
Molecular Pathways Regulating Macrovascular Pathology and Vascular Smooth Muscle Cells Phenotype in Type 2 Diabetes


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Heterogeneity of Vascular Smooth Muscle Cells and T2DM
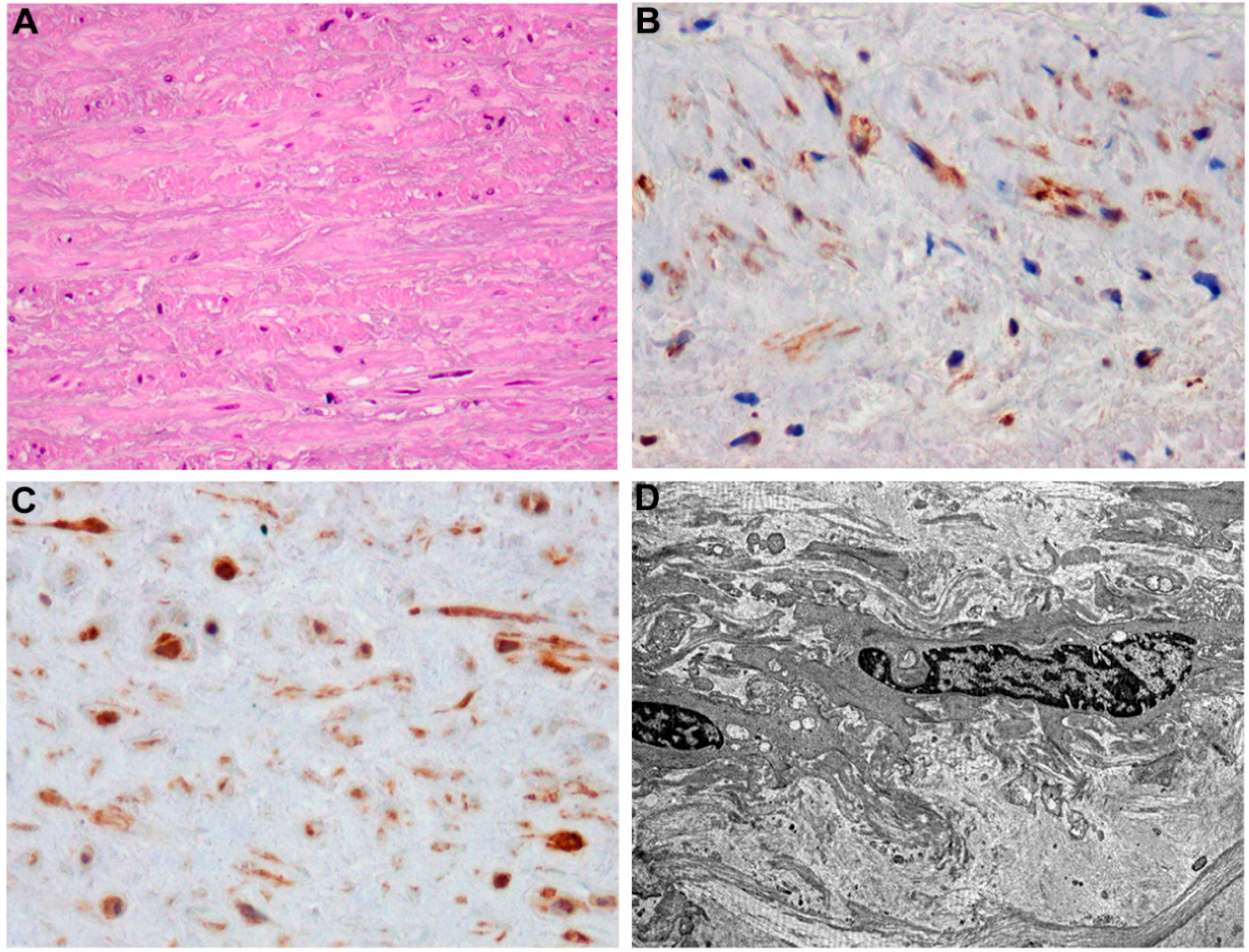
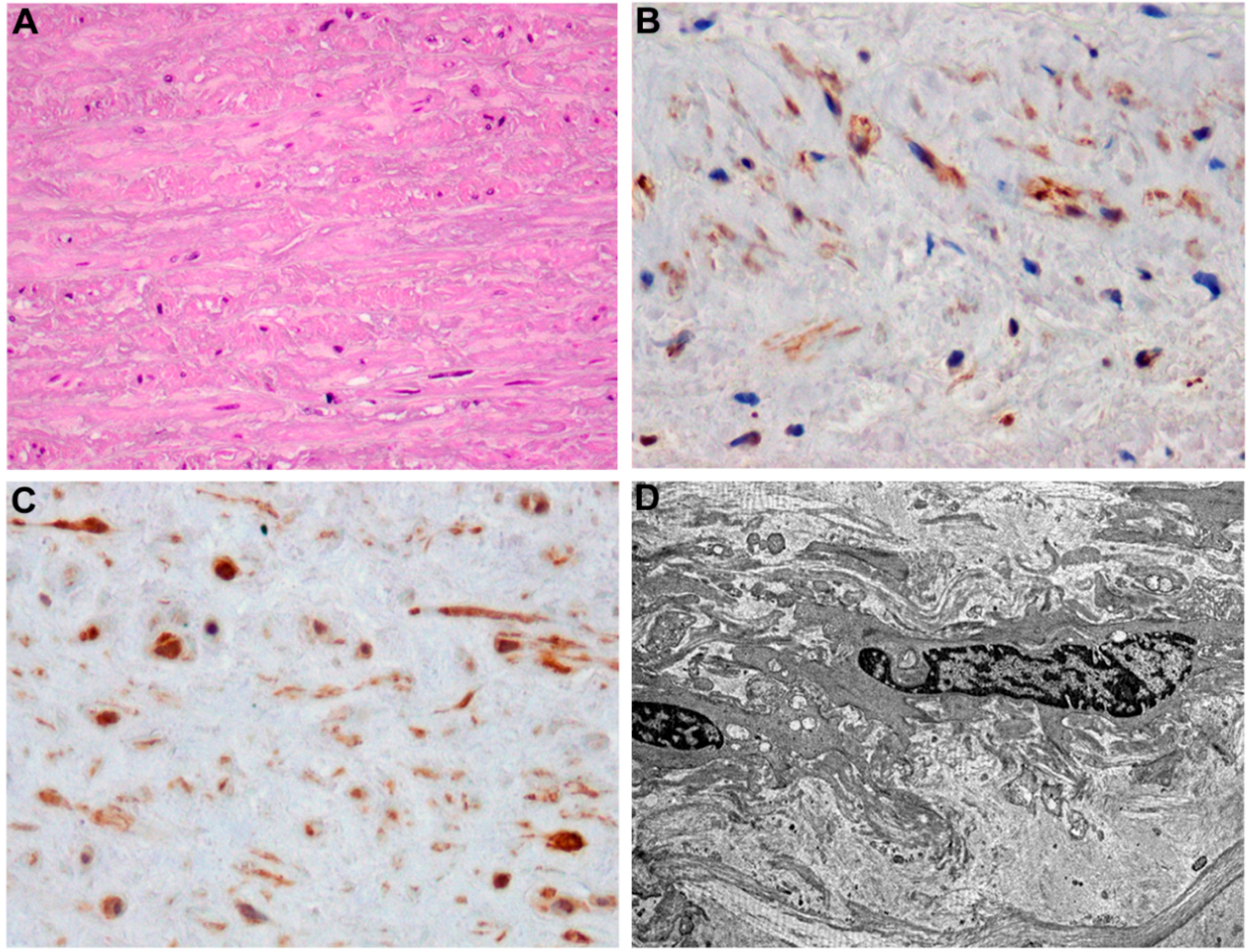
3. T2DM-Induced Macrovascular Disease: Different Players
4. VSMCs and T2DM-Related Atherogenic Risk Factors
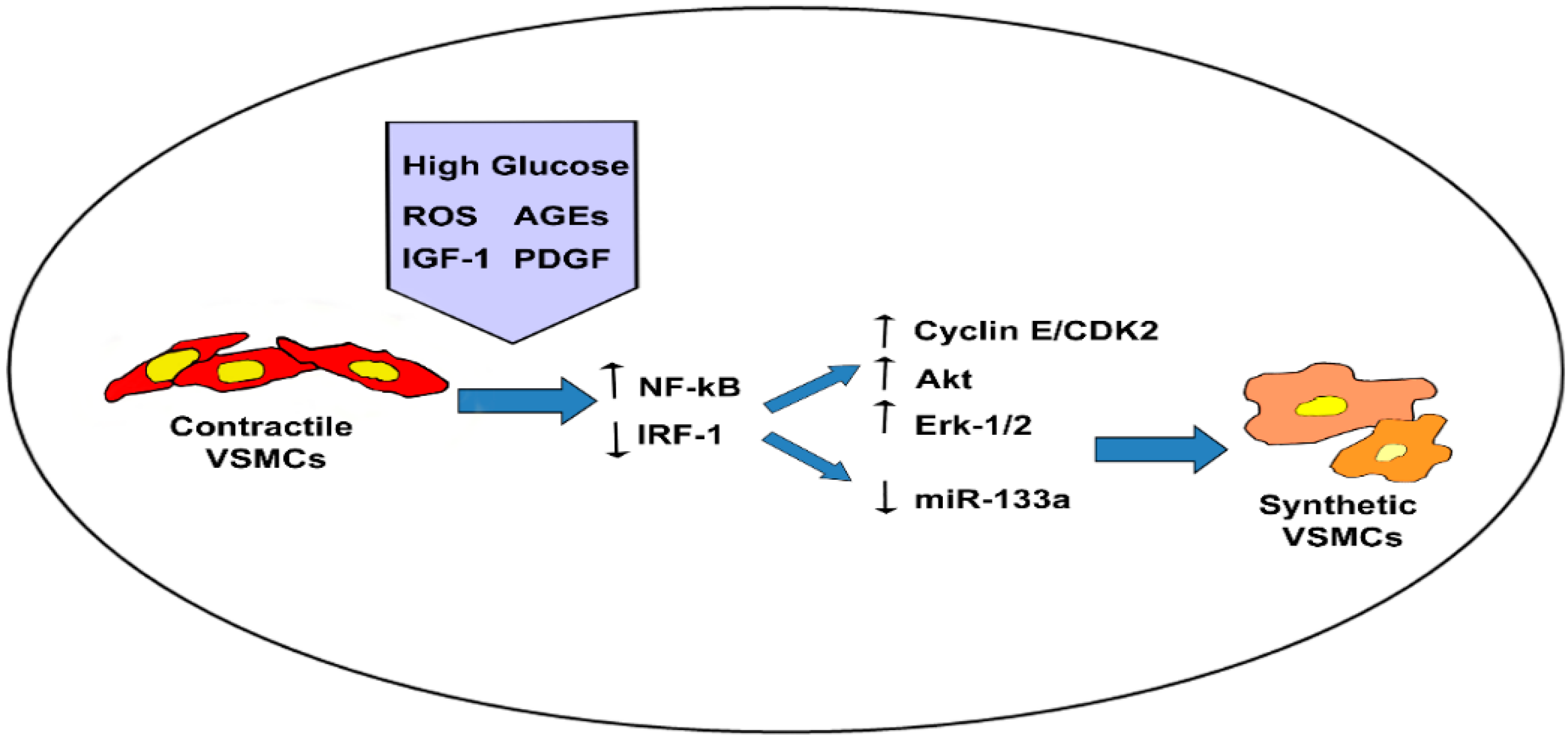
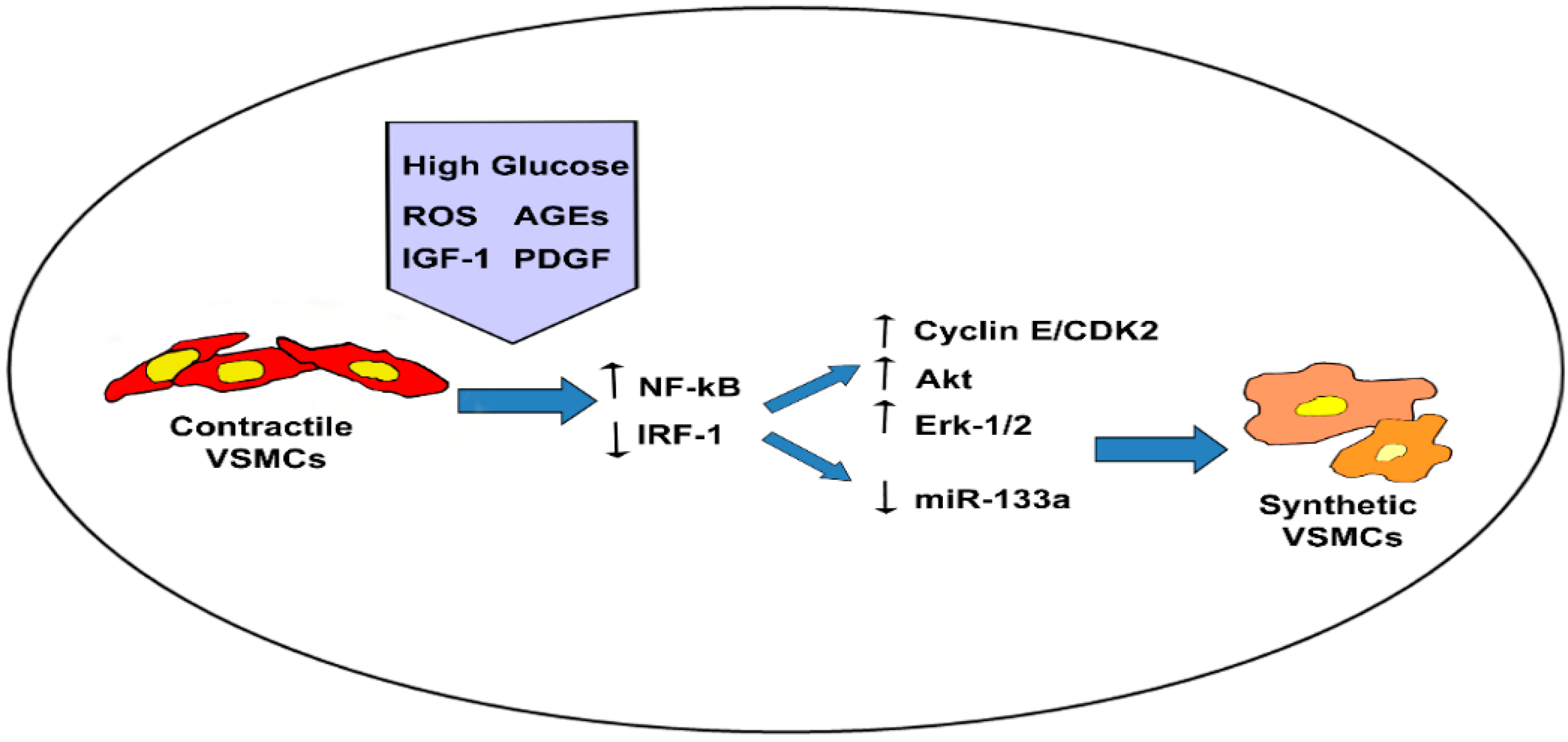
4.1. Hyperglycemia
4.2. Advanced Glycation End-Product
4.3. Hyperinsulinemia and Insulin Growth Factor
5. MicroRNAs and Diabetic Macrovascular Pathology
6. Adipose Tissue and Diabetes-Related Macrovascular Dysfunction
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yach, D.; Stucler, D.; Brownell, K.D. Epidemiologic and economic consequence of the global epidemics of obesity and diabetes. Nat. Med. 2006, 12, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Ussery-Hall, A.; Griffin-Blake, S.; Easton, A.; Vigeant, J.; Balluz, L.; Garvin, W.; Greenlund, K. Prevalence of selected risk behaviors and chronic disease and conditions—Step communities, United States, 2006–2007. MMWR Surveill. Summ. 2010, 59, 1–3. [Google Scholar] [PubMed]
- Orasanu, G.; Plutzky, J. The pathologic continuum of diabetic vascular disease. J. Am. Coll. Cardiol. 2009, 53, S35–S42. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G.; Kocher, O.; Bloom, W.S.; Vandekerckhove, J.; Weber, K. Actin expression in smooth muscle cells of rat aortic intimal thickening, human atheromatous plaque, and cultured rat aortic media. J. Clin. Investig. 1984, 73, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Campbell, J.H. Smooth muscle phenotypic changes in arterial wall homeostasis: Implications for the pathogenesis of atherosclerosis. Exp. Mol. Pathol. 1985, 42, 139–162. [Google Scholar] [CrossRef]
- Walker, L.N.; Bowen-Pope, D.F.; Ross, R.; Reidy, M.A. Production of platelet-derived growth factor-like molecules by cultured arterial smooth muscle cells accompanies proliferation after arterial injury. Proc. Natl. Acad. Sci. USA 1986, 83, 7311–7315. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.R.; Owens, G.K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, A.; Pucci, S.; Ciucci, A.; Pichiorri, F.; Ferlosio, A.; Spagnoli, L.G. Modulation of clusterin isoforms is associated with all-trans retinoic acid-induced proliferative arrest and apoptosis of intimal smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Thyberg, J.; Moskalewski, S. Microtubules and the organization of the Golgi complex. Exp. Cell Res. 1985, 159, 1–16. [Google Scholar] [CrossRef]
- Thyberg, J.; Blomgren, K.; Hedin, U.; Dryjski, M. Phenotypic modulation of smooth muscle cells during the formation of neointimal thickenings in the rat carotid artery after balloon injury: An electron-microscopic and stereological study. Cell Tissue Res. 1995, 281, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, A.; Calzetta, L.; Doldo, E.; Tarquini, C.; Matera, M.G.; Passeri, D. Brain natriuretic peptide modulates calcium homeostasis and epidermal growth factor receptor gene signalling in asthmatic airways smooth muscle cells. Pulm. Pharmacol. Ther. 2015, 31, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Arnaboldi, L.; Orlandi, A.; Yokoyama, K.; Gree, R.; Granata, A.; Hachem, A.; Paoletti, R.; Gelb, M.H.; Corsini, A. Effect of S(–) perillic acid on protein prenylation and arterial smooth muscle cell proliferation. Biochem. Pharmacol. 2001, 62, 1637–1645. [Google Scholar] [CrossRef]
- Orlandi, A.; Marcellini, M.; Pesce, D.; Calvani, M.; Spagnoli, L.G. Propionyl-l-carnitine reduces intimal hyperplasia after injury in normocholesterolemic rabbit carotidartery by modulating proliferation and caspase 3-dependent apoptosis of vascular smooth muscle cells. Atherosclerosis 2002, 160, 81–89. [Google Scholar] [CrossRef]
- Orlandi, A.; Spagnoli, L.G.; Marino, B.; Mauriello, A.; de Angelis, C.; Ramacci, M.T. Propionyl-l-carnitine prevents the progression of atherosclerotic lesions in aged hyperlipemic rabbits. Atherosclerosis 1995, 114, 29–44. [Google Scholar]
- Orlandi, A.; Francesconi, A.; Cocchia, D.; Corsini, A.; Spagnoli, L.G. Phenotypic heterogeneity influences apoptotic susceptibility to retinoic acid and cis-platinum of rat arterial smooth muscle cells in vitro: Implications for the evolution of experimental intimal thickening. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, A.; Bennett, M. Progenitor cell-derived smooth muscle cells in vascular disease. Biochem. Pharmacol. 2010, 79, 1706–1713. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, A.; Ehrlich, H.P.; Ropraz, P.; Spagnoli, L.G.; Gabbiani, G. Rat aortic smooth muscle cells isolated from different layers and at different times after endothelial denudation show distinct biological features in vitro. Arterioscler. Thromb. 1994, 14, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, A.; Ropraz, P.; Gabbiani, G. Proliferative activity and α-smooth muscle actin expression in cultured rat aortic smooth muscle cells are differently modulated by transforming growth factor-β1 and heparin. Exp. Cell Res. 1994, 214, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Ropraz, P.; Verin, V.; Camenzind, E.; Geinoz, A.; Pepper, M.S.; Gabbiani, G.; Bochaton-Piallat, M.L. Heterogeneity of smooth muscle cell populations cultured from pig coronary artery. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sims, S.; Jiao, Y.; Chow, L.H.; Pickering, J.G. Evidence from a novel human cell clone that adult vascular smooth muscle cells can convert reversibly between noncontractile and contractile phenotypes. Circ. Res. 1999, 85, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, A.; Ferlosio, A.; Gabbiani, G.; Spagnoli, L.G.; Ehrlich, P.H. Phenotypic heterogeneity influences the behavior of rat aortic smooth muscle cells in collagen lattice. Exp. Cell Res. 2005, 311, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Forst, T.; Hohberg, C.; Pfutzner, A. Cardiovascular effects of disturbed insulin activity in metabolic syndrome and in type 2 diabetic patients. Horm. Metab. Res. 2009, 41, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.E.; Riches, K. The vascular smooth muscle cell: A therapeutic target in type 2 diabetes? Clin. Sci. 2013, 125, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Faries, P.L.; Rohan, D.I.; Takahara, H.; Wyers, M.C.; Contreras, M.A.; Quist, W.C.; King, G.L.; Logerfo, F.W. Human vascular smooth muscle cells of diabetic origin exhibit increased proliferation, adhesion, and migration. J. Vasc. Surg. 2001, 33, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Madi, H.A.; Riches, K.; Warburton, P.; O’Regan, D.J.; Turner, N.A.; Porter, K.E. Inherent differences in morphology, proliferation, and migration in saphenous vein smooth muscle cells cultured from nondiabetic and type 2 diabetic patients. Am. J. Physiol. Cell Physiol. 2009, 297, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Piga, R.; Naito, Y.; Kokura, S.; Handa, O.; Yoshikawa, T. Short-term high glucose exposure induces monocyte-endothelial cells adhesion and transmigration by increasing VCAM-1 and MCP-1 expression in human aortic endothelial cells. Atherosclerosis 2007, 193, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Schmidt, A.M.; Anderson, G.M.; Zhang, J.; Brett, J.; Zou, Y.S.; Pinsky, D.; Stern, D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J. Biol. Chem. 1994, 269, 9889–9897. [Google Scholar] [PubMed]
- Otsuka, A.; Azuma, K.; Iesaki, T.; Sato, F.; Hirose, T.; Shimizu, T.; Tanaka, Y.; Daida, H.; Kawamori, R.; Watada, H. Temporary hyperglycaemia provokes monocyte adhesion to endothelial cells in rat thoracic aorta. Diabetologia 2005, 48, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Scioli, M.G.; Bielli, A.; Arcuri, G.; Ferlosio, A.; Orlandi, A. Ageing and microvasculature. Vasc. Cell 2014, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Tada, A.; Kanmani, P.; Watanabe, H.; Aso, H.; Suda, Y.; Nochi, T.; Miyazawa, K.; Yoda, K.; He, F.; et al. Advanced application of porcine intramuscular adipocytes for evaluating anti-adipogenic and anti-inflammatory activities of immunobiotics. PLoS ONE 2015, 10, e0119644. [Google Scholar] [CrossRef] [PubMed]
- Stasi, M.A.; Scioli, M.G.; Arcuri, G.; Mattera, G.G.; Lombardo, K.; Marcellini, M.; Riccioni, T.; de Falco, S.; Pisano, C.; Spagnoli, L.G.; et al. Propionyl-l-carnitine improves postischemic blood flow recovery and arteriogenetic revascularization and reduces endothelial NADPH-oxidase 4-mediated superoxide production. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Scioli, M.G.; Cervelli, V.; Arcuri, G.; Gentile, P.; Doldo, E.; Bielli, A.; Bonanno, E.; Orlandi, A. High insulin-induced down-regulation of Erk-1/IGF-1R/FGFR-1 signaling is required for oxidative stress-mediated apoptosis of adipose-derived stem cells. J. Cell. Physiol. 2014, 229, 2077–2087. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, A.; Francesconi, A.; Marcellini, M.; Ferlosio, A.; Spagnoli, L.G. Role of ageing and coronary atherosclerosis in the development of cardiac fibrosis in the rabbit. Cardiovasc. Res. 2004, 64, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.K.; Devaraj, S.; Yang, T.; Jialal, I. α-Tocopherol decreases superoxide anion release in human monocytes under hyperglycemic conditions via inhibition of protein kinase C-α. Diabetes 2002, 51, 3049–3054. [Google Scholar] [CrossRef] [PubMed]
- Spagnoli, L.G.; Orlandi, A.; Santeusanio, G. Foam cells of the rabbit atherosclerotic plaque arrested in metaphase by colchicine show a macrophage phenotype. Atherosclerosis 1991, 88, 87–92. [Google Scholar] [CrossRef]
- Orlandi, A.; Francesconi, A.; Marcellini, M.; di Lascio, A.; Spagnoli, L.G. Propionyl-l-carnitine reduces proliferation and potentiates Bax-related apoptosis of aortic intimal smooth muscle cells by modulating nuclear factor-κB activity. J. Biol. Chem. 2007, 282, 4932–4942. [Google Scholar] [CrossRef] [PubMed]
- Campagnolo, L.; Costanza, G.; Francesconi, A.; Arcuri, G.; Moscatelli, I.; Orlandi, A. Sortilin expression is essential for pro-nerve growth factor-induced apoptosis of rat vascular smooth muscle cells. PLoS ONE 2014, 9, e84969. [Google Scholar] [CrossRef] [PubMed]
- Scioli, M.G.; Bielli, A.; Agostinelli, S.; Tarquini, C.; Arcuri, G.; Ferlosio, A.; Costanza, G.; Doldo, E.; Orlandi, A. Antioxidant treatment prevents serum deprivation- and TNF-α-induced endothelial dysfunction through the inhibition of NADPH oxidase 4 and the restoration of β-oxidation. J. Vasc. Res. 2014, 51, 327–337. [Google Scholar] [PubMed]
- Abo, A.; Pick, E.; Hall, A.; Totty, N.; Teahan, C.G.; Segal, A.W. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 1991, 353, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, C.; Aretini, A.; Marino, G.; Bettarini, U.; Poulet, R.; Maffei, A.; Sbroggiò, M.; Pastore, L.; Gentile, M.T.; Notte, A.; et al. Selective Rac-1 inhibition protects from diabetes-induced vascular injury. Circ. Res. 2006, 98, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.A. Role of nitric oxide in diabetic complications. Am. J. Ther. 2005, 12, 499–502. [Google Scholar] [CrossRef] [PubMed]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Teshima, Y.; Takahashi, N.; Nishio, S.; Saito, S.; Kondo, H.; Fukui, A.; Aoki, K.; Yufu, K.; Nakagawa, M.; Saikawa, T. Production of reactive oxygen species in the diabetic heart. Roles of mitochondria and NADPH oxidase. Circ. J. 2014, 78, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res. 2010, 107, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, B.; Suominen, L. Accumulation and mobilization of cholesteryl esters in cultured human fibroblasts exposed to free cholesterol-rich phospholipid vesicles. Atherosclerosis 1985, 56, 345–358. [Google Scholar] [CrossRef]
- Liu, X.; Luo, F.; Pan, K.; Wu, W.; Chen, H. High glucose upregulates connective tissue growth factor expression in human vascular smooth muscle cells. BMC Cell Biol. 2007, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Spagnoli, L.G.; Orlandi, A.; Mauriello, A.; Santeusanio, G.; de Angelis, C.; Lucreziotti, R.; Ramacci, M.T. Aging and atherosclerosis in the rabbit: 1. Distribution, prevalence and morphology of atherosclerotic lesions. Atherosclerosis 1991, 89, 11–24. [Google Scholar] [CrossRef]
- Orlandi, A.; Mauriello, A.; Marino, B.; Spagnoli, L.G. Age-related modifications of aorta and coronaries in the rabbit: A morphological and morphometrical assessment. Arch. Gerontol. Geriatr. 1993, 17, 37–53. [Google Scholar] [CrossRef]
- Orlandi, A.; Marcellini, M.; Spagnoli, L.G. Aging influences development and progression of early aortic atherosclerotic lesions in cholesterol-fed rabbits. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Spagnoli, L.G.; Mauriello, A.; Orlandi, A.; Sangiorgi, G.; Bonanno, E. Age-related changes affecting atherosclerotic risk. Potential for pharmacological intervention. Drugs Aging 1996, 8, 275–298. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, A.; Bochaton-Piallat, M.L.; Gabbiani, G.; Spagnoli, L.G. Aging, smooth muscle cells and vascular pathobiology: Implications for atherosclerosis. Atherosclerosis 2006, 188, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Ferlosio, A.; Arcuri, G.; Doldo, E.; Scioli, M.G.; de Falco, S.; Spagnoli, L.G.; Orlandi, A. Age-related increase of stem marker expression influences vascular smooth muscle cell properties. Atherosclerosis 2012, 224, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, A.; di Lascio, A.; Francesconi, A.; Scioli, M.G.; Arcuri, G.; Ferlosio, A.; Spagnoli, L.G. Stem cell marker expression and proliferation and apoptosis of vascular smooth muscle cells. Cell Cycle 2008, 7, 3889–3897. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, A.; Ferlosio, A.; Arcuri, G.; Scioli, M.G.; de Falco, S.; Spagnoli, L.G. Flt-1 expression influences apoptotic susceptibility of vascular smooth muscle cells through the NF-κB/IAP-1 pathway. Cardiovasc. Res. 2010, 85, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, A. The contribution of resident vascular stem cells to arterial pathology. J. Stem Cells 2015, 8, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Wassler, M.; Zhang, L.; Li, Y.; Wang, J.; Zhang, Y.; Shelat, H.; Williams, J.; Geng, Y.J. MicroRNA-133a regulates insulin-like growth factor-1 receptor expression and vascular smooth muscle cell proliferation in murine atherosclerosis. Atherosclerosis 2014, 232, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Ferlosio, A.; Orlandi, A. Diabetes and aging: A different phenotypic commitment of circulating and resident stem cells? Acta Diabetol. 2012, 49, 493–494. [Google Scholar] [CrossRef] [PubMed]
- Chait, A.; Bornfeldt, K.E. Diabetes and atherosclerosis: Is there a role for hyperglycemia? J. Lipid Res. 2009, 50, S335–S339. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Mao, X.; Ji, Q.; Lang, M.; Li, S.; Peng, Y.; Zhou, W.; Xiong, B.; Zeng, Q. Inhibition of IFN regulatory factor-1 down-regulate Th1 cell function in patients with acute coronary syndrome. J. Clin. Immunol. 2010, 30, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhang, Z.; Gong, K.; Zhao, P.; Qin, J.; Liu, N. Inhibition of reactive oxygen species/extracellular signal-regulated kinases pathway by pioglitazone attenuates advanced glycation end products-induced proliferation of vascular smooth muscle cells in rats. Biol. Pharm. Bull. 2011, 34, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, L.; Chen, C.; Chi, Y.-L.; Yang, X.-Q.; Xu, Y.; Li, X.-T.; Guo, S.-L.; Xiong, S.-H.; Shen, M.R.; et al. Interferon regulatory factor-1 together with reactive oxygen species promotes the acceleration of cell cycle progression by up-regulating the cyclin E and CDK2 genes during high glucose-induced proliferation of vascular smooth muscle cells. Cardiovasc. Diabetol. 2013, 12, 147. [Google Scholar] [CrossRef] [PubMed]
- McGinn, S.; Poronnik, P.; Gallery, E.D.; Pollock, C.A. The effects of high glucose and atorvastatin on endothelial cell matrix production. Diabetes Med. 2004, 21, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Huang, X.R.; Zhu, H.J.; Johnson, R.; Lan, H.Y. Role of TGF-β signaling in extracellular matrix production under high glucose conditions. Kidney Int. 2003, 63, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.M.; Lee, D.H.; Kim, M.; Kang, Y.J. High glucose induces connective tissue growth factor expression and extracellular matrix accumulation in rat aorta vascular smooth muscle cells via extracellular signal-regulated kinase 1/2. Korean J. Physiol. Pharmacol. 2013, 17, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Bourcier, T.; Sukhova, G.; Libby, P. The nuclear factor κB signaling pathway participates in dysregulation of vascular smooth muscle cells in vitro and in human atherosclerosis. J. Biol. Chem. 1997, 272, 15817–15824. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Togo, M.; Hara, M.; Miyata, T.; Han, K.; Maekawa, H.; Ohno, M.; Hashimoto, Y.; Kurokawa, K.; Watanabe, T. Advanced glycation endproducts stimulate mitogen-activated protein kinase and proliferation in rabbit vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1997, 239, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Raman, K.G.; Lee, K.J.; Lu, Y.; Ferran, L.J., Jr.; Chow, W.S.; Stern, D.; Schmidt, A.M. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 1998, 4, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Li, Y.M.; Imani, F.; Wojciechowicz, D.; Yang, Z.; Liu, F.T.; Cerami, A. Identification of galectin-3 as a high-affinity binding protein for advanced glycation end products (AGE): A new member of the AGE-receptor complex. Mol. Med. 1995, 1, 634–646. [Google Scholar] [PubMed]
- Seki, N.; Hashimoto, N.; Sano, H.; Horiuchi, S.; Yagui, K.; Makino, H.; Saito, Y. Mechanisms involved in the stimulatory effect of advanced glycation end products on growth of rat aortic smooth muscle cells. Metabolism 2003, 52, 1558–1563. [Google Scholar] [CrossRef] [PubMed]
- Bornfeldt, K.E.; Raines, E.W.; Nakano, T.; Graves, L.M.; Krebs, E.G.; Ross, R. Insulin-like growth factor-I and platelet-derived growth factor-BB induce directed migration of human arterial smooth muscle cells via signaling pathways that are distinct from those of proliferation. J. Clin. Investig. 1994, 93, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Pfeifle, B.; Hamann, H.; Fussganger, R.; Ditschuneit, H. Insulin as a growth regulator of arterial smooth muscle cells: Effect of insulin of I.G.F.I. Diabetes Metab. 1987, 13, 326–330. [Google Scholar]
- Wang, C.C.; Gurevich, I.; Draznin, B. Insulin affects vascular smooth muscle cell phenotype and migration via distinct signaling pathways. Diabetes 2003, 52, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- Mughal, R.S.; Scragg, J.L.; Lister, P.; Warburton, P.; Riches, K.; O’Regan, D.J.; Ball, S.G.; Turner, N.A.; Porter, K.E. Cellular mechanisms by which proinsulin C-peptide prevents insulin-induced neointima formation in human saphenous vein. Diabetologia 2010, 53, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- Kotlyar, A.A.; Vered, Z.; Goldberg, I.; Chouraqui, P.; Nas, D.; Fridman, E.; Chen-Levy, Z.; Fytlovich, S.; Sangiorgi, G.; Spagnoli, L.G.; et al. Insulin-like growth factor I and II preserve myocardial structure in postinfarct swine. Heart 2001, 86, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Hitomi, H.; Shah, A.; Alexander, R.W.; Griendling, K.K. Mechanisms of reactive oxygen species-dependent downregulation of insulin receptor substrate-1 by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Ketsawatsomkron, P.; Stepp, D.W.; Fulton, D.J.; Marrero, M.B. Molecular mechanism of angiotensin II-induced insulin resistance in aortic vascular smooth muscle cells: Roles of protein tyrosine phosphatase-1B. Vascul. Pharmacol. 2010, 53, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Schiffrin, E.L. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol. Rev. 2000, 52, 639–672. [Google Scholar] [PubMed]
- Von der Thüsen, J.H.; Borensztajn, K.S.; Moimas, S.; van Heiningen, S.; Teeling, P.; van Berkel, T.J.; Biessen, E.A. IGF-1 has plaque-stabilizing effects in atherosclerosis by altering vascular smooth muscle cell phenotype. Am. J. Pathol. 2011, 178, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Johansson, G.S.; Arnqvist, H.J. Insulin and IGF-I action on insulin receptors, IGF-I receptors, and hybrid insulin/IGF-I receptors in vascular smooth muscle cells. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1124–E1130. [Google Scholar] [CrossRef] [PubMed]
- Engberding, N.; San Martín, A.; Martin-Garrido, A.; Koga, M.; Pounkova, L.; Lyons, E.; Lassègue, B.; Griendling, K.K. Insulin-like growth factor-1 receptor expression masks the antiinflammatory and glucose uptake capacity of insulin in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Cervelli, V.; Scioli, M.G.; Gentile, P.; Doldo, E.; Bonanno, E.; Spagnoli, L.G.; Orlandi, A. Platelet-rich plasma greatly potentiates insulin-induced adipogenic differentiation of human adipose-derived stem cells through a serine/threonine kinase Akt-dependent mechanism and promotes clinical fat graft maintenance. Stem Cells Transl. Med. 2012, 1, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Blackstock, C.D.; Higashi, Y.; Sukhanov, S.; Shai, S.Y.; Stefanovic, B.; Tabony, A.M.; Yoshida, T.; Delafontaine, P. Insulin-like growth factor-1 increases synthesis of collagen type I via induction of the mRNA-binding protein LARP6 expression and binding to the 5' stem-loop of COL1a1 and COL1a2 mRNA. J. Biol. Chem. 2014, 289, 7264–7274. [Google Scholar] [CrossRef] [PubMed]
- Cifarelli, V.; Luppi, P.; Tse, H.M.; He, J.; Piganelli, J.; Trucco, M. Human proinsulin C-peptide reduces high glucose-induced proliferation and NF-κB activation in vascular smooth muscle cells. Atherosclerosis 2008, 201, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Kasuya, Y.; Hamada, Y.; Nakashima, E.; Naruse, K.; Yasuda, Y.; Kato, K.; Hotta, N. Glucose-induced hyperproliferation of cultured rat aortic smooth muscle cells through polyol pathway hyperactivity. Diabetologia 2001, 44, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, L.A.; Poot, M.; Gerrity, R.G.; Bornfeldt, K.E. Diabetes accelerates smooth muscle accumulation in lesions of atherosclerosis: Lack of direct growth-promoting effects of high glucose levels. Diabetes 2001, 50, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Gao, Y.J.; Sharma, A.M. Do cardiac and perivascular adipose tissue play a role in atherosclerosis? Curr. Diabetes Rep. 2008, 8, 20–24. [Google Scholar] [CrossRef]
- Henrichot, E.; Juge-Aubry, C.E.; Pernin, A.; Pache, J.C.; Velebit, V.; Dayer, J.M.; Meda, P.; Chizzolini, C.; Meier, C.A. Production of chemokines by perivascular adipose tissue: A role in the pathogenesis of atherosclerosis? Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2594–2599. [Google Scholar] [CrossRef] [PubMed]
- Lamers, D.; Schlich, R.; Greulich, S.; Sasson, S.; Sell, H.; Eckel, J. Oleic acid and adipokines synergize in inducing proliferation and inflammatory signalling in human vascular smooth muscle cells. J. Cell Mol. Med. 2011, 15, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Funahashi, T.; Arita, Y.; Takahashi, M.; Matsuda, M.; Okamoto, Y.; Iwahashi, H.; Kuriyama, H.; Ouchi, N.; Maeda, K.; et al. Plasma concentrations of a novel, adiposespecific protein, adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Schlich, R.; Willems, M.; Greulich, S.; Ruppe, F.; Knoefel, W.T.; Ouwens, D.M.; Maxhera, B.; Lichtenberg, A.; Eckel, J.; Sell, H. VEGF in the crosstalk between human adipocytes and smooth muscle cells: Depot-specific release from visceral and perivascular adipose tissue. Mediators Inflamm. 2013, 2013, 982458. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, V.; Vesci, L.; Capasso, O.; Esposito, M.T.; Riccioni, T.; Pastore, L.; Orlandi, A.; Pisano, C.; de Falco, S. A placental growth factor variant unable to recognize vascular endothelial growth factor (VEGF) receptor-1 inhibits VEGF-dependent tumor angiogenesis via heterodimerization. Cancer Res. 2010, 70, 1804–1813. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, V.; Lepore, L.; Marcellini, M.; dal Piaz, F.; Tudisco, L.; Ponticelli, S.; Lund, F.W.; Roepstorff, P.; Orlandi, A.; Pisano, C.; et al. The biflavonoid amentoflavone inhibits neovascularization preventing the activity of proangiogenic vascular endothelial growth factors. J. Biol. Chem. 2011, 286, 19641–19651. [Google Scholar] [CrossRef] [PubMed]
- Belvisi, L.; Riccioni, T.; Marcellini, M.; Vesci, L.; Chiarucci, I.; Efrati, D.; Potenza, D.; Scolastico, C.; Manzoni, L.; Lombardo, K.; et al. Biological and molecular properties of a new αvβ3/αvβ5 integrin antagonist. Mol. Cancer Ther. 2005, 4, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Cicatiello, V.; Apicella, I.; Tudisco, L.; Tarallo, V.; Formisano, L.; Sandomenico, A.; Kim, Y.; Bastos-Carvalho, A.; Orlandi, A.; Ambati, J.; et al. Powerful anti-tumor and anti-angiogenic activity of a new anti-vascular endothelial growth factor receptor 1 peptide in colorectal cancer models. Oncotarget 2015, 6, 10563–10576. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.K.; Lam, K.S.; Wang, Y.; Huang, Y.; Carling, D.; Wu, D.; Wong, C.; Xu, A. Adiponectin-induced endothelial nitric oxide synthase activation and nitric oxide production are mediated by APPL1 in endothelial cells. Diabetes 2007, 56, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Ridnour, L.A.; Thomas, D.D.; Mancardi, D.; Espey, M.G.; Miranda, K.M.; Paolocci, N.; Feelisch, M.; Fukuto, J.; Wink, D.A. The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol. Chem. 2004, 385, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, F.; Eto, M.; de Paolis, P.; van der Loo, B.; Bachschmid, M.; Ullrich, V.; Kouroedov, A.; Delli Gatti, C.; Joch, H.; Volpe, M.; et al. High glucose causes upregulation of cyclooxygenase-2 and alters prostanoid profile in human endothelial cells: Role of protein kinase C and reactive oxygen species. Circulation 2003, 107, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casella, S.; Bielli, A.; Mauriello, A.; Orlandi, A. Molecular Pathways Regulating Macrovascular Pathology and Vascular Smooth Muscle Cells Phenotype in Type 2 Diabetes. Int. J. Mol. Sci. 2015, 16, 24353-24368. https://doi.org/10.3390/ijms161024353
Casella S, Bielli A, Mauriello A, Orlandi A. Molecular Pathways Regulating Macrovascular Pathology and Vascular Smooth Muscle Cells Phenotype in Type 2 Diabetes. International Journal of Molecular Sciences. 2015; 16(10):24353-24368. https://doi.org/10.3390/ijms161024353
Chicago/Turabian StyleCasella, Sara, Alessandra Bielli, Alessandro Mauriello, and Augusto Orlandi. 2015. "Molecular Pathways Regulating Macrovascular Pathology and Vascular Smooth Muscle Cells Phenotype in Type 2 Diabetes" International Journal of Molecular Sciences 16, no. 10: 24353-24368. https://doi.org/10.3390/ijms161024353
APA StyleCasella, S., Bielli, A., Mauriello, A., & Orlandi, A. (2015). Molecular Pathways Regulating Macrovascular Pathology and Vascular Smooth Muscle Cells Phenotype in Type 2 Diabetes. International Journal of Molecular Sciences, 16(10), 24353-24368. https://doi.org/10.3390/ijms161024353