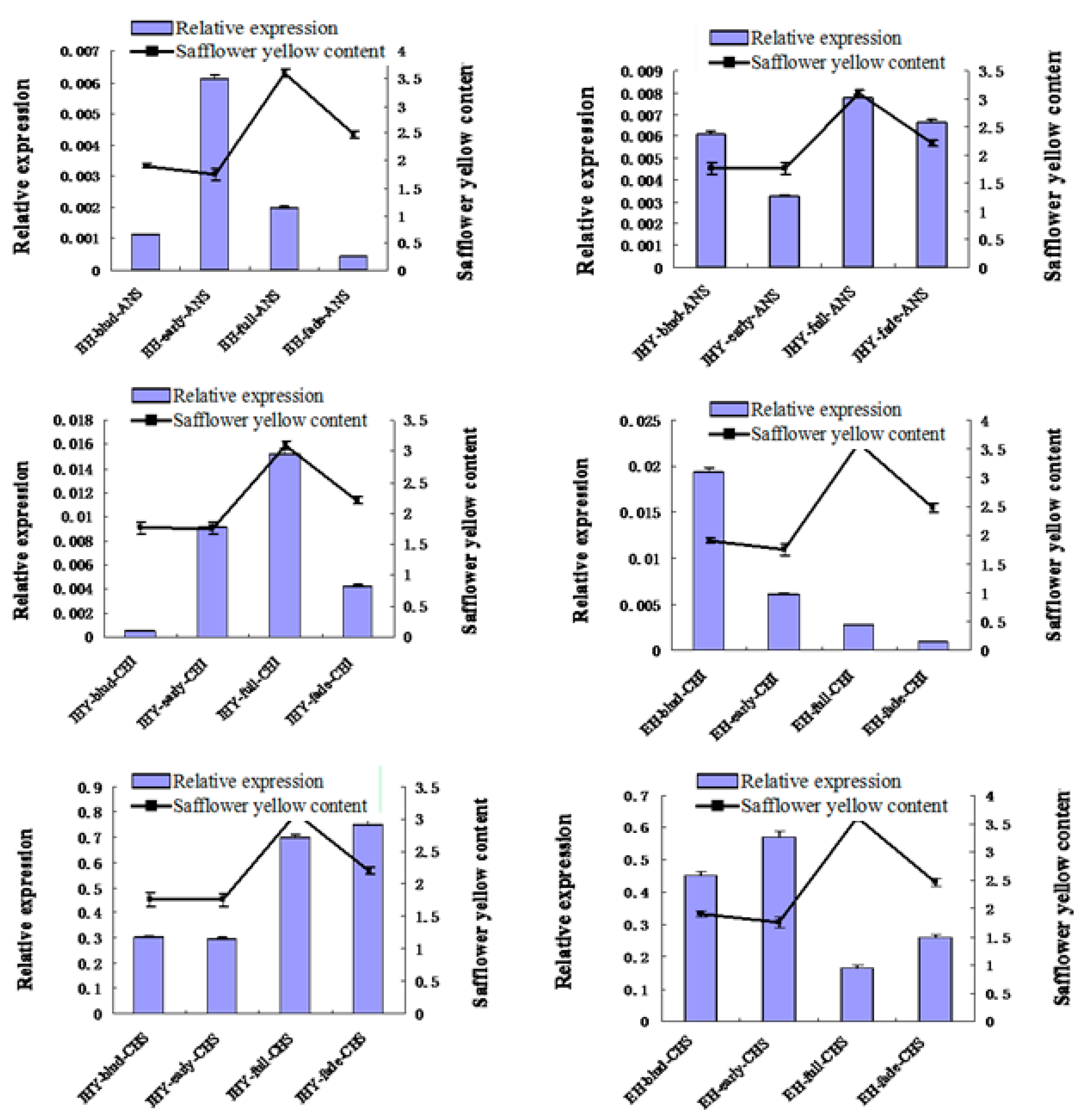
2.7. Validation and Expression Analysis of Putative Flavonoid Synthesis Genes from the Safflower Transcriptome
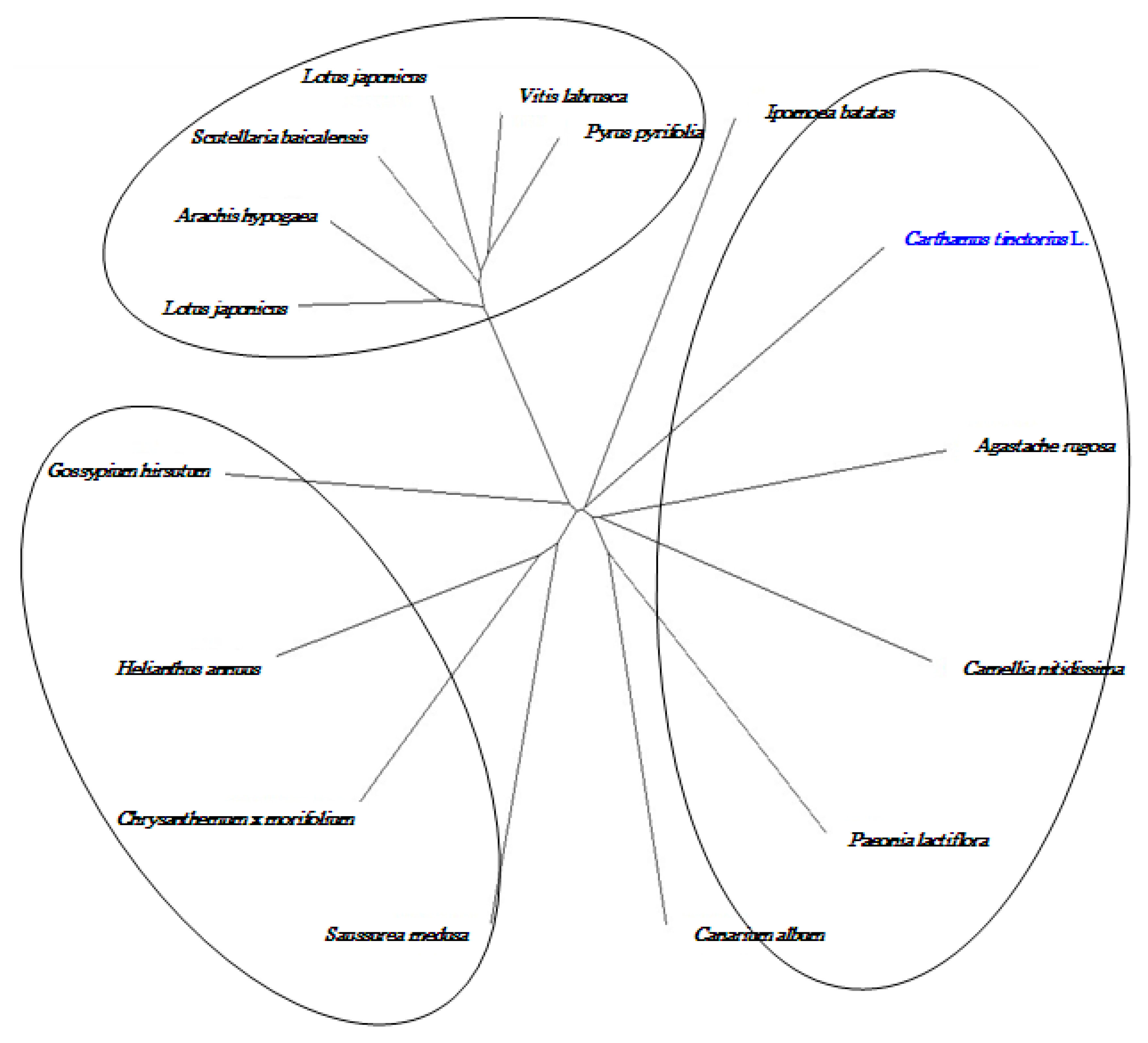
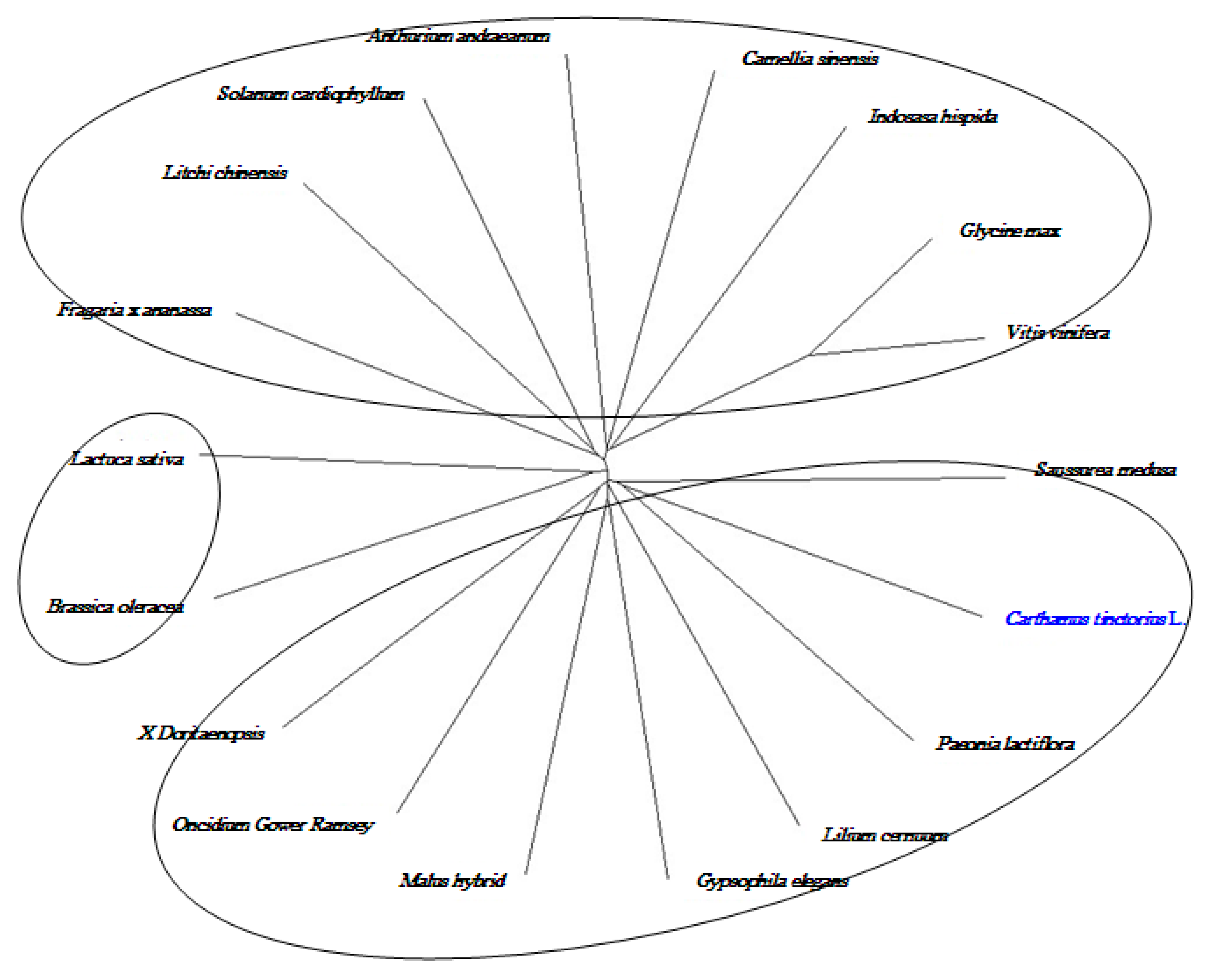
2.8. Cloning and Sequence Analysis of the Full-Length cDNA of Key Gene in Flavonoid Synthesis
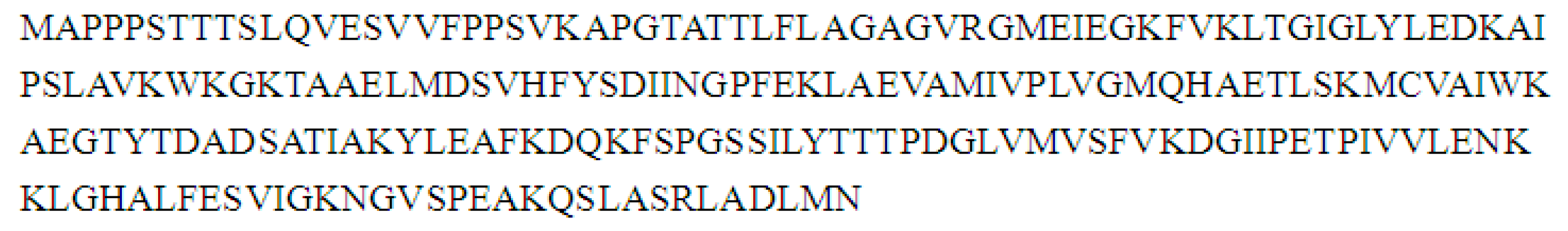
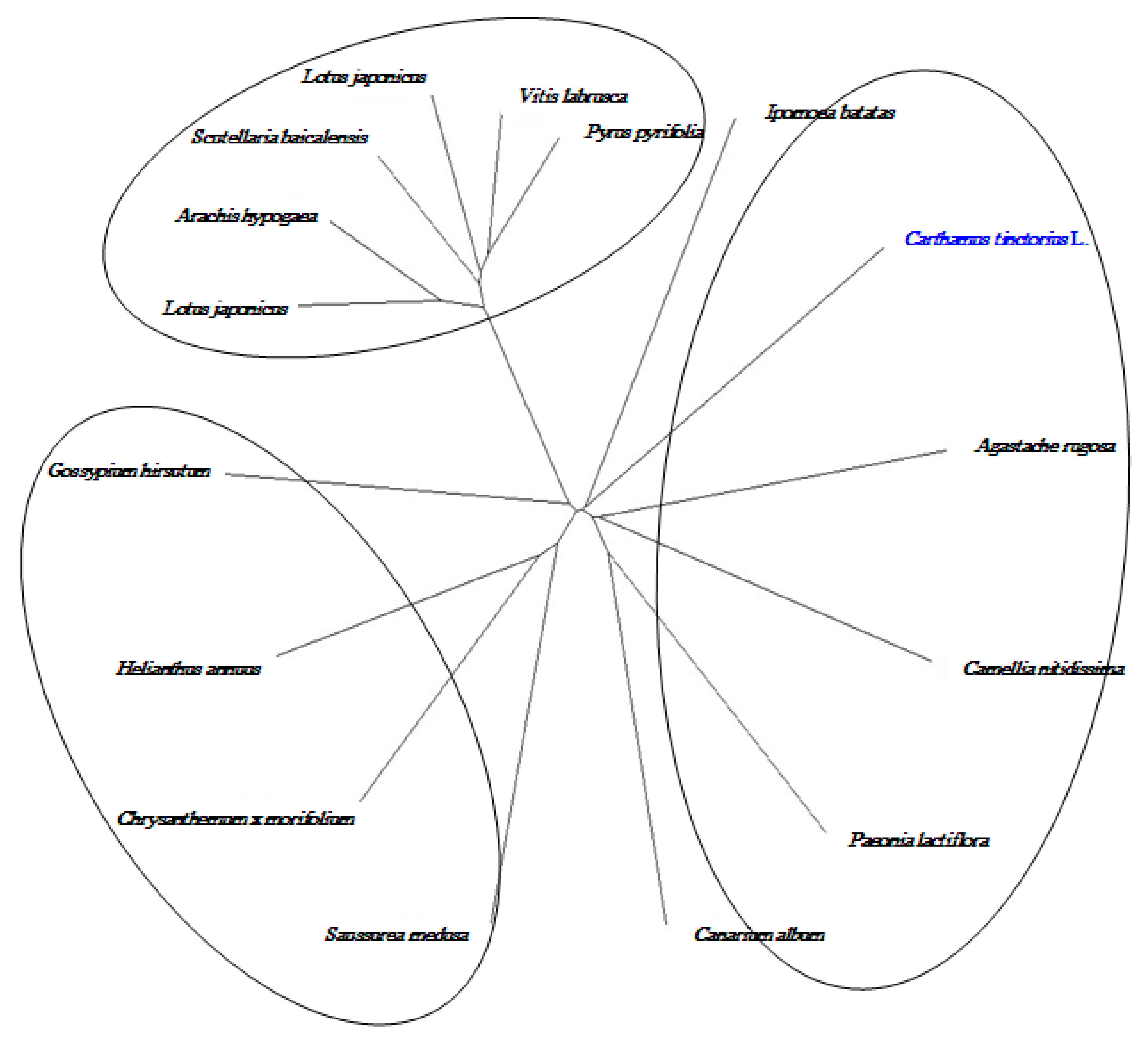

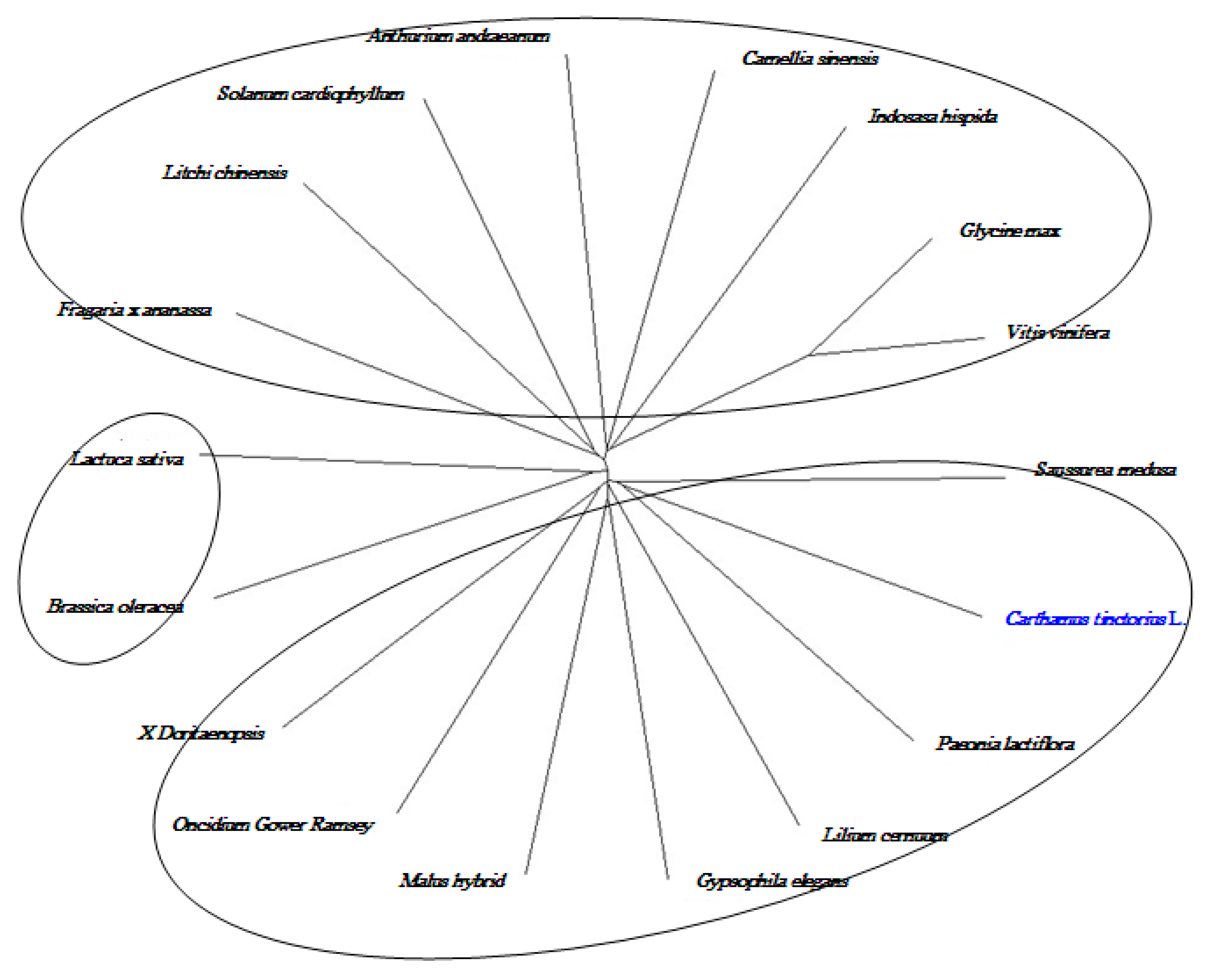
3. Discussion
4. Experimental Section
4.1. Sample Preparation and RNA Extraction
4.2. De Novo Assembly and Sequence Analysis
4.3. Comparative Analysis between Two Samples
4.4. Functional Annotation and Analysis of Pathway Enrichment
4.5. qRT-PCR Validation of 454 Pyrosequencing
4.6. Analysis of Putative Flavonoid Synthesis Genes in the Safflower Transcriptome
4.7. qRT-PCR Analysis and the Isolation of Important Genes in Flavonoid Synthesis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bozan, B.; Temelli, F. Chemical composition and oxidative stability of flax, safflower and poppy seed and seed oils. Bioresour. Technol. 2008, 99, 6354–6359. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.S.; Han, J.Y.; Kim, J.H.; Hwang, J.K. Inhibitory effects of active compounds isolated from safflower(Carthamus tinctorius L.) seeds for melanogenesis. Biol. Pharm. Bull. 2004, 27, 1976–1978. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, P.; Tang, C.; Wang, Y.; Li, Y.; Zhang, H. Antinociceptive and anti-inflammatory activities of extract and two isolated flavonoids of Carthamus. tinctorius L. J. Ethnopharmacol. 2014, 151, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Tang, L.; Xu, Y.; Zhou, G.; Wang, Z. Towards a better understanding of medicinal uses of Carthamus tinctorius L. in traditional Chinese medicine: A phytochemical and pharmacological review. J. Ethnopharmacol. 2014, 151, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Kazuma, K.; Takahashi, T.; Sato, K.; Takeuchi, H.; Matsumoto, T.; Okuno, T. Quinochalcones and flavonoids from fresh florets in different cultivars of Carthamus. tinctorius L. Biosci. Biotechnol. Biochem. 2000, 64, 1588–1599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Nagatsu, A.; Watanabe, T.; Sakakibara, J.; Okuyama, H. Antioxidative compounds isolated from safflower (Carthamus. tinctorius L.) oil cake. Chem. Pharm. Bull. 1997, 45, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Li, J.R.; Wu, W. Study on the antioxidative effect of Safflor Yellow. Zhongguo Zhong Yao Za Zhi 2004, 29, 447–449. [Google Scholar] [PubMed]
- Chen, L.; Xiang, Y.; Kong, L.; Zhang, X.; Sun, B.; Wei, X.; Liu, H. Hydroxysafflor yellow A protects against cerebral ischemia-reperfusion injury by anti-apoptotic effect through PI3K/Akt/GSK3β pathway in rat. Neurochem. Res. 2013, 38, 2268–2275. [Google Scholar] [CrossRef] [PubMed]
- Zang, B.X.; Jin, M.; Si, N.; Zhang, Y.; Wu, W.; Piao, Y.Z. Antagonistic effect of hydroxysaffor yellow A on the platelet activating factor receptor. Acta Pharm. Sin. 2002, 37, 696–699. [Google Scholar]
- Zhang, N.; Xing, M.; Wang, Y.; Liang, H.; Yang, Z.; Shi, F.; Cheng, Y. Hydroxysafflor yellow A improves learning and memory in a rat model of vascular dementia by increasing VEGF and NR1 in the hippocampus. Neurosci. Bull. 2014, 30, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.J.; Ding, H.H.; Deng, J.Y.; Pan, H.; Wang, L.J.; Li, N.S. The inhibition of preadipocyte differentiation and adipogenesis by Zinc-α2-glycoprotein treatment in 3T3-L1cells. J. Diabetes Investig. 2013, 4, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Zhou, Z.M.; Chen, P. Evidence that hydroxysafflor yellow A protects the heart against ischaemia–reperfusion injury by inhibiting mitochondrial perme ability transition pore opening. Clin. Exp. Pharmacol. Physiol. 2008, 35, 211–216. [Google Scholar] [PubMed]
- Guo, Y.; Wang, Y.; Huang, X.; Lv, H.; Fan, R.; Huang, W.; Gan, P.; Liu, W.; Yan, K.; Xia, Z.; et al. Determination of hydroxysafflor yellow A in biological fluids of patients with traumatic brain injury by UPLC-ESI-MS/MS after injection of Xuebijing. Biomed. Chromatogr. 2014, 28, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.J.; Wang, L.J.; Wang, X.Q.; Pan, H.; Li, N.S.; Yang, H.B.; Jin, M.; Zang, B.X.; Gong, F.Y. Hormone-sensitive lipase is involved in the action of hydroxysafflor yellow A (HYSA) inhibiting adipogenesis of 3T3-L1cells. Fitoterapia 2014, 93, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Hale, M.; McCormick, C.; Jackson, J.; DeWoody, J.A. Next-generation pyrosequencing of gonad transcriptomes in the polyploidy lake sturgeon (Acipenser fulvescens): The relat ive merits of normalization and rarefaction in gene discovery. BMC Genom. 2009, 10, 203. [Google Scholar] [CrossRef] [PubMed]
- Bellin, D.; Ferrarini, A.; Chimento, A.; Kaiser, O.; Levenkova, N.; Bouffard, P.; Delledonne, M. Combining next-generation pyrosequencing with microarray for large scale expression analysis in non-model species. BMC Genom. 2009, 10, 555. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Ma, X.; Mo, C.; Wilson, I.W.; Song, C.; Zhao, H.; Yang, Y.; Fu, W.; Qiu, D. An efficient approach to finding Siraitia grosvenorii triterpene biosynthetic genes by RNA-seq and digital gene expression analysis. BMC Genom. 2011, 12, 343. [Google Scholar] [CrossRef] [PubMed]
- Novaes, E.; Drost, D.R.; Farmerie, W.G.; Pappas, G.J., Jr.; Grattapaglia, D.; Sederoff, R.R.; Kirst, M. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genom. 2008, 9, 312. [Google Scholar] [CrossRef] [PubMed]
- Monnier, A.; Liverani, S.; Bouvet, R.; Jesson, B.; Smith, J.Q.; Mosser, J.; Corellou, F.; Bouget, F.Y. Orchestrated transcription of biological processes in the marine picoeukaryote Ostreococcus exposed tolight/dark cycles. BMC Genom. 2010, 11, 192. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dong, Y.; Yang, J.; Liu, X.; Wang, Y.; Yao, N.; Guan, L.; Wang, N.; Wu, J.; Li, X. De novo transcriptome of safflower and the identification of putative genes for oleosin and the biosynthesis of flavonoids. PLoS ONE 2012, 7, 30987. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, F.; Wang, N.; Dong, Y.; Liu, Q.; Zhao, L.; Chen, H.; Liu, W.; Yin, H.; Zhang, X.; et al. Transcriptome exploration in Leymus chinensis under saline-alkaline treatment using 454 pyrosequencing. PLoS ONE 2013, 8, 53632. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, J.M.; Leamon, J.H. The development and impact of 454 sequencing. Nat. Biotechnol. 2008, 26, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, T.; Hayashizaki, Y.; Daub, C.O. TagDust—A program to eliminate artifacts from next generation sequencing data. Bioinformatics 2009, 25, 2839–2840. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.A.; Lin, C.C.; Wang, C.D.; Wu, H.B.; Hwang, P.I. An optimized procedure greatly improves EST vector contamination removal. BMC Genom. 2007, 8, 416. [Google Scholar] [CrossRef] [PubMed]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.J.; Müller, W.E.; Wetter, T.; Suhai, S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Pearl, S.A.; Bowers, J.E.; Reyes-Chin-Wo, S.; Michelmore, R.W.; Burke, J.M. Genetic analysis of safflower domestication. BMC Plant Biol. 2014, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Franks, R.G.; Liu, X.; Kang, M.; Keebler, J.E.; Schaff, J.E.; Huang, H.W.; Xiang, Q.Y. De novo sequencing, characterization, and comparison of inflorescence transcriptomes of Cornus canadensisand C. florida (Cornaceae). PLoS ONE 2013, 8, 82674. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alagna, F.; D'Agostino, N.; Torchia, L.; Servili, M.; Rao, R.; Pietrella, M.; Giuliano, G.; Chiusano, M.L.; Baldoni, L.; Perrotta, G. Comparative 454 pyrosequencing of transcripts fromtwo olive genotypes during fruit development. BMC Genom. 2009, 10, 399. [Google Scholar] [CrossRef] [PubMed]
- Waud, M.; Busschaert, P.; Ruyters, S.; Jacquemyn, H.; Lievens, B. Impact of primer choice on characterization of orchid mycorrhizal communities using 454 pyrosequencing. Mol. Ecol. Resour. 2014, 14, 679–699. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, D.; Sinha, R.; Hazra, S.; Datta, R.; Chattopadhyay, S. De novo transcriptome analysis using 454 pyrosequencing of the Himalayan Mayapple, Podophyllum hexandrum. BMC Genom. 2013, 14, 748. [Google Scholar] [CrossRef] [PubMed]
- Sheveleva, A.; Kudryavtseva, A.; Speranskaya, A.; Belenikin, M.; Melnikova, N.; Chirkov, S. Complete genome sequence of a novel Plum pox virus strain W isolate determined by 454 pyrosequencing. Virus Genes 2013, 47, 385–388. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Xu, X.; Li, Y.; Li, C.; Zhu, Y.; Yan, H.; Sun, Z.; Sun, C.; Song, J.; Bi, Y.; et al. Transcriptome analysis of buds and leaves using454 pyrosequencing to discover genes associated with the biosynthesis of active ingredients in Lonicera. japonica Thunb. PLoS ONE 2013, 8, 62922. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Park, J.H.; Cho, G.T.; Lee, H.S.; Baek, H.J.; Lee, S.Y.; Chung, J.W. Development and molecular characterization of 55 novel polymorphic cDNA-SSR markers in faba bean (Vicia faba L.) using 454 pyrosequencing. Molecules 2013, 18, 1844–1856. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Bashandy, H.; Ainasoja, M.; Kontturi, J.; Pietiäinen, M.; Laitinen, R.A.; Albert, V.A.; Valkonen, J.P.; Elomaa, P.; Teeri, T.H. Functional diversification of duplicated chalcone synthase genes in anthocyanin biosynthesis of Gerbera hybrida. New Phytol. 2014, 201, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Muir, S.R.; Collins, G.J.; Robinson, S.; Hughes, S.; Bovy, A.; Ric De Vos, C.H.; van Tunen, A.J.; Verhoeyen, M.E. Overexpression of petunia chalcone isomerase in tomato results in fruit containing increased levels of flavonols. Nat. Biotechnol. 2001, 19, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chen, X.; Ye, Y.; He, W.; Shen, Y. Cloning and Expression Analysis of Chalcone Isomerase Gene from Narcissus tazetta var. chinensis. Chin. J. Trop. Crops 2011, 32, 2287–2292. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yang, X.; Sun, P.; Tong, W.; Hu, S. The First Illumina-Based De Novo Transcriptome Sequencing and Analysis of Safflower Flowers. PLoS ONE 2012, 7, 38653. [Google Scholar]
- Conesa, A.; Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Salomonis, N.; Hanspers, K.; Zambon, A.C.; Vranizan, K.; Lawlor, S.C.; Dahlquist, K.D.; Doniger, S.W.; Stuart, J.; Conklin, B.R.; Pico, A.R. GenMAPP 2: New features and resources for pathway analysis. BMC Bioinform. 2007, 8, 217. [Google Scholar] [CrossRef] [PubMed]
- Jarosova, J.; Kundu, J.K. Validation of reference genes as internal control for studying viral infections in cereals by quantitative real-time RT-PCR. BMC Plant Biol. 2010, 10, 146. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Dong, Y.; Yao, N.; Zhang, Y.; Wang, N.; Cui, X.; Li, X.; Wang, Y.; Wang, F.; Yang, J.; et al. De Novo Sequencing and Analysis of the Safflower Transcriptome to Discover Putative Genes Associated with Safflor Yellow in Carthamus tinctorius L. Int. J. Mol. Sci. 2015, 16, 25657-25677. https://doi.org/10.3390/ijms161025657
Liu X, Dong Y, Yao N, Zhang Y, Wang N, Cui X, Li X, Wang Y, Wang F, Yang J, et al. De Novo Sequencing and Analysis of the Safflower Transcriptome to Discover Putative Genes Associated with Safflor Yellow in Carthamus tinctorius L. International Journal of Molecular Sciences. 2015; 16(10):25657-25677. https://doi.org/10.3390/ijms161025657
Chicago/Turabian StyleLiu, Xiuming, Yuanyuan Dong, Na Yao, Yu Zhang, Nan Wang, Xiyan Cui, Xiaowei Li, Yanfang Wang, Fawei Wang, Jing Yang, and et al. 2015. "De Novo Sequencing and Analysis of the Safflower Transcriptome to Discover Putative Genes Associated with Safflor Yellow in Carthamus tinctorius L." International Journal of Molecular Sciences 16, no. 10: 25657-25677. https://doi.org/10.3390/ijms161025657
APA StyleLiu, X., Dong, Y., Yao, N., Zhang, Y., Wang, N., Cui, X., Li, X., Wang, Y., Wang, F., Yang, J., Guan, L., Du, L., Li, H., & Li, X. (2015). De Novo Sequencing and Analysis of the Safflower Transcriptome to Discover Putative Genes Associated with Safflor Yellow in Carthamus tinctorius L. International Journal of Molecular Sciences, 16(10), 25657-25677. https://doi.org/10.3390/ijms161025657