
Ca2+-Dependent Regulations and Signaling in Skeletal Muscle: From Electro-Mechanical Coupling to Adaptation




Abstract
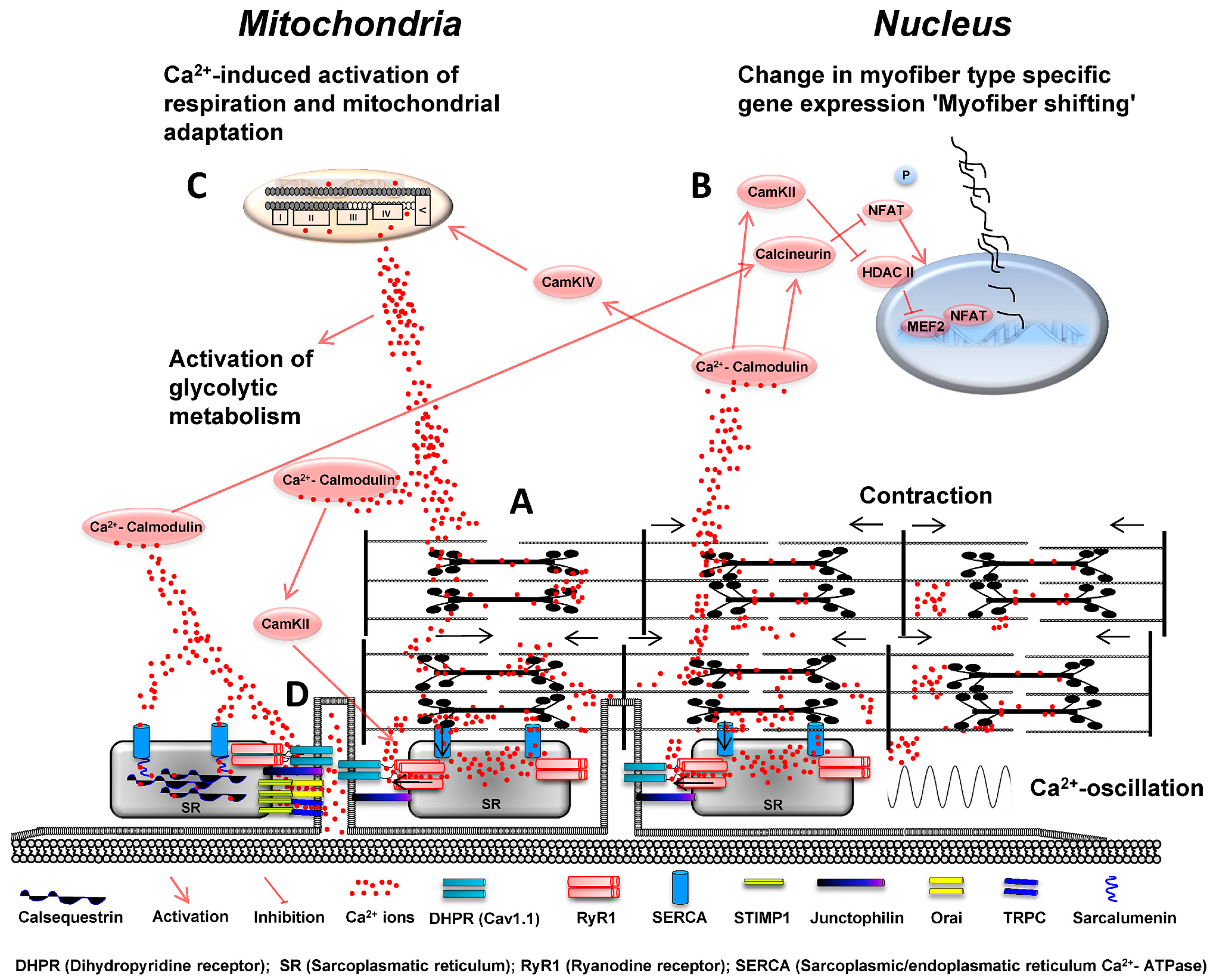
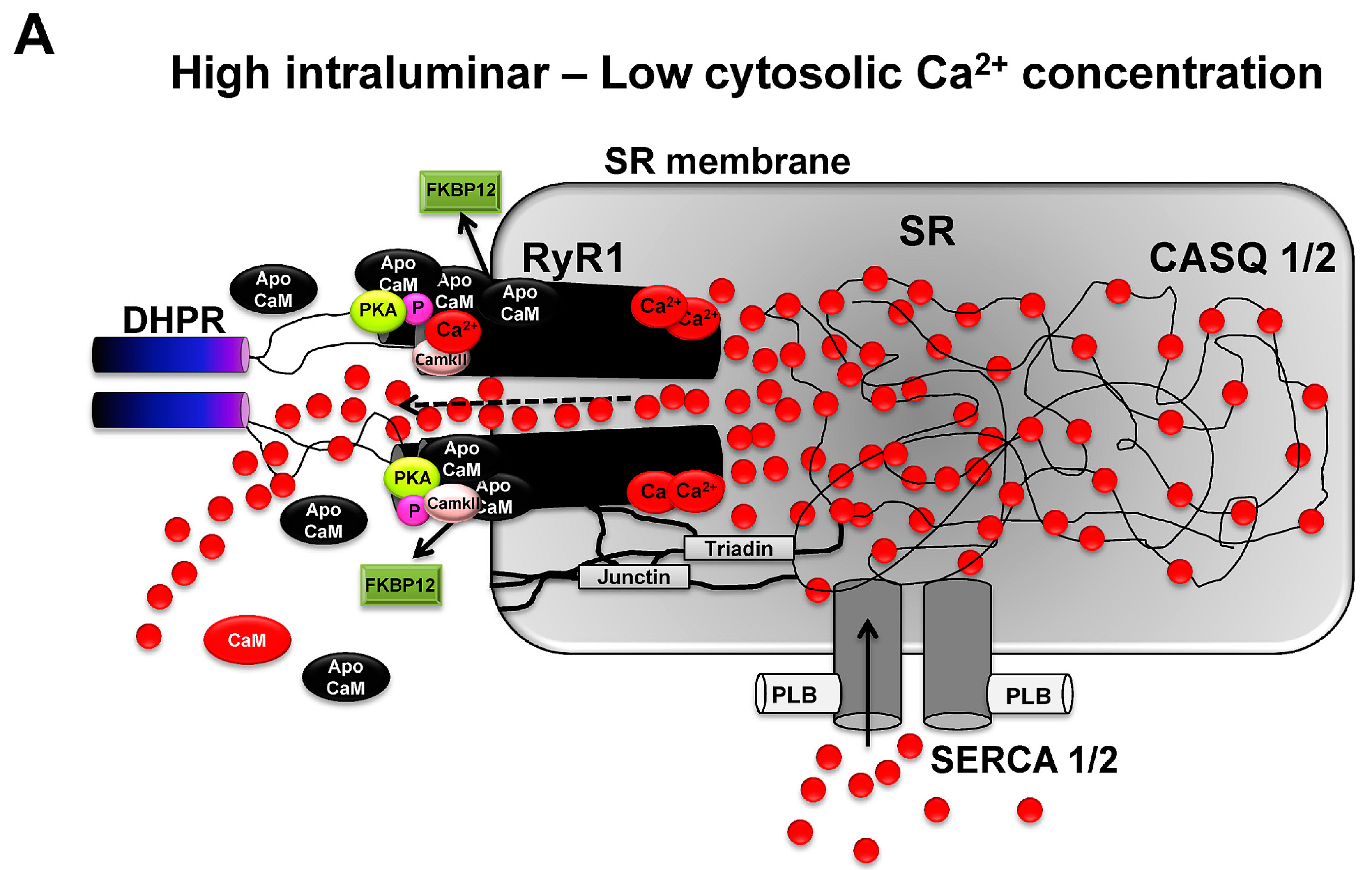
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Electromechanical Coupling in Skeletal Muscle
3. Ca2+ Ions and the Regulation of Electro-Mechanical Coupling
3.1. Ca2+ Ions and the Initiation of Presynaptic Action Potentials
3.2. Ca2+ Storage and Localization in Skeletal Muscle
3.3. Regulation of Ca2+ Homeostasis via Store-Operated Calcium Entry (SOCE) and Molecules Regulating SR Integrity
3.4. Regulation of Ca2+ Release via Voltage and Coupled Gating
3.5. Regulation of RyR1 Activity and Ca2+ Release via Interacting Molecules and Channel Modification
3.6. Modulation of RyR1-Induced Ca2+ Release via Ca2+ Ions and Calmodulin
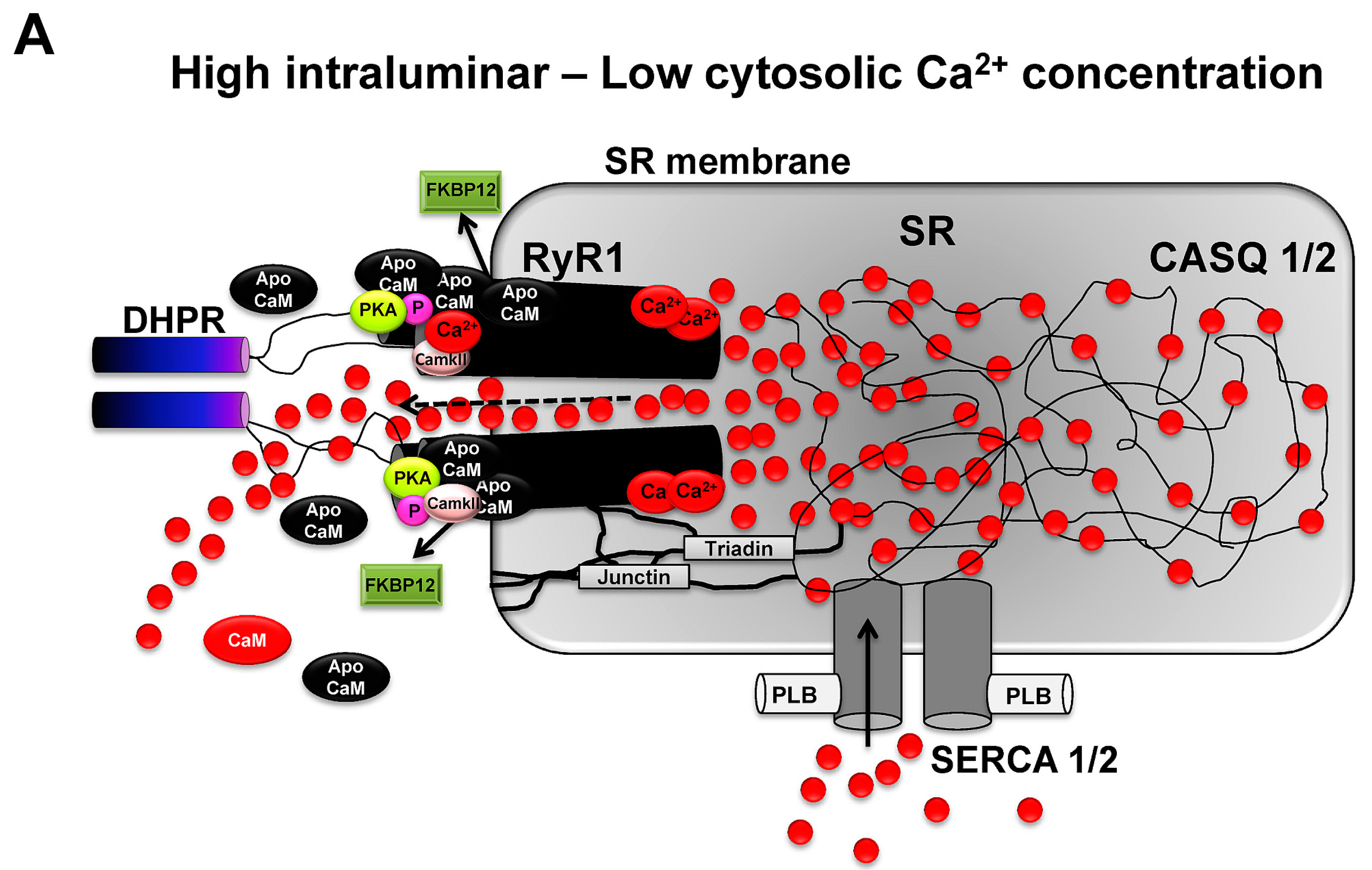
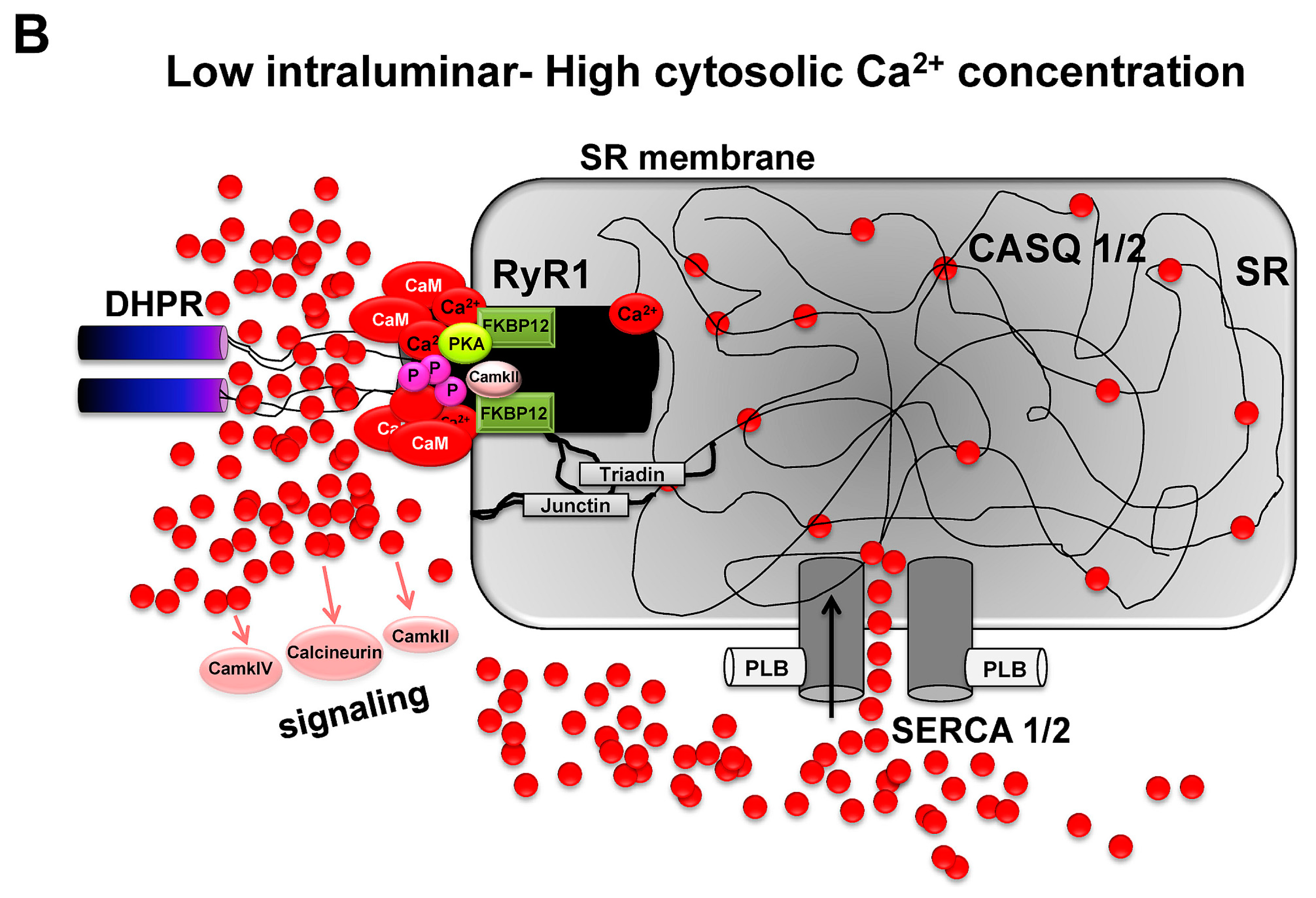
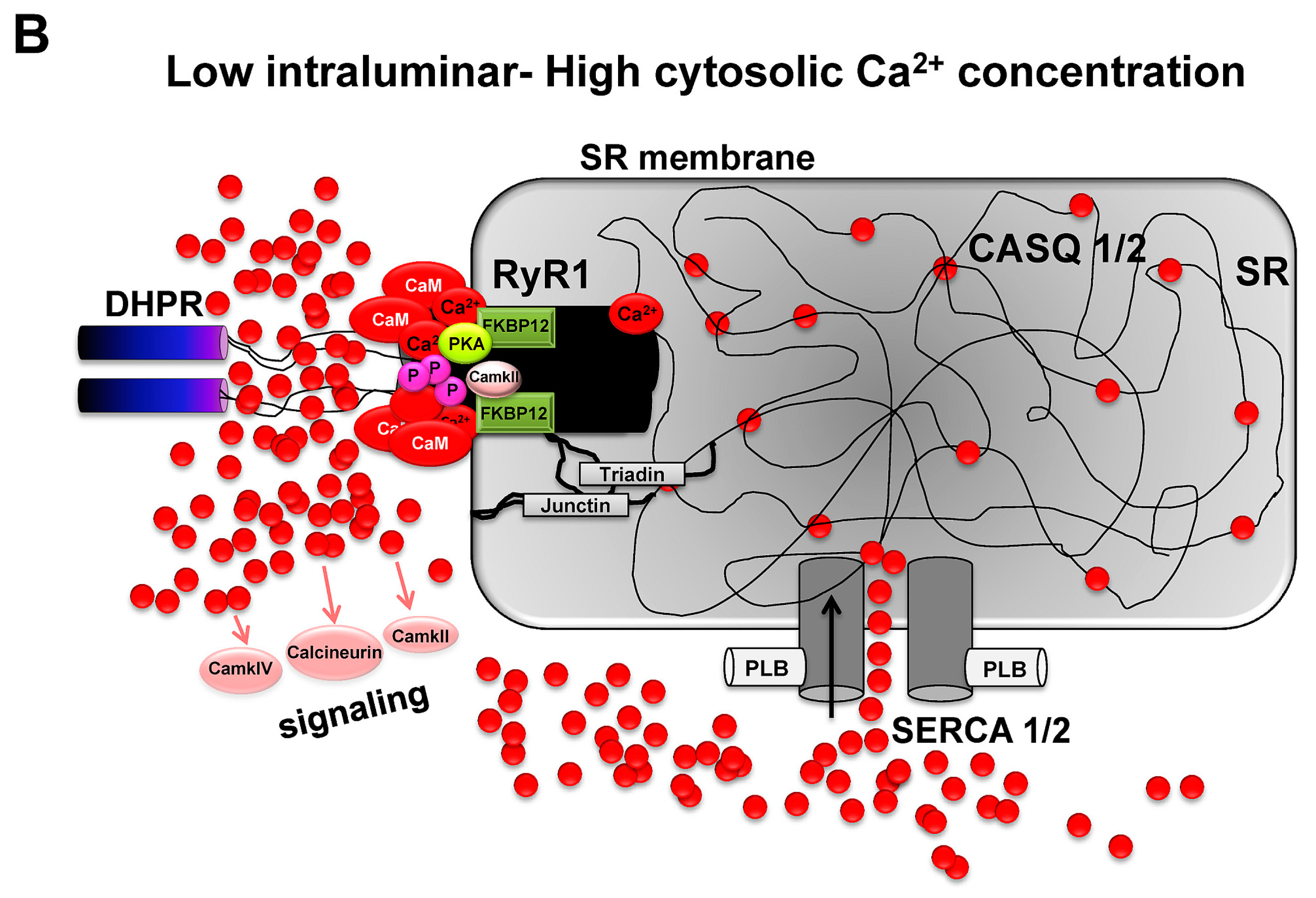
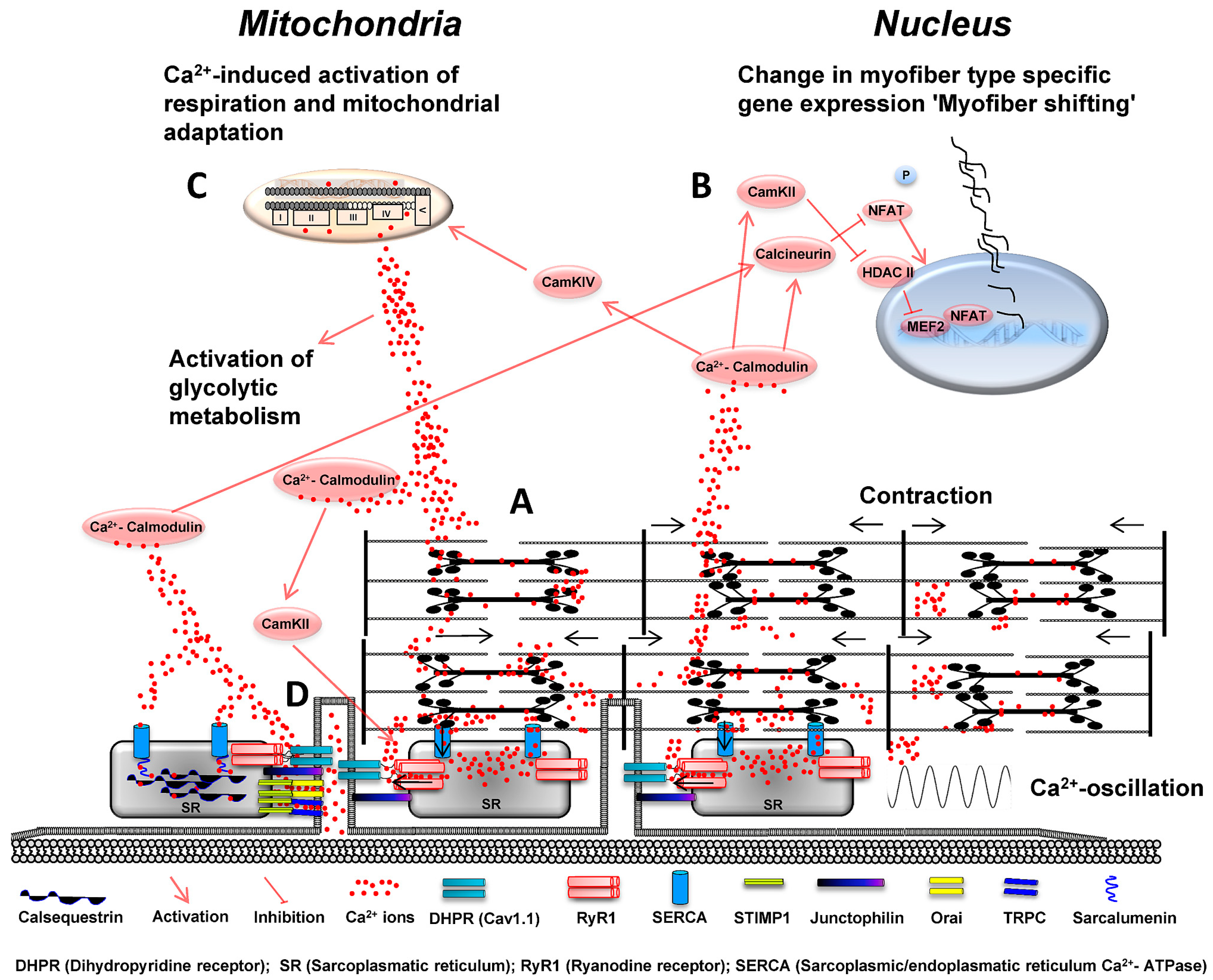
3.7. Ca2+-Dependent Control of Crossbridge Cycling and Force Generation
3.8. Ca2+ Ions and the Regulation of Force Development via Modulation of Regulatory Light Chain Phosphorylation
4. Ca2+ Ions and the Regulation of Skeletal Muscle Relaxation
Regulation of Ca2+ Reuptake into the SR
5. Ca2+ Ions and the Regulation of Energy Metabolism
5.1. Ca2+ Ions and the Regulation of Glycolysis
5.2. Ca2+ Ion-Dependent Regulation of Mitochondrial Function
6. Ca2+ Ion Mediated Regulation of Muscle Plasticity
6.1. Ca2+ Ions and Skeletal Muscle Hypertrophy
6.2. Ca2+-Dependent Activation of Protein Degradation via Calpains
6.3. Ca2+-Dependent Activation of Protein Synthesis
6.4. Mitochondrial Adaptation via CaMK IV and PGC-1α
7. Further Key Ca2+-Signaling in Skeletal Muscle
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nardone, A.; Romano, C.; Schieppati, M. Selective recruitment of high-threshold human motor units during voluntary isotonic lengthening of active muscles. J. Physiol. 1989, 409, 451–471. [Google Scholar] [CrossRef] [PubMed]
- Nardone, A.; Schieppati, M. Shift of activity from slow to fast muscle during voluntary lengthening contractions of the triceps surae muscles in humans. J. Physiol. 1988, 395, 363–381. [Google Scholar] [CrossRef] [PubMed]
- MacIntosh, B.R.; Holash, R.J.; Renaud, J.M. Skeletal muscle fatigue—Regulation of excitation-contraction coupling to avoid metabolic catastrophe. J. Cell Sci. 2012, 125, 2105–2114. [Google Scholar] [CrossRef] [PubMed]
- Sine, S.M. End-plate acetylcholine receptor: Structure, mechanism, pharmacology, and disease. Physiol. Rev. 2012, 92, 1189–1234. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, M.W.; Brinkmeier, H.; Muntener, M. Calcium ion in skeletal muscle: Its crucial role for muscle function, plasticity, and disease. Physiol. Rev. 2000, 80, 1215–1265. [Google Scholar] [PubMed]
- Zucker, R.S. Calcium and transmitter release. J. Physiol. Paris 1993, 87, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.; Slater, C.R.; Bewick, G.S. Synaptic vesicle dynamics in rat fast and slow motor nerve terminals. J. Neurosci. 1999, 19, 2511–2521. [Google Scholar] [PubMed]
- Wood, S.J.; Slater, C.R. Safety factor at the neuromuscular junction. Prog. Neurobiol. 2001, 64, 393–429. [Google Scholar] [CrossRef] [PubMed]
- Capes, E.M.; Loaiza, R.; Valdivia, H.H. Ryanodine receptors. Skelet. Muscle 2011, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Felix, R.; Gurnett, C.A.; de Waard, M.; Campbell, K.P. Dissection of functional domains of the voltage-dependent Ca2+ channel α2δ subunit. J. Neurosci. 1997, 17, 6884–6891. [Google Scholar] [PubMed]
- Milner, R.E.; Famulski, K.S.; Michalak, M. Calcium binding proteins in the sarcoplasmic/endoplasmic reticulum of muscle and nonmuscle cells. Mol. Cell. Biochem. 1992, 112, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fohr, U.G.; Weber, B.R.; Muntener, M.; Staudenmann, W.; Hughes, G.J.; Frutiger, S.; Banville, D.; Schafer, B.W.; Heizmann, C.W. Human α and β parvalbumins: Structure and tissue-specific expression. Eur. J. Biochem. 1993, 215, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, T.; Beam, K.G.; Powell, J.A.; Numa, S. Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature 1988, 336, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Froemming, G.R.; Ohlendieck, K. Oligomerisation of Ca2+-regulatory membrane components involved in the excitation-contraction-relaxation cycle during postnatal development of rabbit skeletal muscle. Biochim. Biophys. Acta 1998, 1387, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Damiani, E.; Margreth, A. Characterization study of the ryanodine receptor and of calsequestrin isoforms of mammalian skeletal muscles in relation to fibre types. J. Muscle Res. Cell Motil. 1994, 15, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Leberer, E.; Hartner, K.T.; Pette, D. Postnatal development of Ca2+-sequestration by the sarcoplasmic reticulum of fast and slow muscles in normal and dystrophic mice. Eur. J. Biochem. 1988, 174, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Leberer, E.; Charuk, J.H.; Green, N.M.; MacLennan, D.H. Molecular cloning and expression of cDNA encoding a lumenal calcium binding glycoprotein from sarcoplasmic reticulum. Proc. Natl. Acad. Sci. USA 1989, 86, 6047–6051. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Ashley, R.H. The structure, function, and cellular regulation of ryanodine-sensitive Ca2+ release channels. Int. Rev. Cytol. 1998, 183, 185–270. [Google Scholar] [PubMed]
- Dowling, P.; Doran, P.; Ohlendieck, K. Drastic reduction of sarcalumenin in Dp427 (dystrophin of 427 kDa)-deficient fibres indicates that abnormal calcium handling plays a key role in muscular dystrophy. Biochem. J. 2004, 379, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Minamisawa, S.; Shimura, M.; Komazaki, S.; Kume, H.; Zhang, M.; Matsumura, K.; Nishi, M.; Saito, M.; Saeki, Y.; et al. Impaired Ca2+ store functions in skeletal and cardiac muscle cells from sarcalumenin-deficient mice. J. Biol. Chem. 2005, 280, 3500–3506. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yoshida, M.; Brotto, L.; Takeshima, H.; Weisleder, N.; Hirata, Y.; Nosek, T.M.; Ma, J.; Brotto, M. Enhanced resistance to fatigue and altered calcium handling properties of sarcalumenin knockout mice. Physiol. Genomics 2005, 23, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Westerblad, H.; Allen, D.G. Changes of myoplasmic calcium concentration during fatigue in single mouse muscle fibers. J. Gen. Physiol. 1991, 98, 615–635. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.A.; Head, S.I.; Bakker, A.J.; Stephenson, D.G. Resting calcium concentrations in isolated skeletal muscle fibres of dystrophic mice. J. Physiol. 1990, 428, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Head, S.I. Membrane potential, resting calcium and calcium transients in isolated muscle fibres from normal and dystrophic mice. J. Physiol. 1993, 469, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Hasselbach, W. Structural and enzymatic properties of the calcium transporting membranes of the sarcoplasmic reticulum. Ann. N. Y. Acad. Sci. 1966, 137, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Sacchetto, R.; Margreth, A.; Pelosi, M.; Carafoli, E. Colocalization of the dihydropyridine receptor, the plasma-membrane calcium ATPase isoform 1 and the sodium/calcium exchanger to the junctional-membrane domain of transverse tubules of rabbit skeletal muscle. Eur. J. Biochem. 1996, 237, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, N.; Ogawa, Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J. Physiol. 2001, 533, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Stiber, J.; Hawkins, A.; Zhang, Z.S.; Wang, S.; Burch, J.; Graham, V.; Ward, C.C.; Seth, M.; Finch, E.; Malouf, N.; et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell. Biol. 2008, 10, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Kiviluoto, S.; Decuypere, J.P.; de, S.H.; Missiaen, L.; Parys, J.B.; Bultynck, G. STIM1 as a key regulator for Ca2+ homeostasis in skeletal-muscle development and function. Skelet. Muscle 2011, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H.; Cherednichenko, G.; Pessah, I.N.; Allen, P.D. Functional coupling between TRPC3 and RyR1 regulates the expressions of key triadic proteins. J. Biol. Chem. 2006, 281, 10042–10048. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Erxleben, C.; Yildirim, E.; Abramowitz, J.; Armstrong, D.L.; Birnbaumer, L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 4682–4687. [Google Scholar] [CrossRef] [PubMed]
- Thornton, A.M.; Zhao, X.; Weisleder, N.; Brotto, L.S.; Bougoin, S.; Nosek, T.M.; Reid, M.; Hardin, B.; Pan, Z.; Ma, J.; et al. Store-operated Ca2+ entry (SOCE) contributes to normal skeletal muscle contractility in young but not in aged skeletal muscle. Aging (Albany NY) 2011, 3, 621–634. [Google Scholar]
- Egan, B.; Zierath, J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Feske, S.; Srikanth, S.; Hogan, P.G.; Rao, A. Signalling to transcription: Store-operated Ca2+ entry and NFAT activation in lymphocytes. Cell Calcium 2007, 42, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Balog, E.M. Excitation-contraction coupling and minor triadic proteins in low-frequency fatigue. Exerc. Sport Sci. Rev. 2010, 38, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Manring, H.; Abreu, E.; Brotto, L.; Weisleder, N.; Brotto, M. Novel excitation-contraction coupling related genes reveal aspects of muscle weakness beyond atrophy-new hopes for treatment of musculoskeletal diseases. Front. Physiol. 2014, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, H.; Komazaki, S.; Nishi, M.; Iino, M.; Kangawa, K. Junctophilins: A novel family of junctional membrane complex proteins. Mol. Cell 2000, 6, 11–22. [Google Scholar] [PubMed]
- Golini, L.; Chouabe, C.; Berthier, C.; Cusimano, V.; Fornaro, M.; Bonvallet, R.; Formoso, L.; Giacomello, E.; Jacquemond, V.; Sorrentino, V. Junctophilin 1 and 2 proteins interact with the l-type Ca2+ channel dihydropyridine receptors (DHPRs) in skeletal muscle. J. Biol. Chem. 2011, 286, 43717–43725. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Komazaki, S.; Sasamoto, K.; Yoshida, M.; Nishi, M.; Kitamura, K.; Takeshima, H. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J. Cell Biol. 2001, 154, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Komazaki, S.; Nishi, M.; Kangawa, K.; Takeshima, H. Immunolocalization of mitsugumin29 in developing skeletal muscle and effects of the protein expressed in amphibian embryonic cells. Dev. Dyn. 1999, 215, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Komazaki, S.; Nishi, M.; Takeshima, H.; Nakamura, H. Abnormal formation of sarcoplasmic reticulum networks and triads during early development of skeletal muscle cells in mitsugumin29-deficient mice. Dev. Growth Differ. 2001, 43, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Park, C.S.; Woo, J.S.; Kim, D.H.; Ma, J.; Lee, E.H. Mitsugumin 53 attenuates the activity of sarcoplasmic reticulum Ca2+-ATPase 1a (SERCA1a) in skeletal muscle. Biochem. Biophys. Res. Commun. 2012, 428, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Protasi, F. Structural interaction between RYRs and DHPRs in calcium release units of cardiac and skeletal muscle cells. Front. Biosci. 2002, 7, d650–d658. [Google Scholar] [CrossRef] [PubMed]
- Valdivia, H.H.; Coronado, R. Inhibition of dihydropyridine-sensitive calcium channels by the plant alkaloid ryanodine. FEBS Lett. 1989, 244, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010. [Google Scholar] [CrossRef]
- Gaburjakova, M.; Gaburjakova, J.; Reiken, S.; Huang, F.; Marx, S.O.; Rosemblit, N.; Marks, A.R. FKBP12 binding modulates ryanodine receptor channel gating. J. Biol. Chem. 2001, 276, 16931–16935. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V. Sarcoplasmic reticulum: Structural determinants and protein dynamics. Int. J. Biochem. Cell Biol. 2011, 43, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Franzini-Armstrong, C.; Ferguson, D.G.; Champ, C. Discrimination between fast- and slow-twitch fibres of guinea pig skeletal muscle using the relative surface density of junctional transverse tubule membrane. J. Muscle Res. Cell Motil. 1988, 9, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Close, R.I. Dynamic properties of mammalian skeletal muscles. Physiol. Rev. 1972, 52, 129–197. [Google Scholar] [PubMed]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011. [Google Scholar] [CrossRef]
- Marx, S.O.; Ondrias, K.; Marks, A.R. Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors). Science 1998, 281, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Lederer, W.J. Calcium sparks. Physiol. Rev. 2008, 88, 1491–1545. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T. Modulation of ryanodine receptor Ca2+ channels (Review). Mol. Med. Rep. 2010, 3, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Sencer, S.; Hamilton, S.L. Calmodulin modulation of proteins involved in excitation-contraction coupling. Front. Biosci. 2002, 7, d1583–d1589. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Laver, D.R. Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites. Clin. Exp. Pharmacol. Physiol. 2007, 34, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.G.; Lamb, G.D.; Stephenson, G.M. Events of the excitation-contraction-relaxation (E-C-R) cycle in fast- and slow-twitch mammalian muscle fibres relevant to muscle fatigue. Acta Physiol. Scand. 1998, 162, 229–245. [Google Scholar] [CrossRef] [PubMed]
- Reiken, S.; Lacampagne, A.; Zhou, H.; Kherani, A.; Lehnart, S.E.; Ward, C.; Huang, F.; Gaburjakova, M.; Gaburjakova, J.; Rosemblit, N.; et al. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: Defective regulation in heart failure. J. Cell Biol. 2003, 160, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.C.; Betzenhauser, M.J.; Reiken, S.; Umanskaya, A.; Shiomi, T.; Marks, A.R. Stress-induced increase in skeletal muscle force requires protein kinase A phosphorylation of the ryanodine receptor. J. Physiol. 2012, 590, 6381–6387. [Google Scholar] [CrossRef] [PubMed]
- Matalon, S.; Hardiman, K.M.; Jain, L.; Eaton, D.C.; Kotlikoff, M.; Eu, J.P.; Sun, J.; Meissner, G.; Stamler, J.S. Regulation of ion channel structure and function by reactive oxygen-nitrogen species. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L1184–L1189. [Google Scholar] [PubMed]
- Suhr, F.; Gehlert, S.; Grau, M.; Bloch, W. Skeletal muscle function during exercise-fine-tuning of diverse subsystems by nitric oxide. Int. J. Mol. Sci. 2013, 14, 7109–7139. [Google Scholar] [CrossRef] [PubMed]
- Eu, J.P.; Sun, J.; Xu, L.; Stamler, J.S.; Meissner, G. The skeletal muscle calcium release channel: Coupled O2 sensor and NO signaling functions. Cell 2000, 102, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Chaube, R.; Hess, D.T.; Wang, Y.J.; Plummer, B.; Sun, Q.A.; Laurita, K.; Stamler, J.S. Regulation of the skeletal muscle ryanodine receptor/Ca2+-release channel RyR1 by S-palmitoylation. J. Biol. Chem. 2014, 289, 8612–8619. [Google Scholar] [CrossRef] [PubMed]
- Gill, C.O.; Jones, S.D. Evaluation of a commercial process for collection and cooling of beef offals by a temperature function integration technique. Int. J. Food Microbiol. 1992, 15, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, R.L.; Vaughan, M. Direct comparison of Ca2+ requirements for calmodulin interaction with and activation of protein phosphatase. Proc. Natl. Acad. Sci. USA 1986, 83, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Stull, J.T. Ca2+-dependent cell signaling through calmodulin-activated protein phosphatase and protein kinases minireview series. J. Biol. Chem. 2001, 276, 2311–2312. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Molecular diversity of myofibrillar proteins: Gene regulation and functional significance. Physiol. Rev. 1996, 76, 371–423. [Google Scholar] [PubMed]
- McKillop, D.F.; Geeves, M.A. Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys. J. 1993, 65, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, S.K.; Kerrick, W.G. Characterization of the effects of Mg2+ on Ca2+- and Sr2+-activated tension generation of skinned skeletal muscle fibers. J. Gen. Physiol. 1975, 66, 427–444. [Google Scholar] [CrossRef] [PubMed]
- Fryer, M.W.; Owen, V.J.; Lamb, G.D.; Stephenson, D.G. Effects of creatine phosphate and P(i) on Ca2+ movements and tension development in rat skinned skeletal muscle fibres. J. Physiol. 1995, 482, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Westerblad, H.; Allen, D.G. The effects of intracellular injections of phosphate on intracellular calcium and force in single fibres of mouse skeletal muscle. Pflugers Arch. 1996, 431, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Lamb, G.D.; Westerblad, H. Skeletal muscle fatigue: Cellular mechanisms. Physiol. Rev. 2008, 88, 287–332. [Google Scholar] [CrossRef] [PubMed]
- Bozzo, C.; Spolaore, B.; Toniolo, L.; Stevens, L.; Bastide, B.; Cieniewski-Bernard, C.; Fontana, A.; Mounier, Y.; Reggiani, C. Nerve influence on myosin light chain phosphorylation in slow and fast skeletal muscles. FEBS J. 2005, 272, 5771–5785. [Google Scholar] [CrossRef] [PubMed]
- Ryder, J.W.; Lau, K.S.; Kamm, K.E.; Stull, J.T. Enhanced skeletal muscle contraction with myosin light chain phosphorylation by a calmodulin-sensing kinase. J. Biol. Chem. 2007, 282, 20447–20454. [Google Scholar] [CrossRef] [PubMed]
- Stull, J.T.; Kamm, K.E.; Vandenboom, R. Myosin light chain kinase and the role of myosin light chain phosphorylation in skeletal muscle. Arch. Biochem. Biophys. 2011, 510, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Tubman, L.A.; MacIntosh, B.R.; Maki, W.A. Myosin light chain phosphorylation and posttetanic potentiation in fatigued skeletal muscle. Pflugers Arch. 1996, 431, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Kensler, R.W.; Yang, Z.; Stull, J.T.; Sweeney, H.L. Myosin light chain phosphorylation affects the structure of rabbit skeletal muscle thick filaments. Biophys. J. 1996, 71, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of contraction in striated muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [PubMed]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, W.A.; Stephenson, D.G. Effects of ADP on sarcoplasmic reticulum function in mechanically skinned skeletal muscle fibres of the rat. J. Physiol. 2001, 532, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Vangheluwe, P.; Schuermans, M.; Zador, E.; Waelkens, E.; Raeymaekers, L.; Wuytack, F. Sarcolipin and phospholamban mRNA and protein expression in cardiac and skeletal muscle of different species. Biochem. J. 2005, 389, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, V.A.; Bombardier, E.; Vigna, C.; Devji, T.; Bloemberg, D.; Gamu, D.; Gramolini, A.O.; Quadrilatero, J.; Tupling, A.R. Co-expression of SERCA isoforms, phospholamban and sarcolipin in human skeletal muscle fibers. PLoS One 2013, 8, e84304. [Google Scholar] [CrossRef] [PubMed]
- Asahi, M.; Kurzydlowski, K.; Tada, M.; MacLennan, D.H. Sarcolipin inhibits polymerization of phospholamban to induce superinhibition of sarco(endo)plasmic reticulum Ca2+-ATPases (SERCAs). J. Biol. Chem. 2002, 277, 26725–26728. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Kirchberger, M.A. Significance of the membrane protein phospholamban in cyclic AMP-mediated regulation of calcium transport by sarcoplasmic reticulum. Recent Adv. Stud. Cardiac. Struct. Metab. 1976, 11, 265–272. [Google Scholar] [PubMed]
- Strehler, E.E.; Zacharias, D.A. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol. Rev. 2001, 81, 21–50. [Google Scholar] [PubMed]
- Baker, J.S.; McCormick, M.C.; Robergs, R.A. Interaction among skeletal muscle metabolic energy systems during intense exercise. J. Nutr. Metab. 2010. [Google Scholar] [CrossRef]
- Schonekess, B.O.; Brindley, P.G.; Lopaschuk, G.D. Calcium regulation of glycolysis, glucose oxidation, and fatty acid oxidation in the aerobic and ischemic heart. Can. J. Physiol. Pharmacol. 1995, 73, 1632–1640. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. The role of calcium ions, calmodulin and troponin in the regulation of phosphorylase kinase from rabbit skeletal muscle. Eur. J. Biochem. 1980, 111, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Sola-Penna, M.; Da, S.D.; Coelho, W.S.; Marinho-Carvalho, M.M.; Zancan, P. Regulation of mammalian muscle type 6-phosphofructo-1-kinase and its implication for the control of the metabolism. IUBMB Life 2010, 62, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Korzeniewski, B. Regulation of oxidative phosphorylation through parallel activation. Biophys. Chem. 2007, 129, 93–110. [Google Scholar] [CrossRef] [PubMed]
- McMillin, J.B.; Madden, M.C. The role of calcium in the control of respiration by muscle mitochondria. Med. Sci. Sports Exerc. 1989, 21, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 2013, 52, 2793–2809. [Google Scholar] [CrossRef] [PubMed]
- Territo, P.R.; Mootha, V.K.; French, S.A.; Balaban, R.S. Ca2+ activation of heart mitochondrial oxidative phosphorylation: Role of the F0/F1-ATPase. Am. J. Physiol. Cell Physiol. 2000, 278, C423–C435. [Google Scholar] [PubMed]
- Rattray, B.; Thompson, M.; Ruell, P.; Caillaud, C. Specific training improves skeletal muscle mitochondrial calcium homeostasis after eccentric exercise. Eur. J. Appl. Physiol. 2013, 113, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Madsen, K.; Ertbjerg, P.; Djurhuus, M.S.; Pedersen, P.K. Calcium content and respiratory control index of skeletal muscle mitochondria during exercise and recovery. Am. J. Physiol. 1996, 271, E1044–E1050. [Google Scholar] [PubMed]
- Picard, M.; Csukly, K.; Robillard, M.E.; Godin, R.; Ascah, A.; Bourcier-Lucas, C.; Burelle, Y. Resistance to Ca2+-induced opening of the permeability transition pore differs in mitochondria from glycolytic and oxidative muscles. Am. J. Physiol Regul. Integr. Comp. Physiol. 2008, 295, R659–R668. [Google Scholar] [CrossRef] [PubMed]
- Semsarian, C.; Wu, M.J.; Ju, Y.K.; Marciniec, T.; Yeoh, T.; Allen, D.G.; Harvey, R.P.; Graham, R.M. Skeletal muscle hypertrophy is mediated by a Ca2+-dependent calcineurin signalling pathway. Nature 1999, 400, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell. Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, G.R. Calcium, calcineurin, and the control of transcription. J. Biol. Chem. 2001, 276, 2313–2316. [Google Scholar] [CrossRef] [PubMed]
- Musaro, A.; McCullagh, K.J.; Naya, F.J.; Olson, E.N.; Rosenthal, N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature 1999, 400, 581–585. [Google Scholar] [CrossRef]
- Talmadge, R.J.; Otis, J.S.; Rittler, M.R.; Garcia, N.D.; Spencer, S.R.; Lees, S.J.; Naya, F.J. Calcineurin activation influences muscle phenotype in a muscle-specific fashion. BMC. Cell. Biol. 2004, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, K.; Yamaguchi, A. Molecular mechanisms in aging and current strategies to counteract sarcopenia. Curr. Aging Sci. 2010, 3, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, K.; Akiho, M.; Nakashima, H.; Nakao, R.; Hirata, M.; Inashima, S.; Yamaguchi, A.; Yasuhara, M. Cyclosporin A modulates cellular localization of MEF2C protein and blocks fiber hypertrophy in the overloaded soleus muscle of mice. Acta Neuropathol. 2008, 115, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Friday, B.B.; Mitchell, P.O.; Kegley, K.M.; Pavlath, G.K. Calcineurin initiates skeletal muscle differentiation by activating MEF2 and MyoD. Differentiation 2003, 71, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Friday, B.B.; Horsley, V.; Pavlath, G.K. Calcineurin activity is required for the initiation of skeletal muscle differentiation. J. Cell Biol. 2000, 149, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, F.S.; Dellavalle, A.; Diaz-Manera, J.; Messina, G.; Cossu, G. Repairing skeletal muscle: Regenerative potential of skeletal muscle stem cells. J. Clin. Investig. 2010, 120, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Verdijk, L.B.; Gleeson, B.G.; Jonkers, R.A.; Meijer, K.; Savelberg, H.H.; Dendale, P.; van Loon, L.J. Skeletal muscle hypertrophy following resistance training is accompanied by a fiber type-specific increase in satellite cell content in elderly men. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.M.; Verburg, E.; Lamb, G.D. Ca2+ activation of diffusible and bound pools of μ-calpain in rat skeletal muscle. J. Physiol. 2006, 576, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Richard, I.; Broux, O.; Allamand, V.; Fougerousse, F.; Chiannilkulchai, N.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995, 81, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Westerblad, H. Role of phosphate and calcium stores in muscle fatigue. J. Physiol. 2001, 536, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Baylor, S.M.; Hollingworth, S. Sarcoplasmic reticulum calcium release compared in slow-twitch and fast-twitch fibres of mouse muscle. J. Physiol. 2003, 551, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The calpain system. Physiol. Rev. 2003, 83, 731–801. [Google Scholar] [PubMed]
- Melloni, E.; Michetti, M.; Salamino, F.; Minafra, R.; Pontremoli, S. Modulation of the calpain autoproteolysis by calpastatin and phospholipids. Biochem. Biophys. Res. Commun. 1996, 229, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Pontremoli, S.; Viotti, P.L.; Michetti, M.; Sparatore, B.; Salamino, F.; Melloni, E. Identification of an endogenous activator of calpain in rat skeletal muscle. Biochem. Biophys. Res. Commun. 1990, 171, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Kar, N.C.; Pearson, C.M. A calcium-activated neutral protease in normal and dystrophic human muscle. Clin. Chim. Acta 1976, 73, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.J.; Mellgren, R.L. Overexpression of a calpastatin transgene in mdx muscle reduces dystrophic pathology. Hum. Mol. Genet. 2002, 11, 2645–2655. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, F.; Carnac, G.; Marcilhac, A.; Benyamin, Y. m-Calpain implication in cell cycle during muscle precursor cell activation. Exp. Cell Res. 2004, 298, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Temm-Grove, C.J.; Wert, D.; Thompson, V.F.; Allen, R.E.; Goll, D.E. Microinjection of calpastatin inhibits fusion in myoblasts. Exp. Cell Res. 1999, 247, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Nikawa, T.; Ikemoto, M.; Sakai, T.; Kano, M.; Kitano, T.; Kawahara, T.; Teshima, S.; Rokutan, K.; Kishi, K. Effects of a soy protein diet on exercise-induced muscle protein catabolism in rats. Nutrition 2002, 18, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Langlais, P.J.; Huang, P.C.; Gabay, S. The involvement of monoaminergic mechanisms in a chemically induced hyperkinetic syndrome in the rat. Psychopharmacol. Bull. 1975, 11, 38–39. [Google Scholar] [PubMed]
- Schiaffino, S.; Mammucari, C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: Insights from genetic models. Skelet. Muscle 2011, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Weigl, L.G. Lost in translation: Regulation of skeletal muscle protein synthesis. Curr. Opin. Pharmacol. 2012, 12, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Shah, O.J.; Anthony, J.C.; Kimball, S.R.; Jefferson, L.S. 4E-BP1 and S6K1: Translational integration sites for nutritional and hormonal information in muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E715–E729. [Google Scholar]
- Spangenburg, E.E. Changes in muscle mass with mechanical load: Possible cellular mechanisms. Appl. Physiol. Nutr. Metab. 2009, 34, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Ducret, T.; Vandebrouck, C.; Cao, M.L.; Lebacq, J.; Gailly, P. Functional role of store-operated and stretch-activated channels in murine adult skeletal muscle fibres. J. Physiol. 2006, 575, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Gulati, P.; Gaspers, L.D.; Dann, S.G.; Joaquin, M.; Nobukuni, T.; Natt, F.; Kozma, S.C.; Thomas, A.P.; Thomas, G. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008, 7, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Krokhmal, A.I. Prevention of complications of adenomectomy. Vrach. Delo 1986, 7, 120–122. [Google Scholar] [PubMed]
- Rose, A.J.; Bisiani, B.; Vistisen, B.; Kiens, B.; Richter, E.A. Skeletal muscle eEF2 and 4EBP1 phosphorylation during endurance exercise is dependent on intensity and muscle fiber type. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R326–R333. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Ojuka, E.O.; Jones, T.E.; Han, D.H.; Chen, M.; Holloszy, J.O. Raising Ca2+ in L6 myotubes mimics effects of exercise on mitochondrial biogenesis in muscle. FASEB J. 2003, 17, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, T.; Ribar, T.J.; Williams, R.S.; Yan, Z. Skeletal muscle adaptation in response to voluntary running in Ca2+/calmodulin-dependent protein kinase IV-deficient mice. Am. J. Physiol. Cell Physiol. 2004, 287, C1311–C1319. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Ribar, T.J.; Cummings, D.E.; Burton, K.A.; McKnight, G.S.; Means, A.R. Spermiogenesis and exchange of basic nuclear proteins are impaired in male germ cells lacking Camk4. Nat. Genet. 2000, 25, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Ren, J.M.; Young, L.H.; Pypaert, M.; Mu, J.; Birnbaum, M.J.; Shulman, G.I. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. USA 2002, 99, 15983–15987. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.J.; Hargreaves, M. Exercise increases Ca2+-calmodulin-dependent protein kinase II activity in human skeletal muscle. J. Physiol. 2003, 553, 303–309. [Google Scholar]
- Wu, H.; Kanatous, S.B.; Thurmond, F.A.; Gallardo, T.; Isotani, E.; Bassel-Duby, R.; Williams, R.S. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science 2002, 296, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Spiegelman, B.M. The role of exercise and PGC-1α in inflammation and chronic disease. Nature 2008, 454, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.R.; Olson, E.N.; Richardson, J.A.; Yang, Q.; Humphries, C.; Shelton, J.M.; Wu, H.; Zhu, W.; Bassel-Duby, R.; Williams, R.S. A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev. 1998, 12, 2499–2509. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.; Carson, B.P.; Garcia-Roves, P.M.; Chibalin, A.V.; Sarsfield, F.M.; Barron, N.; McCaffrey, N.; Moyna, N.M.; Zierath, J.R.; O’Gorman, D.J. Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor coactivator-1 mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J. Physiol. 2010, 588, 1779–1790. [Google Scholar] [PubMed]
- Kusuhara, K.; Madsen, K.; Jensen, L.; Hellsten, Y.; Pilegaard, H. Calcium signalling in the regulation of PGC-1α, PDK4 and HKII mRNA expression. Biol. Chem. 2007, 388, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J.; Kiilerich, K.; Pilegaard, H. PGC-1α-mediated adaptations in skeletal muscle. Pflugers Arch. 2010, 460, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Tothova, J.; Blaauw, B.; Pallafacchina, G.; Rudolf, R.; Argentini, C.; Reggiani, C.; Schiaffino, S. NFATc1 nucleocytoplasmic shuttling is controlled by nerve activity in skeletal muscle. J. Cell Sci. 2006, 119, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Calabria, E.; Ciciliot, S.; Moretti, I.; Garcia, M.; Picard, A.; Dyar, K.A.; Pallafacchina, G.; Tothova, J.; Schiaffino, S.; Murgia, M. NFAT isoforms control activity-dependent muscle fiber type specification. Proc. Natl. Acad. Sci. USA 2009, 106, 13335–13340. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.S.; Shi, X.; Voelker, K.A.; Grange, R.W.; Garcia, J.A.; Hammer, R.E.; Garry, D.J. Foxj3 transcriptionally activates MEF2C and regulates adult skeletal muscle fiber type identity. Dev. Biol. 2010, 337, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S. Fibre types in skeletal muscle: A personal account. Acta Physiol. 2010, 199, 451–463. [Google Scholar] [CrossRef]
- Klee, C.B.; Ren, H.; Wang, X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J. Biol. Chem. 1998, 273, 13367–13370. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, H.; Olson, E.N.; Williams, R.S. Remodeling muscles with calcineurin. Bioessays 2000, 22, 1049. [Google Scholar] [CrossRef] [PubMed]
- Tavi, P.; Westerblad, H. The role of in vivo Ca2+ signals acting on Ca2+-calmodulin-dependent proteins for skeletal muscle plasticity. J. Physiol. 2011, 589, 5021–5031. [Google Scholar] [CrossRef] [PubMed]
- Stemmer, P.M.; Klee, C.B. Dual calcium ion regulation of calcineurin by calmodulin and calcineurin B. Biochemistry 1994, 33, 6859–6866. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cseresnyes, Z.; Randall, W.R.; Schneider, M.F. Activity-dependent nuclear translocation and intranuclear distribution of NFATc in adult skeletal muscle fibers. J. Cell Biol. 2001, 155, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Randall, W.R.; Schneider, M.F. Activity-dependent and -independent nuclear fluxes of HDAC4 mediated by different kinases in adult skeletal muscle. J. Cell Biol. 2005, 168, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.C.; Geiger, P.C.; Han, D.H.; Jones, T.E.; Holloszy, J.O. Calcium induces increases in peroxisome proliferator-activated receptor gamma coactivator-1alpha and mitochondrial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation. J. Biol. Chem. 2007, 282, 18793–18799. [Google Scholar] [CrossRef] [PubMed]
- Seto, J.T.; Quinlan, K.G.; Lek, M.; Zheng, X.F.; Garton, F.; MacArthur, D.G.; Hogarth, M.W.; Houweling, P.J.; Gregorevic, P.; Turner, N.; et al. ACTN3 genotype influences muscle performance through the regulation of calcineurin signaling. J. Clin. Investig. 2013, 123, 4255–4263. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, D.G.; North, K.N. A gene for speed? The evolution and function of α-actinin-3. Bioessays 2004, 26, 786–795. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, D.G.; Seto, J.T.; Raftery, J.M.; Quinlan, K.G.; Huttley, G.A.; Hook, J.W.; Lemckert, F.A.; Kee, A.J.; Edwards, M.R.; Berman, Y.; et al. Loss of ACTN3 gene function alters mouse muscle metabolism and shows evidence of positive selection in humans. Nat. Genet. 2007, 39, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; MacArthur, D.G.; Gulbin, J.P.; Hahn, A.G.; Beggs, A.H.; Easteal, S.; North, K. ACTN3 genotype is associated with human elite athletic performance. Am. J. Hum. Genet. 2003, 73, 627–631. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gehlert, S.; Bloch, W.; Suhr, F. Ca2+-Dependent Regulations and Signaling in Skeletal Muscle: From Electro-Mechanical Coupling to Adaptation. Int. J. Mol. Sci. 2015, 16, 1066-1095. https://doi.org/10.3390/ijms16011066
Gehlert S, Bloch W, Suhr F. Ca2+-Dependent Regulations and Signaling in Skeletal Muscle: From Electro-Mechanical Coupling to Adaptation. International Journal of Molecular Sciences. 2015; 16(1):1066-1095. https://doi.org/10.3390/ijms16011066
Chicago/Turabian StyleGehlert, Sebastian, Wilhelm Bloch, and Frank Suhr. 2015. "Ca2+-Dependent Regulations and Signaling in Skeletal Muscle: From Electro-Mechanical Coupling to Adaptation" International Journal of Molecular Sciences 16, no. 1: 1066-1095. https://doi.org/10.3390/ijms16011066
APA StyleGehlert, S., Bloch, W., & Suhr, F. (2015). Ca2+-Dependent Regulations and Signaling in Skeletal Muscle: From Electro-Mechanical Coupling to Adaptation. International Journal of Molecular Sciences, 16(1), 1066-1095. https://doi.org/10.3390/ijms16011066