Erythropoietin Action in Stress Response, Tissue Maintenance and Metabolism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
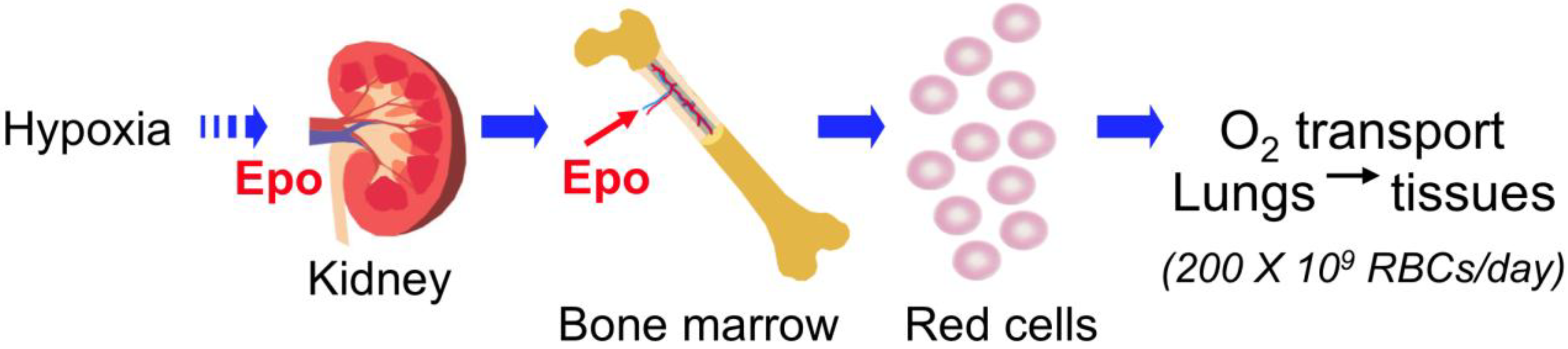
2. Erythropoietin (EPO) Regulation
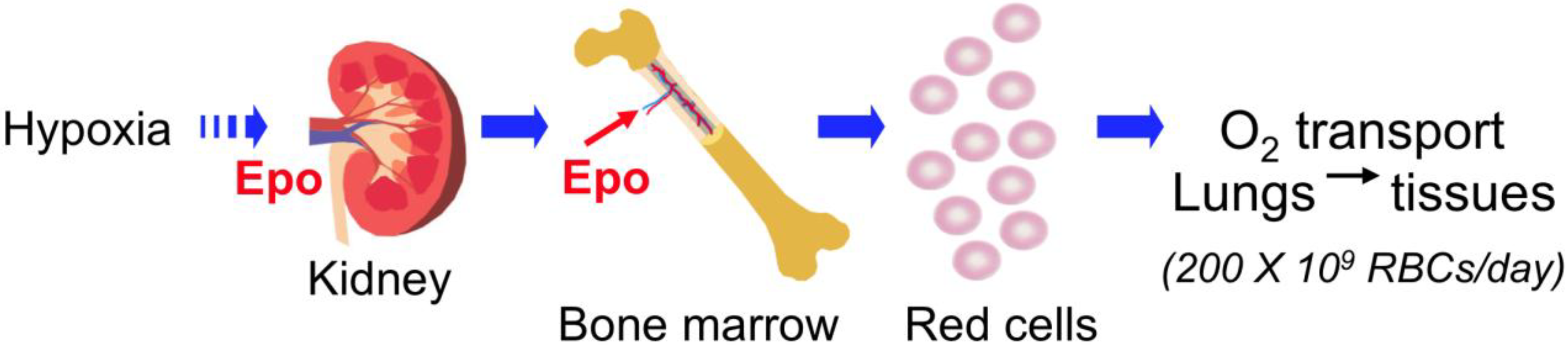
2.1. EPO Production in Fetal Liver and Adult Kidney
2.2. Regulation of EPO Gene Expression by Hypoxia Inducible Factor (HIF)
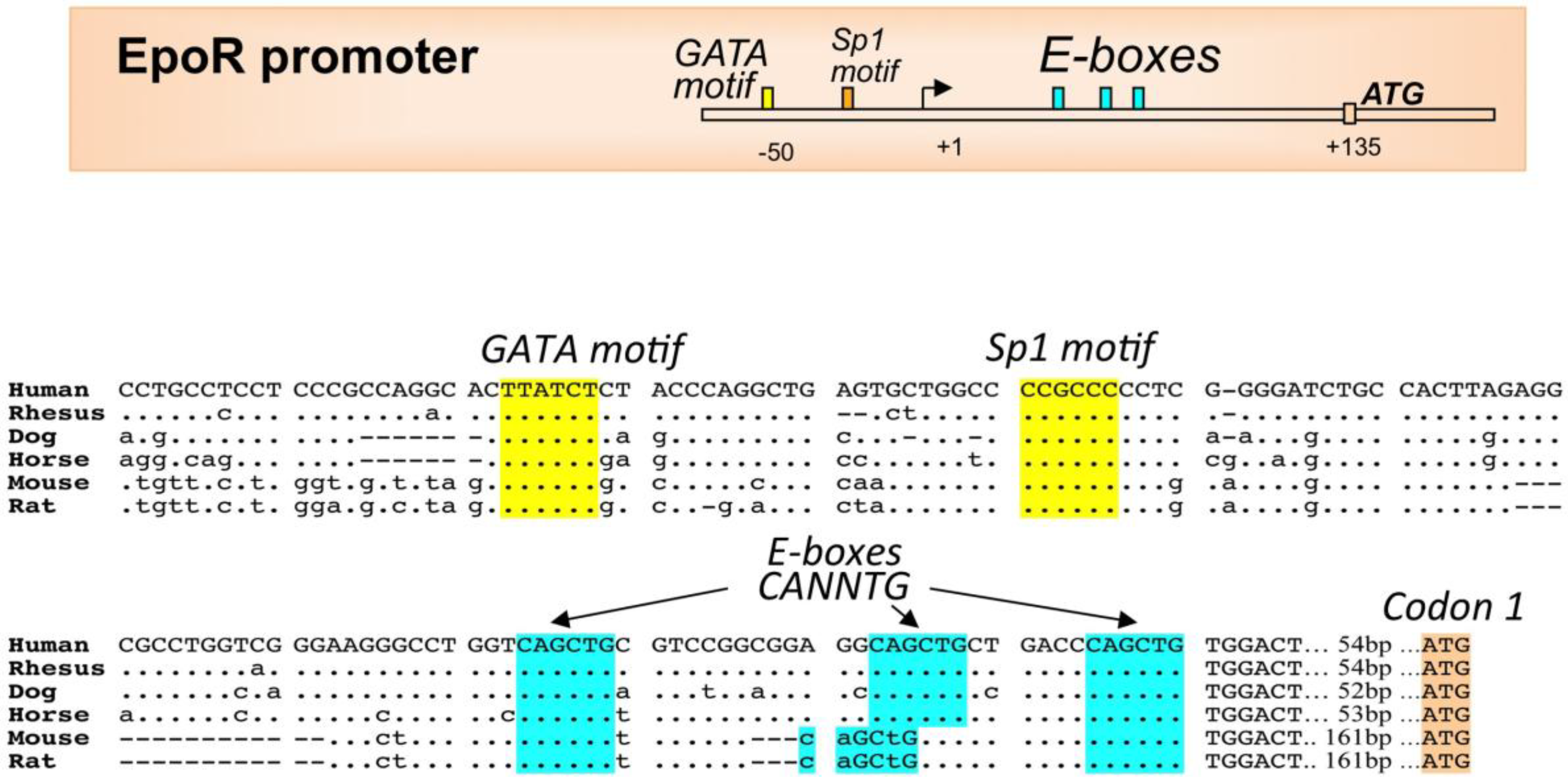
2.3. EPO Regulation by Other Transcription Factors
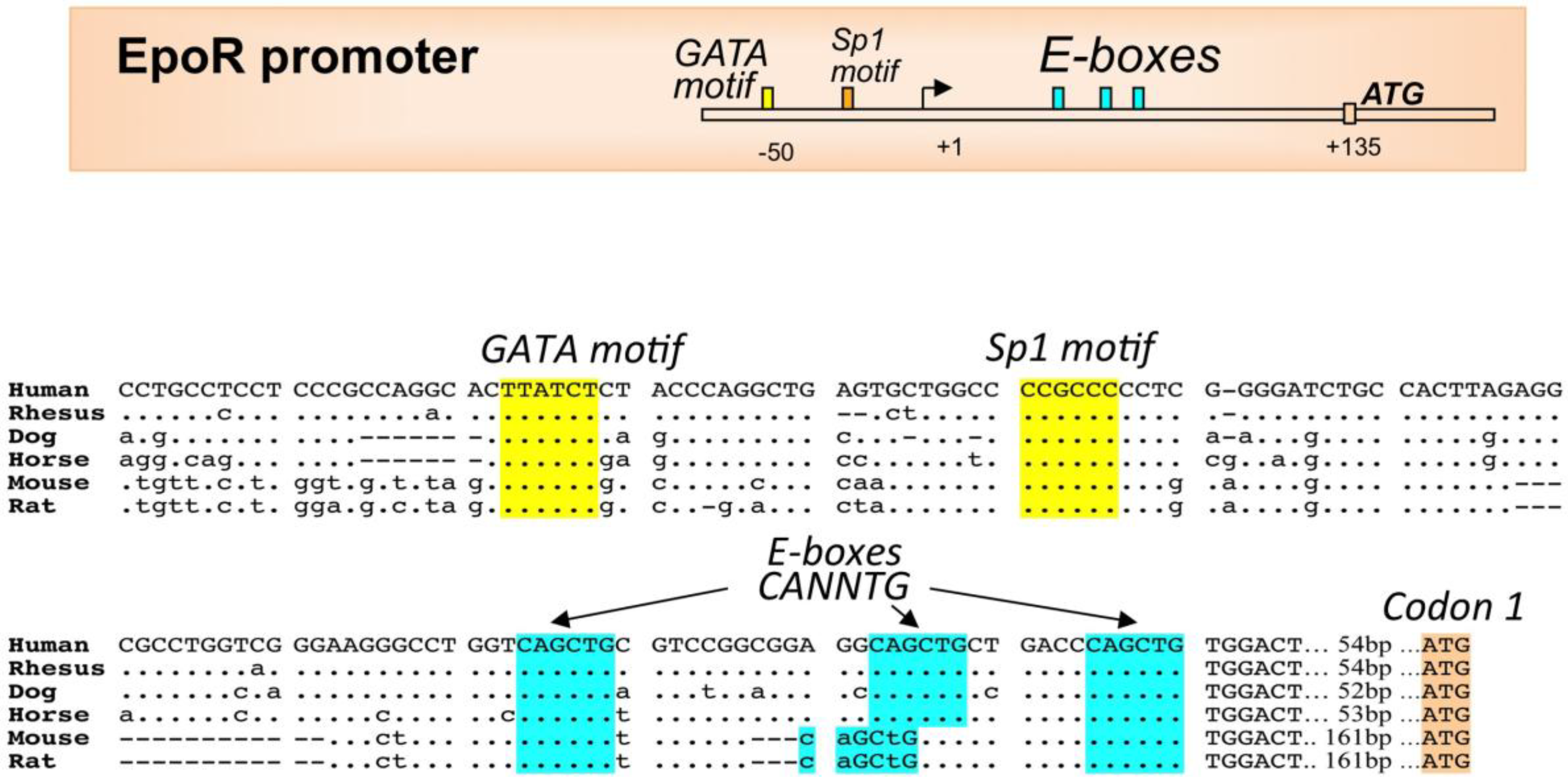
3. EPO Receptor Expression in Erythroid Cells
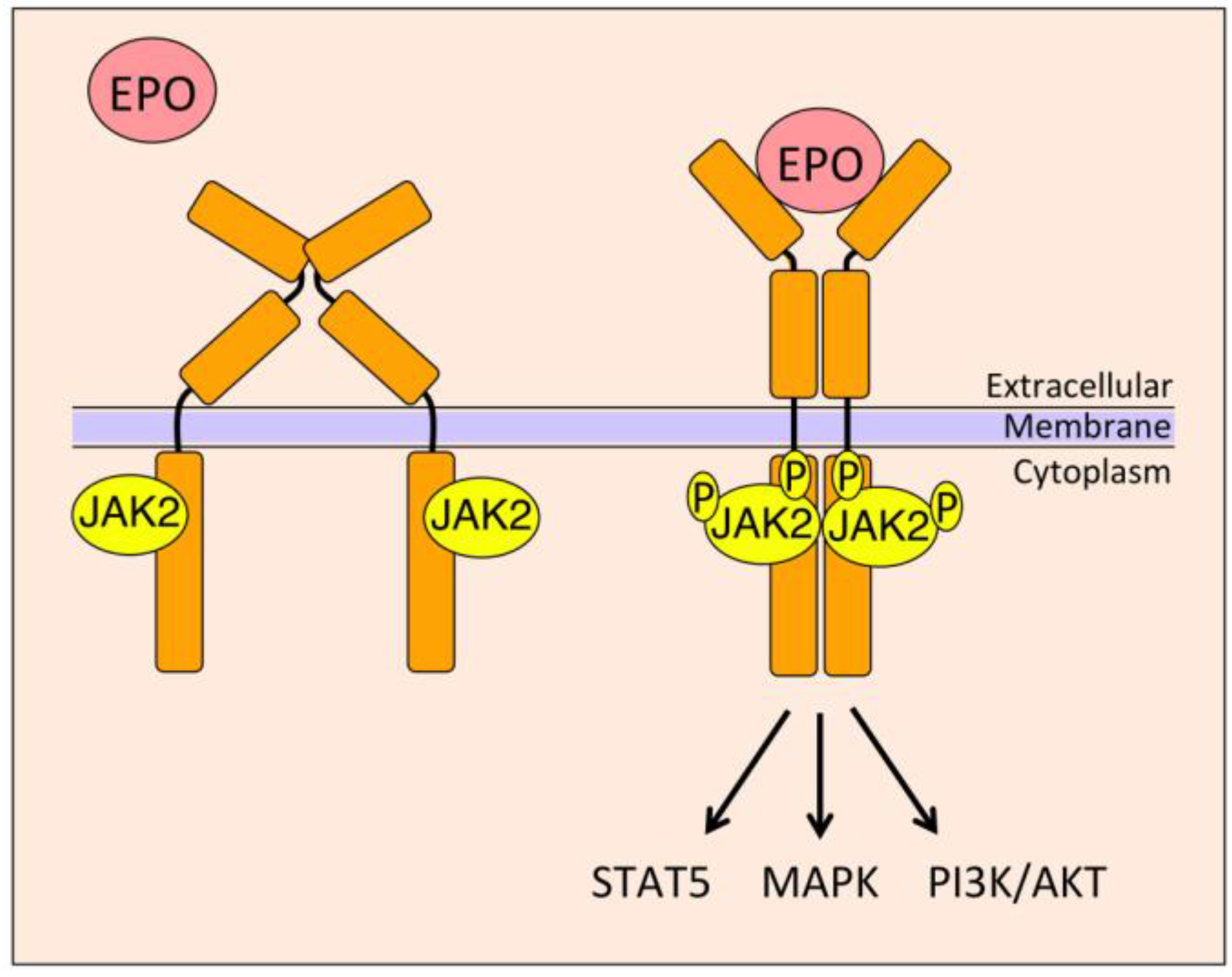
4. EPO Signal Transduction
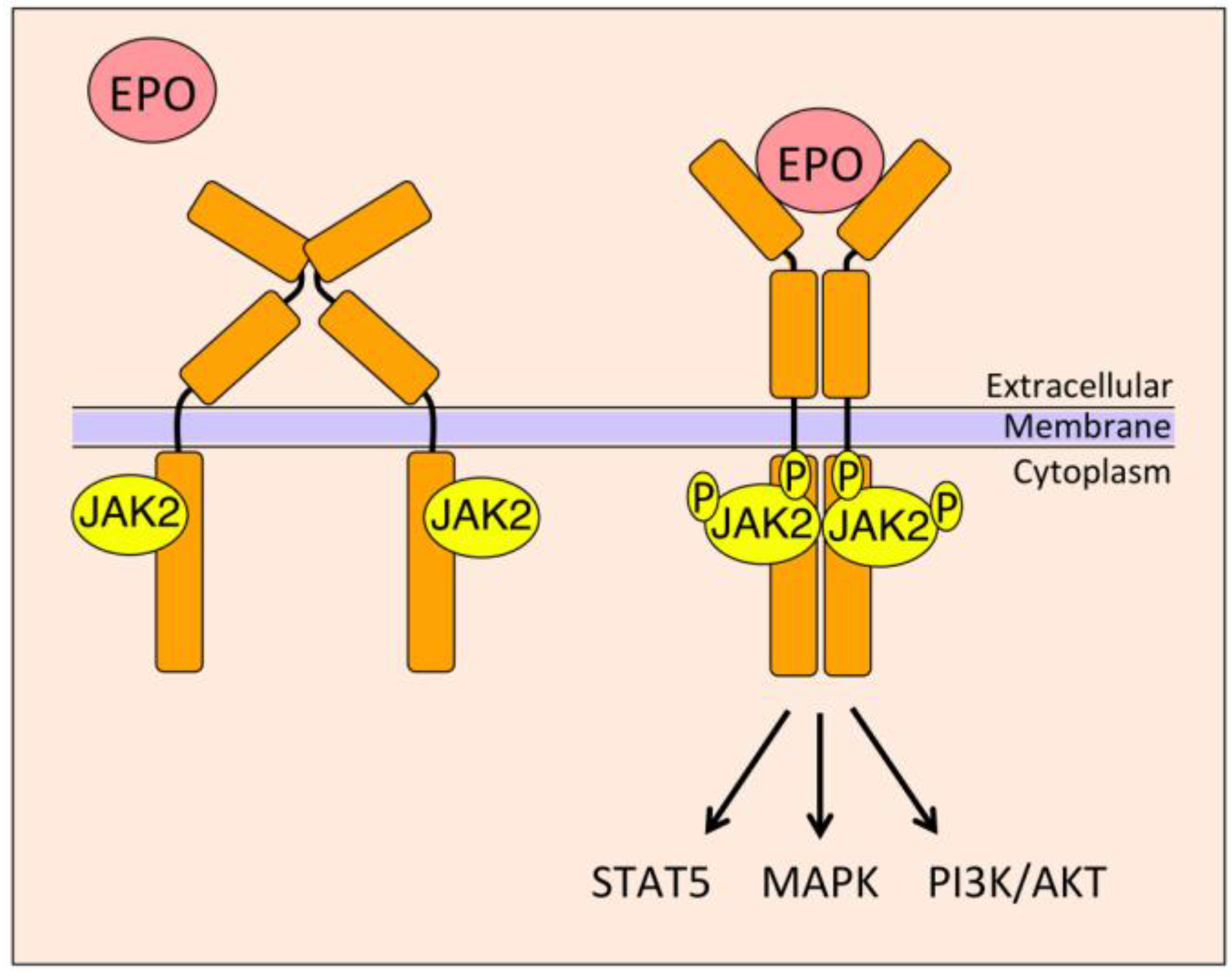
4.1. Janus Kinase 2 (JAK2)/Signal Transducer and Activator of Transcription 5 (STAT5)
4.2. Mitogen-Activated Protein Kinase (MAPK)
4.3. Phosphoinositide 3-Kinase (PI3K)
4.4. Signal Transducer and Activator of Transcription 3 (STAT3)
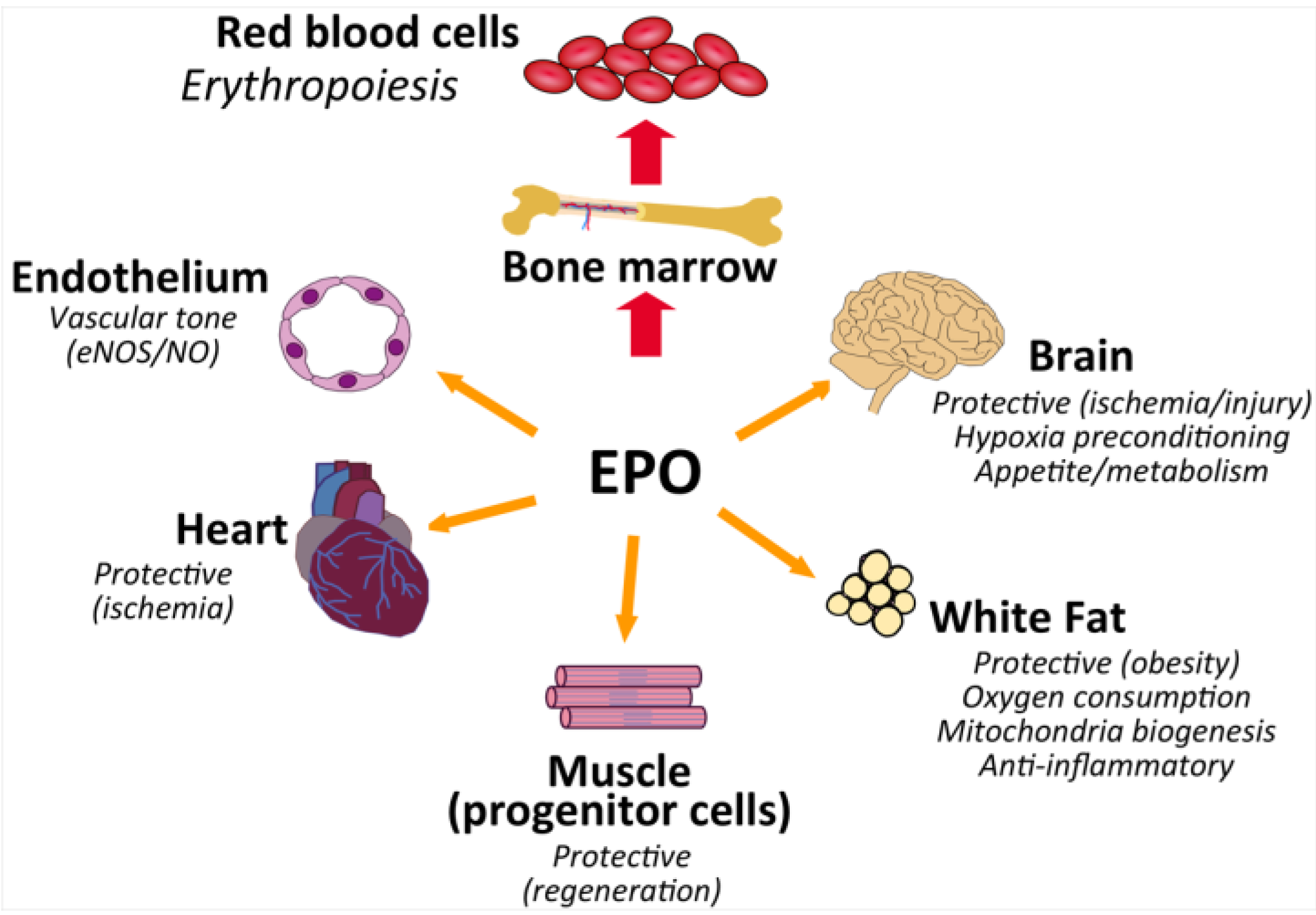
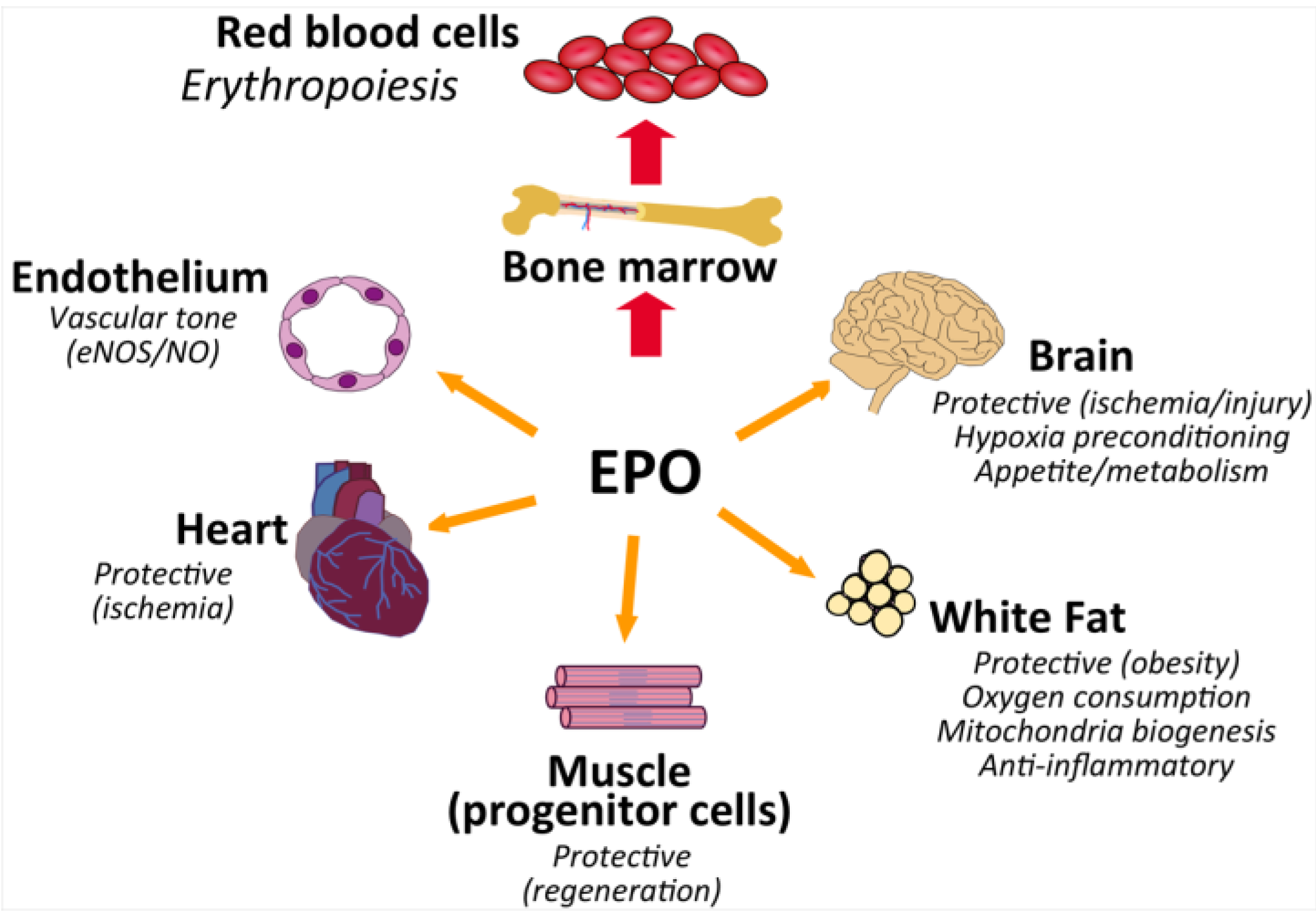
5. EPO Action in Non-Hematopoietic Tissue
5.1. Endothelial Response
5.2. EPO and Heart
5.3. Brain and Neural Protection
5.4. Skeletal Muscle and Wound Healing
6. EPO Regulation of Metabolism and Obesity
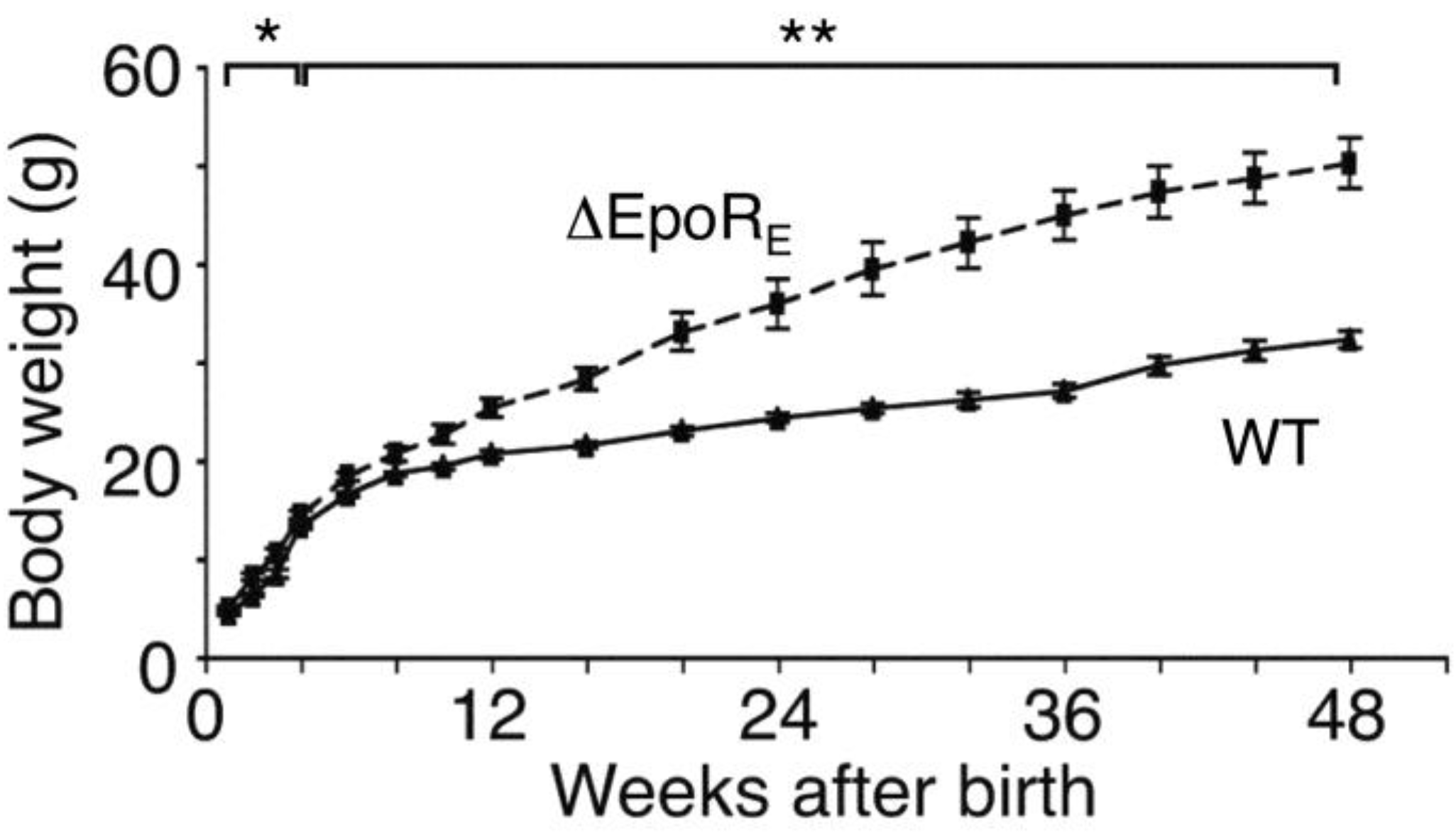
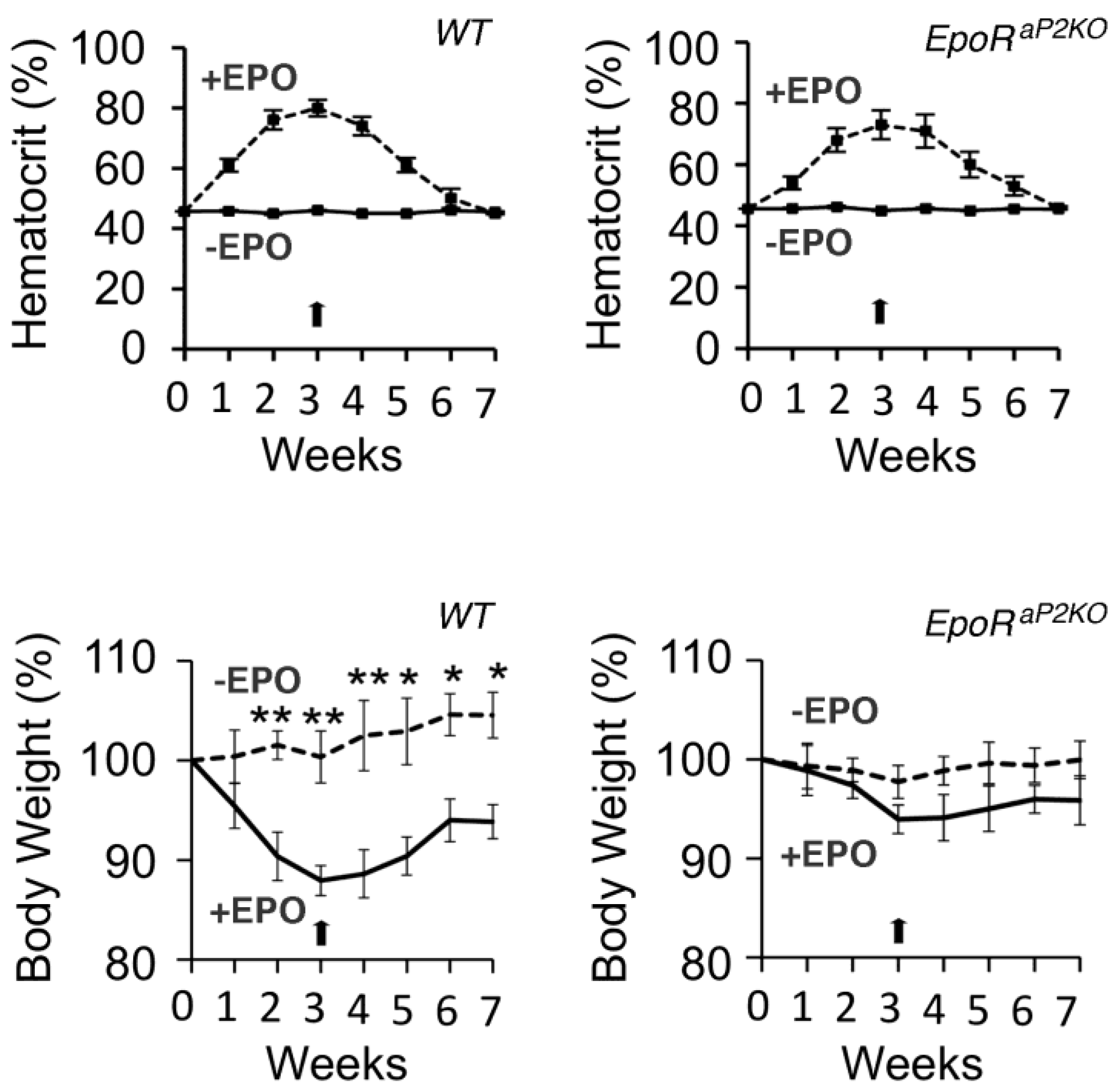
6.1. Phenotype of Mice with EpoR Restricted to Erythroid Tissue
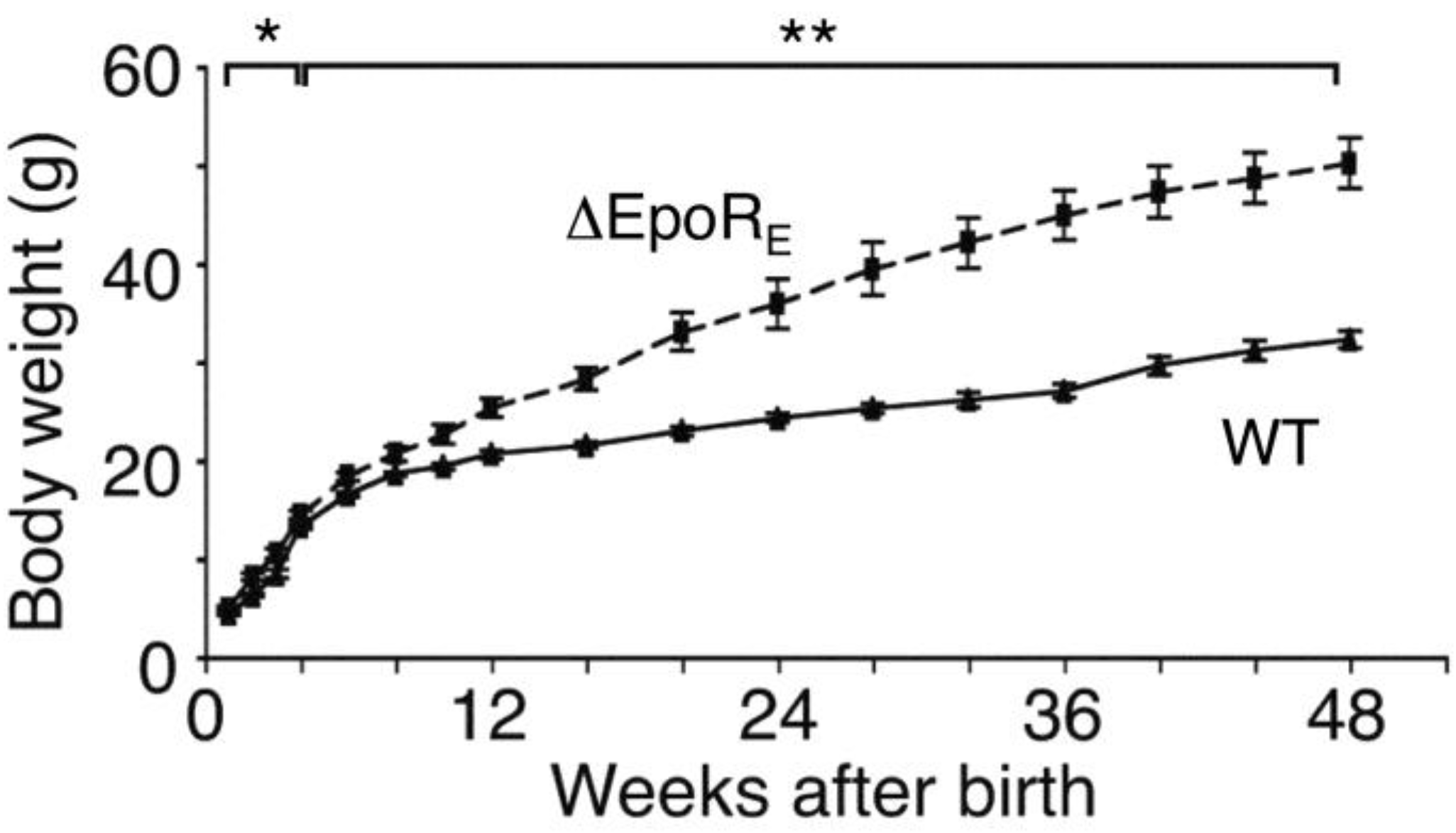
6.2. EPO Treatment in Genetic Mouse Models of Obesity
6.3. Mice with Chronic Elevation of EPO
6.4. Fat Specific Deletion of EPO Receptor (EpoR)
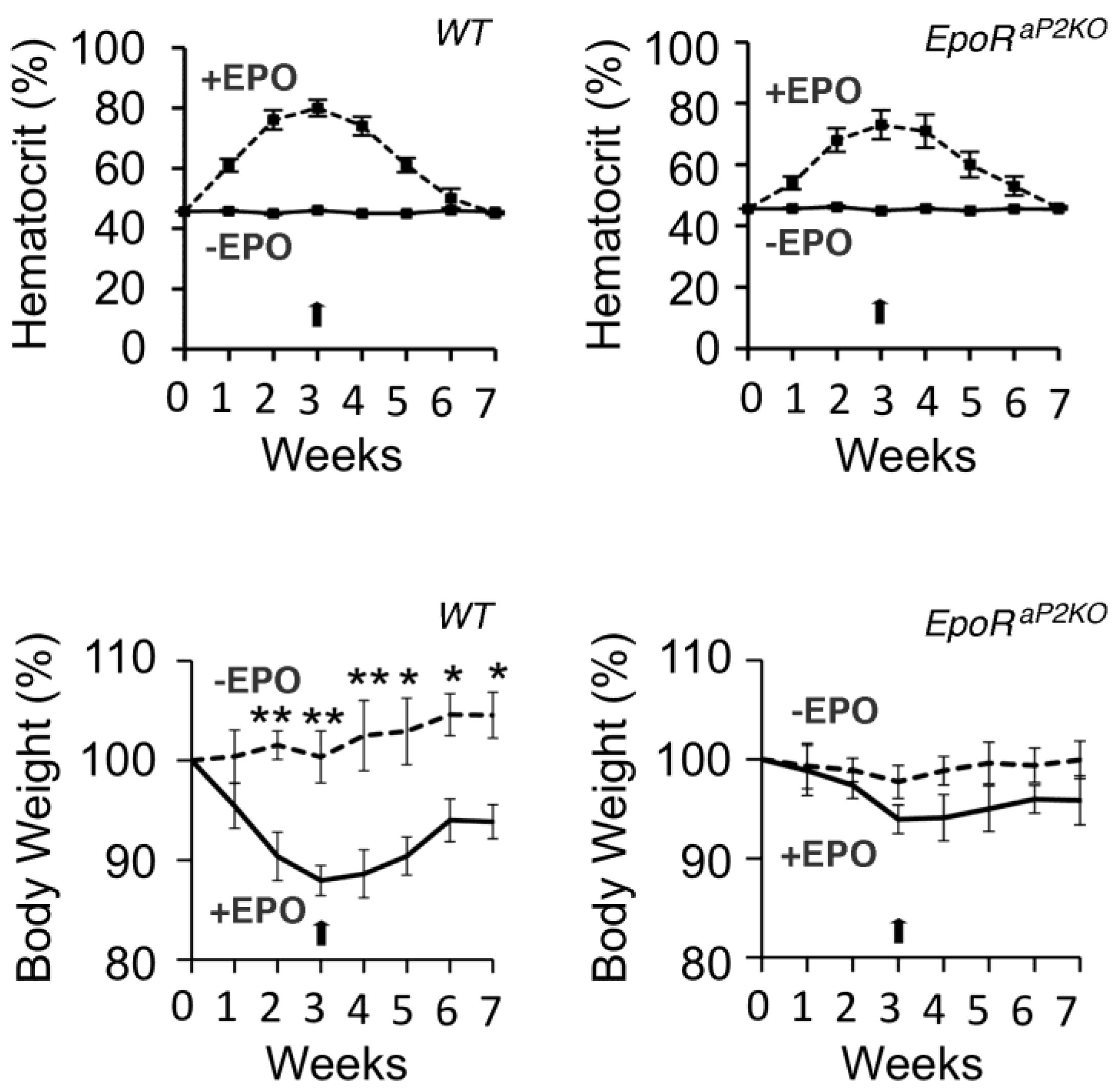
6.5. Pancreatic Islet Cell Response in Mice Treated with EPO
6.6. Metabolic Response to EPO Treatment in Patients
7. EPO Regulation of Inflammation
7.1. Central Nervous System
7.2. Cardiovascular
7.3. Gut and Liver
7.4. Macrophages
8. EPO Therapy and Associated Adverse Events
8.1. Chronic Kidney Disease
8.2. EPO and Cancer
8.3. Stroke Study
8.4. Cardiovascular Studies
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bunn, H.F. Erythropoietin. Cold Spring Harb. P erspect. Med. 2013, 3, a011619. [Google Scholar]
- Wu, H.; Liu, X.; Jaenisch, R.; Lodish, H.F. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 1995, 83, 59–67. [Google Scholar] [CrossRef]
- Lin, C.S.; Lim, S.K.; D’Agati, V.; Costantini, F. Differential effects of an erythropoietin receptor gene disruption on primitive and definitive erythropoiesis. Genes Dev. 1996, 10, 154–164. [Google Scholar] [CrossRef]
- Aapro, M.; Jelkmann, W.; Constantinescu, S.N.; Leyland-Jones, B. Effects of erythropoietin receptors and erythropoiesis-stimulating agents on disease progression in cancer. Br. J. Cancer 2012, 106, 1249–1258. [Google Scholar] [CrossRef]
- Jelkmann, W.; Elliott, S. Erythropoietin and the vascular wall: The controversy continues. Nutr. Metab. Cardiovasc. Dis. 2013, 23, S37–S43. [Google Scholar]
- Recny, M.A.; Scoble, H.A.; Kim, Y. Structural characterization of natural human urinary and recombinant DNA-derived erythropoietin. Identification of des-arginine 166 erythropoietin. J. Biol. Chem. 1987, 262, 17156–17163. [Google Scholar]
- Lin, F.K.; Suggs, S.; Lin, C.H.; Browne, J.K.; Smalling, R.; Egrie, J.C.; Chen, K.K.; Fox, G.M.; Martin, F.; Stabinsky, Z.; et al. Cloning and expression of the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1985, 82, 7580–7584. [Google Scholar] [CrossRef]
- Jacobs, K.; Shoemaker, C.; Rudersdorf, R.; Neill, S.D.; Kaufman, R.J.; Mufson, A.; Seehra, J.; Jones, S.S.; Hewick, R.; Fritsch, E.F.; et al. Isolation and characterization of genomic and cDNA clones of human erythropoietin. Nature 1985, 313, 806–810. [Google Scholar] [CrossRef]
- Wasley, L.C.; Timony, G.; Murtha, P.; Stoudemire, J.; Dorner, A.J.; Caro, J.; Krieger, M.; Kaufman, R.J. The importance of N- and O-linked oligosaccharides for the biosynthesis and in vitro and in vivo biologic activities of erythropoietin. Blood 1991, 77, 2624–2632. [Google Scholar]
- Dame, C.; Fahnenstich, H.; Freitag, P.; Hofmann, D.; Abdul-Nour, T.; Bartmann, P.; Fandrey, J. Erythropoietin mRNA expression in human fetal and neonatal tissue. Blood 1998, 92, 3218–3225. [Google Scholar]
- Zanjani, E.D.; Ascensao, J.L.; McGlave, P.B.; Banisadre, M.; Ash, R.C. Studies on the liver to kidney switch of erythropoietin production. J. Clin. Investig. 1981, 67, 1183–1188. [Google Scholar] [CrossRef]
- Obara, N.; Suzuki, N.; Kim, K.; Nagasawa, T.; Imagawa, S.; Yamamoto, M. Repression via the GATA box is essential for tissue-specific erythropoietin gene expression. Blood 2008, 111, 5223–5232. [Google Scholar] [CrossRef]
- Koury, S.T.; Bondurant, M.C.; Koury, M.J. Localization of erythropoietin synthesizing cells in murine kidneys by in situ hybridization. Blood 1988, 71, 524–527. [Google Scholar]
- Maxwell, P.H.; Osmond, M.K.; Pugh, C.W.; Heryet, A.; Nicholls, L.G.; Tan, C.C.; Doe, B.G.; Ferguson, D.J.; Johnson, M.H.; Ratcliffe, P.J. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int. 1993, 44, 1149–1162. [Google Scholar] [CrossRef]
- Kochling, J.; Curtin, P.T.; Madan, A. Regulation of human erythropoietin gene induction by upstream flanking sequences in transgenic mice. Br. J. Haematol. 1998, 103, 960–968. [Google Scholar] [CrossRef]
- Bondurant, M.C.; Koury, M.J. Anemia induces accumulation of erythropoietin mRNA in the kidney and liver. Mol. Cell. Biol. 1986, 6, 2731–2733. [Google Scholar]
- Eckardt, K.U.; Koury, S.T.; Tan, C.C.; Schuster, S.J.; Kaissling, B.; Ratcliffe, P.J.; Kurtz, A. Distribution of erythropoietin producing cells in rat kidneys during hypoxic hypoxia. Kidney Int. 1993, 43, 815–823. [Google Scholar] [CrossRef]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar]
- Semenza, G.L.; Koury, S.T.; Nejfelt, M.K.; Gearhart, J.D.; Antonarakis, S.E. Cell-type-specific and hypoxia-inducible expression of the human erythropoietin gene in transgenic mice. Proc. Natl. Acad. Sci. USA 1991, 88, 8725–8729. [Google Scholar]
- Semenza, G.L. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood 2009, 114, 2015–2019. [Google Scholar] [CrossRef]
- Rankin, E.B.; Biju, M.P.; Liu, Q.; Unger, T.L.; Rha, J.; Johnson, R.S.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J. Clin. Investig. 2007, 117, 1068–1077. [Google Scholar] [CrossRef]
- Ang, S.O.; Chen, H.; Hirota, K.; Gordeuk, V.R.; Jelinek, J.; Guan, Y.; Liu, E.; Sergueeva, A.I.; Miasnikova, G.Y.; Mole, D.; et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 2002, 32, 614–621. [Google Scholar] [CrossRef]
- Percy, M.J.; Zhao, Q.; Flores, A.; Harrison, C.; Lappin, T.R.J.; Maxwell, P.H.; McMullin, M.F.; Lee, F.S. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 654–659. [Google Scholar] [CrossRef]
- Imagawa, S.; Yamamoto, M.; Miura, Y. Negative regulation of the erythropoietin gene expression by the GATA transcription factors. Blood 1997, 89, 1430–1439. [Google Scholar]
- La Ferla, K.; Reimann, C.; Jelkmann, W.; Hellwig-Bürgel, T. Inhibition of erythropoietin gene expression signaling involves the transcription factors GATA-2 and NF-κB. FASEB J. 2002, 16, 1811–1813. [Google Scholar]
- Dame, C.; Sola, M.C.; Lim, K.-C.; Leach, K.M.; Fandrey, J.; Ma, Y.; Knöpfle, G.; Engel, J.D.; Bungert, J. Hepatic erythropoietin gene regulation by GATA-4. J. Biol. Chem. 2004, 279, 2955–2961. [Google Scholar]
- Noguchi, C.T.; Bae, K.S.; Chin, K.; Wada, Y.; Schechter, A.N.; Hankins, W.D. Cloning of the human erythropoietin receptor gene. Blood 1991, 78, 2548–2556. [Google Scholar]
- Rogers, H.M.; Yu, X.; Wen, J.; Smith, R.; Fibach, E.; Noguchi, C.T. Hypoxia alters progression of the erythroid program. Exp. Hematol. 2008, 36, 17–27. [Google Scholar] [CrossRef]
- Ogilvie, M.; Yu, X.; Nicolas-Metral, V.; Pulido, S.M.; Liu, C.; Ruegg, U.T.; Noguchi, C.T. Erythropoietin stimulates proliferation and interferes with differentiation of myoblasts. J. Biol. Chem. 2000, 275, 39754–39761. [Google Scholar]
- Wang, L.; Teng, R.; di, L.; Rogers, H.; Wu, H.; Kopp, J.B.; Noguchi, C.T. PPARα and Sirt1 mediate erythropoietin action in increasing metabolic activity and browning of white adipocytes to protect against obesity and metabolic disorders. Diabetes 2013, 62, 4122–4131. [Google Scholar] [CrossRef]
- Beck, I.; Ramirez, S.; Weinmann, R.; Caro, J. Enhancer element at the 3'-flanking region controls transcriptional response to hypoxia in the human erythropoietin gene. J. Biol. Chem. 1991, 266, 15563–15566. [Google Scholar]
- Ebert, B.L.; Bunn, H.F. Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol. Cell. Biol. 1998, 18, 4089–4096. [Google Scholar]
- Makita, T.; Hernandez-Hoyos, G.; Chen, T.H.; Wu, H.; Rothenberg, E.V.; Sucov, H.M. A developmental transition in definitive erythropoiesis: Erythropoietin expression is sequentially regulated by retinoic acid receptors and HNF4. Genes Dev. 2001, 15, 889–901. [Google Scholar] [CrossRef]
- Alberta, J.A.; Springett, G.M.; Rayburn, H.; Natoli, T.A.; Loring, J.; Kreidberg, J.A.; Housman, D. Role of the WT1 tumor suppressor in murine hematopoiesis. Blood 2003, 101, 2570–2574. [Google Scholar] [CrossRef]
- Dame, C.; Kirschner, K.M.; Bartz, K.V.; Wallach, T.; Hussels, C.S.; Scholz, H. Wilms tumor suppressor, Wt1, is a transcriptional activator of the erythropoietin gene. Blood 2006, 107, 4282–4290. [Google Scholar] [CrossRef]
- Sánchez-Elsner, T.; Ramírez, J.R.; Sanz-Rodriguez, F.; Varela, E.; Bernabéu, C.; Botella, L.M.; Rodriguez-Sanz, F. A cross-talk between hypoxia and TGF-β orchestrates erythropoietin gene regulation through SP1 and Smads. J. Mol. Biol. 2004, 336, 9–24. [Google Scholar] [CrossRef]
- D’Andrea, A.D.; Lodish, H.F.; Wong, G.G. Expression cloning of the murine erythropoietin receptor. Cell 1989, 57, 277–285. [Google Scholar] [CrossRef]
- Broudy, V.C.; Lin, N.; Brice, M.; Nakamoto, B.; Papayannopoulou, T. Erythropoietin receptor characteristics on primary human erythroid cells. Blood 1991, 77, 2583–2590. [Google Scholar]
- Chiba, T.; Ikawa, Y.; Todokoro, K. GATA-1 transactivates erythropoietin receptor gene, and erythropoietin receptor-mediated signals enhance GATA-1 gene expression. Nucleic Acids Res. 1991, 19, 3843–3848. [Google Scholar] [CrossRef]
- Fujiwara, T.; O’Geen, H.; Keles, S.; Blahnik, K.; Linnemann, A.K.; Kang, Y.A.; Choi, K.; Farnham, P.J.; Bresnick, E.H. Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Mol. Cell 2009, 36, 667–681. [Google Scholar] [CrossRef]
- Chin, K.; Oda, N.; Shen, K.; Noguchi, C.T. Regulation of transcription of the human erythropoietin receptor gene by proteins binding to GATA-1 and Sp1 motifs. Nucleic Acids Res. 1995, 23, 3041–3049. [Google Scholar] [CrossRef]
- Rogers, H.; Wang, L.; Yu, X.; Alnaeeli, M.; Cui, K.; Zhao, K.; Bieker, J.J.; Prchal, J.; Huang, S.; Weksler, B.; et al. T-cell acute leukemia 1 (TAL1) regulation of erythropoietin receptor and association with excessive erythrocytosis. J. Biol. Chem. 2012, 287, 36720–36731. [Google Scholar]
- Anagnostou, A.; Liu, Z.; Steiner, M.; Chin, K.; Lee, E.S.; Kessimian, N.; Noguchi, C.T. Erythropoietin receptor mRNA expression in human endothelial cells. Proc. Natl. Acad. Sci. USA 1994, 91, 3974–3978. [Google Scholar]
- Morishita, E.; Masuda, S.; Nagao, M.; Yasuda, Y.; Sasaki, R. Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience 1997, 76, 105–116. [Google Scholar]
- Yu, X.; Shacka, J.J.; Eells, J.B.; Suarez-Quian, C.; Przygodzki, R.M.; Beleslin-Cokic, B.; Lin, C.S.; Nikodem, V.M.; Hempstead, B.; Flanders, K.C.; et al. Erythropoietin receptor signalling is required for normal brain development. Development 2002, 129, 505–516. [Google Scholar]
- Wang, L.; Jia, Y.; Rogers, H.; Wu, Y.P.; Huang, S.; Noguchi, C.T. GATA-binding protein 4 (GATA-4) and T-cell acute leukemia 1 (TAL1) regulate myogenic differentiation and erythropoietin response via cross-talk with Sirtuin1 (Sirt1). J. Biol. Chem. 2012, 287, 30157–30169. [Google Scholar]
- Funakoshi-Tago, M.; Pelletier, S.; Moritake, H.; Parganas, E.; Ihle, J.N. Jak2 FERM domain interaction with the erythropoietin receptor regulates Jak2 kinase activity. Mol. Cell. Biol. 2008, 28, 1792–1801. [Google Scholar] [CrossRef]
- Livnah, O.; Stura, E.A.; Middleton, S.A.; Johnson, D.L.; Jolliffe, L.K.; Wilson, I.A. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science 1999, 283, 987–990. [Google Scholar] [CrossRef]
- Quelle, F.W.; Wang, D.; Nosaka, T.; Thierfelder, W.E.; Stravopodis, D.; Weinstein, Y.; Ihle, J.N. Erythropoietin induces activation of STAT5 through association with specific tyrosines on the receptor that are not required for a mitogenic response. Mol. Cell. Biol. 1996, 16, 1622–1631. [Google Scholar]
- Zhao, W.; Kitidis, C.; Fleming, M.D.; Lodish, H.F.; Ghaffari, S. Erythropoietin stimulates phosphorylation and activation of GATA-1 via the PI3-kinase/AKT signaling pathway. Blood 2006, 107, 907–915. [Google Scholar]
- Grover, A.; Mancini, E.; Moore, S.; Mead, A.J.; Atkinson, D.; Rasmussen, K.D.; O’Carroll, D.; Jacobsen, S.E.; Nerlov, C. Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate. J. Exp. Med. 2014, 211, 181–188. [Google Scholar] [CrossRef]
- Sawyer, S.T.; Penta, K. Association of JAK2 and STAT5 with erythropoietin receptors. Role of receptor phosphorylation in erythropoietin signal transduction. J. Biol. Chem. 1996, 271, 32430–32437. [Google Scholar] [CrossRef]
- Silva, M.; Benito, A.; Sanz, C.; Prosper, F.; Ekhterae, D.; Nunez, G.; Fernandez-Luna, J.L. Erythropoietin can induce the expression of bcl-xL through STAT5 in erythropoietin-dependent progenitor cell lines. J. Biol. Chem. 1999, 274, 22165–22169. [Google Scholar]
- Socolovsky, M.; Nam, H.; Fleming, M.D.; Haase, V.H.; Brugnara, C.; Lodish, H.F. Ineffective erythropoiesis in STAT5a(−/−)5b(−/−) mice due to decreased survival of early erythroblasts. Blood 2001, 98, 3261–3273. [Google Scholar] [CrossRef]
- Mason, J.M.; Beattie, B.K.; Liu, Q.; Dumont, D.J.; Barber, D.L. The SH2 inositol 5-phosphatase Ship1 is recruited in an SH2-dependent manner to the erythropoietin receptor. J. Biol. Chem. 2000, 275, 4398–4406. [Google Scholar]
- Chen, C.; Sytkowski, A.J. Erythropoietin activates two distinct signaling pathways required for the initiation and the elongation of c-MYC. J. Biol. Chem. 2001, 276, 38518–38526. [Google Scholar] [CrossRef]
- Sui, X.; Krantz, S.B.; You, M.; Zhao, Z. Synergistic activation of MAP kinase (ERK1/2) by erythropoietin and stem cell factor is essential for expanded erythropoiesis. Blood 1998, 92, 1142–1149. [Google Scholar]
- Matsuzaki, T.; Aisaki, K.; Yamamura, Y.; Noda, M.; Ikawa, Y. Induction of erythroid differentiation by inhibition of Ras/ERK pathway in a friend murine leukemia cell line. Oncogene 2000, 19, 1500–1508. [Google Scholar] [CrossRef]
- Miura, Y.; Miura, O.; Ihle, J.N.; Aoki, N. Activation of the mitogen-activated protein kinase pathway by the erythropoietin receptor. J. Biol. Chem. 1994, 269, 29962–29969. [Google Scholar]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. AKT promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Damen, J.E.; Mui, A.L.; Puil, L.; Pawson, T.; Krystal, G. Phosphatidylinositol 3-kinase associates, via its Src homology 2 domains, with the activated erythropoietin receptor. Blood 1993, 81, 3204–3210. [Google Scholar]
- He, T.C.; Zhuang, H.; Jiang, N.; Waterfield, M.D.; Wojchowski, D.M. Association of the p85 regulatory subunit of phosphatidylinositol 3-kinase with an essential erythropoietin receptor subdomain. Blood 1993, 82, 3530–3538. [Google Scholar]
- Wickrema, A.; Uddin, S.; Sharma, A.; Chen, F.; Alsayed, Y.; Ahmad, S.; Sawyer, S.T.; Krystal, G.; Yi, T.; Nishada, K.; et al. Engagement of Gab1 and Gab2 in erythropoietin signaling. J. Biol. Chem. 1999, 274, 24469–24474. [Google Scholar] [CrossRef]
- Shigematsu, H.; Iwasaki, H.; Otsuka, T.; Ohno, Y.; Arima, F.; Niho, Y. Role of the vav proto-oncogene product (Vav) in erythropoietin-mediated cell proliferation and phosphatidylinositol 3-kinase activity. J. Biol. Chem. 1997, 272, 14334–14340. [Google Scholar]
- Jaster, R.; Bittorf, T.; Brock, J. Involvement of phosphatidylinositol 3-kinase in the mediation of erythropoietin-induced activation of p70S6k. Cell Signal. 1997, 9, 175–179. [Google Scholar] [CrossRef]
- Wierenga, A.T.; Vogelzang, I.; Eggen, B.J.; Vellenga, E. Erythropoietin-induced serine 727 phosphorylation of STAT3 in erythroid cells is mediated by a MEK-, ERK-, and MSK1-dependent pathway. Exp. Hematol. 2003, 31, 398–405. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Sinn, D.I.; Jung, K.H.; Kim, E.H.; Kim, S.J.; Kim, J.M.; Ko, S.Y.; Kim, M.; Roh, J.K. Erythropoietin reduces perihematomal inflammation and cell death with eNOS and STAT3 activations in experimental intracerebral hemorrhage. J. Neurochem. 2006, 96, 1728–1739. [Google Scholar] [CrossRef]
- Zhao, J.; Li, G.; Zhang, Y.; Su, X.; Hang, C. The potential role of JAK2/STAT3 pathway on the anti-apoptotic effect of recombinant human erythropoietin (rhEPO) after experimental traumatic brain injury of rats. Cytokine 2011, 56, 343–350. [Google Scholar] [CrossRef]
- Kretz, A.; Happold, C.J.; Marticke, J.K.; Isenmann, S. Erythropoietin promotes regeneration of adult CNS neurons via Jak2/Stat3 and PI3K/AKT pathway activation. Mol. Cell. Neurosci. 2005, 29, 569–579. [Google Scholar] [CrossRef]
- Alnaeeli, M.; Raaka, B.M.; Gavrilova, O.; Teng, R.; Chanturiya, T.; Noguchi, C.T. Erythropoietin signaling: A novel regulator of white adipose tissue inflammation during diet-induced obesity. Diabetes 2014. [Google Scholar] [CrossRef]
- Ghezzi, P.; Bernaudin, M.; Bianchi, R.; Blomgren, K.; Brines, M.; Campana, W.; Cavaletti, G.; Cerami, A.; Chopp, M.; Coleman, T.; et al. Erythropoietin: Not just about erythropoiesis. Lancet 2010, 375, 2142. [Google Scholar]
- Sinclair, A.M.; Coxon, A.; McCaffery, I.; Kaufman, S.; Paweletz, K.; Liu, L.; Busse, L.; Swift, S.; Elliott, S.; Begley, C.G. Functional erythropoietin receptor is undetectable in endothelial, cardiac, neuronal, and renal cells. Blood 2010, 115, 4264–4272. [Google Scholar] [CrossRef]
- Anagnostou, A.; Lee, E.S.; Kessimian, N.; Levinson, R.; Steiner, M. Erythropoietin has a mitogenic and positive chemotactic effect on endothelial cells. Proc. Natl. Acad. Sci. USA 1990, 87, 5978–5982. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Erythropoietin is a novel vascular protectant through activation of AKT1 and mitochondrial modulation of cysteine proteases. Circulation 2002, 106, 2973–2979. [Google Scholar] [CrossRef]
- Hou, J.; Wang, S.; Shang, Y.C.; Chong, Z.Z.; Maiese, K. Erythropoietin employs cell longevity pathways of SIRT1 to foster endothelial vascular integrity during oxidant stress. Curr. Neurovasc. Res. 2011, 8, 220–235. [Google Scholar] [CrossRef]
- Beleslin-Cokic, B.B.; Cokic, V.P.; Yu, X.; Weksler, B.B.; Schechter, A.N.; Noguchi, C.T. Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood 2004, 104, 2073–2080. [Google Scholar] [CrossRef]
- Beleslin-Cokic, B.B.; Cokic, V.P.; Wang, L.; Piknova, B.; Teng, R.; Schechter, A.N.; Noguchi, C.T. Erythropoietin and hypoxia increase erythropoietin receptor and nitric oxide levels in lung microvascular endothelial cells. Cytokine 2011, 54, 129–135. [Google Scholar] [CrossRef]
- Ruschitzka, F.T.; Wenger, R.H.; Stallmach, T.; Quaschning, T.; de Wit, C.; Wagner, K.; Labugger, R.; Kelm, M.; Noll, G.; Rulicke, T.; et al. Nitric oxide prevents cardiovascular disease and determines survival in polyglobulic mice overexpressing erythropoietin. Proc. Natl. Acad. Sci. USA 2000, 97, 11609–11613. [Google Scholar]
- Kanagy, N.L.; Perrine, M.F.; Cheung, D.K.; Walker, B.R. Erythropoietin administration in vivo increases vascular nitric oxide synthase expression. J. Cardiovasc. Pharmacol. 2003, 42, 527–533. [Google Scholar] [CrossRef]
- Quaschning, T.; Ruschitzka, F.; Stallmach, T.; Shaw, S.; Morawietz, H.; Goettsch, W.; Hermann, M.; Slowinski, T.; Theuring, F.; Hocher, B.; et al. Erythropoietin-induced excessive erythrocytosis activates the tissue endothelin system in mice. FASEB J. 2003, 17, 259–261. [Google Scholar]
- Kertesz, N.; Wu, J.; Chen, T.H.; Sucov, H.M.; Wu, H. The role of erythropoietin in regulating angiogenesis. Dev. Biol. 2004, 276, 101–110. [Google Scholar]
- Suzuki, N.; Ohneda, O.; Takahashi, S.; Higuchi, M.; Mukai, H.Y.; Nakahata, T.; Imagawa, S.; Yamamoto, M. Erythroid-specific expression of the erythropoietin receptor rescued its null mutant mice from lethality. Blood 2002, 100, 2279–2288. [Google Scholar] [CrossRef]
- Satoh, K.; Kagaya, Y.; Nakano, M.; Ito, Y.; Ohta, J.; Tada, H.; Karibe, A.; Minegishi, N.; Suzuki, N.; Yamamoto, M.; et al. Important role of endogenous erythropoietin system in recruitment of endothelial progenitor cells in hypoxia-induced pulmonary hypertension in mice. Circulation 2006, 113, 1442–1450. [Google Scholar] [CrossRef]
- Carraway, M.S.; Suliman, H.B.; Jones, W.S.; Chen, C.W.; Babiker, A.; Piantadosi, C.A. Erythropoietin activates mitochondrial biogenesis and couples red cell mass to mitochondrial mass in the heart. Circ. Res. 2010, 106, 1722–1730. [Google Scholar] [CrossRef]
- Teng, R.; Calvert, J.W.; Sibmooh, N.; Piknova, B.; Suzuki, N.; Sun, J.; Martinez, K.; Yamamoto, M.; Schechter, A.N.; Lefer, D.J.; et al. Acute erythropoietin cardioprotection is mediated by endothelial response. Basic Res. Cardiol. 2011, 106, 343–354. [Google Scholar] [CrossRef]
- Santhanam, A.V.; Smith, L.A.; Nath, K.A.; Katusic, Z.S. In vivo stimulatory effect of erythropoietin on endothelial nitric oxide synthase in cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H781–H786. [Google Scholar] [CrossRef]
- Bahlmann, F.H.; Song, R.; Boehm, S.M.; Mengel, M.; von Wasielewski, R.; Lindschau, C.; Kirsch, T.; de Groot, K.; Laudeley, R.; Niemczyk, E.; et al. Low-dose therapy with the long-acting erythropoietin analogue darbepoetin alpha persistently activates endothelial AKT and attenuates progressive organ failure. Circulation 2004, 110, 1006–1012. [Google Scholar] [CrossRef]
- Yasuda, Y.; Masuda, S.; Chikuma, M.; Inoue, K.; Nagao, M.; Sasaki, R. Estrogen-dependent production of erythropoietin in uterus and its implication in uterine angiogenesis. J. Biol. Chem. 1998, 273, 25381–25387. [Google Scholar] [CrossRef]
- Mukundan, H.; Resta, T.C.; Kanagy, N.L. 17β-Estradiol decreases hypoxic induction of erythropoietin gene expression. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R496–R504. [Google Scholar]
- Wu, H.; Lee, S.H.; Gao, J.; Liu, X.; Iruela-Arispe, M.L. Inactivation of erythropoietin leads to defects in cardiac morphogenesis. Development 1999, 126, 3597–3605. [Google Scholar]
- Banerjee, D.; Rodriguez, M.; Nag, M.; Adamson, J.W. Exposure of endothelial cells to recombinant human erythropoietin induces nitric oxide synthase activity. Kidney Int. 2000, 57, 1895–1904. [Google Scholar] [CrossRef]
- Burger, D.; Lei, M.; Geoghegan-Morphet, N.; Lu, X.; Xenocostas, A.; Feng, Q. Erythropoietin protects cardiomyocytes from apoptosis via up-regulation of endothelial nitric oxide synthase. Cardiovasc. Res. 2006, 72, 51–59. [Google Scholar] [CrossRef]
- Rui, T.; Feng, Q.; Lei, M.; Peng, T.; Zhang, J.; Xu, M.; Abel, E.D.; Xenocostas, A.; Kvietys, P.R. Erythropoietin prevents the acute myocardial inflammatory response induced by ischemia/reperfusion via induction of AP-1. Cardiovasc. Res. 2005, 65, 719–727. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Chin, K.; Noguchi, C.T. Tissue specific expression of human erythropoietin receptor in transgenic mice. Dev. Biol. 1994, 166, 159–169. [Google Scholar] [CrossRef]
- Liu, C.; Shen, K.; Liu, Z.; Noguchi, C.T. Regulated human erythropoietin receptor expression in mouse brain. J. Biol. Chem. 1997, 272, 32395–32400. [Google Scholar] [CrossRef]
- Masuda, S.; Okano, M.; Yamagishi, K.; Nagao, M.; Ueda, M.; Sasaki, R. A novel site of erythropoietin production. Oxygen-dependent production in cultured rat astrocytes. J. Biol. Chem. 1994, 269, 19488–19493. [Google Scholar]
- Marti, H.H.; Gassmann, M.; Wenger, R.H.; Kvietikova, I.; Morganti-Kossmann, M.C.; Kossmann, T.; Trentz, O.; Bauer, C. Detection of erythropoietin in human liquor: Intrinsic erythropoietin production in the brain. Kidney Int. 1997, 51, 416–418. [Google Scholar] [CrossRef]
- Bernaudin, M.; Bellail, A.; Marti, H.H.; Yvon, A.; Vivien, D.; Duchatelle, I.; Mackenzie, E.T.; Petit, E. Neurons and astrocytes express EPO mRNA: Oxygen-sensing mechanisms that involve the redox-state of the brain. Glia 2000, 30, 271–278. [Google Scholar] [CrossRef]
- Marti, H.H.; Wenger, R.H.; Rivas, L.A.; Straumann, U.; Digicaylioglu, M.; Henn, V.; Yonekawa, Y.; Bauer, C.; Gassmann, M. Erythropoietin gene expression in human, monkey and murine brain. Eur. J. Neurosci. 1996, 8, 666–676. [Google Scholar] [CrossRef]
- Chavez, J.C.; Baranova, O.; Lin, J.; Pichiule, P. The transcriptional activator hypoxia inducible factor 2 (HIF-2/EPAS-1) regulates the oxygen-dependent expression of erythropoietin in cortical astrocytes. J. Neurosci. 2006, 26, 9471–9481. [Google Scholar] [CrossRef]
- Knabe, W.; Knerlich, F.; Washausen, S.; Kietzmann, T.; Siren, A.L.; Brunnett, G.; Kuhn, H.J.; Ehrenreich, H. Expression patterns of erythropoietin and its receptor in the developing midbrain. Anat. Embryol. 2004, 207, 503–512. [Google Scholar] [CrossRef]
- Knabe, W.; Siren, A.L.; Ehrenreich, H.; Kuhn, H.J. Expression patterns of erythropoietin and its receptor in the developing spinal cord and dorsal root ganglia. Anat. Embryol. 2005, 210, 209–219. [Google Scholar] [CrossRef]
- Tsai, P.T.; Ohab, J.J.; Kertesz, N.; Groszer, M.; Matter, C.; Gao, J.; Liu, X.; Wu, H.; Carmichael, S.T. A critical role of erythropoietin receptor in neurogenesis and post-stroke recovery. J. Neurosci. 2006, 26, 1269–1274. [Google Scholar] [CrossRef]
- Digicaylioglu, M.; Bichet, S.; Marti, H.H.; Wenger, R.H.; Rivas, L.A.; Bauer, C.; Gassmann, M. Localization of specific erythropoietin binding sites in defined areas of the mouse brain. Proc. Natl. Acad. Sci. USA 1995, 92, 3717–3720. [Google Scholar] [CrossRef]
- Prass, K.; Scharff, A.; Ruscher, K.; Lowl, D.; Muselmann, C.; Victorov, I.; Kapinya, K.; Dirnagl, U.; Meisel, A. Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke 2003, 34, 1981–1986. [Google Scholar] [CrossRef]
- Chikuma, M.; Masuda, S.; Kobayashi, T.; Nagao, M.; Sasaki, R. Tissue-specific regulation of erythropoietin production in the murine kidney, brain, and uterus. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1242–E1248. [Google Scholar]
- Studer, L.; Csete, M.; Lee, S.H.; Kabbani, N.; Walikonis, J.; Wold, B.; McKay, R. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J. Neurosci. 2000, 20, 7377–7383. [Google Scholar]
- Shingo, T.; Sorokan, S.T.; Shimazaki, T.; Weiss, S. Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J. Neurosci. 2001, 21, 9733–9743. [Google Scholar]
- Chen, Z.Y.; Asavaritikrai, P.; Prchal, J.T.; Noguchi, C.T. Endogenous erythropoietin signaling is required for normal neural progenitor cell proliferation. J. Biol. Chem. 2007, 282, 25875–25883. [Google Scholar]
- Juul, S.E.; Yachnis, A.T.; Rojiani, A.M.; Christensen, R.D. Immunohistochemical localization of erythropoietin and its receptor in the developing human brain. Pediatr. Dev. Pathol. 1999, 2, 148–158. [Google Scholar] [CrossRef]
- Juul, S.E.; Anderson, D.K.; Li, Y.; Christensen, R.D. Erythropoietin and erythropoietin receptor in the developing human central nervous system. Pediatr. Res. 1998, 43, 40–49. [Google Scholar]
- Chin, K.; Yu, X.; Beleslin-Cokic, B.; Liu, C.; Shen, K.; Mohrenweiser, H.W.; Noguchi, C.T. Production and processing of erythropoietin receptor transcripts in brain. Brain Res. Mol. Brain Res. 2000, 81, 29–42. [Google Scholar] [CrossRef]
- Juul, S.E.; Yachnis, A.T.; Christensen, R.D. Tissue distribution of erythropoietin and erythropoietin receptor in the developing human fetus. Early Hum. Dev. 1998, 52, 235–249. [Google Scholar] [CrossRef]
- Juul, S.E.; Stallings, S.A.; Christensen, R.D. Erythropoietin in the cerebrospinal fluid of neonates who sustained CNS injury. Pediatr. Res. 1999, 46, 543–547. [Google Scholar] [CrossRef]
- Sakanaka, M.; Wen, T.C.; Matsuda, S.; Masuda, S.; Morishita, E.; Nagao, M.; Sasaki, R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc. Natl. Acad. Sci. USA 1998, 95, 4635–4640. [Google Scholar]
- Bernaudin, M.; Nedelec, A.S.; Divoux, D.; MacKenzie, E.T.; Petit, E.; Schumann-Bard, P. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J. Cereb. Blood Flow Metab. 2002, 22, 393–403. [Google Scholar]
- Wang, L.; Zhang, Z.; Wang, Y.; Zhang, R.; Chopp, M. Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke 2004, 35, 1732–1737. [Google Scholar] [CrossRef]
- Malhotra, S.; Savitz, S.I.; Ocava, L.; Rosenbaum, D.M. Ischemic preconditioning is mediated by erythropoietin through PI-3 kinase signaling in an animal model of transient ischemic attack. J. Neurosci. Res. 2006, 83, 19–27. [Google Scholar] [CrossRef]
- Leconte, C.; Tixier, E.; Freret, T.; Toutain, J.; Saulnier, R.; Boulouard, M.; Roussel, S.; Schumann-Bard, P.; Bernaudin, M. Delayed hypoxic postconditioning protects against cerebral ischemia in the mouse. Stroke 2009, 40, 3349–3355. [Google Scholar] [CrossRef]
- Teng, R.; Gavrilova, O.; Suzuki, N.; Chanturiya, T.; Schimel, D.; Hugendubler, L.; Mammen, S.; Yver, D.R.; Cushman, S.W.; Mueller, E.; et al. Disrupted erythropoietin signalling promotes obesity and alters hypothalamus proopiomelanocortin production. Nat. Commun. 2011, 2, 520. [Google Scholar]
- Sadamoto, Y.; Igase, K.; Sakanaka, M.; Sato, K.; Otsuka, H.; Sakaki, S.; Masuda, S.; Sasaki, R. Erythropoietin prevents place navigation disability and cortical infarction in rats with permanent occlusion of the middle cerebral artery. Biochem. Biophys. Res. Commun. 1998, 253, 26–32. [Google Scholar] [CrossRef]
- Bernaudin, M.; Marti, H.H.; Roussel, S.; Divoux, D.; Nouvelot, A.; MacKenzie, E.T.; Petit, E. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 1999, 19, 643–651. [Google Scholar]
- Lewczuk, P.; Hasselblatt, M.; Kamrowski-Kruck, H.; Heyer, A.; Unzicker, C.; Siren, A.L.; Ehrenreich, H. Survival of hippocampal neurons in culture upon hypoxia: Effect of erythropoietin. Neuroreport 2000, 11, 3485–3488. [Google Scholar] [CrossRef]
- Digicaylioglu, M.; Lipton, S.A. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-κB signalling cascades. Nature 2001, 412, 641–647. [Google Scholar] [CrossRef]
- Wang, C.Y.; Mayo, M.W.; Korneluk, R.G.; Goeddel, D.V.; Baldwin, A.S., Jr. NF-κB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281, 1680–1683. [Google Scholar] [CrossRef]
- De Smaele, E.; Zazzeroni, F.; Papa, S.; Nguyen, D.U.; Jin, R.; Jones, J.; Cong, R.; Franzoso, G. Induction of GADD45β by NF-κB downregulates pro-apoptotic JNK signalling. Nature 2001, 414, 308–313. [Google Scholar] [CrossRef]
- Chen, C.; Edelstein, L.C.; Gelinas, C. The Rel/NF-κB family directly activates expression of the apoptosis inhibitor Bcl-xL. Mol. Cell. Biol. 2000, 20, 2687–2695. [Google Scholar] [CrossRef]
- Brines, M.; Grasso, G.; Fiordaliso, F.; Sfacteria, A.; Ghezzi, P.; Fratelli, M.; Latini, R.; Xie, Q.W.; Smart, J.; Su-Rick, C.J.; et al. Erythropoietin mediates tissue protection through an erythropoietin and common β-subunit heteroreceptor. Proc. Natl. Acad. Sci. USA 2004, 101, 14907–14912. [Google Scholar] [CrossRef]
- Sanchez, P.E.; Navarro, F.P.; Fares, R.P.; Nadam, J.; Georges, B.; Moulin, C.; le Cavorsin, M.; Bonnet, C.; Ryvlin, P.; Belmeguenai, A.; et al. Erythropoietin receptor expression is concordant with erythropoietin but not with common β chain expression in the rat brain throughout the life span. J. Comp. Neurol. 2009, 514, 403–414. [Google Scholar] [CrossRef]
- Um, M.; Gross, A.W.; Lodish, H.F. A “classical” homodimeric erythropoietin receptor is essential for the antiapoptotic effects of erythropoietin on differentiated neuroblastoma SH-SY5Y and pheochromocytoma PC-12 cells. Cell Signal. 2007, 19, 634–645. [Google Scholar] [CrossRef]
- Wang, S.; Wu, Z.; Chiang, P.; Fink, D.J.; Mata, M. Vector-mediated expression of erythropoietin improves functional outcome after cervical spinal cord contusion injury. Gene Ther. 2012, 19, 907–914. [Google Scholar]
- Wang, L.; Zhang, Z.G.; Zhang, R.L.; Gregg, S.R.; Hozeska-Solgot, A.; LeTourneau, Y.; Wang, Y.; Chopp, M. Matrix metalloproteinase 2 (MMP2) and MMP9 secreted by erythropoietin-activated endothelial cells promote neural progenitor cell migration. J. Neurosci. 2006, 26, 5996–6003. [Google Scholar] [CrossRef]
- Gorio, A.; Gokmen, N.; Erbayraktar, S.; Yilmaz, O.; Madaschi, L.; Cichetti, C.; di Giulio, A.M.; Vardar, E.; Cerami, A.; Brines, M. Recombinant human erythropoietin counteracts secondary injury and markedly enhances neurological recovery from experimental spinal cord trauma. Proc. Natl. Acad. Sci. USA 2002, 99, 9450–9455. [Google Scholar] [CrossRef]
- Pinzon, A.; Marcillo, A.; Pabon, D.; Bramlett, H.M.; Bunge, M.B.; Dietrich, W.D. A re-assessment of erythropoietin as a neuroprotective agent following rat spinal cord compression or contusion injury. Exp. Neurol. 2008, 213, 129–136. [Google Scholar] [CrossRef]
- Cerri, G.; Montagna, M.; Madaschi, L.; Merli, D.; Borroni, P.; Baldissera, F.; Gorio, A. Erythropoietin effect on sensorimotor recovery after contusive spinal cord injury: An electrophysiological study in rats. Neuroscience 2012, 219, 290–301. [Google Scholar] [CrossRef]
- Kuang, S.; Kuroda, K.; le Grand, F.; Rudnicki, M.A. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 2007, 129, 999–1010. [Google Scholar] [CrossRef]
- Jia, Y.; Warin, R.; Yu, X.; Epstein, R.; Noguchi, C.T. Erythropoietin signaling promotes transplanted progenitor cell survival. FASEB J. 2009, 23, 3089–3099. [Google Scholar] [CrossRef]
- Christensen, B.; Lundby, C.; Jessen, N.; Nielsen, T.S.; Vestergaard, P.F.; Moller, N.; Pilegaard, H.; Pedersen, S.B.; Kopchick, J.J.; Jorgensen, J.O. Evaluation of functional erythropoietin receptor status in skeletal muscle in vivo: Acute and prolonged studies in healthy human subjects. PLoS One 2012, 7, e31857. [Google Scholar]
- Lundby, C.; Hellsten, Y.; Jensen, M.B.; Munch, A.S.; Pilegaard, H. Erythropoietin receptor in human skeletal muscle and the effects of acute and long-term injections with recombinant human erythropoietin on the skeletal muscle. J. Appl. Physiol. 2008, 104, 1154–1160. [Google Scholar] [CrossRef]
- Lamon, S.; Zacharewicz, E.; Stephens, A.N.; Russell, A.P. EPO-receptor is present in mouse C2C12 and human primary skeletal muscle cells but EPO does not influence myogenesis. Physiol. Rep. 2014, 2, e00256. [Google Scholar]
- Jia, Y.; Suzuki, N.; Yamamoto, M.; Gassmann, M.; Noguchi, C.T. Endogenous erythropoietin signaling facilitates skeletal muscle repair and recovery following pharmacologically induced damage. FASEB J. 2012, 26, 2847–2858. [Google Scholar] [CrossRef]
- Rezaeian, F.; Wettstein, R.; Egger, J.F.; Sandmann, F.; Rucker, M.; Tobalem, M.; Vollmar, B.; Menger, M.D.; Harder, Y. Erythropoietin-induced upregulation of endothelial nitric oxide synthase but not vascular endothelial growth factor prevents musculocutaneous tissue from ischemic damage. Lab. Investig. 2010, 90, 40–51. [Google Scholar] [CrossRef]
- Kato, S.; Amano, H.; Ito, Y.; Eshima, K.; Aoyama, N.; Tamaki, H.; Sakagami, H.; Satoh, Y.; Izumi, T.; Majima, M. Effect of erythropoietin on angiogenesis with the increased adhesion of platelets to the microvessels in the hind-limb ischemia model in mice. J. Pharmacol. Sci. 2010, 112, 167–175. [Google Scholar] [CrossRef]
- Rotter, R.; Menshykova, M.; Winkler, T.; Matziolis, G.; Stratos, I.; Schoen, M.; Bittorf, T.; Mittlmeier, T.; Vollmar, B. Erythropoietin improves functional and histological recovery of traumatized skeletal muscle tissue. J. Orthop. Res. 2008, 26, 1618–1626. [Google Scholar] [CrossRef]
- Wang, L.; Jia, Y.; Rogers, H.; Suzuki, N.; Gassmann, M.; Wang, Q.; McPherron, A.C.; Kopp, J.B.; Yamamoto, M.; Noguchi, C.T. Erythropoietin contributes to slow oxidative muscle fiber specification via PGC-1α and AMPK activation. Int. J. Biochem. Cell Biol. 2013, 45, 1155–1164. [Google Scholar] [CrossRef]
- Mak, R.H. Metabolic effects of erythropoietin in patients on peritoneal dialysis. Pediatr. Nephrol. 1998, 12, 660–665. [Google Scholar] [CrossRef]
- Mak, R.H. Effect of recombinant human erythropoietin on insulin, amino acid, and lipid metabolism in uremia. J. Pediatr. 1996, 129, 97–104. [Google Scholar] [CrossRef]
- Bofill, C.; Joven, J.; Bages, J.; Vilella, E.; Sans, T.; Cavalle, P.; Miralles, R.; Llobet, J.; Camps, J. Response to repeated phlebotomies in patients with non-insulin-dependent diabetes mellitus. Metabolism 1994, 43, 614–620. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Baskin, D.G. Leptin and the brain: Then and now. J. Clin. Investig. 2013, 123, 2344–2345. [Google Scholar] [CrossRef]
- Galeano, M.; Altavilla, D.; Cucinotta, D.; Russo, G.T.; Calo, M.; Bitto, A.; Marini, H.; Marini, R.; Adamo, E.B.; Seminara, P.; et al. Recombinant human erythropoietin stimulates angiogenesis and wound healing in the genetically diabetic mouse. Diabetes 2004, 53, 2509–2517. [Google Scholar] [CrossRef]
- Loeffler, I.; Ruster, C.; Franke, S.; Liebisch, M.; Wolf, G. Erythropoietin ameliorates podocyte injury in advanced diabetic nephropathy in the db/db mouse. Am. J. Physiol. Renal Physiol. 2013, 305, F911–F918. [Google Scholar] [CrossRef]
- Menne, J.; Park, J.K.; Shushakova, N.; Mengel, M.; Meier, M.; Fliser, D. The continuous erythropoietin receptor activator affects different pathways of diabetic renal injury. J. Am. Soc. Nephrol. 2007, 18, 2046–2053. [Google Scholar] [CrossRef]
- Shushakova, N.; Park, J.K.; Menne, J.; Fliser, D. Chronic erythropoietin treatment affects different molecular pathways of diabetic cardiomyopathy in mouse. Eur. J. Clin. Investig. 2009, 39, 755–760. [Google Scholar] [CrossRef]
- Choi, D.; Schroer, S.A.; Lu, S.Y.; Wang, L.; Wu, X.; Liu, Y.; Zhang, Y.; Gaisano, H.Y.; Wagner, K.U.; Wu, H.; et al. Erythropoietin protects against diabetes through direct effects on pancreatic beta cells. J. Exp. Med. 2010, 207, 2831–2842. [Google Scholar] [CrossRef]
- Campfield, L.A.; Smith, F.J.; Guisez, Y.; Devos, R.; Burn, P. Recombinant mouse OB protein: Evidence for a peripheral signal linking adiposity and central neural networks. Science 1995, 269, 546–549. [Google Scholar]
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar]
- Pelleymounter, M.A.; Cullen, M.J.; Baker, M.B.; Hecht, R.; Winters, D.; Boone, T.; Collins, F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science 1995, 269, 540–543. [Google Scholar]
- Katz, O.; Stuible, M.; Golishevski, N.; Lifshitz, L.; Tremblay, M.L.; Gassmann, M.; Mittelman, M.; Neumann, D. Erythropoietin treatment leads to reduced blood glucose levels and body mass: Insights from murine models. J. Endocrinol. 2010, 205, 87–95. [Google Scholar]
- Soliz, J.; Soulage, C.; Hermann, D.M.; Gassmann, M. Acute and chronic exposure to hypoxia alters ventilatory pattern but not minute ventilation of mice overexpressing erythropoietin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1702–R1710. [Google Scholar] [CrossRef]
- Soliz, J.; Joseph, V.; Soulage, C.; Becskei, C.; Vogel, J.; Pequignot, J.M.; Ogunshola, O.; Gassmann, M. Erythropoietin regulates hypoxic ventilation in mice by interacting with brainstem and carotid bodies. J. Physiol. 2005, 568, 559–571. [Google Scholar] [CrossRef]
- Schuler, B.; Vogel, J.; Grenacher, B.; Jacobs, R.A.; Arras, M.; Gassmann, M. Acute and chronic elevation of erythropoietin in the brain improves exercise performance in mice without inducing erythropoiesis. FASEB J. 2012, 26, 3884–3890. [Google Scholar] [CrossRef]
- Dale, E.A.; Satriotomo, I.; Mitchell, G.S. Cervical spinal erythropoietin induces phrenic motor facilitation via extracellular signal-regulated protein kinase and AKT signaling. J. Neurosci. 2012, 32, 5973–5983. [Google Scholar] [CrossRef]
- Hojman, P.; Brolin, C.; Gissel, H.; Brandt, C.; Zerahn, B.; Pedersen, B.K.; Gehl, J. Erythropoietin over-expression protects against diet-induced obesity in mice through increased fat oxidation in muscles. PLoS One 2009, 4, e5894. [Google Scholar]
- Surwit, R.S.; Kuhn, C.M.; Cochrane, C.; McCubbin, J.A.; Feinglos, M.N. Diet-induced type II diabetes in C57BL/6J mice. Diabetes 1988, 37, 1163–1167. [Google Scholar] [CrossRef]
- Surwit, R.S.; Feinglos, M.N.; Rodin, J.; Sutherland, A.; Petro, A.E.; Opara, E.C.; Kuhn, C.M.; Rebuffe-Scrive, M. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metabolism 1995, 44, 645–651. [Google Scholar] [CrossRef]
- Toye, A.A.; Lippiat, J.D.; Proks, P.; Shimomura, K.; Bentley, L.; Hugill, A.; Mijat, V.; Goldsworthy, M.; Moir, L.; Haynes, A.; et al. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 2005, 48, 675–686. [Google Scholar] [CrossRef]
- Luk, C.T.; Shi, S.Y.; Choi, D.; Cai, E.P.; Schroer, S.A.; Woo, M. In vivo knockdown of adipocyte erythropoietin receptor does not alter glucose or energy homeostasis. Endocrinology 2013, 154, 3652–3659. [Google Scholar] [CrossRef]
- Bianchi, R.; Buyukakilli, B.; Brines, M.; Savino, C.; Cavaletti, G.; Oggioni, N.; Lauria, G.; Borgna, M.; Lombardi, R.; Cimen, B.; et al. Erythropoietin both protects from and reverses experimental diabetic neuropathy. Proc. Natl. Acad. Sci. USA 2004, 101, 823–828. [Google Scholar] [CrossRef]
- Scott, S.D. Dose conversion from recombinant human erythropoietin to darbepoetin alfa: Recommendations from clinical studies. Pharmacotherapy 2002, 22, 160S–165S. [Google Scholar] [CrossRef]
- Macdougall, I.C. CERA (Continuous Erythropoietin Receptor Activator): A new erythropoiesis-stimulating agent for the treatment of anemia. Curr. Hematol. Rep. 2005, 4, 436–440. [Google Scholar]
- Allegra, V.; Martimbianco, L.; Vasile, A. Lipid and apolipoprotein patterns during erythropoietin therapy: Roles of erythropoietin, route of administration, and diet. Nephrol. Dial. Transpl. 1997, 12, 924–932. [Google Scholar] [CrossRef]
- Prata, M.M.; Madeira, C.; Vicente, O.; Miguel, M.J. Lipid profile in haemodialysis patients treated with recombinant human erythropoietin. Nephrol. Dial. Transpl. 1998, 13, 2345–2347. [Google Scholar] [CrossRef]
- Christensen, B.; Vendelbo, M.H.; Krusenstjerna-Hafstrøm, T.; Madsen, M.; Pedersen, S.B.; Jessen, N.; Møller, N.; Jørgensen, J.O.L. Erythropoietin administration acutely stimulates resting energy expenditure in healthy young men. J. Appl. Physiol. 2012, 112, 1114–1121. [Google Scholar] [CrossRef]
- Millot, S.; Andrieu, V.; Letteron, P.; Lyoumi, S.; Hurtado-Nedelec, M.; Karim, Z.; Thibaudeau, O.; Bennada, S.; Charrier, J.L.; Lasocki, S.; et al. Erythropoietin stimulates spleen BMP4-dependent stress erythropoiesis and partially corrects anemia in a mouse model of generalized inflammation. Blood 2010, 116, 6072–6081. [Google Scholar] [CrossRef]
- Villa, P.; Bigini, P.; Mennini, T.; Agnello, D.; Laragione, T.; Cagnotto, A.; Viviani, B.; Marinovich, M.; Cerami, A.; Coleman, T.R.; et al. Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J. Exp. Med. 2003, 198, 971–975. [Google Scholar] [CrossRef]
- Yatsiv, I.; Grigoriadis, N.; Simeonidou, C.; Stahel, P.F.; Schmidt, O.I.; Alexandrovitch, A.G.; Tsenter, J.; Shohami, E. Erythropoietin is neuroprotective, improves functional recovery, and reduces neuronal apoptosis and inflammation in a rodent model of experimental closed head injury. FASEB J. 2005, 19, 1701–1703. [Google Scholar]
- Mausberg, A.K.; Meyer Zu Horste, G.; Dehmel, T.; Stettner, M.; Lehmann, H.C.; Sheikh, K.A.; Kieseier, B.C. Erythropoietin ameliorates rat experimental autoimmune neuritis by inducing transforming growth factor-β in macrophages. PLoS One 2011, 6, e26280. [Google Scholar] [CrossRef]
- Liu, X.; Shen, J.; Jin, Y.; Duan, M.; Xu, J. Recombinant human erythropoietin (rhEPO) preconditioning on nuclear factor-kappa B (NF-κB) activation & proinflammatory cytokines induced by myocardial ischaemia-reperfusion. Indian J. Med. Res. 2006, 124, 343–354. [Google Scholar]
- Hirose, S.; Takahashi, M.; Ogawa, R.; Morimoto, H.; Izawa, A.; Sato, H.; Ise, H.; Hongo, M.; Ikeda, U. Erythropoietin attenuates the development of experimental autoimmune myocarditis. Cardiovasc. Drugs Ther. 2007, 21, 17–27. [Google Scholar]
- Schreiber, S.; Howaldt, S.; Schnoor, M.; Nikolaus, S.; Bauditz, J.; Gasche, C.; Lochs, H.; Raedler, A. Recombinant erythropoietin for the treatment of anemia in inflammatory bowel disease. N. Engl. J. Med. 1996, 334, 619–623. [Google Scholar] [CrossRef]
- Nairz, M.; Schroll, A.; Moschen, A.R.; Sonnweber, T.; Theurl, M.; Theurl, I.; Taub, N.; Jamnig, C.; Neurauter, D.; Huber, L.A.; et al. Erythropoietin contrastingly affects bacterial infection and experimental colitis by inhibiting nuclear factor-κB-inducible immune pathways. Immunity 2011, 34, 61–74. [Google Scholar] [CrossRef]
- Zhu, L.; Jin, W.; Pan, H.; Hu, Z.; Zhou, J.; Hang, C.; Shi, J. Erythropoietin inhibits the increase of intestinal labile zinc and the expression of inflammatory mediators after traumatic brain injury in rats. J. Trauma 2009, 66, 730–736. [Google Scholar] [CrossRef]
- Meng, R.; Zhu, D.; Bi, Y.; Yang, D.; Wang, Y. Erythropoietin inhibits gluconeogenesis and inflammation in the liver and improves glucose intolerance in high-fat diet-fed mice. PLoS One 2013, 8, e53557. [Google Scholar]
- Sepodes, B.; Maio, R.; Pinto, R.; Sharples, E.; Oliveira, P.; McDonald, M.; Yaqoob, M.; Thiemermann, C.; Mota-Filipe, H. Recombinant human erythropoietin protects the liver from hepatic ischemia-reperfusion injury in the rat. Transpl. Int. 2006, 19, 919–926. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Chow, A.; Huggins, M.; Ahmed, J.; Hashimoto, D.; Lucas, D.; Kunisaki, Y.; Pinho, S.; Leboeuf, M.; Noizat, C.; van Rooijen, N.; et al. CD169+ macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat. Med. 2013, 19, 429–436. [Google Scholar] [CrossRef]
- Lu, K.Y.; Ching, L.C.; Su, K.H.; Yu, Y.B.; Kou, Y.R.; Hsiao, S.H.; Huang, Y.C.; Chen, C.Y.; Cheng, L.C.; Pan, C.C.; et al. Erythropoietin suppresses the formation of macrophage foam cells: Role of liver X receptor α. Circulation 2010, 121, 1828–1837. [Google Scholar] [CrossRef]
- Drueke, T.B.; Locatelli, F.; Clyne, N.; Eckardt, K.U.; Macdougall, I.C.; Tsakiris, D.; Burger, H.U.; Scherhag, A. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N. Engl. J. Med. 2006, 355, 2071–2084. [Google Scholar] [CrossRef]
- Singh, A.K.; Szczech, L.; Tang, K.L.; Barnhart, H.; Sapp, S.; Wolfson, M.; Reddan, D. Correction of anemia with epoetin alfa in chronic kidney disease. N. Engl. J. Med. 2006, 355, 2085–2098. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Burdmann, E.A.; Chen, C.Y.; Cooper, M.E.; de Zeeuw, D.; Eckardt, K.U.; Feyzi, J.M.; Ivanovich, P.; Kewalramani, R.; Levey, A.S.; et al. A trial of darbepoetin α in type 2 diabetes and chronic kidney disease. N. Engl. J. Med. 2009, 361, 2019–2032. [Google Scholar] [CrossRef]
- Szczech, L.A.; Barnhart, H.X.; Inrig, J.K.; Reddan, D.N.; Sapp, S.; Califf, R.M.; Patel, U.D.; Singh, A.K. Secondary analysis of the CHOIR trial epoetin-α dose and achieved hemoglobin outcomes. Kidney Int. 2008, 74, 791–798. [Google Scholar] [CrossRef]
- Solomon, S.D.; Uno, H.; Lewis, E.F.; Eckardt, K.U.; Lin, J.; Burdmann, E.A.; de Zeeuw, D.; Ivanovich, P.; Levey, A.S.; Parfrey, P.; et al. Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N. Engl. J. Med. 2010, 363, 1146–1155. [Google Scholar] [CrossRef]
- Szczech, L.A.; Barnhart, H.X.; Sapp, S.; Felker, G.M.; Hernandez, A.; Reddan, D.; Califf, R.M.; Inrig, J.K.; Patel, U.D.; Singh, A.K. A secondary analysis of the CHOIR trial shows that comorbid conditions differentially affect outcomes during anemia treatment. Kidney Int. 2010, 77, 239–246. [Google Scholar] [CrossRef]
- Littlewood, T.J.; Bajetta, E.; Nortier, J.W.; Vercammen, E.; Rapoport, B. Effects of epoetin α on hematologic parameters and quality of life in cancer patients receiving nonplatinum chemotherapy: Results of a randomized, double-blind, placebo-controlled trial. J. Clin. Oncol. 2001, 19, 2865–2874. [Google Scholar]
- Vansteenkiste, J.; Pirker, R.; Massuti, B.; Barata, F.; Font, A.; Fiegl, M.; Siena, S.; Gateley, J.; Tomita, D.; Colowick, A.B.; et al. Double-blind, placebo-controlled, randomized phase III trial of darbepoetin alfa in lung cancer patients receiving chemotherapy. J. Natl. Cancer Inst. 2002, 94, 1211–1220. [Google Scholar] [CrossRef]
- Hedenus, M.; Adriansson, M.; San Miguel, J.; Kramer, M.H.; Schipperus, M.R.; Juvonen, E.; Taylor, K.; Belch, A.; Altes, A.; Martinelli, G.; et al. Efficacy and safety of darbepoetin α in anaemic patients with lymphoproliferative malignancies: A randomized, double-blind, placebo-controlled study. Br. J. Haematol. 2003, 122, 394–403. [Google Scholar] [CrossRef]
- Leyland-Jones, B. Breast cancer trial with erythropoietin terminated unexpectedly. Lancet Oncol. 2003, 4, 459–460. [Google Scholar] [CrossRef]
- Henke, M.; Laszig, R.; Rube, C.; Schafer, U.; Haase, K.D.; Schilcher, B.; Mose, S.; Beer, K.T.; Burger, U.; Dougherty, C.; et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: Randomised, double-blind, placebo-controlled trial. Lancet 2003, 362, 1255–1260. [Google Scholar] [CrossRef]
- Bennett, C.L.; Silver, S.M.; Djulbegovic, B.; Samaras, A.T.; Blau, C.A.; Gleason, K.J.; Barnato, S.E.; Elverman, K.M.; Courtney, D.M.; McKoy, J.M.; et al. Venous thromboembolism and mortality associated with recombinant erythropoietin and darbepoetin administration for the treatment of cancer-associated anemia. JAMA 2008, 299, 914–924. [Google Scholar] [CrossRef]
- Rizzo, J.D.; Somerfield, M.R.; Hagerty, K.L.; Seidenfeld, J.; Bohlius, J.; Bennett, C.L.; Cella, D.F.; Djulbegovic, B.; Goode, M.J.; Jakubowski, A.A.; et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Hematology/American Society of Clinical Oncology clinical practice guideline update. [CrossRef]
- Sargin, D.; Friedrichs, H.; El-Kordi, A.; Ehrenreich, H. Erythropoietin as neuroprotective and neuroregenerative treatment strategy: Comprehensive overview of 12 years of preclinical and clinical research. Best Pract. Res. Clin. Anaesthesiol. 2010, 24, 573–594. [Google Scholar] [CrossRef]
- Ehrenreich, H.; Hasselblatt, M.; Dembowski, C.; Cepek, L.; Lewczuk, P.; Stiefel, M.; Rustenbeck, H.H.; Breiter, N.; Jacob, S.; Knerlich, F.; et al. Erythropoietin therapy for acute stroke is both safe and beneficial. Mol. Med. 2002, 8, 495–505. [Google Scholar]
- Ehrenreich, H.; Fischer, B.; Norra, C.; Schellenberger, F.; Stender, N.; Stiefel, M.; Sirén, A.-L.; Paulus, W.; Nave, K.-A.; Gold, R.; et al. Exploring recombinant human erythropoietin in chronic progressive multiple sclerosis. Brain J. Neurol. 2007, 130, 2577–2588. [Google Scholar] [CrossRef]
- Wustenberg, T.; Begemann, M.; Bartels, C.; Gefeller, O.; Stawicki, S.; Hinze-Selch, D.; Mohr, A.; Falkai, P.; Aldenhoff, J.B.; Knauth, M.; et al. Recombinant human erythropoietin delays loss of gray matter in chronic schizophrenia. Mol. Psychiatry 2011, 16, 26–36. [Google Scholar] [CrossRef]
- Kristensen, P.L.; Pedersen-Bjergaard, U.; Kjær, T.W.; Olsen, N.V.; Dela, F.; Holst, J.J.; Faber, J.; Tarnow, L.; Thorsteinsson, B. Influence of erythropoietin on cognitive performance during experimental hypoglycemia in patients with type 1 diabetes mellitus: A randomized cross-over trial. PLoS One 2013, 8, e59672. [Google Scholar]
- Pang, L.; Bian, M.; Zang, X.-X.; Wu, Y.; Xu, D.-H.; Dong, N.; Wang, Z.-H.; Yan, B.-L.; Wang, D.-W.; Zhao, H.-J.; et al. Neuroprotective effects of erythropoietin in patients with carbon monoxide poisoning. J. Biochem. Mol. Toxicol. 2013, 27, 266–271. [Google Scholar] [CrossRef]
- Miskowiak, K.W.; Vinberg, M.; Harmer, C.J.; Ehrenreich, H.; Kessing, L.V. Erythropoietin: A candidate treatment for mood symptoms and memory dysfunction in depression. Psychopharmacology 2012, 219, 687–698. [Google Scholar] [CrossRef]
- Ehrenreich, H.; Weissenborn, K.; Prange, H.; Schneider, D.; Weimar, C.; Wartenberg, K.; Schellinger, P.D.; Bohn, M.; Becker, H.; Wegrzyn, M.; et al. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke 2009, 40, e647–e656. [Google Scholar] [CrossRef]
- Ehrenreich, H.; Kastner, A.; Weissenborn, K.; Streeter, J.; Sperling, S.; Wang, K.K.; Worthmann, H.; Hayes, R.L.; von Ahsen, N.; Kastrup, A.; et al. Circulating damage marker profiles support a neuroprotective effect of erythropoietin in ischemic stroke patients. Mol. Med. 2011, 17, 1306–1310. [Google Scholar]
- Andropoulos, D.B.; Brady, K.; Easley, R.B.; Dickerson, H.A.; Voigt, R.G.; Shekerdemian, L.S.; Meador, M.R.; Eisenman, C.A.; Hunter, J.V.; Turcich, M.; et al. Erythropoietin neuroprotection in neonatal cardiac surgery: A phase I/II safety and efficacy trial. J. Thorac. Cardiovasc. Surg. 2013, 146, 124–131. [Google Scholar] [CrossRef]
- Joyeux-Faure, M.; Durand, M.; Bedague, D.; Protar, D.; Incagnoli, P.; Paris, A.; Ribuot, C.; Levy, P.; Chavanon, O. Evaluation of the effect of one large dose of erythropoietin against cardiac and cerebral ischemic injury occurring during cardiac surgery with cardiopulmonary bypass: A randomized double-blind placebo-controlled pilot study. Fundam. Clin. Pharmacol. 2012, 26, 761–770. [Google Scholar] [CrossRef]
- Cramer, S.C.; Hill, M.D. Human choriogonadotropin and epoetin alfa in acute ischemic stroke patients (REGENESIS-LED trial). Int. J. Stroke 2014, 9, 321–327. [Google Scholar] [CrossRef]
- Taniguchi, N.; Nakamura, T.; Sawada, T.; Matsubara, K.; Furukawa, K.; Hadase, M.; Nakahara, Y.; Nakamura, T.; Matsubara, H. Erythropoietin prevention trial of coronary restenosis and cardiac remodeling after ST-elevated acute myocardial infarction (EPOC-AMI): A pilot, randomized, placebo-controlled study. Circ. J. 2010, 74, 2365–2371. [Google Scholar] [CrossRef]
- Ferrario, M.; Arbustini, E.; Massa, M.; Rosti, V.; Marziliano, N.; Raineri, C.; Campanelli, R.; Bertoletti, A.; de Ferrari, G.M.; Klersy, C.; et al. High-dose erythropoietin in patients with acute myocardial infarction: A pilot, randomised, placebo-controlled study. Int. J. Cardiol. 2011, 147, 124–131. [Google Scholar] [CrossRef]
- Najjar, S.S.; Rao, S.V.; Melloni, C.; Raman, S.V.; Povsic, T.J.; Melton, L.; Barsness, G.W.; Prather, K.; Heitner, J.F.; Kilaru, R.; et al. Intravenous erythropoietin in patients with ST-segment elevation myocardial infarction: REVEAL: A randomized controlled trial. JAMA 2011, 305, 1863–1872. [Google Scholar] [CrossRef]
- Prunier, F.; Biere, L.; Gilard, M.; Boschat, J.; Mouquet, F.; Bauchart, J.J.; Charbonnier, B.; Genee, O.; Guerin, P.; Warin-Fresse, K.; et al. Single high-dose erythropoietin administration immediately after reperfusion in patients with ST-segment elevation myocardial infarction: Results of the erythropoietin in myocardial infarction trial. Am. Heart J. 2012, 163, 200–207. [Google Scholar]
- Voors, A.A.; Belonje, A.M.; Zijlstra, F.; Hillege, H.L.; Anker, S.D.; Slart, R.H.; Tio, R.A.; van’t Hof, A.; Jukema, J.W.; Peels, H.O.; et al. A single dose of erythropoietin in ST-elevation myocardial infarction. Eur. Heart J. 2010, 31, 2593–2600. [Google Scholar] [CrossRef]
- Ott, I.; Schulz, S.; Mehilli, J.; Fichtner, S.; Hadamitzky, M.; Hoppe, K.; Ibrahim, T.; Martinoff, S.; Massberg, S.; Laugwitz, K.L.; et al. Erythropoietin in patients with acute ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention: A randomized, double-blind trial. Circ. Cardiovasc. Interv. 2010, 3, 408–413. [Google Scholar] [CrossRef]
- Moon, C.; Krawczyk, M.; Paik, D.; Lakatta, E.G.; Talan, M.I. Cardioprotection by recombinant human erythropoietin following acute experimental myocardial infarction: Dose response and therapeutic window. Cardiovasc. Drugs Ther. 2005, 19, 243–250. [Google Scholar] [CrossRef]
- Talan, M.I.; Ahmet, I.; Lakatta, E.G. Did clinical trials in which erythropoietin failed to reduce acute myocardial infarct size miss a narrow therapeutic window. PLoS One 2012, 7, e34819. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, Y.; Wang, L.; Dey, S.; Alnaeeli, M.; Suresh, S.; Rogers, H.; Teng, R.; Noguchi, C.T. Erythropoietin Action in Stress Response, Tissue Maintenance and Metabolism. Int. J. Mol. Sci. 2014, 15, 10296-10333. https://doi.org/10.3390/ijms150610296
Zhang Y, Wang L, Dey S, Alnaeeli M, Suresh S, Rogers H, Teng R, Noguchi CT. Erythropoietin Action in Stress Response, Tissue Maintenance and Metabolism. International Journal of Molecular Sciences. 2014; 15(6):10296-10333. https://doi.org/10.3390/ijms150610296
Chicago/Turabian StyleZhang, Yuanyuan, Li Wang, Soumyadeep Dey, Mawadda Alnaeeli, Sukanya Suresh, Heather Rogers, Ruifeng Teng, and Constance Tom Noguchi. 2014. "Erythropoietin Action in Stress Response, Tissue Maintenance and Metabolism" International Journal of Molecular Sciences 15, no. 6: 10296-10333. https://doi.org/10.3390/ijms150610296
APA StyleZhang, Y., Wang, L., Dey, S., Alnaeeli, M., Suresh, S., Rogers, H., Teng, R., & Noguchi, C. T. (2014). Erythropoietin Action in Stress Response, Tissue Maintenance and Metabolism. International Journal of Molecular Sciences, 15(6), 10296-10333. https://doi.org/10.3390/ijms150610296