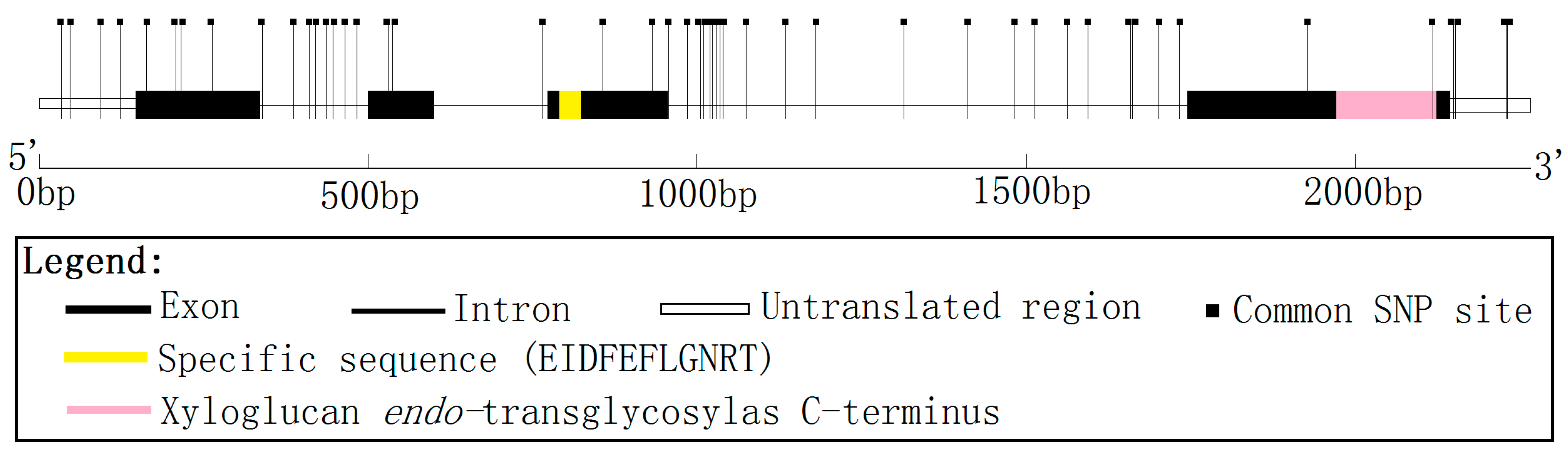
2.1. Isolation of PtoXET16A from P. tomentosa
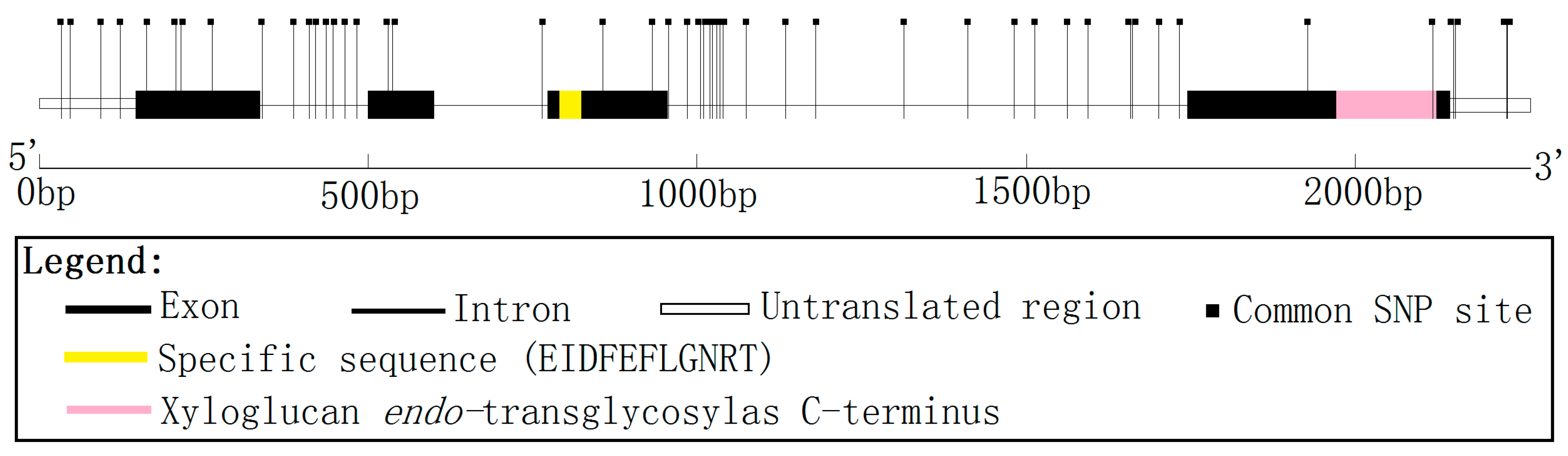
A full-length cDNA encoding a XET16A-like protein was isolated from a cDNA library prepared from mature xylem zone of
P. tomentosa. The cDNA clone (GenBank Accession No. KM267530) is 1141 bp in length, and contains a full-length open reading frame (870 bp), encoding a polypeptide of 290 amino acids with an estimated molecular mass of 33.70 kD and a pI of 7.62 (
http://web.expasy.org/protparam/), flanked by 146 bp of 5' untranslated region (5'UTR) and 122 bp of 3'UTR (
Figure 1). Alignment of the full-length cDNA sequence to the genomic sequence showed that
PtoXET16A has three introns and four exons (
Figure 1). Identification of protein domains, families and functional sites by matches to the Prosite database (
http://prosite.expasy.org/prosite.html) and analysis of the protein sequence for Pfam matches (
http://pfam.sanger.ac.uk/) showed that the predicted protein has the active site of glycosyl hydrolase family 16 EIDFEFLGNRT (at residues 107–117) (
Figure 1) and an XET
C-terminal sequence in the fourth exon (at residues 234–284) (
Figure 1).
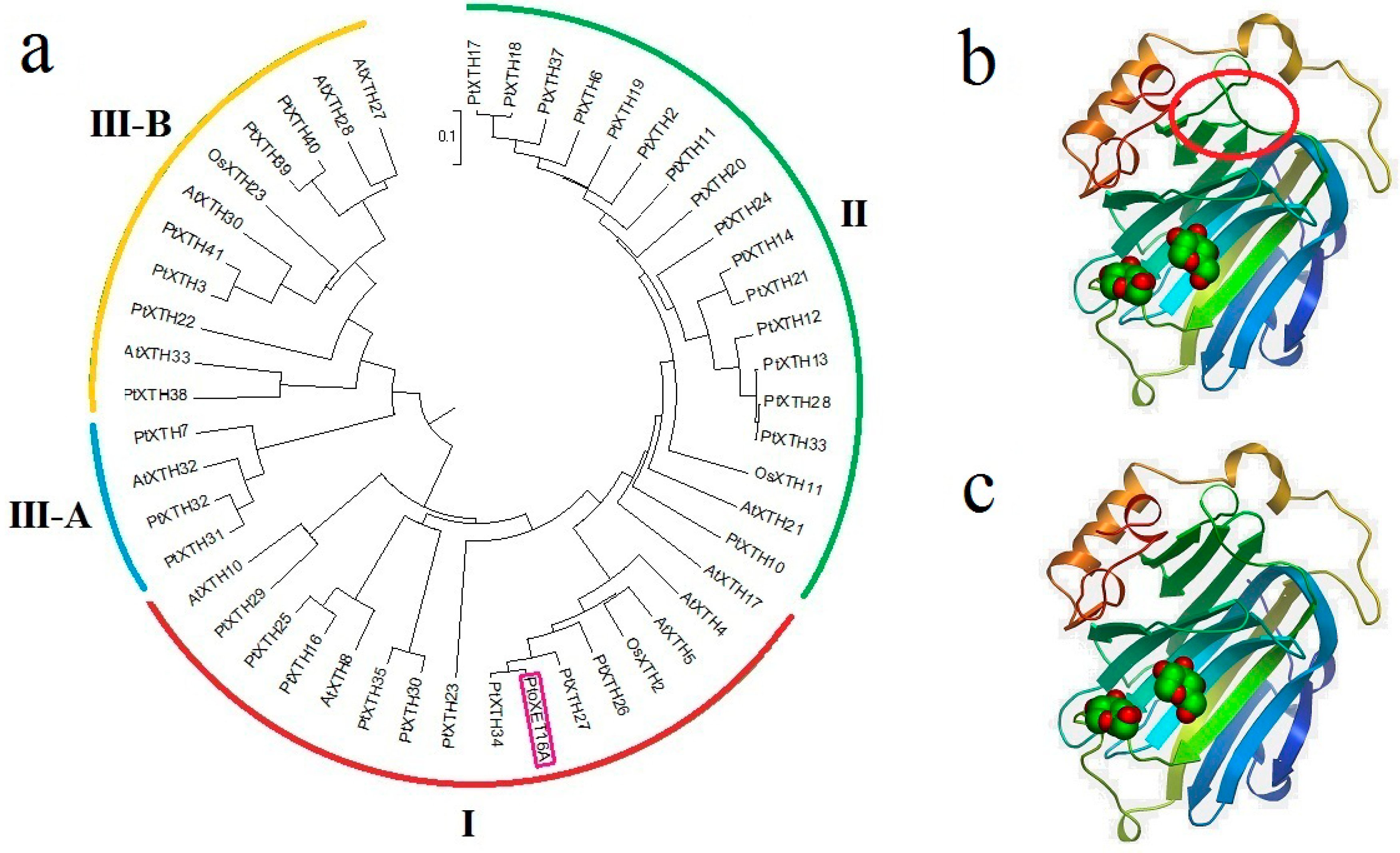
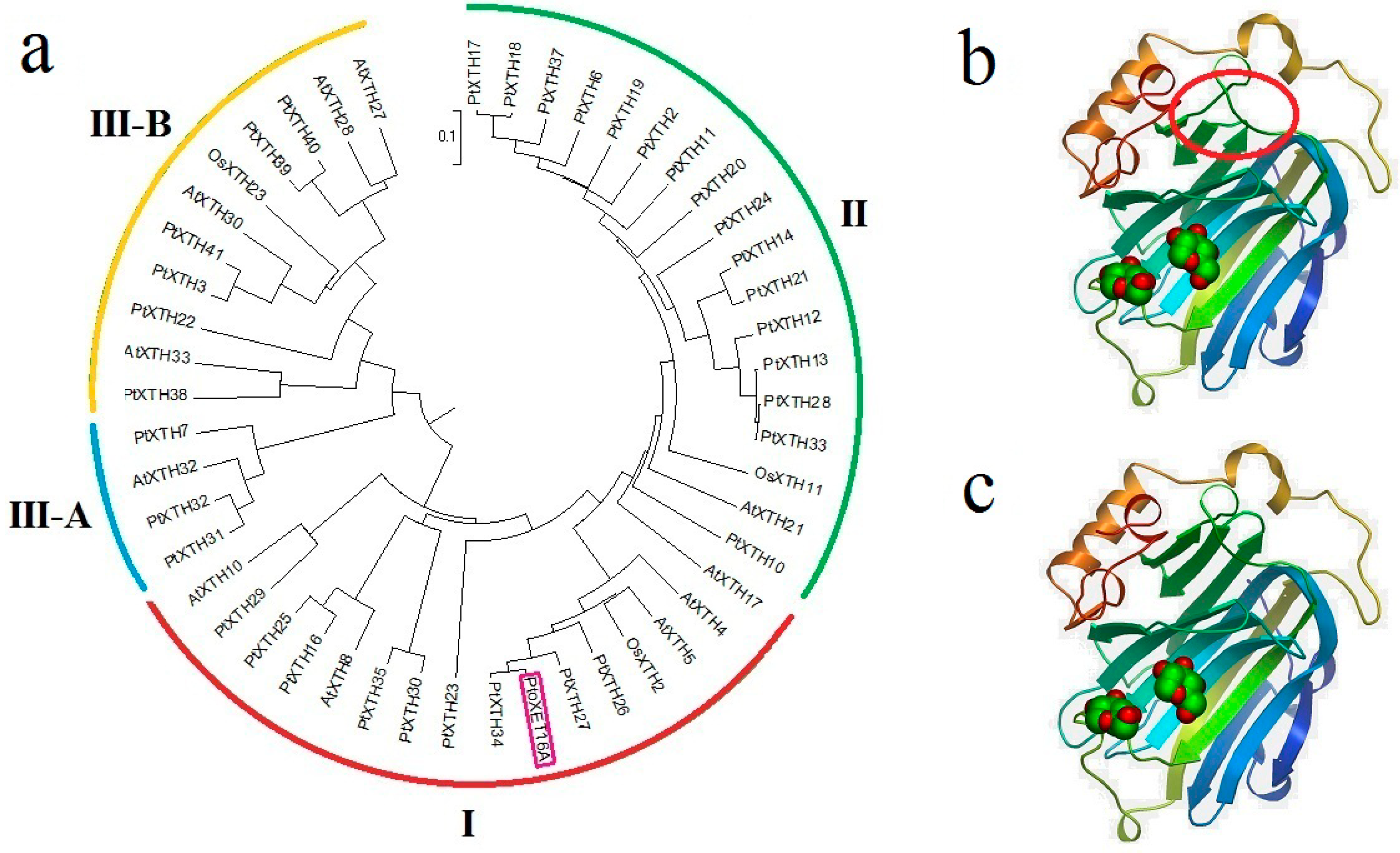
The molecular phylogeny of
XTH gene products includes three major branches (I/II, IIIA and IIIB) (
Figure 2). Of these, the largest cluster confirmed previous studies that suggested merging groups I and II. This analysis indicates that
PtoXET16A belongs to group I. A BLASTP search with PtoXET16A as the query sequence revealed that the PtoXET16A protein shares 98% identity with PttXET16-34 (AAN87142), 79% identity with AtXTH5 (AT5G13870) and 76% with OsXTH2 (Os11g0539200) (
Figure 2,
Table S1). The alignment shows that PtoXET16A lacks four amino acids (YIIV) that are present in the XET16As from other species. The tertiary structure predicted using Swissmodel (
http://swissmodel.expasy.org/), showed that PtoXET16A and PttXET16-34 have similar structures. However, the amino acids missing in PtoXET16A but present in PttXET16-34 did produce a structural difference in one region (
Figure 2).
Figure 1.
Genomic organization of
PtoXET16A. Exons are shown as boxes and introns as lines. Positions of common SNP markers are shown as vertical lines. The active site of glycosyl hydrolases family 16 EIDFEFLGNRT (at residues 107–117) and a xyloglucan
endo-transglycosylase (XET)
C-terminus in fourth exon (at residues 234–284), identified by analysis of protein sequence for Pfam matches (
http://pfam.sanger.ac.uk/), are shown.
Figure 1.
Genomic organization of
PtoXET16A. Exons are shown as boxes and introns as lines. Positions of common SNP markers are shown as vertical lines. The active site of glycosyl hydrolases family 16 EIDFEFLGNRT (at residues 107–117) and a xyloglucan
endo-transglycosylase (XET)
C-terminus in fourth exon (at residues 234–284), identified by analysis of protein sequence for Pfam matches (
http://pfam.sanger.ac.uk/), are shown.
2.2. Analysis of PtoXET16A Expression
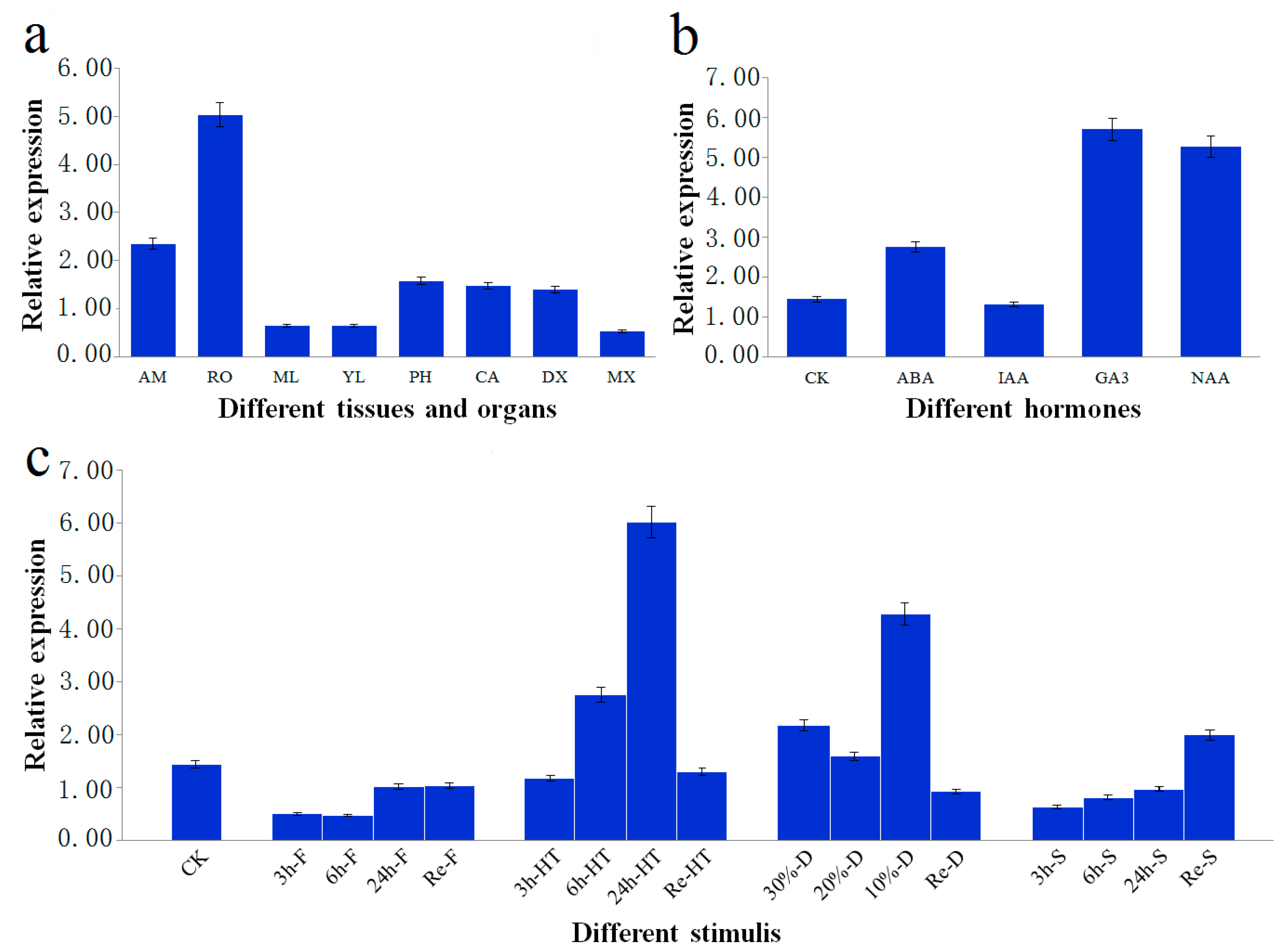
We determined to what extent
PtoXET16A exhibits tissue-specific expression in
P.
tomentosa. Levels of
PtoXET16A mRNA in various poplar tissues, including apical meristem, root, phloem, cambium, developing xylem, mature xylem, young leaf and mature leaf, were measured by quantitative real time-PCR (RT-PCR) with gene-specific primers and
Actin as an internal control (
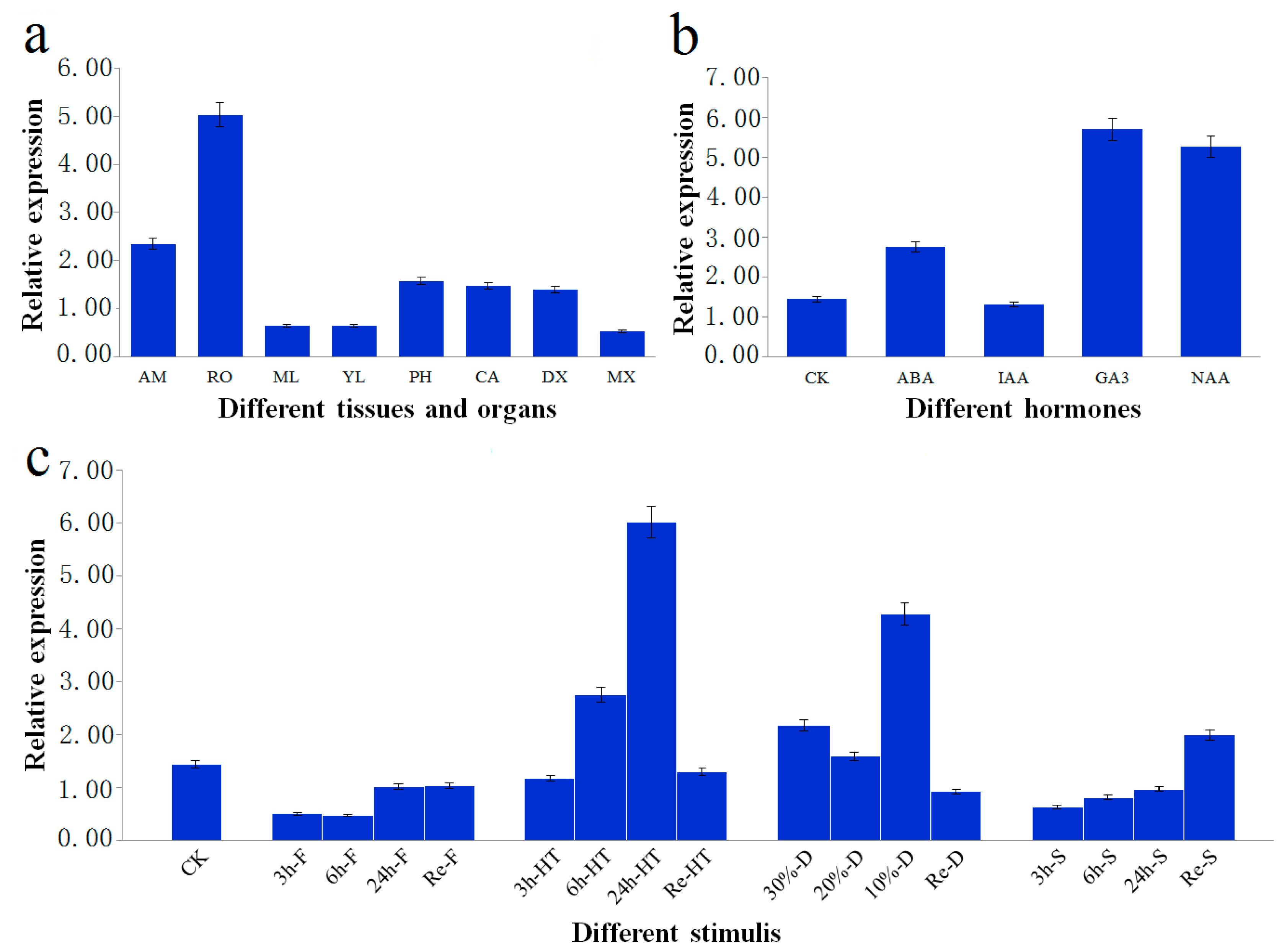
Figure 3a).
PtoXET16A mRNA was the most abundant in root (5.033 ± 0.012), followed by phloem (1.573 ± 0.002), cambium (1.471 ± 0.009), and developing xylem (1.392 ± 0.006). In contrast, relatively lower abundances of
PtoXET16A mRNA were detected in mature leaf (0.647 ± 0.013), young leaf (0.637 ± 0.002) and mature xylem (0.530 ± 0.016). These observations indicated that
PtoXET16A shows preferential expression in vascular tissues, suggesting that
PtoXET16A plays an important role in wood formation.
We further tested whether hormone treatments induced
PtoXET16A expression, testing abscisic acid (ABA), indoleacetic acid (IAA), GA, and naphthylacetic acetic acid (NAA) (
Figure 3b) in
P.
tomentosa. The results revealed that the expression of
PtoXET16A was induced by treatment with most plant hormones, except for IAA (
Figure 3b). Of these treatments, the expression level of
PtoXET16A following GA treatment was more than four times higher than the controls, indicating that the expression of
PtoXET16A could be strongly regulated by GA.
Figure 2.
A rooted phylogenetic tree and three-dimensional structures of
XTH gene products. (
a) A rooted phylogenetic tree of PtoXET16A and other predicted products of
XTH genes. The similarity to other
XTH gene products was calculated using the UPGMA program. Full-length protein sequences were used for the comparison and the gene models used are listed in
Table S1. The phylogenetic tree presents predicted protein sequences for the
XTH family of
P. trichocarpa, numbered according to Geisler-Lee
et al. [
9],
Arabidopsis
thaliana XTH proteins, numbered according to Yokoyama and Nishitani [
7], and
Oryza sativa XTH gene products, numbered according to Yokoyama
et al. [
8]; (
b) Three-dimensional structures of PtoXET16A constructed using Swissmodel (
http://swissmodel.expasy.org/); (
c) Three-dimensional structures of PttXET16-34, constructed using Swissmodel (
http://swissmodel.expasy.org/). The polypeptide chain is colored from blue (
N terminus) to red (
C terminus). The red circle shows the location of four missing amino acids (YIIV) compared with PttXET16-34.
Figure 2.
A rooted phylogenetic tree and three-dimensional structures of
XTH gene products. (
a) A rooted phylogenetic tree of PtoXET16A and other predicted products of
XTH genes. The similarity to other
XTH gene products was calculated using the UPGMA program. Full-length protein sequences were used for the comparison and the gene models used are listed in
Table S1. The phylogenetic tree presents predicted protein sequences for the
XTH family of
P. trichocarpa, numbered according to Geisler-Lee
et al. [
9],
Arabidopsis
thaliana XTH proteins, numbered according to Yokoyama and Nishitani [
7], and
Oryza sativa XTH gene products, numbered according to Yokoyama
et al. [
8]; (
b) Three-dimensional structures of PtoXET16A constructed using Swissmodel (
http://swissmodel.expasy.org/); (
c) Three-dimensional structures of PttXET16-34, constructed using Swissmodel (
http://swissmodel.expasy.org/). The polypeptide chain is colored from blue (
N terminus) to red (
C terminus). The red circle shows the location of four missing amino acids (YIIV) compared with PttXET16-34.
![]()
We further tested whether
PtoXET16A was inducible by different abiotic stresses (
Figure 3c). Similar expression patterns were observed in plants exposed to freezing, heat and high-salinity stresses, in which the relative
PtoXET16A mRNA levels gradually increased over the course of the stress treatment (
Figure 3c). Compared with the control, the relative expression of
PtoXET16A was repressed in freezing and high-salinity stresses; conversely,
PtoXET16A expression was significantly induced in heat and drought conditions. When the plants recovered,
PtoXET16A expression returned to the level of the control (
Figure 3c). We also found that the strongest relative expression of
PtoXET16A was in response to high-temperature stress (
Figure 3c). Under drought conditions, the highest expression was observed at 10% soil water content (4.260 ± 0.011), followed by 30% soil water content (2.171 ± 0.007) (
Figure 3c). These results indicated that
PtoXET16A expression is sensitive to heat and drought stimuli.
Figure 3.
Relative transcript levels of PtoXET16A. The error bars represent ± standard deviation. (a) Relative transcript levels of PtoXET16A in P. tomentosa tissues and organs. AM, apical meristem; RO, root; ML, mature leaves; YL, young leaves; PH, phloem; CA, cambium; DX, developing xylem; MX, mature xylem; (b) Relative transcript levels of PtoXET16A before and after different treatments in P. tomentosa. CK, control check; ABA, abscisic acid; IAA, indoleacetic acid; GA, gibberellin acid; NAA, naphthylacetic acetic acid. (c) Expression analysis in P. tomentosa of PtoXET16A in response to abiotic stresses; CK, control check; Re, recovered condition; F, freezing stress; HT, high-temperature stress; D, drought condition; S, high-salinity stress. Samples were exposed to 150 mM NaCl, 4 °C and 42 °C for 3, 6, and 24 h for high-salinity, freezing stress and high temperature stress treatments, respectively. Drought condition was induced by withholding soil water content to 30%, 20%, and 10% of their original content at room temperature. The treated plants were then transferred to pots under normal growing conditions for 24 h to recover from cold, heat, drought and high salinity, which were denoted as Re-F, Re-HT, Re-D, Re-S, respectively. As control, samples without treatments were used.
Figure 3.
Relative transcript levels of PtoXET16A. The error bars represent ± standard deviation. (a) Relative transcript levels of PtoXET16A in P. tomentosa tissues and organs. AM, apical meristem; RO, root; ML, mature leaves; YL, young leaves; PH, phloem; CA, cambium; DX, developing xylem; MX, mature xylem; (b) Relative transcript levels of PtoXET16A before and after different treatments in P. tomentosa. CK, control check; ABA, abscisic acid; IAA, indoleacetic acid; GA, gibberellin acid; NAA, naphthylacetic acetic acid. (c) Expression analysis in P. tomentosa of PtoXET16A in response to abiotic stresses; CK, control check; Re, recovered condition; F, freezing stress; HT, high-temperature stress; D, drought condition; S, high-salinity stress. Samples were exposed to 150 mM NaCl, 4 °C and 42 °C for 3, 6, and 24 h for high-salinity, freezing stress and high temperature stress treatments, respectively. Drought condition was induced by withholding soil water content to 30%, 20%, and 10% of their original content at room temperature. The treated plants were then transferred to pots under normal growing conditions for 24 h to recover from cold, heat, drought and high salinity, which were denoted as Re-F, Re-HT, Re-D, Re-S, respectively. As control, samples without treatments were used.
![]()
2.3. SNP Diversity and Genotyping
To identify SNPs in
PtoXET16A, the approximately 2266 bp genomic region of
PtoXET16A was amplified and sequenced from 43 unrelated individuals, representing almost the entire natural range of
P.
tomentosa.
Table 1 summarizes the statistical analysis of nucleotide polymorphisms over different regions of
PtoXET16A. Across the samples, 134 SNPs were detected in the whole gene at a frequency of approximately one SNP every 17 bp (
Table 1). Forty-three of these SNPs occurred in exons, and included 18 missense and 25 nonsense mutations (
Table 1). All together, 49 of 134 SNPs (38.1%) were considered as common (frequency > 0.10). In general, the
PtoXET16A locus has high nucleotide diversity (π), with π = 0.01266 and θ
w = 0.01392, respectively (
Table 1). More specifically, estimates of nucleotide diversity, π, for the different gene regions ranged from 0.00239 (intron 2) to 0.02461 (intron 1), and θ
w varied between 0.00142 (intron 2) and 0.01985 (intron 1). Within coding regions, the non-synonymous nucleotide substitution rate (π
nonsyn) was markedly lower than π
syn, with a π
nonsyn/π
syn ratio of 0.2554 < 1.0, suggesting that diversity at the synonymous sites of exon regions resulted from strong purifying selection (
Table 1). The 49 common SNPs were successfully genotyped across 426 trees in the association population by using locked nucleic acid technology.
Genetic differentiation within and among three geographically independent climatic regions was examined using the nucleotide diversity data from
PtoXET16A (
Table 2). Levels of nucleotide variation (measured using π) in the three climatic regions varied, but showed similar patterns of π
tot, π
sil, π
s and π
n (
Table 2). These observations suggested that the level of selective constraint was similar between the three climatic regions. Tajima’s D was positive in the southern, northeastern and northwestern climatic regions but negative in the
P. tomentosa population as a whole; however, no significant departures from the neutral expectation were observed (
Table 2). The Fu and Li’s D statistical tests were positive for the northeastern and northwestern populations, but were negative for the southern region and the
P. tomentosa population as a whole, revealing the existence of an excess of low-frequency mutations for this gene region in the
P. tomentosa species-wide samples (
Table 2).
Table 1.
Nucleotide polymorphisms at the PtoXET16A locus.
Table 1.
Nucleotide polymorphisms at the PtoXET16A locus.
| Region | No. of bp | No. of Polymorphic Sites | Percentage Polymorphism | Nucleotide Diversity |
|---|
| π | θw |
|---|
| 5'UTR | 146 | 5 | 3.42 | 0.01350 | 0.00792 |
| Exon 1 | 190 | 12 | 6.32 | 0.01226 | 0.01460 |
| Synonymous | 45.10 | 3 | 6.65 | 0.01290 | 0.01537 |
| Non-synonymous | 143.9 | 9 | 6.25 | 0.01216 | 0.01446 |
| Intron 1 | 163 | 14 | 8.59 | 0.02461 | 0.01985 |
| Exon 2 | 101 | 4 | 3.96 | 0.00947 | 0.00915 |
| Synonymous | 23.00 | 4 | 17.39 | 0.04160 | 0.04019 |
| Non-synonymous | 76.00 | 0 | 0.00 | 0.00000 | 0.00000 |
| Intron 2 | 173 | 1 | 0.58 | 0.00239 | 0.00142 |
| Exon 3 | 182 | 2 | 1.10 | 0.00433 | 0.00254 |
| Synonymous | 38.28 | 1 | 2.61 | 0.00885 | 0.00604 |
| Non-synonymous | 141.72 | 1 | 0.71 | 0.00317 | 0.00163 |
| Intron 3 | 789 | 61 | 7.73 | 0.01746 | 0.01851 |
| Exon 4 | 400 | 25 | 6.25 | 0.00548 | 0.01445 |
| Synonymous | 83.60 | 10 | 11.96 | 0.01682 | 0.02765 |
| Non-synonymous | 312.40 | 15 | 4.80 | 0.00252 | 0.01110 |
| 3'UTR | 122 | 10 | 8.20 | 0.01810 | 0.01894 |
| Total | 2266 | 134 | 5.91 | 0.01266 | 0.01392 |
| Synonymous | 190.98 | 18 | 9.43 | 0.01719 | 0.02178 |
| Non-synonymous | 679.02 | 25 | 3.68 | 0.00439 | 0.00851 |
Table 2.
Summary of nucleotide variation in PtoXET16A in P. tomentosa natural populations from three climatic regions.
Table 2.
Summary of nucleotide variation in PtoXET16A in P. tomentosa natural populations from three climatic regions.
| Population | N | πtot | πsil | πs | πn | Tajima’s D | Fu and Li’s D |
|---|
| Northeastern region | 14 | 0.01380 | 0.01786 | 0.01747 | 0.00447 | 0.95307 | 0.51753 |
| Southern region | 15 | 0.01309 | 0.01657 | 0.01814 | 0.00505 | 0.71556 | −0.31283 |
| Northwestern region | 14 | 0.01166 | 0.01509 | 0.01670 | 0.00377 | 0.48336 | 0.02633 |
| Total | 43 | 0.01266 | 0.01625 | 0.01719 | 0.00439 | −0.33198 | −2.48533 |
2.4. Linkage Disequilibrium and Phenotype-Genotype Associations
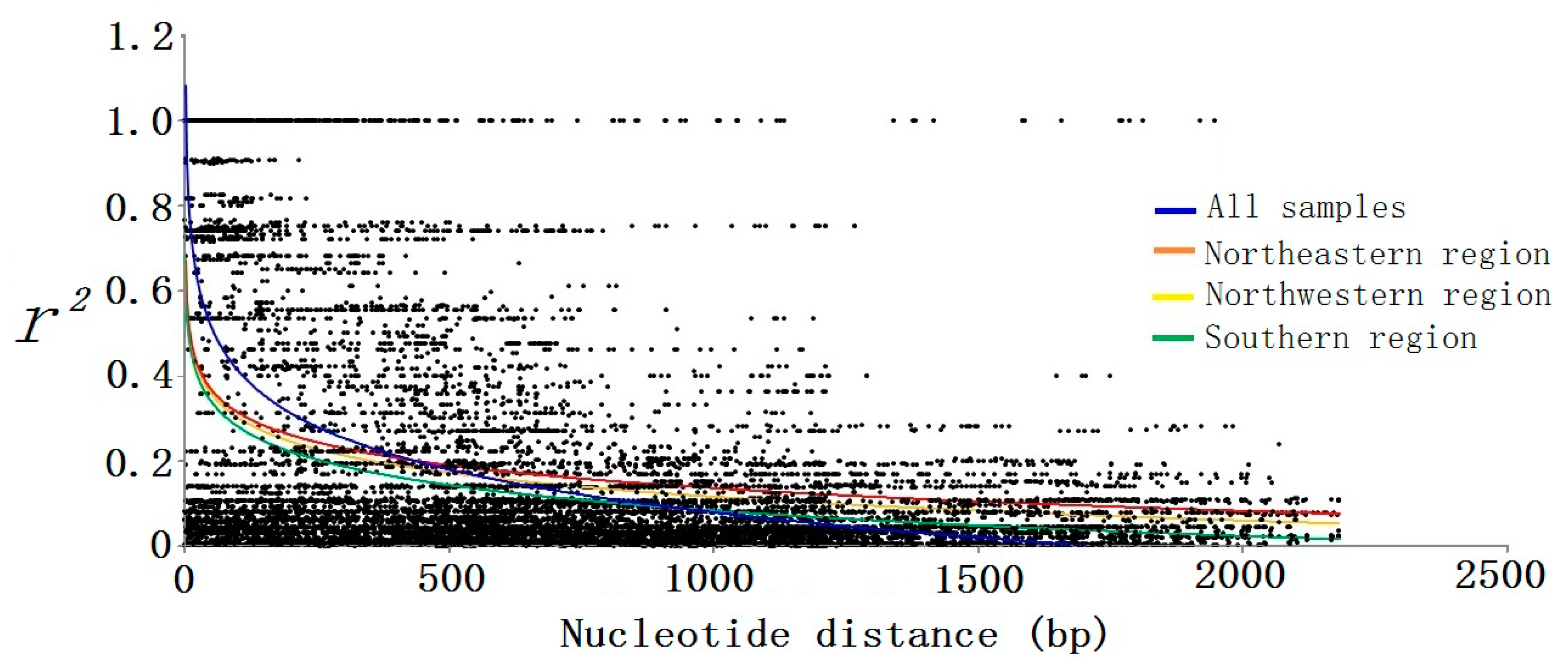
The nonlinear regression shows a clear and rapid decline of LD with distance in base pairs within
PtoXET16A (
r2 ≥ 0.1, within 900 bp), indicating that LD of the SNP loci did not extend over the entire gene region (
Figure 4). Within-group analyses of LD showed a similar decline in samples from the southern region, with the
r2 values declining to 0.1 within 900 bp. Nevertheless, we observed a higher level of LD within samples from the northeastern and northwestern regions, with the
r2 values declining to 0.1 within approximately 1700 bp. These results revealed that the northeastern and northwestern regions seem to have experienced similar histories and the southern region had a higher evolutionary rate. Associations between 30 SNPs and 10 growth and wood quality traits were tested by using the mixed linear model (MLM) in TASSEL version 2.1 (Buckler lab, New York, NY, USA, 2010). The MLM identified 37 significant markers (
p < 0.05), but correction for false discovery rate (FDR) (
FDR < 0.05) reduced this to 13. These associations were identified in the exon, intron, and 3'UTR regions of
PtoXET16A (
Table 3).
Figure 4.
The decay of short-range linkage disequilibrium within PtoXET16A for all samples and each climatic region. We sequenced the PtoXET16A regions from a panel of 43 unrelated individuals (15 from the southern region, 14 from the northwestern region, and 14 from the northeastern region). Pairwise correlations between SNPs are plotted against the physical distance between the SNPs in base pairs. The curves describe the nonlinear regression of r2 onto the physical distance in base pairs.
Figure 4.
The decay of short-range linkage disequilibrium within PtoXET16A for all samples and each climatic region. We sequenced the PtoXET16A regions from a panel of 43 unrelated individuals (15 from the southern region, 14 from the northwestern region, and 14 from the northeastern region). Pairwise correlations between SNPs are plotted against the physical distance between the SNPs in base pairs. The curves describe the nonlinear regression of r2 onto the physical distance in base pairs.
Table 3.
SNP markers significantly associated with growth and wood properties in the association population.
Table 3.
SNP markers significantly associated with growth and wood properties in the association population.
| Trait | Marker | Position | Mutation | p-Value | FDR | r2 (%) |
|---|
| Lignin content | SNP6 | Exon 1 | [G:C] ns | <0.001 | <0.001 | 10.95 |
| SNP16 | Intron 3 | [C:T] | <0.001 | 0.009 | 5.37 |
| SNP29 | 3'UTR | [G:C] | <0.001 | 0.003 | 6.16 |
| D | SNP15 | Exon 3 | [C:T] ns | <0.001 | 0.012 | 4.27 |
| V | SNP15 | Exon 3 | [C:T] ns | <0.001 | 0.012 | 4.25 |
| Fiber length | SNP21 | Intron 3 | [C:T] | <0.001 | 0.001 | 5.70 |
| SNP22 | Intron 3 | [G:T] | <0.001 | 0.001 | 5.66 |
| SNP23 | Intron 3 | [C:T] | <0.001 | 0.020 | 3.77 |
| SNP29 | 3'UTR | [G:C] | 0.001 | 0.031 | 3.40 |
| Fiber width | SNP27 | Exon 4 | [A:G] s | 0.001 | 0.031 | 3.65 |
| MFA | SNP14 | Intron 2 | [C:T] | 0.001 | 0.031 | 3.56 |
| SNP21 | Intron 3 | [C:T] | 0.002 | 0.038 | 3.40 |
2.4.1. Single-SNP-Based Association
In total, twelve associations representing nine significant SNPs were identified (
Table 3). Of these, SNP15, a missense mutation in exon 3, resulted in an encoded amino acid change from Tyr to His, and was significantly associated with two traits, including diameter at breast height and stem volume (
Table 3). SNP6, another missense mutation in exon 1, resulted in an encoded amino acid change from Ala to Pro, was significantly associated with lignin content and explained 10.95% of the phenotypic variance (
Table 3). SNP21 in intron 3, a synonymous mutation, was found to be associated with fiber length and microfiber angle, and SNP29 from the 3'UTR showed genetic association with fiber length and lignin content (
Table 3). Markedly, SNP16, one splice variation in the second base of intron 3, resulted in splice junction (splice donor) from GT to GC, was significantly associated with lignin content and explained 5.37% of the phenotypic variance. All together, these SNP loci explained a small proportion of the phenotypic variance, with the individual effects ranging from 3.40% to 10.95% (
Table 3). These small SNP effects are in accordance with polygenic quantitative models of complex traits.
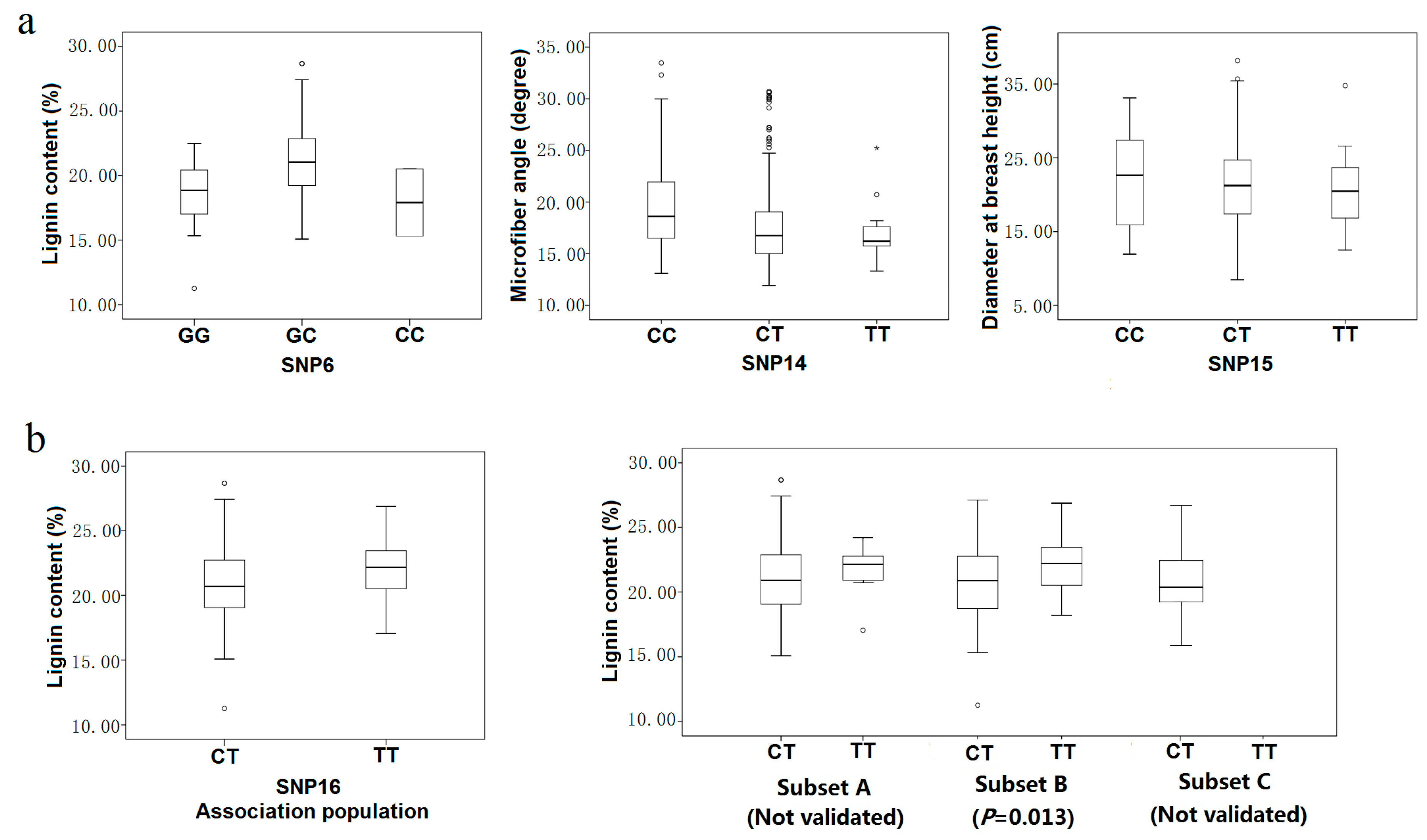
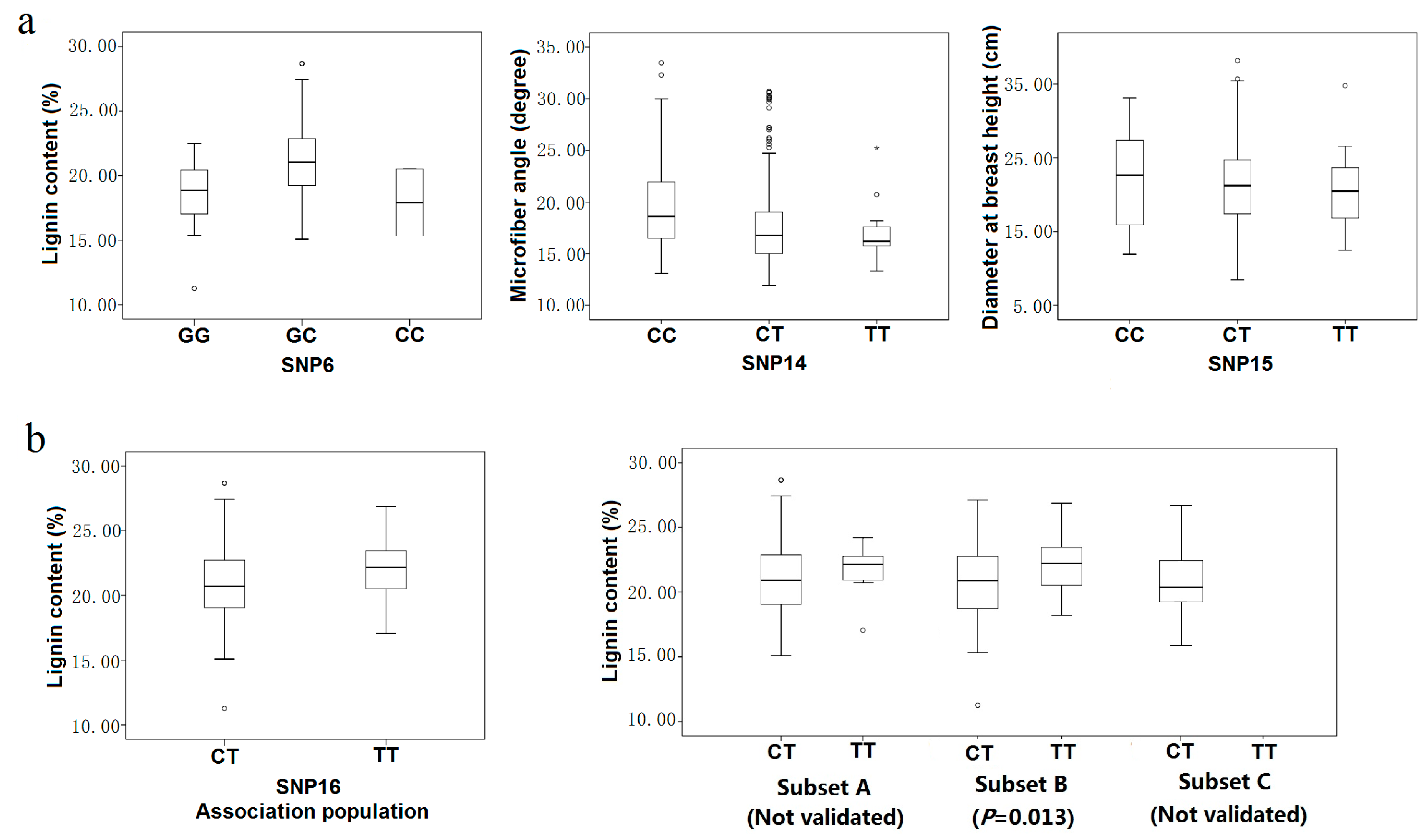
Most of the associations were consistent with modes of gene action other than codominance (
Table 4). Five of the 12 marker-trait pairs for which dominance and additive effects could be calculated were consistent with over- or underdominance (|d/a| > 1.25). For example, heterozygotes for SNP6 had higher lignin content, on average, than either homozygote class (18.09% for GG, 21.02% for GC, 17.92% for CC) (
Figure 5a). The remaining seven marker-trait pairs were split between modes of gene action that were partially to fully dominant (0.50 < |d/a| < 1.25) (
Figure 5a), such as SNP14 (20.96 for CC, 17.59 for CT, 17.26 for TT in microfiber angle) or codominant (|d/a| ≤ 0.5), such as SNP15 (22.14 for CC, 21.44 for CT, 21.14 for TT in diameter at breast height) (
Figure 5a).
Table 4.
List of marker effects for significant marker-trait pairs.
Table 4.
List of marker effects for significant marker-trait pairs.
| Trait | SNP | 2a a | d b | d/a | 2a/spc | Frequency d | a e |
|---|
| Lignin content | SNP6 | 1.333 | 2.43 | 3.646 | 0.531 | 0.482 | C | −1.412 |
| SNP16 | 22.019 | 9.7645 | 0.887 | 8.353 | 0.461 | C | 3.524 |
| SNP29 | 5.909 | −2.8905 | −0.978 | 2.241 | 0.335 | G | 1.964 |
| D | SNP15 | 0.994 | −0.211 | −0.425 | 0.177 | 0.485 | T | −0.164 |
| V | SNP15 | 0.029 | −0.0255 | −1.759 | 0.075 | 0.485 | T | 0.003 |
| Fiber length | SNP21 | 0.0214 | 0.0153 | 1.43 | 0.255 | 0.495 | T | −0.012 |
| SNP22 | 0.087 | −0.0225 | −0.517 | 1.036 | 0.494 | G | 0.032 |
| SNP23 | 0.013 | −0.002 | −0.308 | 0.163 | 0.460 | C | −0.003 |
| SNP29 | 0.152 | 0.081 | 1.066 | 1.81 | 0.335 | G | −0.051 |
| Fiber width | SNP27 | 2.189 | 2.2155 | 2.024 | 1.104 | 0.496 | A | −1.624 |
| MFA | SNP14 | 3.703 | −1.5205 | −0.821 | 0.818 | 0.479 | T | −0.385 |
| SNP21 | 1.414 | −1.661 | −2.349 | 0.313 | 0.495 | T | 0.340 |
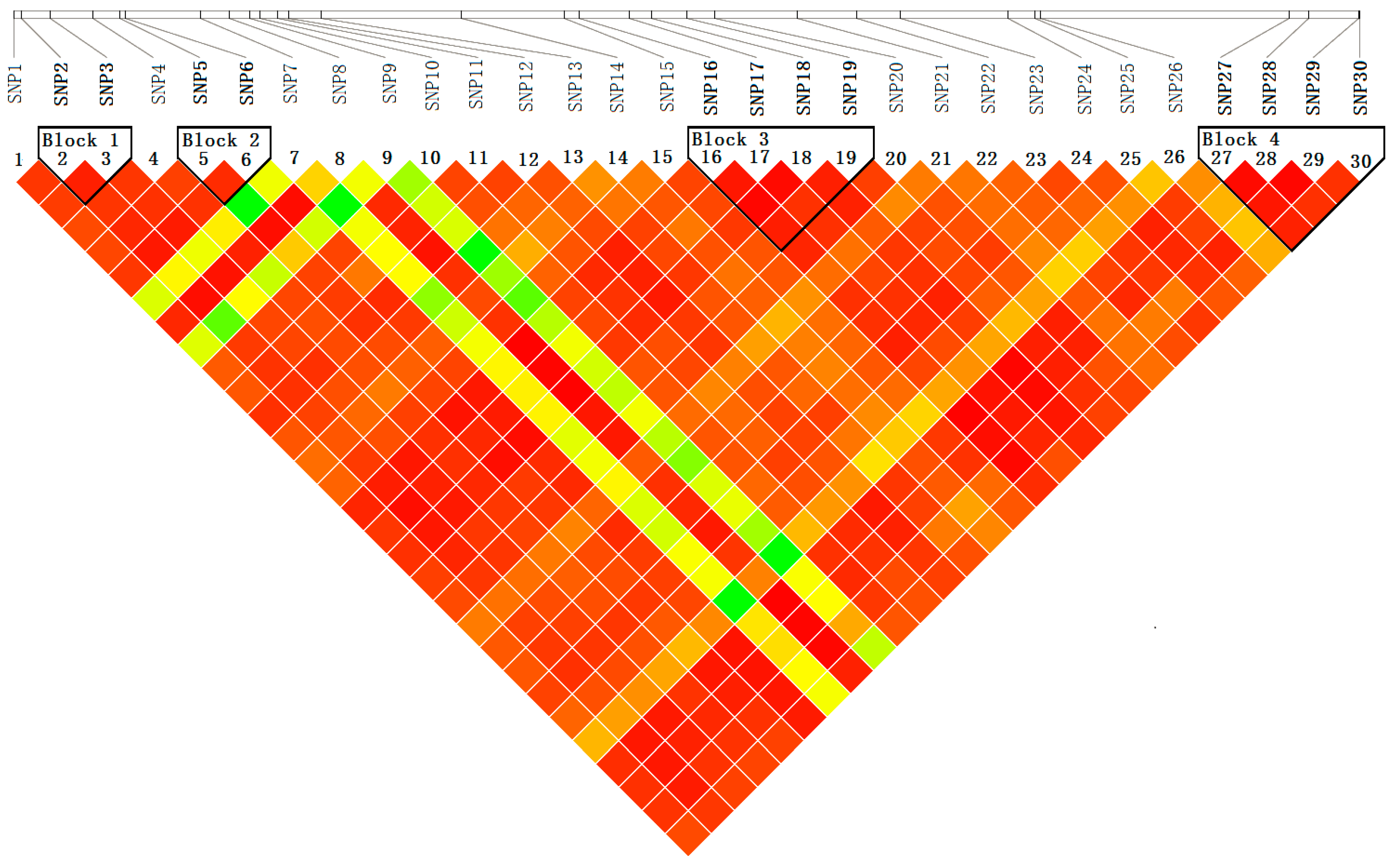
2.4.2. Haplotype-Based Associations
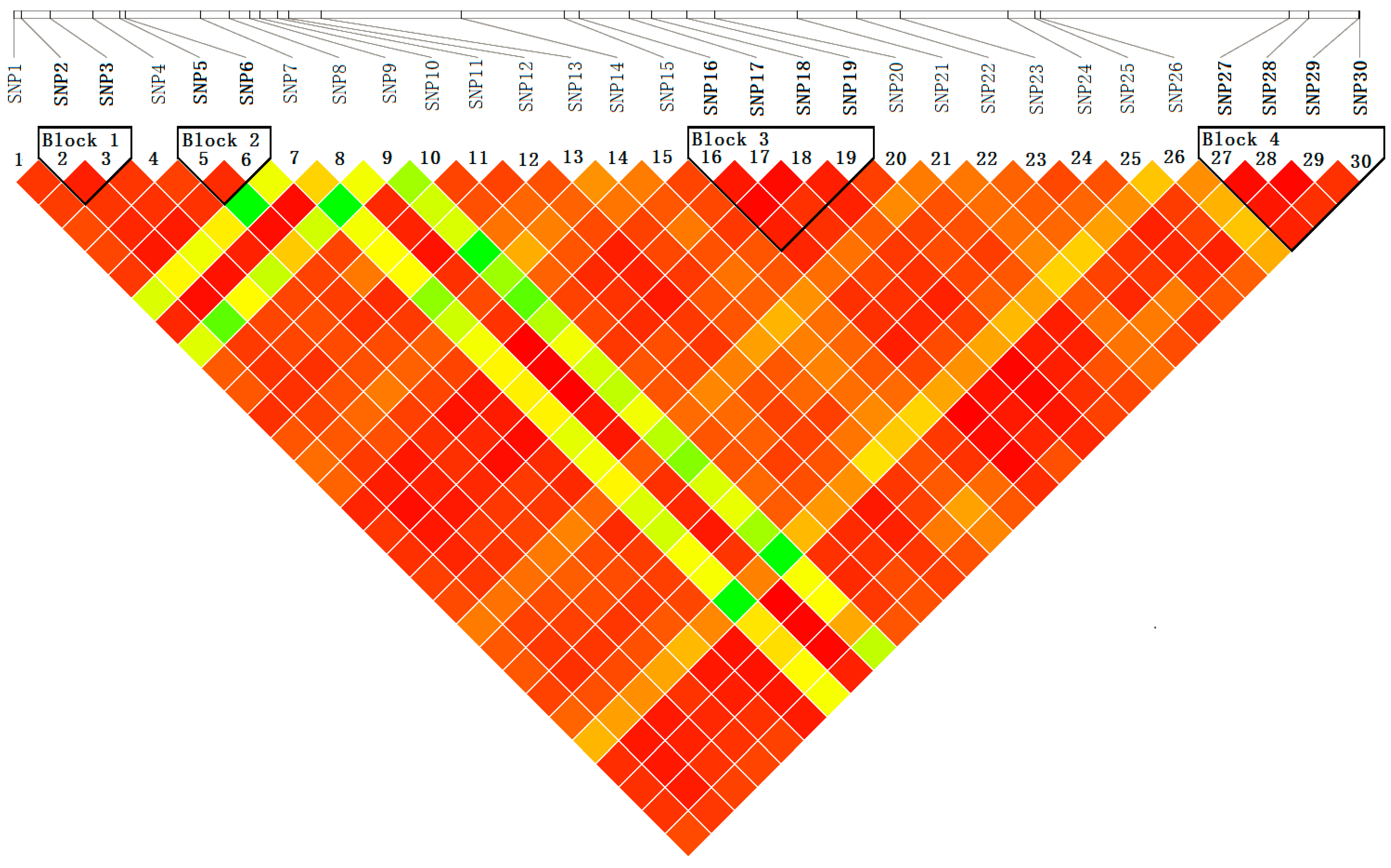
Haplotype analysis by Haploview, using genotype data for 30 SNPs from 426 individuals in the association population, showed four distinct haplotype blocks within
PtoXET16A (
Figure 6). We used a haplotype trend regression test [
28] to identify the haplotypes significantly associated with the 10 growth and wood quality traits. Three common haplotypes (allele frequency > 5%,
p-value < 0.05) were observed with significant effect on these traits. These haplotypes span exon 1, intron 3, exon 4 and the 3'UTR (
Figure 6). The proportion of phenotypic variation explained by these haplotypes ranged from 1.63% to 10.46% (
Table 5). Among them, three haplotypes from SNP5-6 and SNP16-19 were associated with diameter at breast height, with the individual effects ranging from 1.63% to 2.71%. In addition, association between the three haplotypes and stem volume were observed, which explained 3.05%–4.41% of the phenotypic variance (
Table 5). Of these, two haplotypes from SNP5-6 were associated with lignin content and one haplotype from SNP27-30 was associated with microfiber angle (
Table 5).
Figure 5.
Genotypic effect on SNP markers in the association population (a) Three modes of gene action quantified using the ratio of dominant to additive effects estimated from least-square means for each genotypic class. Three modes of gene action were observed as over-dominance (SNP6), full dominance (SNP14) and co-dominance (SNP15); (b) Genotypic effect on SNP6 with lignin content in the discovery population and three subsets (Subset A, 170 individuals from the southern region; Subset B, 91 individuals from the northwestern region; Subset C, 165 from the northeastern region).
Figure 5.
Genotypic effect on SNP markers in the association population (a) Three modes of gene action quantified using the ratio of dominant to additive effects estimated from least-square means for each genotypic class. Three modes of gene action were observed as over-dominance (SNP6), full dominance (SNP14) and co-dominance (SNP15); (b) Genotypic effect on SNP6 with lignin content in the discovery population and three subsets (Subset A, 170 individuals from the southern region; Subset B, 91 individuals from the northwestern region; Subset C, 165 from the northeastern region).
Table 5.
Haplotypes significantly associated with growth and wood property traits.
Table 5.
Haplotypes significantly associated with growth and wood property traits.
| Trait | p (Overall) | r2 (%) | Haplotype | Frequency | Mean | p (ind) |
|---|
| Diameter at breast height | 0.0235 | 1.63 | SNP5-6 | | | |
| | | G-C | 0.0666 | 19.7014 | 0.010 |
| | | T-C | 0.4513 | 21.6812 | 0.010 |
| <0.0001 | 2.71 | SNP16-19 | | | |
| | | C-T-A-T | 0.4062 | 21.0053 | <0.001 |
| Stem volume | 0.0005 | 3.05 | SNP5-6 | | | |
| | | G-C | 0.0666 | 0.4210 | <0.001 |
| | | T-C | 0.4501 | 0.6243 | <0.001 |
| 0.0038 | 4.41 | SNP16-19 | | | |
| | | C-T-A-T | 0.4062 | 0.5638 | <0.001 |
| Lignin content | 0.0013 | 10.46 | SNP5-6 | | | |
| | | G-C | 0.0687 | 19.8953 | 0.003 |
| | | G-G | 0.4744 | 20.9963 | <0.001 |
| Microfiber angle | 0.4737 | 3.86 | SNP27-30 | | | |
| | | G-A-C-T | 0.4871 | 15.235 | 0.050 |
Figure 6.
Pairwise linkage disequilibrium (r2) between SNP markers. The common genotyped SNPs are shown on a schematic of PtoXET16A and the pairwise r2 values are shown by color-coding in the matrix below.
Figure 6.
Pairwise linkage disequilibrium (r2) between SNP markers. The common genotyped SNPs are shown on a schematic of PtoXET16A and the pairwise r2 values are shown by color-coding in the matrix below.
2.4.3. Validation of Association Testing
Three smaller subsets derived from the association population were used to validate the significant single-SNP associations identified in the association population (
Table 6). Eight of the twelve significant marker-trait associations were validated in at least one of the three smaller subsets. In total, six SNP markers (SNP6, SNP14, SNP15, SNP16, SNP21, and SNP29) were significantly associated with five traits, including fiber length, lignin content,
D,
V and
MFA at the threshold of
p < 0.05. The proportion of phenotypic variation explained by the six SNP markers varied from 2.39% to 13.07% (
Table 6). Associations of SNP22, SNP23 and SNP27 with fiber length/width were not validated in any of the three smaller subsets. The failure to validate these three significant associations in this study may arise from the complexity of quantitative traits, the small sample size or other factors.
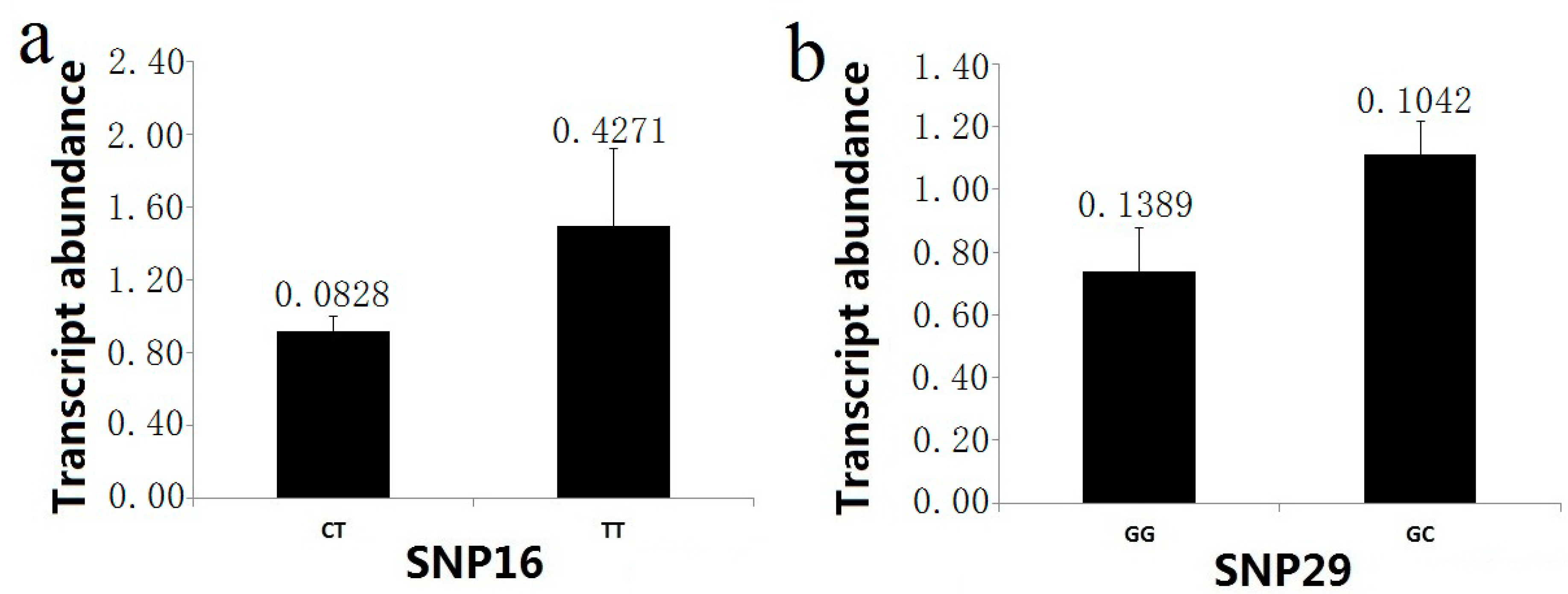
2.5. Transcript Analysis of SNP Genotypes
To identify whether these significant associations affect gene expression at the mRNA level, we validated SNP associations via gene expression analyses. Transcript levels among the different genotypic classes for nine significantly associated SNPs (
Table 3) were compared by RT-PCR with gene-specific primers. The assays used secondary xylem from the 20-year-old trees to quantify the mRNA levels in 30 trees (including almost all genotypes). Measurement of different transcript abundance across three (using analysis of variance, ANOVA) or two genotypic classes (using
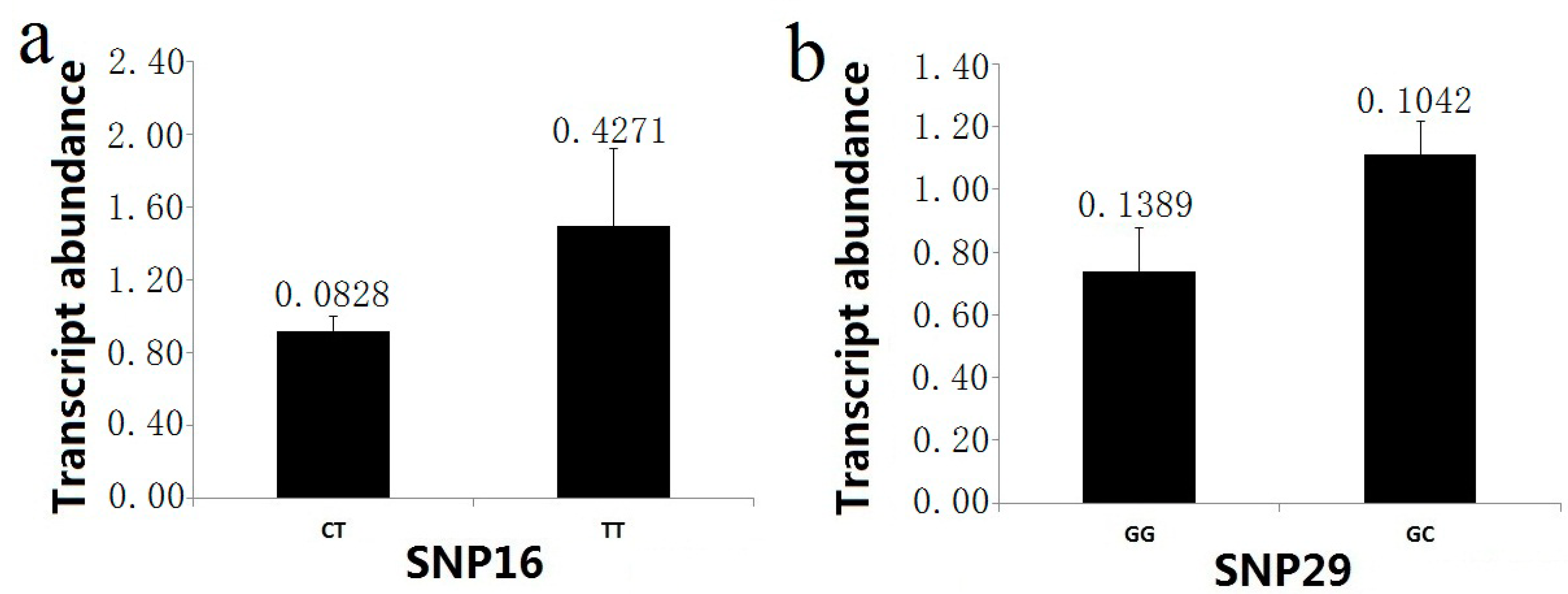
t-test) indicated that two markers (SNP16 and SNP29) showed significant differences in the RNA transcript levels in the association population. For the marker SNP16 (intron 3, IVS3 + 2T>C), the higher abundance of the mRNA (1.4922 ± 0.4271) was found in the TT group, and the transcript level of the CT group was 0.9175 ± 0.0828 (
Figure 7). In examining genotype-specific transcript levels for SNP29 (3'UTR), the heterozygous trees (1.1116 ± 0.1042 in GC group) for this marker showed higher relative mRNA abundance than the homozygous trees (0.7390 ± 0.1389 in GG group) (
Figure 7).
Table 6.
SNP markers significantly associated with growth and wood properties traits in three subsets.
Table 6.
SNP markers significantly associated with growth and wood properties traits in three subsets.
| Trait | Marker | Subset A | Subset B | Subset C |
|---|
| p-Value | r2 (%) | p-Value | r2 (%) | p-Value | r2 (%) |
|---|
| Lignin content | SNP6 | \ | \ | ≤0.001 | 10.35 | ≤0.001 | 2.39 |
| SNP16 | \ | \ | 0.013 | 3.81 | \ | \ |
| D | SNP15 | \ | \ | 0.008 | 5.69 | \ | \ |
| V | SNP15 | \ | \ | 0.008 | 5.69 | \ | \ |
| Fiber length | SNP21 | \ | \ | 0.009 | 5.55 | \ | \ |
| SNP29 | 0.009 | 4.19 | 0.020 | 4.64 | \ | \ |
| MFA | SNP14 | \ | \ | 0.047 | 3.69 | 0.045 | 6.82 |
| SNP21 | \ | \ | ≤0.001 | 13.07 | \ | \ |
Figure 7.
PtoXET16A transcript abundance varies among genotypic classes for significant SNP associations. The error bars represent ± standard deviation. (a) Transcript abundance in the two genotypic classes for SNP16; (b) Transcript abundance in the two genotypic classes for SNP29.
Figure 7.
PtoXET16A transcript abundance varies among genotypic classes for significant SNP associations. The error bars represent ± standard deviation. (a) Transcript abundance in the two genotypic classes for SNP16; (b) Transcript abundance in the two genotypic classes for SNP29.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}