Expression and Effects of High-Mobility Group Box 1 in Cervical Cancer

Abstract
:1. Background
2. Results
2.1. Patient Characteristics
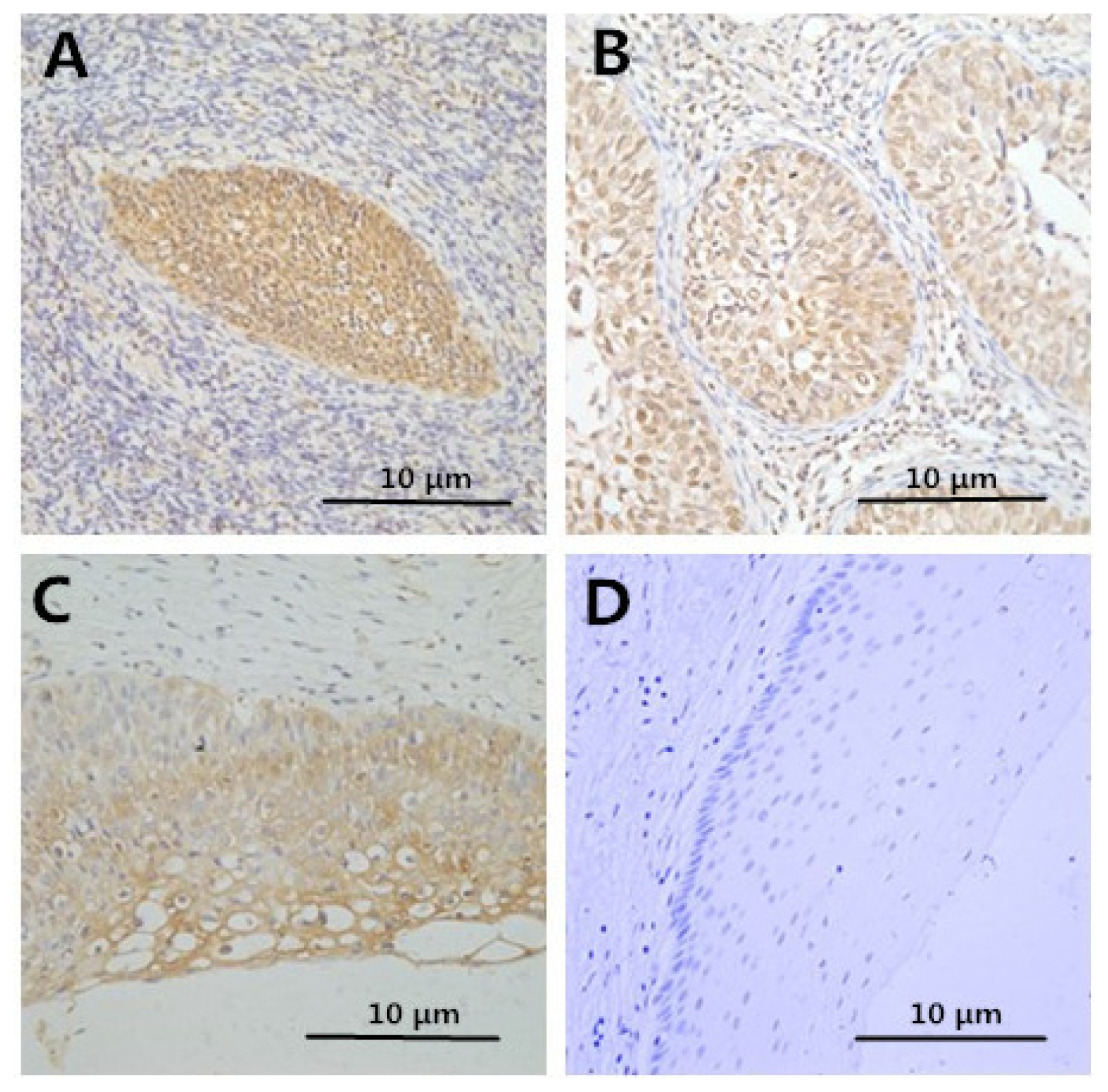
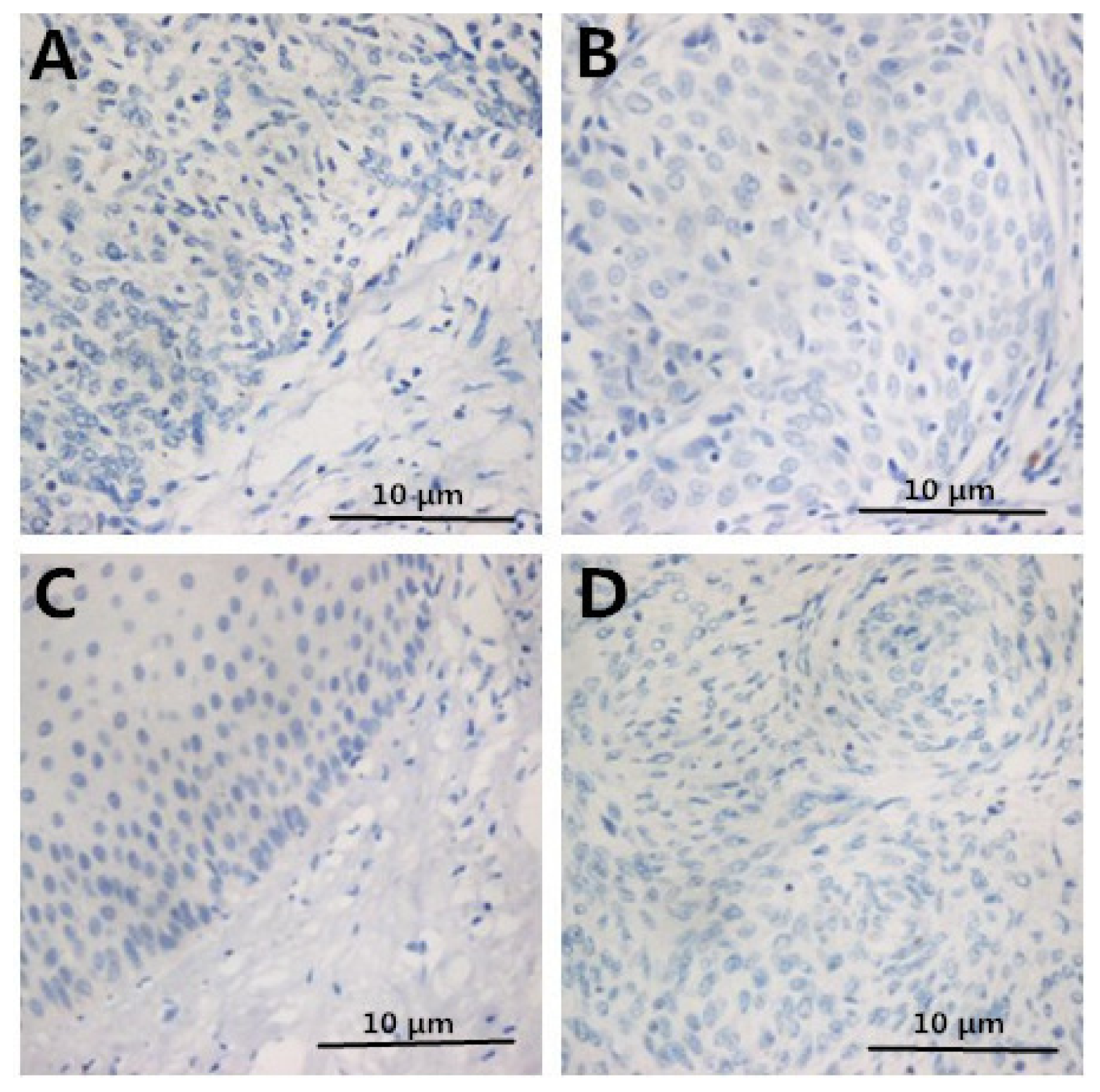
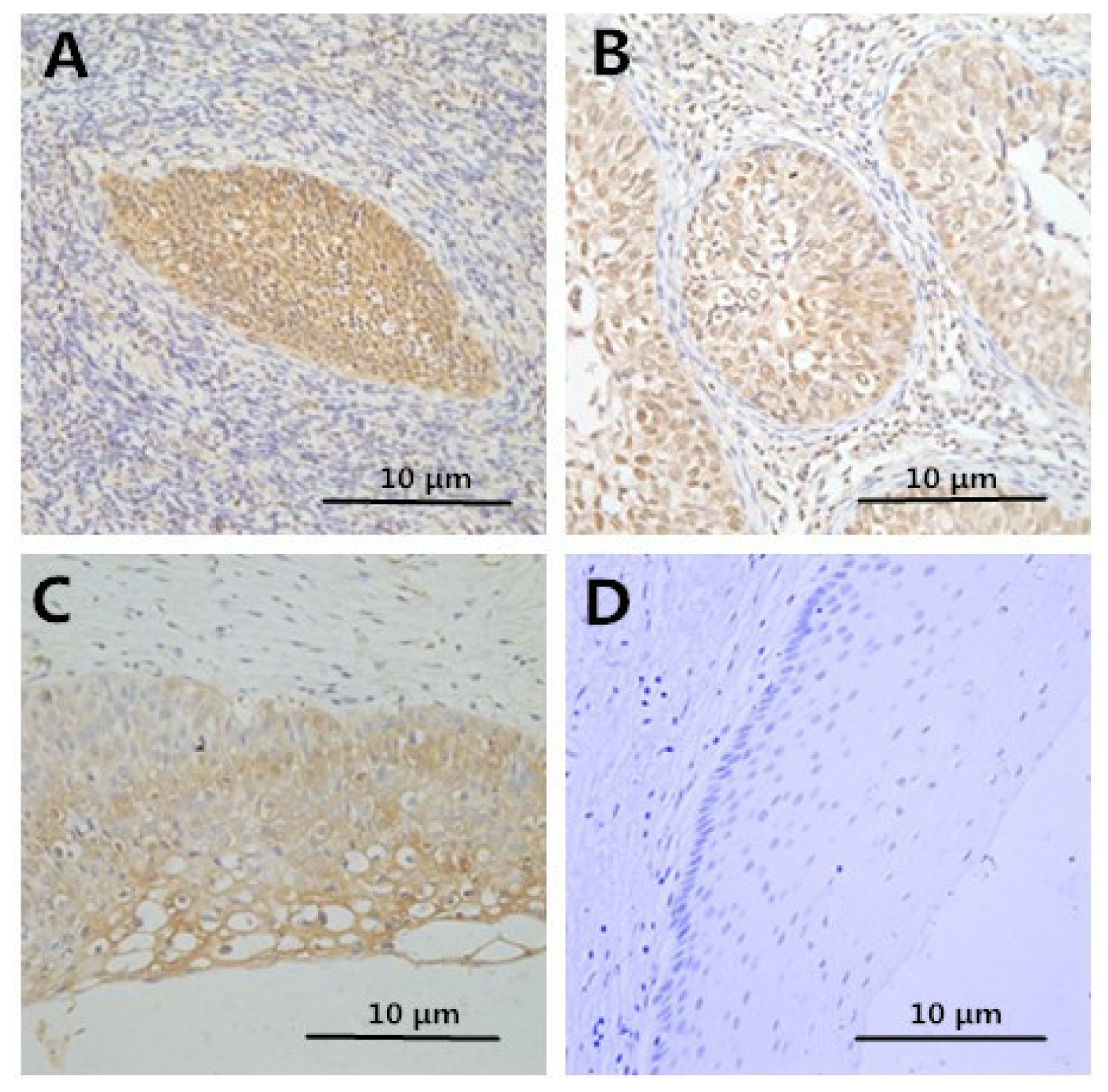
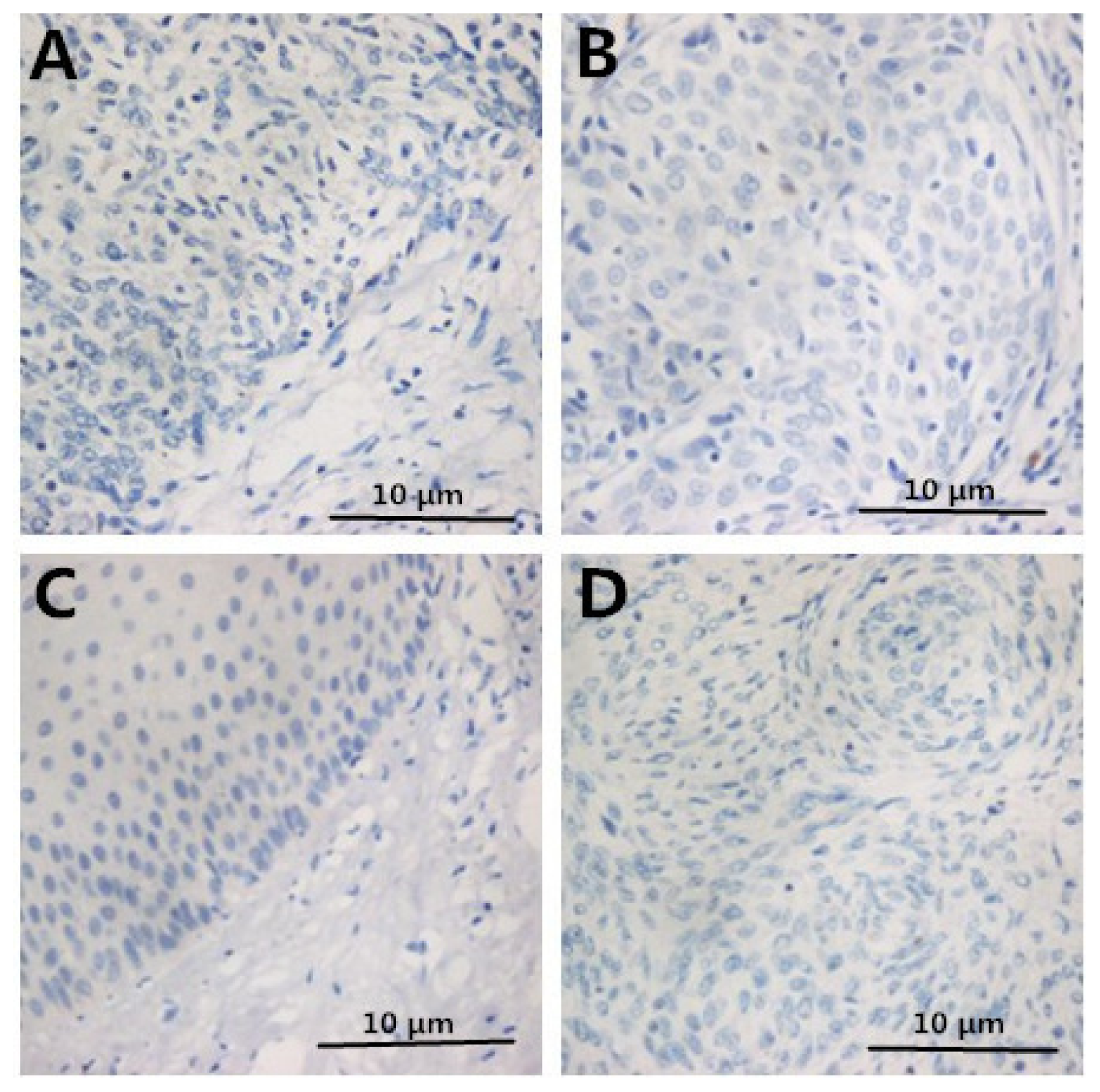
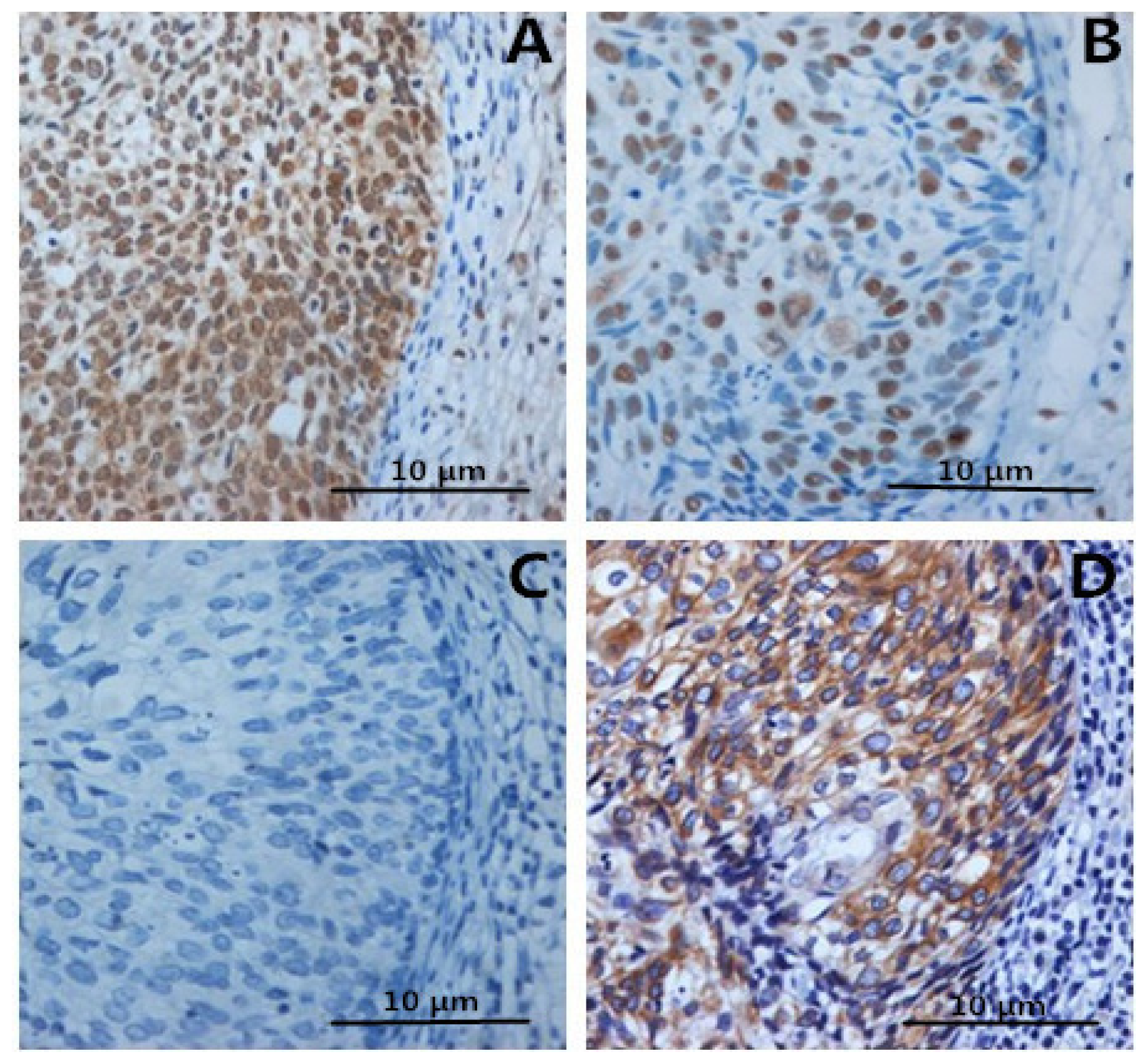
2.2. Immunohistochemistry
2.3. Correlation between Clinicopathologic Factors and HMGB1 Expression
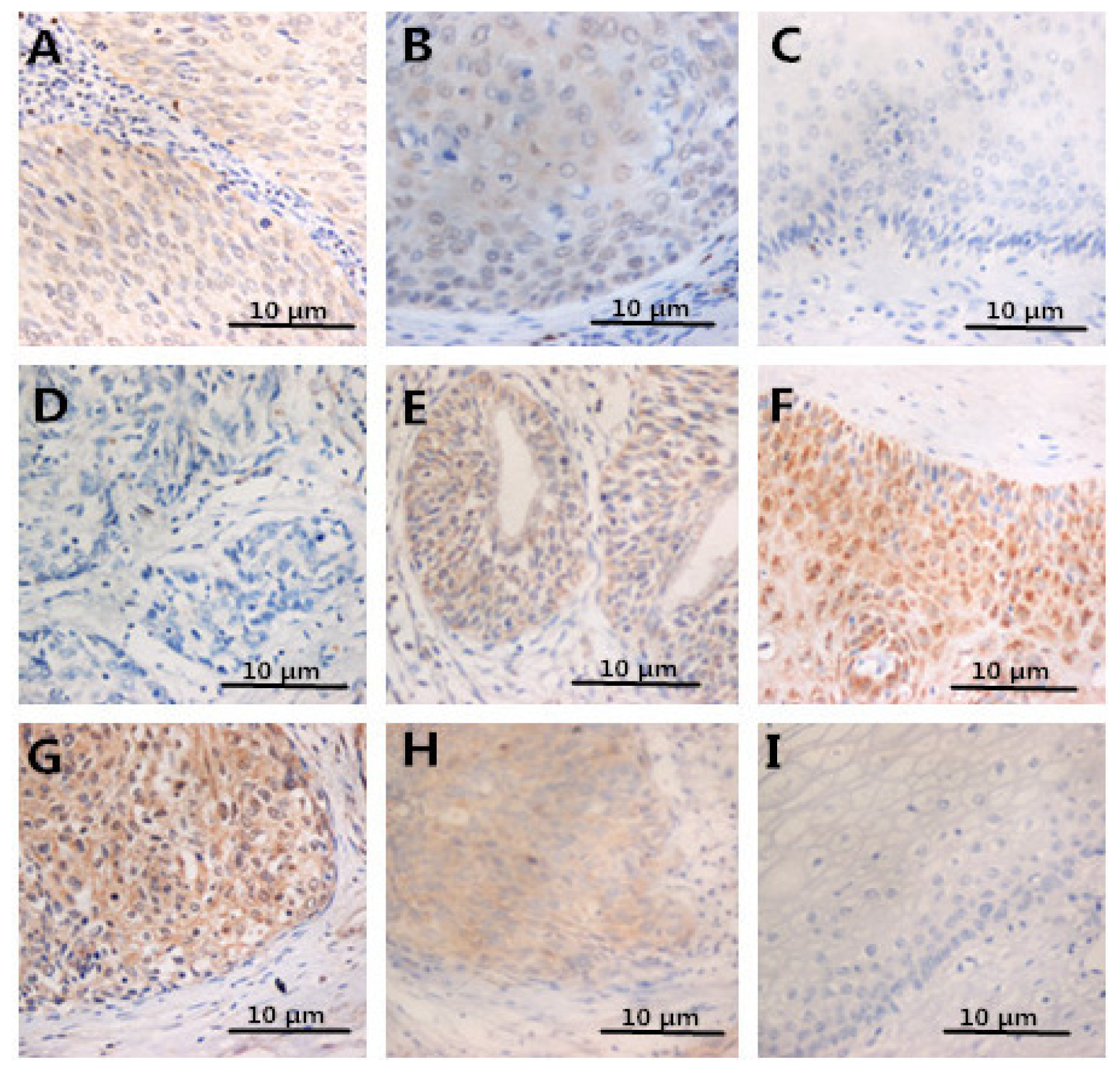
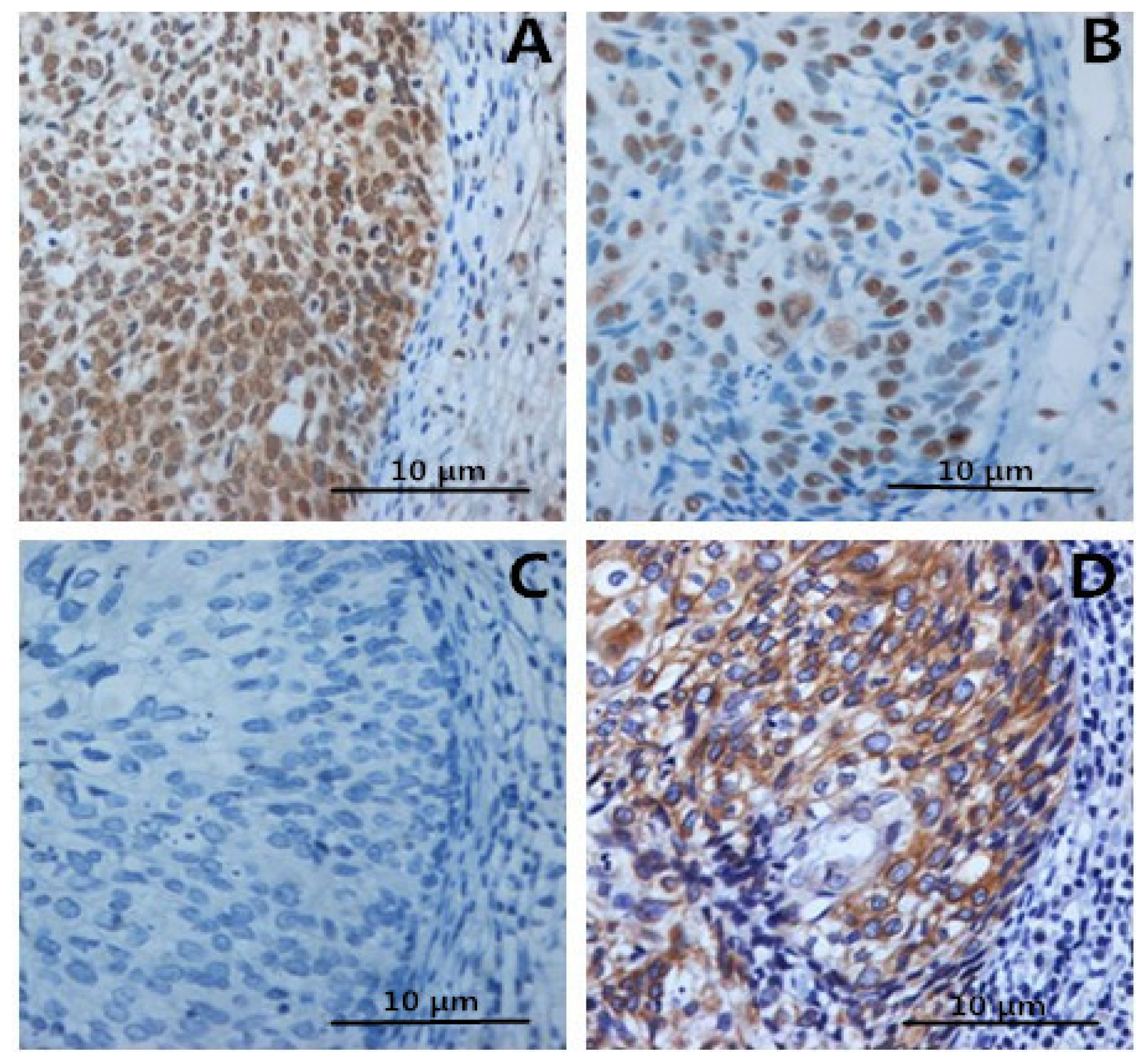
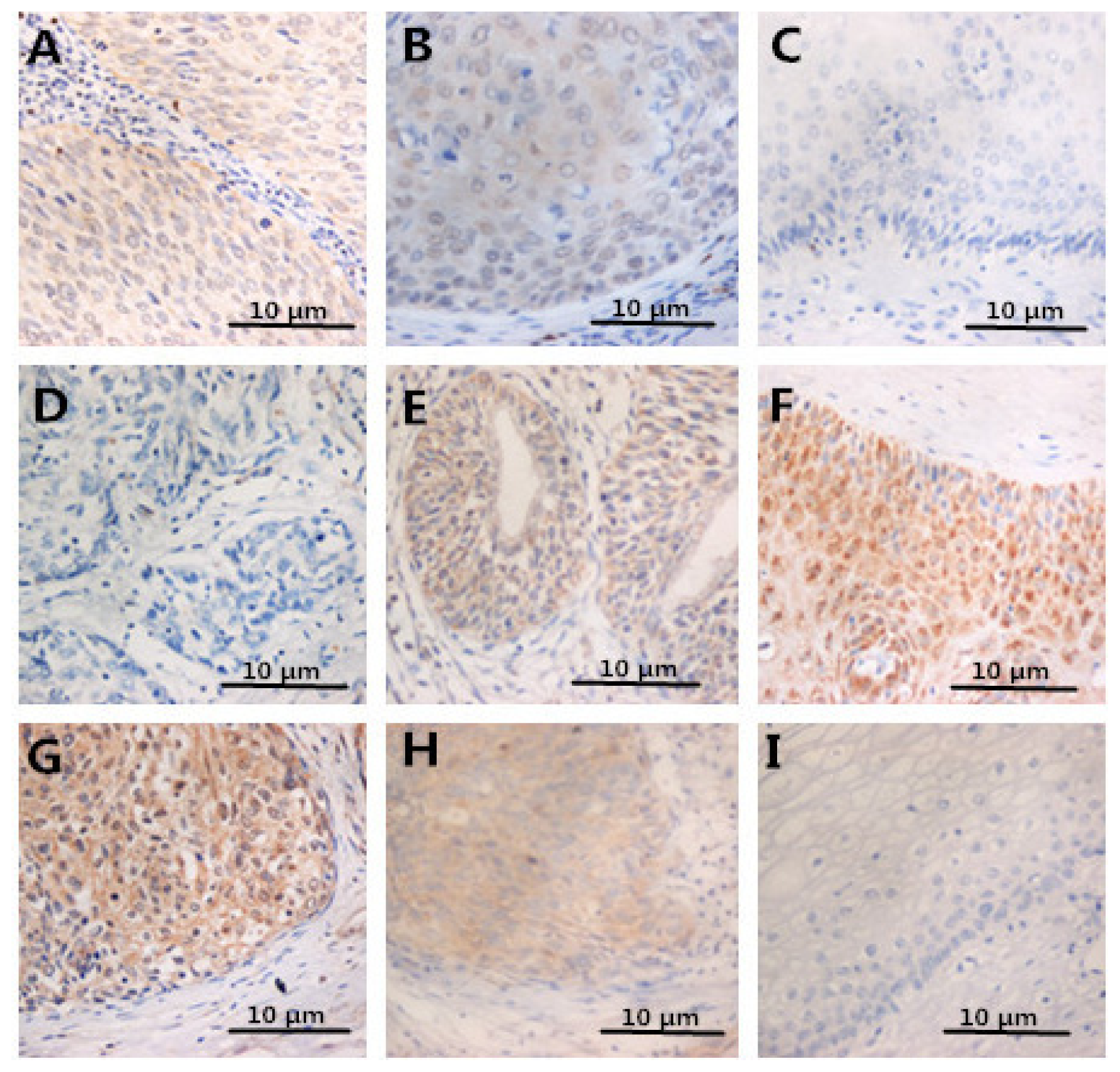
2.4. Expression of FOXP3, IL-2, IL-10 in Specimens
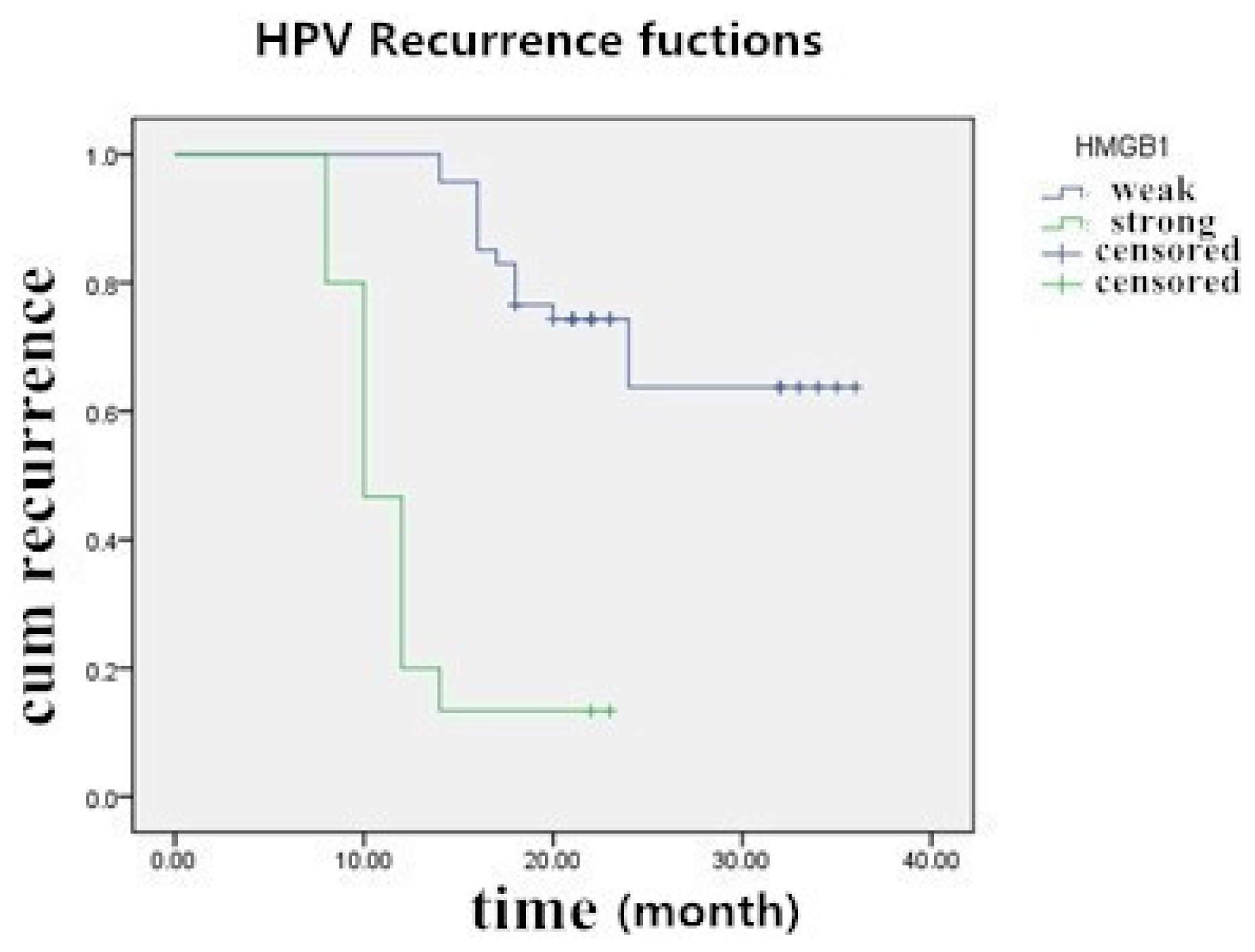
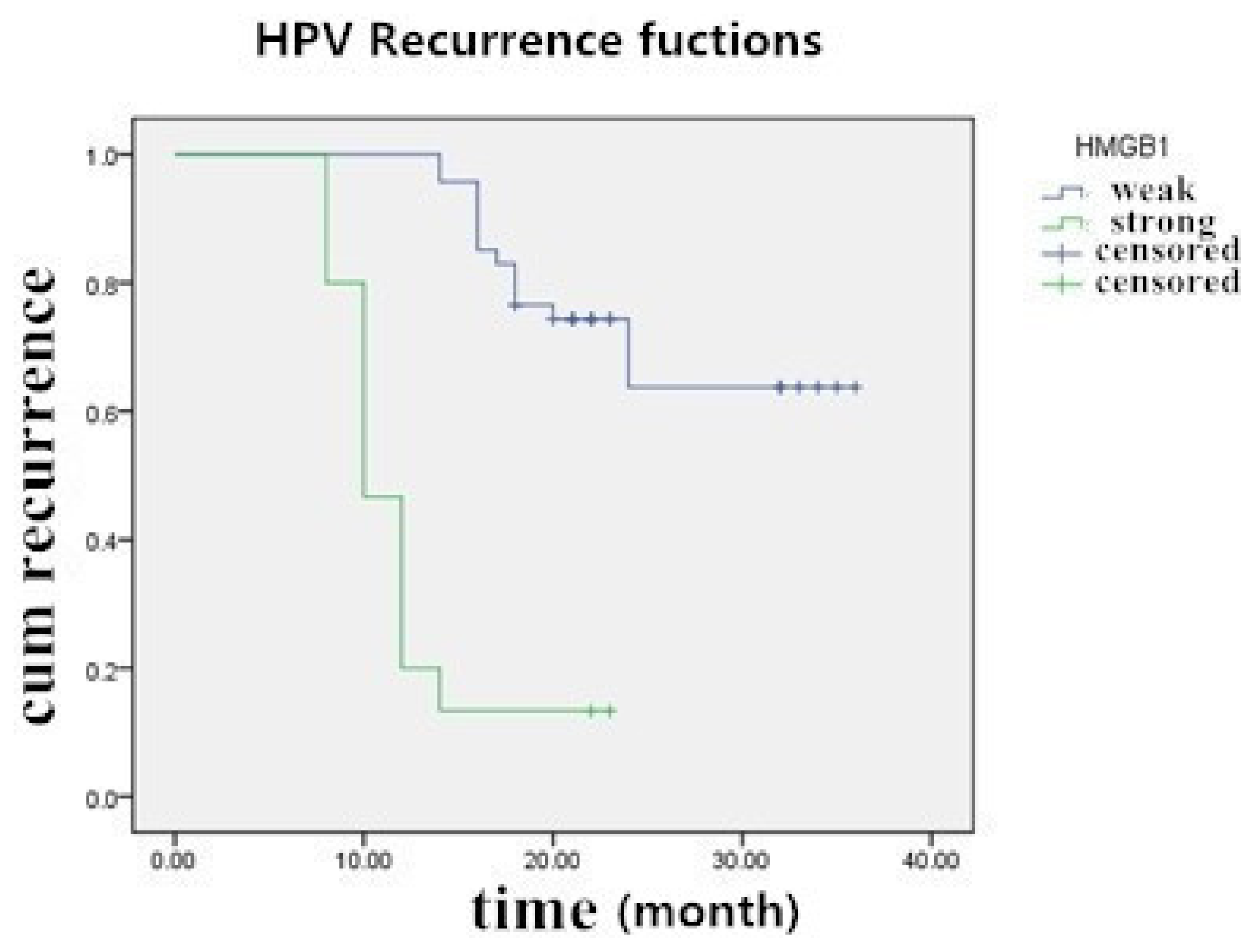
2.5. The Prognostic Significance of HMGB1 Protein in Cervical Carcinoma Tissue Samples
2.6. The Relationship between SCC-Ag and HMGB1 Expression
3. Discussion
4. Materials and Methods
4.1. Specimens
4.2. Immunohistochemistry
4.3. Grading of Immunostaining
4.4. Immunoradiometric Assay
4.5. Statistical Analyses
5. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsThis study was devised by Xiaoao Pang and Yao Zhang. Xiaoao Pang made this draft. Cervical tissues were collected and pathological diagnosis was completed by Chenglin Huang. Heng Wei and Jing Zhang worked on the immunochemical assay. Qingshuang Luo worked on data management and statistical analysis. The final manuscript was read and approved by all the authors.
References
- Hao, Q.; Du, X.Q.; Fu, X.; Tian, J. Expression and clinical significance of HMGB1 and RAGE in cervical squamous cell carcinoma. Zhonghua Zhong Liu Za Zhi 2008, 30, 292–295. [Google Scholar]
- Thomas, J.O.; Travers, A.A. HMG1 and 2, and related “architectural” DNA-binding proteins. Trends Biochem. Sci 2001, 26, 167–174. [Google Scholar]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol 2010, 28, 367–388. [Google Scholar]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., III; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol 2010, 190, 881–892. [Google Scholar]
- Volp, K.; Brezniceanu, M.L.; Bosser, S.; Brabletz, T.; Kirchner, T.; Gottel, D.; Joos, S.; Zörnig, M. Increased expression of high mobility group box 1(HMGB1) is associated with an elevated level of the antiapoptotic c-IAP2 protein in human colon carcinomas. Gut 2006, 55, 234–242. [Google Scholar]
- Brezniceanu, M.L.; Volp, K.; Bosser, S.; Solbach, C.; Lichter, P.; Joos, S.; Zörnig, M. HMGB1 inhibits cell death in yeast and mammalian cells and is abundantly expressed in human breast carcinoma. FASEB J 2003, 17, 1295–1297. [Google Scholar]
- Cheng, B.Q.; Jia, C.Q.; Liu, C.T.; Lu, X.F.; Zhong, N.; Zhang, Z.L.; Fan, W.; Li, Y.Q. Serum high mobility group box chromosomal protein 1 is associated with clinicopathologic features in patients with hepatocellular carcinoma. Dig. Liver Dis 2008, 40, 446–452. [Google Scholar]
- Takada, M.; Hirata, K.; Ajiki, T.; Suzuki, Y.; Kuroda, Y. Expression of receptor for advanced glycation end products (RAGE) and MMP-9 in human pancreatic cancer cells. Hepatogastroenterology 2004, 51, 928–930. [Google Scholar]
- Kuniyasu, H.; Chihara, Y.; Kondo, H.; Ohmori, H.; Ukai, R. Amphoterin induction in prostatic stromal cells by androgen deprivation is associated with metastatic prostate cancer. Oncol. Rep 2003, 10, 1863–1868. [Google Scholar]
- Sheng, X.; Du, X.; Zhang, X.; Li, D.; Lu, C.; Li, Q.; Ma, Z.; Song, Q.; Wang, C. Clinical value of serum HMGB1 levels in early detection of recurrent squamous cell carcinoma of uterine cervix: Comparison with serum SCCA, CYFRA21–1, and CEA levels. Croat. Med. J 2009, 50, 455–464. [Google Scholar]
- Sakaguchi, S.; Miyara, M.; Costantino, C.M.; Hafler, D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol 2010, 10, 490–500. [Google Scholar]
- Molling, J.W.; de Gruijl, T.D.; Glim, J.; Moreno, M.; Rozendaal, L.; Meijer, C.J.L.M.; van den Eertwegh, A.J.M.; Scheper, R.J.; von Blomberg, M.E.; Bontke, H.J. CD4+CD25hi regulatory T-cell frequency correlates with persistence of human papillomavirus type 16 and T helper cell responses in patients with cervical intraepithelial neoplasia. Int. J. Cancer 2007, 121, 1749–1755. [Google Scholar]
- Liu, Z.; Falo, L.D., Jr.; You, Z. Knockdown of HMGB1 in tumor cells attenuates their ability to induce regulatory T cells and uncovers naturally acquired CD8 T cell-dependent antitumor immunity. J. Immunol 2011, 187, 118–125. [Google Scholar]
- Huang, L.F.; Yao, Y.M.; Zhang, L.T.; Dong, N.; Yu, Y.; Sheng, Z.Y. The effect of high-mobility group box 1 protein on activity of regulatory T cells after thermal injury in rats. Shock 2009, 31, 322–329. [Google Scholar]
- Zhang, Y.; Yao, Y.; Huang, L.; Dong, N.; Yu, Y.; Sheng, Z. The Potential Effect and Mechanism of High-Mobility Group Box 1 Protein on Regulatory T Cell-Mediated Immunosuppression. J. Interf. Cytokine Res 2011, 31, 249–257. [Google Scholar]
- Jeong, B.K.; Choi, D.H.; Huh, S.J.; Park, W.; Bae, D.S.; Kim, B.G. The role of squamous cell carcinoma antigen as a prognostic and predictive factor in carcinoma of uterine cervix. Radiat. Oncol. J 2011, 29, 191–198. [Google Scholar]
- Foell, D.; Wittkowski, H.; Roth, J. Mechanisms of disease: A ‘DAMP’ view of inflammatory arthritis. Nat. Rev. Rheumatol 2007, 3, 382–390. [Google Scholar]
- Gilmore, T.D.; Koedood, M.; Piffat, K.A.; White, D.W. Rel/NF-κB/lκB proteins and cancer. Oncogene 1996, 13, 1367–1378. [Google Scholar]
- Chen, Y.; Li, R.; Wang, R.; Liu, Z. The significance of nuclear factor kappa B p65 expression on the vascular endothelial cells of rectum adnocarcinoma of human. Hua Xi Yi Ke Da Xue Xue Bao 2001, 32, 196–199. [Google Scholar]
- O-charbenrat, P.; Wongkajomsil, P.A.; Phys-Evans, P.H.; Eccles, S.A. Signaling pathways required for matrix metalloproteinase-9 induction by betacelluin in head-and-neck squamous carcinoma cells. Int. J. Cancer 2004, 111, 174–183. [Google Scholar]
- Wang, Z.; Larregina, A.T.; Shufesky, W.J.; Perone, M.J.; Montecalvo, A.; Zahorchak, A.F.; Thomson, A.W.; Morelli, A.E. Use of the inhibitory effete of apoptotic cells on dendritic cells for graft survival via T-cell deletion and regulatory T cells. Am. J. Transplant 2006, 6, 1297–1311. [Google Scholar]
- Hazzalin, C.A.; Mahadevan, L.C. MAPK-regulated transcription: A continuously variable gene switch? Nat. Rev. Mol. Cell Biol 2002, 3, 30–40. [Google Scholar]
- Arrighi, J.F.; Rebsamen, M.; Rousset, F.; Kindler, V.; Hauser, C. A critical role for p38 mitogen-activated protein kinase in the maturation of human blood-derived dendritic cells induced by lipopolysaccharide, TNF-alpha, and contact sensitizers. J. Immunol 2001, 166, 3837–3845. [Google Scholar]
- Kusume, A.; Sasahira, T.; Luo, Y.; Isobe, M.; Nakagawa, N.; Tatsumoto, N.; Fujii, K.; Ohmori, H.; Kuniyasu, H. Suppression of dendritic cells by HMGB1 is associated with lymph node metastasis of human colon cancer. Pathobiology 2009, 76, 155–162. [Google Scholar]
- Dumitriu, I.E.; Baruah, P.; Valentinis, B.; Voll, R.E.; Herrmann, M.; Nawroth, P.P.; Arnold, B.; Bianchi, M.E.; Manfredi, A.A.; Rovere-Querini, P. Release of high mobility group box1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J. Immunol 2005, 174, 7506–7515. [Google Scholar]
- Rovere-Querini, P.; Capobianco, A.; Scaffidi, P.; Valentinis, B.; Catalanotti, F.; Giazzon, M.; Dumitriu, I.E.; Müller, S.; Iannacone, M.; Traversari, C.; et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep 2004, 5, 825–830. [Google Scholar]
- Ishii, K.J.; Coban, C.; Akira, A. Manifold meehanisms of Toll-like receptor-ligand recognition. J. Clin. Immunol 2005, 25, 511–521. [Google Scholar]
- DeMarco, R.A.; Fink, M.P.; Lotze, M.T. Monocytes promote natural killer cell interferon gamma production in response to endogenous danger signal HMGB1. Mol. Immunol 2005, 42, 433–444. [Google Scholar]
- Gaarenstroom, K.N.; Kenter, G.G.; Bonfrer, J.M.G.; Korse, C.M.; van deVijver, M.J.; Fleuren, G.-J.; Trimbos, J.B. Can initial serum cyfra 21–1, SCC antigen, and TPA levels in squamous cell cervical cancer predict lymph node metastases or prognosis? Gynecol. Oncol 2000, 77, 164–170. [Google Scholar]
- Ohara, K.; Tanaka, Y.; Tsunoda, H.; Nishida, M.; Sugahara, S.; Itai, Y. Assessment of cervical cancer radio response by serum squamous cell carcinoma antigen and magnetic resonance imaging. Obstet. Gynecol 2002, 100, 781–787. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Number of cases (%) |
|---|---|
| Range | 19~68 (years) |
| Average | 44 (years) |
| Samples | |
| Normal squamous epithelial specimens | 12 (12) |
| Cervical intraepithelial neoplasms (CIN) | 51 (51) |
| CIN I/II | 26 (51) |
| CIN III | 25 (49) |
| Invasive CC (ICC) | 37 (37) |
| Well-differentiated (WICC) | 7 (18.9) |
| Moderately differentiated (MICC) | 24 (64.9) |
| Poorly differentiated (PICC) | 6 (16.2) |
| Histology | |
| Squamous cell carcinoma (SCC) | 31 (83.8) |
| Adenosquamous cell carcinoma (ACC) | 6 (16.2) |
| FIGO stage | |
| Ia | 3 (8.2) |
| Ib | 18 (48.6) |
| IIa | 15 (40.5) |
| IIb | 1 (2.7) |
| Lymph nodes metastasis (LN) | |
| Absent | 26 (70.3) |
| Present | 11 (29.7) |
| Characteristics | n | HMGB1 | p | |||
|---|---|---|---|---|---|---|
| − | + | ++ | +++ | |||
| Normal | 12 | 10 | 2 | 0 | 0 | <0.05 |
| CIN | 51 | 2 | 21 | 28 | 0 | |
| CIN I/II | 26 | 1 | 16 | 9 | 0 | <0.01 |
| CIN III | 25 | 1 | 5 | 19 | 0 | <0.05 |
| ICC | 37 | 0 | 1 | 20 | 16 | |
| WICC | 7 | 0 | 1 | 6 | 0 | |
| MICC | 24 | 0 | 0 | 10 | 4 | 0.717 |
| PICC | 6 | 0 | 0 | 4 | 2 | |
| Histology | ||||||
| SCC | 31 | 0 | 0 | 17 | 14 | 0.353 |
| ACC | 6 | 0 | 1 | 3 | 2 | |
| FIGO stage | ||||||
| I | 21 | 0 | 1 | 19 | 1 | <0.05 |
| II | 16 | 0 | 0 | 1 | 15 | |
| LN metastasis | ||||||
| Absent | 26 | 0 | 1 | 17 | 8 | 0.02 |
| Present | 11 | 0 | 0 | 3 | 8 | |
| Pathological grade | n | HMGB1 | FOXP3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| − | + | ++ | +++ | − | + | ++ | +++ | ||
| Normal | 12 | 10 | 2 | 0 | 0 | 10 | 2 | 0 | 0 |
| CINI/II | 26 | 1 | 16 | 9 | 0 | 2 | 18 | 6 | 0 |
| CINIII | 25 | 1 | 5 | 19 | 0 | 1 | 5 | 17 | 2 |
| ICC | 37 | 0 | 1 | 20 | 16 | 0 | 3 | 19 | 15 |
| r = 0.829, p < 0.05 | |||||||||
| Pathological grade | n | IL-2 | IL-10 | ||||||
| − | + | ++ | +++ | − | + | ++ | +++ | ||
| Normal | 12 | 0 | 0 | 2 | 10 | 10 | 2 | 0 | 0 |
| CINI/II | 26 | 0 | 2 | 18 | 6 | 3 | 10 | 12 | 1 |
| CINIII | 25 | 2 | 19 | 2 | 2 | 0 | 5 | 17 | 3 |
| ICC | 37 | 28 | 6 | 2 | 1 | 0 | 4 | 16 | 17 |
| r = −0.587, p < 0.05 | r = 0.746, p < 0.05 | ||||||||
| HMGB1 | n | SCC-Ag | |
|---|---|---|---|
| <1.5 ng/mL | >1.5 ng/mL | ||
| +/++ | 18 | 13 (72.2) | 5 (27.8) |
| +++ | 14 | 5 (35.7) | 9 (64.3) |
| Total | 32 | 18 | 14 |
| r = 0.517, p < 0.01 | |||
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pang, X.; Zhang, Y.; Wei, H.; Zhang, J.; Luo, Q.; Huang, C.; Zhang, S. Expression and Effects of High-Mobility Group Box 1 in Cervical Cancer. Int. J. Mol. Sci. 2014, 15, 8699-8712. https://doi.org/10.3390/ijms15058699
Pang X, Zhang Y, Wei H, Zhang J, Luo Q, Huang C, Zhang S. Expression and Effects of High-Mobility Group Box 1 in Cervical Cancer. International Journal of Molecular Sciences. 2014; 15(5):8699-8712. https://doi.org/10.3390/ijms15058699
Chicago/Turabian StylePang, Xiaoao, Yao Zhang, Heng Wei, Jing Zhang, Qingshuang Luo, Chenglin Huang, and Shulan Zhang. 2014. "Expression and Effects of High-Mobility Group Box 1 in Cervical Cancer" International Journal of Molecular Sciences 15, no. 5: 8699-8712. https://doi.org/10.3390/ijms15058699
APA StylePang, X., Zhang, Y., Wei, H., Zhang, J., Luo, Q., Huang, C., & Zhang, S. (2014). Expression and Effects of High-Mobility Group Box 1 in Cervical Cancer. International Journal of Molecular Sciences, 15(5), 8699-8712. https://doi.org/10.3390/ijms15058699