Evolving Concepts in the Pathogenesis of NASH: Beyond Steatosis and Inflammation

Abstract
:1. Introduction
2. The Evolution of Hypotheses of NASH Pathogenesis
3. A Central Role for Lipotoxicity and Insulin Resistance
4. The Role of Iron in NAFLD and NASH
5. Progressive NASH: From Inflammation to Fibrosis
6. Cell Death in NASH: A Nexus of Injury and Fibrosis
7. A Role for Hepatocyte Senescence in Disease Progression
8. The Hepatic Inflammatory Response: Implications for NASH
9. Sterile Inflammation, Toll-Like Receptors and Propagation of the Inflammatory Response
10. The Immune Response in NASH: A Th17 Response?
11. Treatment of NASH: Current Therapies and Potential Targets
12. Summary and Conclusions
Acknowledgments
Conflicts of Interest
References
- Clark, J.M. The epidemiology of nonalcoholic fatty liver disease in adults. J. Clin. Gastroenterol 2006, 40, S5–S10. [Google Scholar]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar]
- Cortez-Pinto, H.; Chatham, J.; Chacko, V.P.; Arnold, C.; Rashid, A.; Diehl, A.M. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: A pilot study. JAMA 1999, 282, 1659–1664. [Google Scholar]
- Mari, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell MeTable 2006, 4, 185–198. [Google Scholar]
- Mari, M.; Colell, A.; Morales, A.; Caballero, F.; Moles, A.; Fernandez, A.; Terrones, O.; Basanez, G.; Antonsson, B.; Garcia-Ruiz, C.; et al. Mechanism of mitochondrial glutathione-dependent hepatocellular susceptibility to TNF despite NF-kappaB activation. Gastroenterology 2008, 134, 1507–1520. [Google Scholar]
- Polotsky, V.Y.; Patil, S.P.; Savransky, V.; Laffan, A.; Fonti, S.; Frame, L.A.; Steele, K.E.; Schweizter, M.A.; Clark, J.M.; Torbenson, M.S.; et al. Obstructive sleep apnea, insulin resistance, and steatohepatitis in severe obesity. Am. J. Respir. Crit. Care Med 2009, 179, 228–234. [Google Scholar]
- Musso, G.; Gambino, R.; de Michieli, F.; Durazzo, M.; Pagano, G.; Cassader, M. Adiponectin gene polymorphisms modulate acute adiponectin response to dietary fat: Possible pathogenetic role in NASH. Hepatology 2008, 47, 1167–1177. [Google Scholar]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar]
- D’Albuquerque, L.A.; Gonzalez, A.M.; Wahle, R.C.; de Oliveira Souza, E.; Mancero, J.M.; de Oliveira e Silva, A. Liver transplantation for subacute hepatocellular failure due to massive steatohepatitis after bariatric surgery. Liver Transpl 2008, 14, 881–885. [Google Scholar]
- Monetti, M.; Levin, M.C.; Watt, M.J.; Sajan, M.P.; Marmor, S.; Hubbard, B.K.; Stevens, R.D.; Bain, J.R.; Newgard, C.B.; Farese, R.V., Sr.; et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab 2007, 6, 69–78. [Google Scholar]
- Liao, W.; Hui, T.Y.; Young, S.G.; Davis, R.A. Blocking microsomal triglyceride transfer protein interferes with apoB secretion without causing retention or stress in the ER. J. Lipid Res 2003, 44, 978–985. [Google Scholar]
- Li, Z.Z.; Berk, M.; McIntyre, T.M.; Feldstein, A.E. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: Role of stearoyl-CoA desaturase. J. Biol. Chem 2009, 284, 5637–5644. [Google Scholar]
- Flowers, M.T.; Groen, A.K.; Oler, A.T.; Keller, M.P.; Choi, Y.; Schueler, K.L.; Richards, O.C.; Lan, H.; Miyazaki, M.; Kuipers, F.; et al. Cholestasis and hypercholesterolemia in SCD1-deficient mice fed a low-fat, high-carbohydrate diet. J. Lipid Res 2006, 47, 2668–2680. [Google Scholar]
- Ntambi, J.M.; Miyazaki, M.; Stoehr, J.P.; Lan, H.; Kendziorski, C.M.; Yandell, B.S.; Song, Y.; Cohen, P.; Friedman, J.M.; Attie, A.D. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. USA 2002, 99, 11482–11486. [Google Scholar]
- Jiang, G.; Li, Z.; Liu, F.; Ellsworth, K.; Dallas-Yang, Q.; Wu, M.; Ronan, J.; Esau, C.; Murphy, C.; Szalkowski, D.; et al. Prevention of obesity in mice by antisense oligonucleotide inhibitors of stearoyl-CoA desaturase-1. J. Clin. Investig 2005, 115, 1030–1038. [Google Scholar]
- Rajasekar, P.; Anuradha, C.V. Fructose-induced hepatic gluconeogenesis: Effect of l-carnitine. Life Sci 2007, 80, 1176–1183. [Google Scholar]
- Schenk, S.; Horowitz, J.F. Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J. Clin. Investig 2007, 117, 1690–1698. [Google Scholar]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar]
- Han, M.S.; Park, S.Y.; Shinzawa, K.; Kim, S.; Chung, K.W.; Lee, J.H.; Kwon, C.H.; Lee, K.W.; Park, C.K.; Chung, W.J.; et al. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J. Lipid Res 2008, 49, 84–97. [Google Scholar]
- Tan, M.; Hao, F.; Xu, X.; Chisolm, G.M.; Cui, M.Z. Lysophosphatidylcholine activates a novel PKD2-mediated signaling pathway that controls monocyte migration. Arterioscler. Thromb. Vasc. Biol 2009, 29, 1376–1382. [Google Scholar]
- Malaguarnera, M.; di Rosa, M.; Nicoletti, F.; Malaguarnera, L. Molecular mechanisms involved in NAFLD progression. J. Mol. Med 2009, 87, 679–695. [Google Scholar]
- Gregor, M.G.; Hotamisligil, G.S. Adipocyte stress: The endoplasmic reticulum and metabolic disease. J. Lipid Res 2007, 48, 1905–1914. [Google Scholar]
- Targher, G.; Bertolini, L.; Rodella, S.; Zoppini, G.; Scala, L.; Zenari, L.; Falezza, G. Associations between plasma adiponectin concentrations and liver histology in patients with nonalcoholic fatty liver disease. Clin. Endocrinol 2006, 64, 679–683. [Google Scholar]
- Bluher, M. Adipose tissue dysfunction contributes to obesity related metabolic diseases. Best Pract. Res. Clin. Endocrinol. MeTable 2013, 27, 163–177. [Google Scholar]
- Bluher, M. Adipose tissue dysfunction in obesity. Exp. Clin. Endocrinol. Diabetes 2009, 117, 241–250. [Google Scholar]
- Zhou, Y.P.; Grill, V.E. Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J. Clin. Investig 1994, 93, 870–876. [Google Scholar]
- Bollheimer, L.C.; Skelly, R.H.; Chester, M.W.; McGarry, J.D.; Rhodes, C.J. Chronic exposure to free fatty acid reduces pancreatic beta cell insulin content by increasing basal insulin secretion that is not compensated for by a corresponding increase in proinsulin biosynthesis translation. J. Clin. Investig 1998, 101, 1094–1101. [Google Scholar]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar]
- Kim, F.; Pham, M.; Luttrell, I.; Bannerman, D.D.; Tupper, J.; Thaler, J.; Hawn, T.R.; Raines, E.W.; Schwartz, M.W. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ. Res 2007, 100, 1589–1596. [Google Scholar]
- Cusi, K. Role of insulin resistance and lipotoxicity in non-alcoholic steatohepatitis. Clin. Liver Dis 2009, 13, 545–563. [Google Scholar]
- Arner, P. The adipocyte in insulin resistance: Key molecules and the impact of the thiazolidinediones. Trends Endocrinol. MeTable 2003, 14, 137–145. [Google Scholar]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar]
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008, 322, 1539–1543. [Google Scholar]
- Tilg, H. Adipocytokines in nonalcoholic fatty liver disease: Key players regulating steatosis, inflammation and fibrosis. Curr. Pharm. Des 2010, 16, 1893–1895. [Google Scholar]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med 2005, 11, 183–190. [Google Scholar]
- Nakamura, S.; Takamura, T.; Matsuzawa-Nagata, N.; Takayama, H.; Misu, H.; Noda, H.; Nabemoto, S.; Kurita, S.; Ota, T.; Ando, H.; et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J. Biol. Chem 2009, 284, 14809–14818. [Google Scholar]
- Fessler, M.B.; Rudel, L.L.; Brown, J.M. Toll-like receptor signaling links dietary fatty acids to the metabolic syndrome. Curr. Opin. Lipidol 2009, 20, 379–385. [Google Scholar]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [Google Scholar]
- Li, Z.; Berk, M.; McIntyre, T.M.; Gores, G.J.; Feldstein, A.E. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology 2008, 47, 1495–1503. [Google Scholar]
- O’Brien, J.; Powell, L.W. Non-alcoholic fatty liver disease: Is iron relevant? Hepatol. Int 2012, 6, 332–341. [Google Scholar]
- George, D.K.; Goldwurm, S.; MacDonald, G.A.; Cowley, L.L.; Walker, N.I.; Ward, P.J.; Jazwinska, E.C.; Powell, L.W. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology 1998, 114, 311–318. [Google Scholar]
- Bonkovsky, H.L.; Jawaid, Q.; Tortorelli, K.; LeClair, P.; Cobb, J.; Lambrecht, R.W.; Banner, B.F. Non-alcoholic steatohepatitis and iron: Increased prevalence of mutations of the HFE gene in non-alcoholic steatohepatitis. J. Hepatol 1999, 31, 421–429. [Google Scholar]
- Nelson, J.E.; Bhattacharya, R.; Lindor, K.D.; Chalasani, N.; Raaka, S.; Heathcote, E.J.; Miskovsky, E.; Shaffer, E.; Rulyak, S.J.; Kowdley, K.V. HFE C282Y mutations are associated with advanced hepatic fibrosis in Caucasians with nonalcoholic steatohepatitis. Hepatology 2007, 46, 723–729. [Google Scholar]
- Hernaez, R.; Yeung, E.; Clark, J.M.; Kowdley, K.V.; Brancati, F.L.; Kao, W.H. Hemochromatosis gene and nonalcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol 2011, 55, 1079–1085. [Google Scholar]
- Moirand, R.; Mortaji, A.M.; Loreal, O.; Paillard, F.; Brissot, P.; Deugnier, Y. A new syndrome of liver iron overload with normal transferrin saturation. Lancet 1997, 349, 95–97. [Google Scholar]
- Mendler, M.H.; Turlin, B.; Moirand, R.; Jouanolle, A.M.; Sapey, T.; Guyader, D.; le Gall, J.Y.; Brissot, P.; David, V.; Deugnier, Y. Insulin resistance-associated hepatic iron overload. Gastroenterology 1999, 117, 1155–1163. [Google Scholar]
- Machado, M.; Cortez-Pinto, H. Nash, insulin resistance and iron. Liver Int 2006, 26, 1159–1162. [Google Scholar]
- Kim, M.G.; Choi, W.C. Differential diagnosis of diabetes mellitus caused by liver cirrhosis and other type 2 diabetes mellitus. Korean J. Hepatol 2006, 12, 524–529. [Google Scholar]
- Valenti, L.; Fracanzani, A.L.; Dongiovanni, P.; Bugianesi, E.; Marchesini, G.; Manzini, P.; Vanni, E.; Fargion, S. Iron depletion by phlebotomy improves insulin resistance in patients with nonalcoholic fatty liver disease and hyperferritinemia: Evidence from a case-control study. Am. J. Gastroenterol 2007, 102, 1251–1258. [Google Scholar]
- Vigano, M.; Vergani, A.; Trombini, P.; Paleari, F.; Piperno, A. Insulin resistance influence iron metabolism and hepatic steatosis in type II diabetes. Gastroenterology 2000, 118, 986–987. [Google Scholar]
- Facchini, F.S.; Hua, N.W.; Stoohs, R.A. Effect of iron depletion in carbohydrate-intolerant patients with clinical evidence of nonalcoholic fatty liver disease. Gastroenterology 2002, 122, 931–939. [Google Scholar]
- Morrison, E.D.; Brandhagen, D.J.; Phatak, P.D.; Barton, J.C.; Krawitt, E.L.; el-Serag, H.B.; Gordon, S.C.; Galan, M.V.; Tung, B.Y.; Ioannou, G.N.; et al. Serum ferritin level predicts advanced hepatic fibrosis among U.S. patients with phenotypic hemochromatosis. Ann. Intern. Med 2003, 138, 627–633. [Google Scholar]
- Ruddell, R.G.; Hoang-Le, D.; Barwood, J.M.; Rutherford, P.S.; Piva, T.J.; Watters, D.J.; Santambrogio, P.; Arosio, P.; Ramm, G.A. Ferritin functions as a proinflammatory cytokine via iron-independent protein kinase C zeta/nuclear factor kappaB-regulated signaling in rat hepatic stellate cells. Hepatology 2009, 49, 887–900. [Google Scholar]
- Comert, B.; Mas, M.R.; Erdem, H.; Dinc, A.; Saglamkaya, U.; Cigerim, M.; Kuzhan, O.; Unal, T.; Kocabalkan, F. Insulin resistance in non-alcoholic steatohepatitis. Dig. Liver Dis 2001, 33, 353–358. [Google Scholar]
- Fargion, S.; Mattioli, M.; Fracanzani, A.L.; Sampietro, M.; Tavazzi, D.; Fociani, P.; Taioli, E.; Valenti, L.; Fiorelli, G. Hyperferritinemia, iron overload, and multiple metabolic alterations identify patients at risk for nonalcoholic steatohepatitis. Am. J. Gastroenterol 2001, 96, 2448–2455. [Google Scholar]
- Bugianesi, E.; Manzini, P.; D’Antico, S.; Vanni, E.; Longo, F.; Leone, N.; Massarenti, P.; Piga, A.; Marchesini, G.; Rizzetto, M. Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology 2004, 39, 179–187. [Google Scholar]
- Kahn, S.E.; Zinman, B.; Haffner, S.M.; O’Neill, M.C.; Kravitz, B.G.; Yu, D.; Freed, M.I.; Herman, W.H.; Holman, R.R.; Jones, N.P.; et al. Obesity is a major determinant of the association of C-reactive protein levels and the metabolic syndrome in type 2 diabetes. Diabetes 2006, 55, 2357–2364. [Google Scholar]
- Lecube, A.; Hernandez, C.; Pelegri, D.; Simo, R. Factors accounting for high ferritin levels in obesity. Int. J. Obes 2008, 32, 1665–1669. [Google Scholar]
- Lecube, A.; Hernandez, C.; Simo, R. Glucose abnormalities in non-alcoholic fatty liver disease and chronic hepatitis C virus infection: The role of iron overload. Diabetes/Metab. Res. Rev 2009, 25, 403–410. [Google Scholar]
- Yoneda, M.; Nozaki, Y.; Endo, H.; Mawatari, H.; Iida, H.; Fujita, K.; Yoneda, K.; Takahashi, H.; Kirikoshi, H.; Inamori, M.; et al. Serum ferritin is a clinical biomarker in Japanese patients with nonalcoholic steatohepatitis (NASH) independent of HFE gene mutation. Dig. Dis. Sci 2010, 55, 808–814. [Google Scholar]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar]
- Wallace, K.; Burt, A.D.; Wright, M.C. Liver fibrosis. Biochem. J 2008, 411, 1–18. [Google Scholar]
- Friedman, S.L. Hepatic fibrosis—Overview. Toxicology 2008, 254, 120–129. [Google Scholar]
- Pinzani, M. PDGF and signal transduction in hepatic stellate cells. Front. Biosci 2002, 7, d1720–d1726. [Google Scholar]
- Ikeda, K.; Wakahara, T.; Wang, Y.Q.; Kadoya, H.; Kawada, N.; Kaneda, K. In vitro migratory potential of rat quiescent hepatic stellate cells and its augmentation by cell activation. Hepatology 1999, 29, 1760–1767. [Google Scholar]
- Bonacchi, A.; Romagnani, P.; Romanelli, R.G.; Efsen, E.; Annunziato, F.; Lasagni, L.; Francalanci, M.; Serio, M.; Laffi, G.; Pinzani, M.; et al. Signal transduction by the chemokine receptor CXCR3: Activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J. Biol. Chem 2001, 276, 9945–9954. [Google Scholar]
- Svegliati-Baroni, G.; Saccomanno, S.; van Goor, H.; Jansen, P.; Benedetti, A.; Moshage, H. Involvement of reactive oxygen species and nitric oxide radicals in activation and proliferation of rat hepatic stellate cells. Liver 2001, 21, 1–12. [Google Scholar]
- Zhan, S.S.; Jiang, J.X.; Wu, J.; Halsted, C.; Friedman, S.L.; Zern, M.A.; Torok, N.J. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 2006, 43, 435–443. [Google Scholar]
- Tahashi, Y.; Matsuzaki, K.; Date, M.; Yoshida, K.; Furukawa, F.; Sugano, Y.; Matsushita, M.; Himeno, Y.; Inagaki, Y.; Inoue, K. Differential regulation of TGF-beta signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology 2002, 35, 49–61. [Google Scholar]
- Wanninger, J.; Neumeier, M.; Hellerbrand, C.; Schacherer, D.; Bauer, S.; Weiss, T.S.; Huber, H.; Schaffler, A.; Aslanidis, C.; Scholmerich, J.; et al. Lipid accumulation impairs adiponectin-mediated induction of activin A by increasing TGFbeta in primary human hepatocytes. Biochim. Biophys. Acta 2011, 1811, 626–633. [Google Scholar]
- Svegliati Baroni, G.; D’Ambrosio, L.; Ferretti, G.; Casini, A.; di Sario, A.; Salzano, R.; Ridolfi, F.; Saccomanno, S.; Jezequel, A.M.; Benedetti, A. Fibrogenic effect of oxidative stress on rat hepatic stellate cells. Hepatology 1998, 27, 720–726. [Google Scholar]
- Paradis, V.; Perlemuter, G.; Bonvoust, F.; Dargere, D.; Parfait, B.; Vidaud, M.; Conti, M.; Huet, S.; Ba, N.; Buffet, C.; et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: A potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology 2001, 34, 738–744. [Google Scholar]
- Kinnman, N.; Housset, C. Peribiliary myofibroblasts in biliary type liver fibrosis. Front. Biosci 2002, 7, d496–503. [Google Scholar]
- Kruglov, E.A.; Nathanson, R.A.; Nguyen, T.; Dranoff, J.A. Secretion of MCP-1/CCL2 by bile duct epithelia induces myofibroblastic transdifferentiation of portal fibroblasts. Am. J. Physiol. Gastrointest. Liver Physiol 2006, 290, G765–771. [Google Scholar]
- Guyot, C.; Lepreux, S.; Combe, C.; Doudnikoff, E.; Bioulac-Sage, P.; Balabaud, C.; Desmouliere, A. Hepatic fibrosis and cirrhosis: The (myo)fibroblastic cell subpopulations involved. Int. J. Biochem. Cell Biol 2006, 38, 135–151. [Google Scholar]
- Forbes, S.J.; Russo, F.P.; Rey, V.; Burra, P.; Rugge, M.; Wright, N.A.; Alison, M.R. A significant proportion of myofibroblasts are of bone marrow origin in human liver fibrosis. Gastroenterology 2004, 126, 955–963. [Google Scholar]
- Higashiyama, R.; Moro, T.; Nakao, S.; Mikami, K.; Fukumitsu, H.; Ueda, Y.; Ikeda, K.; Adachi, E.; Bou-Gharios, G.; Okazaki, I.; et al. Negligible contribution of bone marrow-derived cells to collagen production during hepatic fibrogenesis in mice. Gastroenterology 2009, 137, 1459–1466 e1451. [Google Scholar]
- Higashiyama, R.; Inagaki, Y.; Hong, Y.Y.; Kushida, M.; Nakao, S.; Niioka, M.; Watanabe, T.; Okano, H.; Matsuzaki, Y.; Shiota, G.; et al. Bone marrow-derived cells express matrix metalloproteinases and contribute to regression of liver fibrosis in mice. Hepatology 2007, 45, 213–222. [Google Scholar]
- Kisseleva, T.; Uchinami, H.; Feirt, N.; Quintana-Bustamante, O.; Segovia, J.C.; Schwabe, R.F.; Brenner, D.A. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J. Hepatol 2006, 45, 429–438. [Google Scholar]
- Brunt, E.M. Histopathology of non-alcoholic fatty liver disease. Clin. Liver Dis 2009, 13, 533–544. [Google Scholar]
- Schwimmer, J.B.; Behling, C.; Newbury, R.; Deutsch, R.; Nievergelt, C.; Schork, N.J.; Lavine, J.E. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005, 42, 641–649. [Google Scholar]
- Carter-Kent, C.; Yerian, L.M.; Brunt, E.M.; Angulo, P.; Kohli, R.; Ling, S.C.; Xanthakos, S.A.; Whitington, P.F.; Charatcharoenwitthaya, P.; Yap, J.; et al. Nonalcoholic steatohepatitis in children: A multicenter clinicopathological study. Hepatology 2009, 50, 1113–1120. [Google Scholar]
- Skoien, R.; Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; O’Brien, P.E.; Tilg, H.; et al. Heterogeneity of fibrosis patterns in non-alcoholic fatty liver disease supports the presence of multiple fibrogenic pathways. Liver Int 2013, 33, 624–632. [Google Scholar]
- Ekstedt, M.; Franzen, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Unalp, A.; Behling, C.E.; Lavine, J.E.; Neuschwander-Tetri, B.A. NASH Clinical Research Network. Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): A histologic marker of advanced NAFLD-Clinicopathologic correlations from the nonalcoholic steatohepatitis clinical research network. Hepatology 2009, 49, 809–820. [Google Scholar]
- Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; Weltman, M.D.; Tilg, H.; Moschen, A.R.; et al. Progressive fibrosis in nonalcoholic steatohepatitis: Association with altered regeneration and a ductular reaction. Gastroenterology 2007, 133, 80–90. [Google Scholar]
- Anderson, N.; Borlak, J. Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol. Rev 2008, 60, 311–357. [Google Scholar]
- Tetri, L.H.; Basaranoglu, M.; Brunt, E.M.; Yerian, L.M.; Neuschwander-Tetri, B.A. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am. J. Physiol. Gastrointest. Liver Physiol 2008, 295, G987–G995. [Google Scholar]
- Machado, M.V.; Cortez-Pinto, H. Cell death and nonalcoholic steatohepatitis: Where is ballooning relevant? Expert Rev. Gastroenterol. Hepatol 2011, 5, 213–222. [Google Scholar]
- Jaeschke, H.; Gujral, J.S.; Bajt, M.L. Apoptosis and necrosis in liver disease. Liver Int 2004, 24, 85–89. [Google Scholar]
- Canbay, A.; Friedman, S.; Gores, G.J. Apoptosis: The nexus of liver injury and fibrosis. Hepatology 2004, 39, 273–278. [Google Scholar]
- Malhi, H.; Gores, G.J.; Lemasters, J.J. Apoptosis and necrosis in the liver: A tale of two deaths? Hepatology 2006, 43, S31–S44. [Google Scholar]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar]
- Alkhouri, N.; Carter-Kent, C.; Feldstein, A.E. Apoptosis in nonalcoholic fatty liver disease: Diagnostic and therapeutic implications. Expert Rev. Gastroenterol. Hepatol 2011, 5, 201–212. [Google Scholar]
- Bechmann, L.P.; Gieseler, R.K.; Sowa, J.P.; Kahraman, A.; Erhard, J.; Wedemeyer, I.; Emons, B.; Jochum, C.; Feldkamp, T.; Gerken, G.; et al. Apoptosis is associated with CD36/fatty acid translocase upregulation in non-alcoholic steatohepatitis. Liver Int 2010, 30, 850–859. [Google Scholar]
- Feldstein, A.E.; Canbay, A.; Guicciardi, M.E.; Higuchi, H.; Bronk, S.F.; Gores, G.J. Diet associated hepatic steatosis sensitizes to Fas mediated liver injury in mice. J. Hepatol 2003, 39, 978–983. [Google Scholar]
- Malhi, H.; Barreyro, F.J.; Isomoto, H.; Bronk, S.F.; Gores, G.J. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut 2007, 56, 1124–1131. [Google Scholar]
- Volkmann, X.; Fischer, U.; Bahr, M.J.; Ott, M.; Lehner, F.; Macfarlane, M.; Cohen, G.M.; Manns, M.P.; Schulze-Osthoff, K.; Bantel, H. Increased hepatotoxicity of tumor necrosis factor-related apoptosis-inducing ligand in diseased human liver. Hepatology 2007, 46, 1498–1508. [Google Scholar]
- Ribeiro, P.S.; Cortez-Pinto, H.; Sola, S.; Castro, R.E.; Ramalho, R.M.; Baptista, A.; Moura, M.C.; Camilo, M.E.; Rodrigues, C.M. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am. J. Gastroenterol 2004, 99, 1708–1717. [Google Scholar]
- Farrell, G.C.; Larter, C.Z.; Hou, J.Y.; Zhang, R.H.; Yeh, M.M.; Williams, J.; dela Pena, A.; Francisco, R.; Osvath, S.R.; Brooling, J.; et al. Apoptosis in experimental NASH is associated with p53 activation and TRAIL receptor expression. J. Gastroenterol. Hepatol 2009, 24, 443–452. [Google Scholar]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem 2006, 281, 12093–12101. [Google Scholar]
- Wang, Y.; Ausman, L.M.; Russell, R.M.; Greenberg, A.S.; Wang, X.D. Increased apoptosis in high-fat diet-induced nonalcoholic steatohepatitis in rats is associated with c-Jun NH2-terminal kinase activation and elevated proapoptotic Bax. J. Nutr 2008, 138, 1866–1871. [Google Scholar]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [Google Scholar]
- Schattenberg, J.M.; Singh, R.; Wang, Y.; Lefkowitch, J.H.; Rigoli, R.M.; Scherer, P.E.; Czaja, M.J. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology 2006, 43, 163–172. [Google Scholar]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig 2004, 114, 147–152. [Google Scholar]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar]
- Feldstein, A.E.; Gores, G.J. Apoptosis in alcoholic and nonalcoholic steatohepatitis. Front. Biosci 2005, 10, 3093–3099. [Google Scholar]
- Hug, H.; Strand, S.; Grambihler, A.; Galle, J.; Hack, V.; Stremmel, W.; Krammer, P.H.; Galle, P.R. Reactive oxygen intermediates are involved in the induction of CD95 ligand mRNA expression by cytostatic drugs in hepatoma cells. J. Biol. Chem 1997, 272, 28191–28193. [Google Scholar]
- Wei, Y.R.; Wang, D.; Gentile, C.L.; Pagliassotti, M.J. Reduced endoplasmic reticulum luminal calcium links saturated fatty acid-mediated endoplasmic reticulum stress and cell death in liver cells. Mol. Cell. Biochem 2009, 331, 31–40. [Google Scholar]
- Kammoun, H.L.; Hainault, I.; Ferre, P.; Foufelle, F. Nutritional related liver disease: Targeting the endoplasmic reticulum stress. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 575–582. [Google Scholar]
- Gregor, M.F.; Yang, L.; Fabbrini, E.; Mohammed, B.S.; Eagon, J.C.; Hotamisligil, G.S.; Klein, S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes 2009, 58, 693–700. [Google Scholar]
- Cazanave, S.C.; Gores, G.J. Mechanisms and clinical implications of hepatocyte lipoapoptosis. Clin. Lipidol 2010, 5, 71–85. [Google Scholar]
- Deniaud, A.; Sharaf el dein, O.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol 2011, 54, 795–809. [Google Scholar]
- Su, Q.; Tsai, J.; Xu, E.; Qiu, W.; Bereczki, E.; Santha, M.; Adeli, K. Apolipoprotein B100 acts as a molecular link between lipid-induced endoplasmic reticulum stress and hepatic insulin resistance. Hepatology 2009, 50, 77–84. [Google Scholar]
- Wang, D.; Wei, Y.; Pagliassotti, M.J. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology 2006, 147, 943–951. [Google Scholar]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol 2009, 24, 830–840. [Google Scholar]
- Canbay, A.; Taimr, P.; Torok, N.; Higuchi, H.; Friedman, S.; Gores, G.J. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab. Investig 2003, 83, 655–663. [Google Scholar]
- Nakajima, T.; Moriguchi, M.; Katagishi, T.; Sekoguchi, S.; Nishikawa, T.; Takashima, H.; Kimura, H.; Minami, M.; Itoh, Y.; Kagawa, K.; et al. Premature telomere shortening and impaired regenerative response in hepatocytes of individuals with NAFLD. Liver Int 2006, 26, 23–31. [Google Scholar]
- Calado, R.T.; Brudno, J.; Mehta, P.; Kovacs, J.J.; Wu, C.; Zago, M.A.; Chanock, S.J.; Boyer, T.D.; Young, N.S. Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology 2011, 53, 1600–1607. [Google Scholar]
- Aravinthan, A.; Scarpini, C.; Tachtatzis, P.; Verma, S.; Penrhyn-Lowe, S.; Harvey, R.; Davies, S.E.; Allison, M.; Coleman, N.; Alexander, G. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J. Hepatol 2013, 58, 549–556. [Google Scholar]
- Ikeda, H.; Sasaki, M.; Sato, Y.; Harada, K.; Zen, Y.; Mitsui, T.; Nakanuma, Y. Large cell change of hepatocytes in chronic viral hepatitis represents a senescent-related lesion. Hum. Pathol 2009, 40, 1774–1782. [Google Scholar]
- Ben-Porath, I.; Weinberg, R.A. The signals and pathways activating cellular senescence. Int. J. Biochem. Cell Biol 2005, 37, 961–976. [Google Scholar]
- Chen, Q.M. Replicative senescence and oxidant-induced premature senescence. Beyond the control of cell cycle checkpoints. Ann. N. Y. Acad. Sci 2000, 908, 111–125. [Google Scholar]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol 2007, 8, 729–740. [Google Scholar]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol 2010, 5, 99–118. [Google Scholar]
- Adams, P.D. Remodeling of chromatin structure in senescent cells and its potential impact on tumor suppression and aging. Gene 2007, 397, 84–93. [Google Scholar]
- Narita, M.; Nunez, S.; Heard, E.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar]
- Lawless, C.; Wang, C.; Jurk, D.; Merz, A.; Zglinicki, T.; Passos, J.F. Quantitative assessment of markers for cell senescence. Exp. Gerontol 2010, 45, 772–778. [Google Scholar]
- Hornsby, P.J. Senescence and life span. Pflugers Arch 2010, 459, 291–299. [Google Scholar]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol 2004, 6, 168–170. [Google Scholar]
- Evan, G.I.; d’Adda di Fagagna, F. Cellular senescence: Hot or what? Curr. Opin. Genet. Dev 2009, 19, 25–31. [Google Scholar]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar]
- Hamilton, M.L.; van Remmen, H.; Drake, J.A.; Yang, H.; Guo, Z.M.; Kewitt, K.; Walter, C.A.; Richardson, A. Does oxidative damage to DNA increase with age? Proc. Natl. Acad. Sci. USA 2001, 98, 10469–10474. [Google Scholar]
- Ikeyama, S.; Wang, X.T.; Li, J.; Podlutsky, A.; Martindale, J.L.; Kokkonen, G.; van Huizen, R.; Gorospe, M.; Holbrook, N.J. Expression of the pro-apoptotic gene gadd153/chop is elevated in liver with aging and sensitizes cells to oxidant injury. J. Biol. Chem 2003, 278, 16726–16731. [Google Scholar]
- Von Zglinicki, T.; Saretzki, G.; Docke, W.; Lotze, C. Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: A model for senescence? Exp. Cell Res 1995, 220, 186–193. [Google Scholar]
- Packer, L.; Fuehr, K. Low oxygen concentration extends the lifespan of cultured human diploid cells. Nature 1977, 267, 423–425. [Google Scholar]
- Passos, J.F.; von Zglinicki, T. Mitochondria, telomeres and cell senescence. Exp. Gerontol 2005, 40, 466–472. [Google Scholar]
- Von Zglinicki, T.; Pilger, R.; Sitte, N. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic. Biol. Med 2000, 28, 64–74. [Google Scholar]
- Houben, J.M.; Moonen, H.J.; van Schooten, F.J.; Hageman, G.J. Telomere length assessment: Biomarker of chronic oxidative stress? Free Radic. Biol. Med 2008, 44, 235–246. [Google Scholar]
- Ahmed, S.; Passos, J.F.; Birket, M.J.; Beckmann, T.; Brings, S.; Peters, H.; Birch-Machin, M.A.; von Zglinicki, T.; Saretzki, G. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J. Cell Sci 2008, 121, 1046–1053. [Google Scholar]
- Henle, E.S.; Han, Z.; Tang, N.; Rai, P.; Luo, Y.; Linn, S. Sequence-specific DNA cleavage by Fe2+-mediated fenton reactions has possible biological implications. J. Biol. Chem 1999, 274, 962–971. [Google Scholar]
- Kawanishi, S.; Oikawa, S. Mechanism of telomere shortening by oxidative stress. Ann. N. Y. Acad. Sci 2004, 1019, 278–284. [Google Scholar]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol 2010, 6, 347. [Google Scholar]
- Sarkar, D.; Lebedeva, I.V.; Emdad, L.; Kang, D.C.; Baldwin, A.S., Jr.; Fisher, P.B. Human polynucleotide phosphorylase (hPNPaseold-35): A potential link between aging and inflammation. Cancer Res 2004, 64, 7473–7478. [Google Scholar]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar]
- Schnabl, B.; Purbeck, C.A.; Choi, Y.H.; Hagedorn, C.H.; Brenner, D. Replicative senescence of activated human hepatic stellate cells is accompanied by a pronounced inflammatory but less fibrogenic phenotype. Hepatology 2003, 37, 653–664. [Google Scholar]
- Skoien, R.; Powell, E.E.; Melino, M.; Clouston, A.D.; Gabrielli, B.G.; Jonsson, J.R. Fatty acids induce hepatocyte senescence in vitro: Implications for pathogenesis in non-alcoholic stetatohepatitis. Hepatology 2010, 52, 1044A–1045A. [Google Scholar]
- Ren, J.L.; Pan, J.S.; Lu, Y.P.; Sun, P.; Han, J. Inflammatory signaling and cellular senescence. Cell Signal 2009, 21, 378–383. [Google Scholar]
- Heydtmann, M.; Lalor, P.F.; Eksteen, J.A.; Hubscher, S.G.; Briskin, M.; Adams, D.H. CXC chemokine ligand 16 promotes integrin-mediated adhesion of liver-infiltrating lymphocytes to cholangiocytes and hepatocytes within the inflamed human liver. J. Immunol 2005, 174, 1055–1062. [Google Scholar]
- Heydtmann, M.; Adams, D.H. Chemokines in the immunopathogenesis of hepatitis C infection. Hepatology 2009, 49, 676–688. [Google Scholar]
- Efsen, E.; Grappone, C.; DeFranco, R.M.; Milani, S.; Romanelli, R.G.; Bonacchi, A.; Caligiuri, A.; Failli, P.; Annunziato, F.; Pagliai, G.; et al. Up-regulated expression of fractalkine and its receptor CX3CR1 during liver injury in humans. J. Hepatol 2002, 37, 39–47. [Google Scholar]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. Modulation of the microenvironment by senescent biliary epithelial cells may be involved in the pathogenesis of primary biliary cirrhosis. J. Hepatol 2010, 53, 318–325. [Google Scholar]
- Holt, A.P.; Salmon, M.; Buckley, C.D.; Adams, D.H. Immune interactions in hepatic fibrosis. Clin. Liver Dis 2008, 12, 861–882. [Google Scholar]
- Abrignani, S. Bystander activation by cytokines of intrahepatic T cells in chronic viral hepatitis. Semin. Liver Dis 1997, 17, 319–322. [Google Scholar]
- Aravinthan, A.; Pietrosi, G.; Hoare, M.; Jupp, J.; Marshall, A.; Verrill, C.; Davies, S.; Bateman, A.; Sheron, N.; Allison, M.; et al. Hepatocyte expression of the senescence marker p21 is linked to fibrosis and an adverse liver-related outcome in alcohol-related liver disease. PLoS One 2013, 8, e72904. [Google Scholar]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar]
- Yang, S.; Koteish, A.; Lin, H.; Huang, J.; Roskams, T.; Dawson, V.; Diehl, A.M. Oval cells compensate for damage and replicative senescence of mature hepatocytes in mice with fatty liver disease. Hepatology 2004, 39, 403–411. [Google Scholar]
- Zhan, Y.T.; An, W. Roles of liver innate immune cells in nonalcoholic fatty liver disease. World J. Gastroenterol 2010, 16, 4652–4660. [Google Scholar]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis 2010, 30, 245–257. [Google Scholar]
- Matsuoka, M.; Tsukamoto, H. Stimulation of hepatic lipocyte collagen production by Kupffer cell-derived transforming growth factor beta: Implication for a pathogenetic role in alcoholic liver fibrogenesis. Hepatology 1990, 11, 599–605. [Google Scholar]
- Malaguarnera, L.; di Rosa, M.; Zambito, A.M.; dell’Ombra, N.; di Marco, R.; Malaguarnera, M. Potential role of chitotriosidase gene in nonalcoholic fatty liver disease evolution. Am. J. Gastroenterol 2006, 101, 2060–2069. [Google Scholar]
- Malaguarnera, L.; di Rosa, M.; Zambito, A.M.; dell’Ombra, N.; Nicoletti, F.; Malaguarnera, M. Chitotriosidase gene expression in Kupffer cells from patients with non-alcoholic fatty liver disease. Gut 2006, 55, 1313–1320. [Google Scholar]
- Friedman, S.L. Mac the knife? Macrophages- the double-edged sword of hepatic fibrosis. J. Clin. Investig 2005, 115, 29–32. [Google Scholar]
- Hironaka, K.; Sakaida, I.; Matsumura, Y.; Kaino, S.; Miyamoto, K.; Okita, K. Enhanced interstitial collagenase (matrix metalloproteinase-13) production of Kupffer cell by gadolinium chloride prevents pig serum-induced rat liver fibrosis. Biochem. Biophys. Res. Commun 2000, 267, 290–295. [Google Scholar]
- Knittel, T.; Mehde, M.; Kobold, D.; Saile, B.; Dinter, C.; Ramadori, G. Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: Regulation by TNF-alpha and TGF-beta1. J. Hepatol 1999, 30, 48–60. [Google Scholar]
- Chen, C.J.; Kono, H.; Golenbock, D.; Reed, G.; Akira, S.; Rock, K.L. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med 2007, 13, 851–856. [Google Scholar]
- Alkhouri, N.; Morris-Stiff, G.; Campbell, C.; Lopez, R.; Tamimi, T.A.; Yerian, L.; Zein, N.N.; Feldstein, A.E. Neutrophil to lymphocyte ratio: A new marker for predicting steatohepatitis and fibrosis in patients with nonalcoholic fatty liver disease. Liver Int 2012, 32, 297–302. [Google Scholar]
- O'Shea, D.; Cawood, T.J.; O’Farrelly, C.; Lynch, L. Natural killer cells in obesity: Impaired function and increased susceptibility to the effects of cigarette smoke. PLoS One 2010, 5, e8660. [Google Scholar]
- Kahraman, A.; Schlattjan, M.; Kocabayoglu, P.; Yildiz-Meziletoglu, S.; Schlensak, M.; Fingas, C.D.; Wedemeyer, I.; Marquitan, G.; Gieseler, R.K.; Baba, H.A.; et al. Major histocompatibility complex class I-related chains A and B (MIC A/B): A novel role in nonalcoholic steatohepatitis. Hepatology 2010, 51, 92–102. [Google Scholar]
- Bendelac, A.; Savage, P.B.; Teyton, L. The biology of NKT cells. Annu. Rev. Immunol 2007, 25, 297–336. [Google Scholar]
- Safadi, R.; Zigmond, E.; Pappo, O.; Shalev, Z.; Ilan, Y. Amelioration of hepatic fibrosis via beta-glucosylceramide-mediated immune modulation is associated with altered CD8 and NKT lymphocyte distribution. Int. Immunol 2007, 19, 1021–1029. [Google Scholar]
- Nuti, S.; Rosa, D.; Valiante, N.M.; Saletti, G.; Caratozzolo, M.; Dellabona, P.; Barnaba, V.; Abrignani, S. Dynamics of intra-hepatic lymphocytes in chronic hepatitis C: Enrichment for Valpha24+ T cells and rapid elimination of effector cells by apoptosis. Eur. J. Immunol 1998, 28, 3448–3455. [Google Scholar]
- Park, O.; Jeong, W.I.; Wang, L.; Wang, H.; Lian, Z.X.; Gershwin, M.E.; Gao, B. Diverse roles of invariant natural killer T cells in liver injury and fibrosis induced by carbon tetrachloride. Hepatology 2009, 49, 1683–1694. [Google Scholar]
- Syn, W.K.; Oo, Y.H.; Pereira, T.A.; Karaca, G.F.; Jung, Y.; Omenetti, A.; Witek, R.P.; Choi, S.S.; Guy, C.D.; Fearing, C.M.; et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 2010, 51, 1998–2007. [Google Scholar]
- Tajiri, K.; Shimizu, Y.; Tsuneyama, K.; Sugiyama, T. Role of liver-infiltrating CD3+CD56+ natural killer T cells in the pathogenesis of nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol 2009, 21, 673–680. [Google Scholar]
- Xu, C.F.; Yu, C.H.; Li, Y.M.; Xu, L.; Du, J.; Shen, Z. Association of the frequency of peripheral natural killer T cells with nonalcoholic fatty liver disease. World J. Gastroenterol 2007, 13, 4504–4508. [Google Scholar]
- Elinav, E.; Pappo, O.; Sklair-Levy, M.; Margalit, M.; Shibolet, O.; Gomori, M.; Alper, R.; Thalenfeld, B.; Engelhardt, D.; Rabbani, E.; et al. Adoptive transfer of regulatory NKT lymphocytes ameliorates non-alcoholic steatohepatitis and glucose intolerance in ob/ob mice and is associated with intrahepatic CD8 trapping. J. Pathol 2006, 209, 121–128. [Google Scholar]
- Safadi, R.; Ohta, M.; Alvarez, C.E.; Fiel, M.I.; Bansal, M.; Mehal, W.Z.; Friedman, S.L. Immune stimulation of hepatic fibrogenesis by CD8 cells and attenuation by transgenic interleukin-10 from hepatocytes. Gastroenterology 2004, 127, 870–882. [Google Scholar]
- Novobrantseva, T.I.; Majeau, G.R.; Amatucci, A.; Kogan, S.; Brenner, I.; Casola, S.; Shlomchik, M.J.; Koteliansky, V.; Hochman, P.S.; Ibraghimov, A. Attenuated liver fibrosis in the absence of B cells. J. Clin. Investig 2005, 115, 3072–3082. [Google Scholar]
- Mosmann, T. Complexity or coherence? Cytokine secretion by B cells. Nat. Immunol 2000, 1, 465–466. [Google Scholar]
- Harris, D.P.; Haynes, L.; Sayles, P.C.; Duso, D.K.; Eaton, S.M.; Lepak, N.M.; Johnson, L.L.; Swain, S.L.; Lund, F.E. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat. Immunol 2000, 1, 475–482. [Google Scholar]
- Hammerich, L.; Heymann, F.; Tacke, F. Role of IL-17 and Th17 cells in liver diseases. Clin. Dev. Immunol 2011, 2011, 345803. [Google Scholar]
- Imamura, M.; Ogawa, T.; Sasaguri, Y.; Chayama, K.; Ueno, H. Suppression of macrophage infiltration inhibits activation of hepatic stellate cells and liver fibrogenesis in rats. Gastroenterology 2005, 128, 138–146. [Google Scholar]
- Muhanna, N.; Horani, A.; Doron, S.; Safadi, R. Lymphocyte-hepatic stellate cell proximity suggests a direct interaction. Clin. Exp. Immunol 2007, 148, 338–347. [Google Scholar]
- Winau, F.; Hegasy, G.; Weiskirchen, R.; Weber, S.; Cassan, C.; Sieling, P.A.; Modlin, R.L.; Liblau, R.S.; Gressner, A.M.; Kaufmann, S.H. Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity 2007, 26, 117–129. [Google Scholar]
- Parsonage, G.; Filer, A.D.; Haworth, O.; Nash, G.B.; Rainger, G.E.; Salmon, M.; Buckley, C.D. A stromal address code defined by fibroblasts. Trends Immunol 2005, 26, 150–156. [Google Scholar]
- Gao, B.; Seki, E.; Brenner, D.A.; Friedman, S.; Cohen, J.I.; Nagy, L.; Szabo, G.; Zakhari, S. Innate immunity in alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol 2011, 300, G516–G525. [Google Scholar]
- Brenner, D.A.; Seki, E.; Taura, K.; Kisseleva, T.; Deminicis, S.; Iwaisako, K.; Inokuchi, S.; Schnabl, B.; Oesterreicher, C.H.; Paik, Y.H.; et al. Non-alcoholic steatohepatitis-induced fibrosis: Toll-like receptors, reactive oxygen species and Jun N-terminal kinase. Hepatol. Res 2011, 41, 683–686. [Google Scholar]
- Maher, J.J. DAMPs ramp up drug toxicity. J. Clin. Investig 2009, 119, 246–249. [Google Scholar]
- Matzinger, P. Tolerance, danger, and the extended family. Ann. Rev. Immunol 1994, 12, 991–1045. [Google Scholar]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Ann. Rev. Immunol 2010, 28, 321–342. [Google Scholar]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar]
- Park, J.S.; Gamboni-Robertson, F.; He, Q.; Svetkauskaite, D.; Kim, J.Y.; Strassheim, D.; Sohn, J.W.; Yamada, S.; Maruyama, I.; Banerjee, A.; et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell Physiol 2006, 290, C917–C924. [Google Scholar]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol 2011, 29, 707–735. [Google Scholar]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in inflammation and immunity. Cell 2010, 140, 798–804. [Google Scholar]
- Silva, M.T. Secondary necrosis: The natural outcome of the complete apoptotic program. FEBS Lett 2010, 584, 4491–4499. [Google Scholar]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol 2005, 5, 331–342. [Google Scholar]
- Tang, D.; Shi, Y.; Kang, R.; Li, T.; Xiao, W.; Wang, H.; Xiao, X. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J. Leukoc. Biol 2007, 81, 741–747. [Google Scholar]
- Su, G.L.; Klein, R.D.; Aminlari, A.; Zhang, H.Y.; Steinstraesser, L.; Alarcon, W.H.; Remick, D.G.; Wang, S.C. Kupffer cell activation by lipopolysaccharide in rats: Role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology 2000, 31, 932–936. [Google Scholar]
- Kopydlowski, K.M.; Salkowski, C.A.; Cody, M.J.; van Rooijen, N.; Major, J.; Hamilton, T.A.; Vogel, S.N. Regulation of macrophage chemokine expression by lipopolysaccharide in vitro and in vivo. J. Immunol 1999, 163, 1537–1544. [Google Scholar]
- Seki, E.; Tsutsui, H.; Nakano, H.; Tsuji, N.; Hoshino, K.; Adachi, O.; Adachi, K.; Futatsugi, S.; Kuida, K.; Takeuchi, O.; et al. Lipopolysaccharide-induced IL-18 secretion from murine Kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL-12 and IL-1beta. J. Immunol 2001, 166, 2651–2657. [Google Scholar]
- Schwabe, R.F.; Seki, E.; Brenner, D.A. Toll-like receptor signaling in the liver. Gastroenterology 2006, 130, 1886–1900. [Google Scholar]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol 2007, 47, 571–579. [Google Scholar]
- Scott, M.J.; Billiar, T.R. Beta2-integrin-induced p38 MAPK activation is a key mediator in the CD14/TLR4/MD2-dependent uptake of lipopolysaccharide by hepatocytes. J. Biol. Chem 2008, 283, 29433–29446. [Google Scholar]
- Brun, P.; Castagliuolo, I.; Pinzani, M.; Palu, G.; Martines, D. Exposure to bacterial cell wall products triggers an inflammatory phenotype in hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol 2005, 289, G571–G578. [Google Scholar]
- Gabele, E.; Muhlbauer, M.; Dorn, C.; Weiss, T.S.; Froh, M.; Schnabl, B.; Wiest, R.; Scholmerich, J.; Obermeier, F.; Hellerbrand, C. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem. Biophys. Res. Commun 2008, 376, 271–276. [Google Scholar]
- Uhrig, A.; Banafsche, R.; Kremer, M.; Hegenbarth, S.; Hamann, A.; Neurath, M.; Gerken, G.; Limmer, A.; Knolle, P.A. Development and functional consequences of LPS tolerance in sinusoidal endothelial cells of the liver. J. Leukoc. Biol 2005, 77, 626–633. [Google Scholar]
- Martin-Armas, M.; Simon-Santamaria, J.; Pettersen, I.; Moens, U.; Smedsrod, B.; Sveinbjornsson, B. Toll-like receptor 9 (TLR9) is present in murine liver sinusoidal endothelial cells (LSECs) and mediates the effect of CpG-oligonucleotides. J. Hepatol 2006, 44, 939–946. [Google Scholar]
- Chen, X.M.; O’Hara, S.P.; Nelson, J.B.; Splinter, P.L.; Small, A.J.; Tietz, P.S.; Limper, A.H.; LaRusso, N.F. Multiple TLRs are expressed in human cholangiocytes and mediate host epithelial defense responses to Cryptosporidium parvum via activation of NF-kappaB. J. Immunol 2005, 175, 7447–7456. [Google Scholar]
- Shu, S.A.; Lian, Z.X.; Chuang, Y.H.; Yang, G.X.; Moritoki, Y.; Comstock, S.S.; Zhong, R.Q.; Ansari, A.A.; Liu, Y.J.; Gershwin, M.E. The role of CD11c(+) hepatic dendritic cells in the induction of innate immune responses. Clin. Exp. Immunol 2007, 149, 335–343. [Google Scholar]
- Imaeda, A.B.; Watanabe, A.; Sohail, M.A.; Mahmood, S.; Mohamadnejad, M.; Sutterwala, F.S.; Flavell, R.A.; Mehal, W.Z. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J. Clin. Investig 2009, 119, 305–314. [Google Scholar]
- Hoque, R.; Sohail, M.; Malik, A.; Sarwar, S.; Luo, Y.; Shah, A.; Barrat, F.; Flavell, R.; Gorelick, F.; Husain, S.; et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 2011, 141, 358–369. [Google Scholar]
- Busconi, L.; Bauer, J.W.; Tumang, J.R.; Laws, A.; Perkins-Mesires, K.; Tabor, A.S.; Lau, C.; Corley, R.B.; Rothstein, T.L.; Lund, F.E.; et al. Functional outcome of B cell activation by chromatin immune complex engagement of the B cell receptor and TLR9. J. Immunol 2007, 179, 7397–7405. [Google Scholar]
- Barrat, F.J.; Meeker, T.; Gregorio, J.; Chan, J.H.; Uematsu, S.; Akira, S.; Chang, B.; Duramad, O.; Coffman, R.L. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med 2005, 202, 1131–1139. [Google Scholar]
- Bamboat, Z.M.; Balachandran, V.P.; Ocuin, L.M.; Obaid, H.; Plitas, G.; DeMatteo, R.P. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology 2010, 51, 621–632. [Google Scholar]
- Guiducci, C.; Tripodo, C.; Gong, M.; Sangaletti, S.; Colombo, M.P.; Coffman, R.L.; Barrat, F.J. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J. Exp. Med 2010, 207, 2931–2942. [Google Scholar]
- Iyer, S.S.; Pulskens, W.P.; Sadler, J.J.; Butter, L.M.; Teske, G.J.; Ulland, T.K.; Eisenbarth, S.C.; Florquin, S.; Flavell, R.A.; Leemans, J.C.; et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl. Acad. Sci. USA 2009, 106, 20388–20393. [Google Scholar]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 2011, 32, 157–164. [Google Scholar]
- Carp, H. Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J. Exp. Med 1982, 155, 264–275. [Google Scholar]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol 2011, 12, 222–230. [Google Scholar]
- Pereira, A.F.; Pereira, L.B.; Vale, E.C.; Tanure, L.A. Four cases of Muckle-Wells syndrome within the same family. Anais Brasileiros de Dermatologia 2010, 85, 907–911. [Google Scholar]
- Coddou, C.; Yan, Z.; Obsil, T.; Huidobro-Toro, J.P.; Stojilkovic, S.S. Activation and regulation of purinergic P2X receptor channels. Pharmacol. Rev 2011, 63, 641–683. [Google Scholar]
- Di Virgilio, F. Liaisons dangereuses: P2X(7) and the inflammasome. Trends Pharmacol. Sci 2007, 28, 465–472. [Google Scholar]
- Stros, M. HMGB proteins: Interactions with DNA and chromatin. Biochim. Biophys. Acta 2010, 1799, 101–113. [Google Scholar]
- Andersson, U.; Tracey, K.J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev.Immunol 2011, 29, 139–162. [Google Scholar]
- Wang, H.; Yang, H.; Czura, C.J.; Sama, A.E.; Tracey, K.J. HMGB1 as a late mediator of lethal systemic inflammation. Am. J. Respir. Crit. Care Med 2001, 164, 1768–1773. [Google Scholar]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar]
- Tsung, A.; Klune, J.R.; Zhang, X.; Jeyabalan, G.; Cao, Z.; Peng, X.; Stolz, D.B.; Geller, D.A.; Rosengart, M.R.; Billiar, T.R. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med 2007, 204, 2913–2923. [Google Scholar]
- Soehnlein, O.; Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol 2010, 10, 427–439. [Google Scholar]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar]
- Kono, H.; Karmarkar, D.; Iwakura, Y.; Rock, K.L. Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J. Immunol 2010, 184, 4470–4478. [Google Scholar]
- Taylor, K.R.; Trowbridge, J.M.; Rudisill, J.A.; Termeer, C.C.; Simon, J.C.; Gallo, R.L. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J. Biol. Chem 2004, 279, 17079–17084. [Google Scholar]
- Opitz, B.; Hippenstiel, S.; Eitel, J.; Suttorp, N. Extra- and intracellular innate immune recognition in endothelial cells. Thromb. Haemost 2007, 98, 319–326. [Google Scholar]
- Harja, E.; Bu, D.X.; Hudson, B.I.; Chang, J.S.; Shen, X.; Hallam, K.; Kalea, A.Z.; Lu, Y.; Rosario, R.H.; Oruganti, S.; et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J. Clin. Investig 2008, 118, 183–194. [Google Scholar]
- Wong, J.; Johnston, B.; Lee, S.S.; Bullard, D.C.; Smith, C.W.; Beaudet, A.L.; Kubes, P. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J. Clin. Investig 1997, 99, 2782–2790. [Google Scholar]
- Lee, W.Y.; Kubes, P. Leukocyte adhesion in the liver: Distinct adhesion paradigm from other organs. J. Hepatol 2008, 48, 504–512. [Google Scholar]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334 e327. [Google Scholar]
- Csak, T.; Velayudham, A.; Hritz, I.; Petrasek, J.; Levin, I.; Lippai, D.; Catalano, D.; Mandrekar, P.; Dolganiuc, A.; Kurt-Jones, E.; et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol 2011, 300, G433–G441. [Google Scholar]
- Romics, L., Jr.; Mandrekar, P.; Kodys, K.; Velayudham, A.; Drechsler, Y.; Dolganiuc, A.; Szabo, G. Increased lipopolysaccharide sensitivity in alcoholic fatty livers is independent of leptin deficiency and toll-like receptor 4 (TLR4) or TLR2 mRNA expression. Alcohol. Clin. Exp. Res 2005, 29, 1018–1026. [Google Scholar]
- Mencin, A.; Kluwe, J.; Schwabe, R.F. Toll-like receptors as targets in chronic liver diseases. Gut 2009, 58, 704–720. [Google Scholar]
- Baffy, G. Kupffer cells in non-alcoholic fatty liver disease: The emerging view. J. Hepatol 2009, 51, 212–223. [Google Scholar]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar]
- Kagan, J.C.; Medzhitov, R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 2006, 125, 943–955. [Google Scholar]
- Frazier, T.H.; DiBaise, J.K.; McClain, C.J. Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. J. Parent. Enter. Nutr 2011, 35, 14S–20S. [Google Scholar]
- Wigg, A.J.; Roberts-Thomson, I.C.; Dymock, R.B.; McCarthy, P.J.; Grose, R.H.; Cummins, A.G. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001, 48, 206–211. [Google Scholar]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar]
- Li, Z.; Yang, S.; Lin, H.; Huang, J.; Watkins, P.A.; Moser, A.B.; Desimone, C.; Song, X.Y.; Diehl, A.M. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003, 37, 343–350. [Google Scholar]
- Brun, P.; Castagliuolo, I.; di Leo, V.; Buda, A.; Pinzani, M.; Palu, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol 2007, 292, G518–G525. [Google Scholar]
- Solga, S.F.; Diehl, A.M. Non-alcoholic fatty liver disease: Lumen-liver interactions and possible role for probiotics. J. Hepatol 2003, 38, 681–687. [Google Scholar]
- Velayudham, A.; Dolganiuc, A.; Ellis, M.; Petrasek, J.; Kodys, K.; Mandrekar, P.; Szabo, G. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology 2009, 49, 989–997. [Google Scholar]
- Ilan, Y. Leaky gut and the liver: A role for bacterial translocation in nonalcoholic steatohepatitis. World J. Gastroenterol 2012, 18, 2609–2618. [Google Scholar]
- Seo, Y.S.; Shah, V.H. The role of gut-liver axis in the pathogenesis of liver cirrhosis and portal hypertension. Clin. Mol. Hepatol 2012, 18, 337–346. [Google Scholar]
- Seki, E.; Schnabl, B. Role of innate immunity and the microbiota in liver fibrosis: Crosstalk between the liver and gut. J. Physiol 2012, 590, 447–458. [Google Scholar]
- Hartmann, P.; Haimerl, M.; Mazagova, M.; Brenner, D.A.; Schnabl, B. Toll-like receptor 2-mediated intestinal injury and enteric tumor necrosis factor receptor I contribute to liver fibrosis in mice. Gastroenterology 2012, 143, 1330–1340 e1331. [Google Scholar]
- Watanabe, A.; Hashmi, A.; Gomes, D.A.; Town, T.; Badou, A.; Flavell, R.A.; Mehal, W.Z. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology 2007, 46, 1509–1518. [Google Scholar]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig 2006, 116, 3015–3025. [Google Scholar]
- Senn, J.J. Toll-like receptor-2 is essential for the development of palmitate-induced insulin resistance in myotubes. J. Biol. Chem 2006, 281, 26865–26875. [Google Scholar]
- Miura, K.; Seki, E.; Ohnishi, H.; Brenner, D.A. Role of toll-like receptors and their downstream molecules in the development of nonalcoholic fatty liver disease. Gastroenterol. Res. Pract 2010, 2010, 362847. [Google Scholar]
- Seki, E.; Brenner, D.A. Toll-like receptors and adaptor molecules in liver disease: Update. Hepatology 2008, 48, 322–335. [Google Scholar]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med 2011, 17, 179–188. [Google Scholar]
- Kamari, Y.; Shaish, A.; Vax, E.; Shemesh, S.; Kandel-Kfir, M.; Arbel, Y.; Olteanu, S.; Barshack, I.; Dotan, S.; Voronov, E.; et al. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J. Hepatol 2011, 55, 1086–1094. [Google Scholar]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol 2011, 12, 408–415. [Google Scholar]
- Stienstra, R.; Saudale, F.; Duval, C.; Keshtkar, S.; Groener, J.E.; van Rooijen, N.; Staels, B.; Kersten, S.; Muller, M. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology 2010, 51, 511–522. [Google Scholar]
- Netea, M.G.; Nold-Petry, C.A.; Nold, M.F.; Joosten, L.A.; Opitz, B.; van der Meer, J.H.; van de Veerdonk, F.L.; Ferwerda, G.; Heinhuis, B.; Devesa, I.; et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 2009, 113, 2324–2335. [Google Scholar]
- Csak, T.; Dolganiuc, A.; Kodys, K.; Nath, B.; Petrasek, J.; Bala, S.; Lippai, D.; Szabo, G. Mitochondrial antiviral signaling protein defect links impaired antiviral response and liver injury in steatohepatitis in mice. Hepatology 2011, 53, 1917–1931. [Google Scholar]
- Ji, J.; Zhang, L.; Wang, P.; Mu, Y.M.; Zhu, X.Y.; Wu, Y.Y.; Yu, H.; Zhang, B.; Chen, S.M.; Sun, X.Z. Saturated free fatty acid, palmitic acid, induces apoptosis in fetal hepatocytes in culture. Exp. Toxicol. Pathol 2005, 56, 369–376. [Google Scholar]
- Seki, E.; de Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med 2007, 13, 1324–1332. [Google Scholar]
- Nolan, J.P.; Leibowitz, A.I. Endotoxins in liver disease. Gastroenterology 1978, 75, 765–766. [Google Scholar]
- Grinko, I.; Geerts, A.; Wisse, E. Experimental biliary fibrosis correlates with increased numbers of fat-storing and Kupffer cells, and portal endotoxemia. J. Hepatol 1995, 23, 449–458. [Google Scholar]
- Chan, C.C.; Hwang, S.J.; Lee, F.Y.; Wang, S.S.; Chang, F.Y.; Li, C.P.; Chu, C.J.; Lu, R.H.; Lee, S.D. Prognostic value of plasma endotoxin levels in patients with cirrhosis. Scand. J. Gastroenterol 1997, 32, 942–946. [Google Scholar]
- Fukui, H.; Brauner, B.; Bode, J.C.; Bode, C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: Reevaluation with an improved chromogenic assay. J. Hepatol 1991, 12, 162–169. [Google Scholar]
- Nath, B.; Levin, I.; Csak, T.; Petrasek, J.; Mueller, C.; Kodys, K.; Catalano, D.; Mandrekar, P.; Szabo, G. Hepatocyte-specific hypoxia-inducible factor-1alpha is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology 2011, 53, 1526–1537. [Google Scholar]
- Clement, S.; Juge-Aubry, C.; Sgroi, A.; Conzelmann, S.; Pazienza, V.; Pittet-Cuenod, B.; Meier, C.A.; Negro, F. Monocyte chemoattractant protein-1 secreted by adipose tissue induces direct lipid accumulation in hepatocytes. Hepatology 2008, 48, 799–807. [Google Scholar]
- Cosgrove, B.D.; Cheng, C.; Pritchard, J.R.; Stolz, D.B.; Lauffenburger, D.A.; Griffith, L.G. An inducible autocrine cascade regulates rat hepatocyte proliferation and apoptosis responses to tumor necrosis factor-alpha. Hepatology 2008, 48, 276–288. [Google Scholar]
- Petrasek, J.; Dolganiuc, A.; Csak, T.; Kurt-Jones, E.A.; Szabo, G. Type I interferons protect from Toll-like receptor 9-associated liver injury and regulate IL-1 receptor antagonist in mice. Gastroenterology 2011, 140, 697–708 e694. [Google Scholar]
- Rangwala, F.; Guy, C.D.; Lu, J.; Suzuki, A.; Burchette, J.L.; Abdelmalek, M.F.; Chen, W.; Diehl, A.M. Increased production of sonic hedgehog by ballooned hepatocytes. J. Pathol 2011, 224, 401–410. [Google Scholar]
- Syn, W.K.; Witek, R.P.; Curbishley, S.M.; Jung, Y.; Choi, S.S.; Enrich, B.; Omenetti, A.; Agboola, K.M.; Fearing, C.M.; Tilg, H.; et al. Role for hedgehog pathway in regulating growth and function of invariant NKT cells. Eur. J. Immunol 2009, 39, 1879–1892. [Google Scholar]
- Bohinc, B.N.; Diehl, A.M. Mechanisms of disease progression in NASH: New paradigms. Clin. Liver Dis 2012, 16, 549–565. [Google Scholar]
- Syn, W.K.; Choi, S.S.; Liaskou, E.; Karaca, G.F.; Agboola, K.M.; Oo, Y.H.; Mi, Z.; Pereira, T.A.; Zdanowicz, M.; Malladi, P.; et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011, 53, 106–115. [Google Scholar]
- Tacke, F.; Luedde, T.; Trautwein, C. Inflammatory pathways in liver homeostasis and liver injury. Clin. Rev. Allergy Immunol 2009, 36, 4–12. [Google Scholar]
- Argo, C.K.; Northup, P.G.; Al-Osaimi, A.M.; Caldwell, S.H. Systematic review of risk factors for fibrosis progression in non-alcoholic steatohepatitis. J. Hepatol 2009, 51, 371–379. [Google Scholar]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human non-alcoholic fatty liver disease. Hepatology 2013, 59, 1393–1405. [Google Scholar]
- Lefkowitch, J.H.; Haythe, J.H.; Regent, N. Kupffer cell aggregation and perivenular distribution in steatohepatitis. Modern Pathol 2002, 15, 699–704. [Google Scholar]
- Fotiadu, A.; Gagalis, A.; Akriviadis, E.; Kotoula, V.; Sinakos, E.; Karkavelas, G.; Hytiroglou, P. Clinicopathological correlations in a series of adult patients with non-alcoholic fatty liver disease. Pathol. Int 2010, 60, 87–92. [Google Scholar]
- Lalor, P.F.; Edwards, S.; McNab, G.; Salmi, M.; Jalkanen, S.; Adams, D.H. Vascular adhesion protein-1 mediates adhesion and transmigration of lymphocytes on human hepatic endothelial cells. J. Immunol 2002, 169, 983–992. [Google Scholar]
- Middleton, J.; Patterson, A.M.; Gardner, L.; Schmutz, C.; Ashton, B.A. Leukocyte extravasation: Chemokine transport and presentation by the endothelium. Blood 2002, 100, 3853–3860. [Google Scholar]
- Lalor, P.F.; Shields, P.; Grant, A.; Adams, D.H. Recruitment of lymphocytes to the human liver. Immunol. Cell Biol 2002, 80, 52–64. [Google Scholar]
- Curbishley, S.M.; Eksteen, B.; Gladue, R.P.; Lalor, P.; Adams, D.H. CXCR 3 activation promotes lymphocyte transendothelial migration across human hepatic endothelium under fluid flow. Am. J. Pathol 2005, 167, 887–899. [Google Scholar]
- Kudo, S.; Matsuno, K.; Ezaki, T.; Ogawa, M. A novel migration pathway for rat dendritic cells from the blood: Hepatic sinusoids-lymph translocation. J. Exp. Med 1997, 185, 777–784. [Google Scholar]
- Adams, D.H.; Eksteen, B. Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nat. Rev. Immunol 2006, 6, 244–251. [Google Scholar]
- Mosmann, T.R.; Coffman, R.L. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol 1989, 7, 145–173. [Google Scholar]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol 2009, 27, 485–517. [Google Scholar]
- Sandler, N.G.; Mentink-Kane, M.M.; Cheever, A.W.; Wynn, T.A. Global gene expression profiles during acute pathogen-induced pulmonary inflammation reveal divergent roles for Th1 and Th2 responses in tissue repair. J. Immunol 2003, 171, 3655–3667. [Google Scholar]
- Chiaramonte, M.G.; Donaldson, D.D.; Cheever, A.W.; Wynn, T.A. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J. Clin. Investig 1999, 104, 777–785. [Google Scholar]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol 2005, 6, 1123–1132. [Google Scholar]
- Miossec, P.; Korn, T.; Kuchroo, V.K. Interleukin-17 and type 17 helper T cells. N. Engl. J. Med 2009, 361, 888–898. [Google Scholar]
- Hirota, K.; Yoshitomi, H.; Hashimoto, M.; Maeda, S.; Teradaira, S.; Sugimoto, N.; Yamaguchi, T.; Nomura, T.; Ito, H.; Nakamura, T.; et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J. Exp. Med 2007, 204, 2803–2812. [Google Scholar]
- Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003, 52, 65–70. [Google Scholar]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol 2007, 8, 967–974. [Google Scholar]
- Ouyang, W.; Kolls, J.K.; Zheng, Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 2008, 28, 454–467. [Google Scholar]
- Dong, C. Regulation and pro-inflammatory function of interleukin-17 family cytokines. Immunol. Rev 2008, 226, 80–86. [Google Scholar]
- Jones, C.E.; Chan, K. Interleukin-17 stimulates the expression of interleukin-8, growth-related oncogene-alpha, and granulocyte-colony-stimulating factor by human airway epithelial cells. Am. J. Respir. Cell Mol. Biol 2002, 26, 748–753. [Google Scholar]
- Ye, P.; Rodriguez, F.H.; Kanaly, S.; Stocking, K.L.; Schurr, J.; Schwarzenberger, P.; Oliver, P.; Huang, W.; Zhang, P.; Zhang, J.; et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med 2001, 194, 519–527. [Google Scholar]
- Sparna, T.; Retey, J.; Schmich, K.; Albrecht, U.; Naumann, K.; Gretz, N.; Fischer, H.P.; Bode, J.G.; Merfort, I. Genome-wide comparison between IL-17 and combined TNF-alpha/IL-17 induced genes in primary murine hepatocytes. BMC Genomics 2010, 11, 226. [Google Scholar]
- Patel, D.N.; King, C.A.; Bailey, S.R.; Holt, J.W.; Venkatachalam, K.; Agrawal, A.; Valente, A.J.; Chandrasekar, B. Interleukin-17 stimulates C-reactive protein expression in hepatocytes and smooth muscle cells via p38 MAPK and ERK1/2-dependent NF-kappaB and C/EBPbeta activation. J. Biol. Chem 2007, 282, 27229–27238. [Google Scholar]
- Pene, J.; Chevalier, S.; Preisser, L.; Venereau, E.; Guilleux, M.H.; Ghannam, S.; Moles, J.P.; Danger, Y.; Ravon, E.; Lesaux, S.; et al. Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J. Immunol 2008, 180, 7423–7430. [Google Scholar]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A.; et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006, 314, 1461–1463. [Google Scholar]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748. [Google Scholar]
- Abiru, S.; Migita, K.; Maeda, Y.; Daikoku, M.; Ito, M.; Ohata, K.; Nagaoka, S.; Matsumoto, T.; Takii, Y.; Kusumoto, K.; et al. Serum cytokine and soluble cytokine receptor levels in patients with non-alcoholic steatohepatitis. Liver Int 2006, 26, 39–45. [Google Scholar]
- Lemmers, A.; Moreno, C.; Gustot, T.; Marechal, R.; Degre, D.; Demetter, P.; de Nadai, P.; Geerts, A.; Quertinmont, E.; Vercruysse, V.; et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology 2009, 49, 646–657. [Google Scholar]
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol 2011, 166, 281–290. [Google Scholar]
- Harada, K.; Shimoda, S.; Sato, Y.; Isse, K.; Ikeda, H.; Nakanuma, Y. Periductal interleukin-17 production in association with biliary innate immunity contributes to the pathogenesis of cholangiopathy in primary biliary cirrhosis. Clin. Exp. Immunol 2009, 157, 261–270. [Google Scholar]
- Lomonaco, R.; Sunny, N.E.; Bril, F.; Cusi, K. Nonalcoholic fatty liver disease: Current issues and novel treatment approaches. Drugs 2013, 73, 1–14. [Google Scholar]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology 2010, 52, 79–104. [Google Scholar]
- Harrison, S.A.; Fecht, W.; Brunt, E.M.; Neuschwander-Tetri, B.A. Orlistat for overweight subjects with nonalcoholic steatohepatitis: A randomized, prospective trial. Hepatology 2009, 49, 80–86. [Google Scholar]
- Promrat, K.; Kleiner, D.E.; Niemeier, H.M.; Jackvony, E.; Kearns, M.; Wands, J.R.; Fava, J.L.; Wing, R.R. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010, 51, 121–129. [Google Scholar]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar]
- Salamone, F.; Li Volti, G.; Titta, L.; Puzzo, L.; Barbagallo, I.; La Delia, F.; Zelber-Sagi, S.; Malaguarnera, M.; Pelicci, P.G.; Giorgio, M.; et al. Moro orange juice prevents fatty liver in mice. World J. Gastroenterol 2012, 18, 3862–3868. [Google Scholar]
- Mummadi, R.R.; Kasturi, K.S.; Chennareddygari, S.; Sood, G.K. Effect of bariatric surgery on nonalcoholic fatty liver disease: Systematic review and meta-analysis. Clin. Gastroenterol. Hepatol 2008, 6, 1396–1402. [Google Scholar]
- Dixon, J.B.; Bhathal, P.S.; Hughes, N.R.; O’Brien, P.E. Nonalcoholic fatty liver disease: Improvement in liver histological analysis with weight loss. Hepatology 2004, 39, 1647–1654. [Google Scholar]
- Kral, J.G.; Thung, S.N.; Biron, S.; Hould, F.S.; Lebel, S.; Marceau, S.; Simard, S.; Marceau, P. Effects of surgical treatment of the metabolic syndrome on liver fibrosis and cirrhosis. Surgery 2004, 135, 48–58. [Google Scholar]
- Klein, S.; Mittendorfer, B.; Eagon, J.C.; Patterson, B.; Grant, L.; Feirt, N.; Seki, E.; Brenner, D.; Korenblat, K.; McCrea, J. Gastric bypass surgery improves metabolic and hepatic abnormalities associated with nonalcoholic fatty liver disease. Gastroenterology 2006, 130, 1564–1572. [Google Scholar]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med 2010, 362, 1675–1685. [Google Scholar]
- Miller, E.R., 3rd; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med 2005, 142, 37–46. [Google Scholar]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA 2007, 297, 842–857. [Google Scholar]
- Berry, D.; Wathen, J.K.; Newell, M. Bayesian model averaging in meta-analysis: Vitamin E supplementation and mortality. Clin. Trials 2009, 6, 28–41. [Google Scholar]
- Gerss, J.; Kopcke, W. The questionable association of vitamin E supplementation and mortality—Inconsistent results of different meta-analytic approaches. Cell. Mol. Biol 2009, 55 Suppl, OL1111–OL1120. [Google Scholar]
- Ji, H.F.; Shen, L. On the mechanism of action of vitamin E for nonalcoholic steatohepatitis. Hepatology 2011, 53, 1067. [Google Scholar]
- Attar, B.M.; van Thiel, D.H. Current concepts and management approaches in nonalcoholic fatty liver disease. Sci. World J 2013, 2013, 481893. [Google Scholar]
- Molloy, J.W.; Calcagno, C.J.; Williams, C.D.; Jones, F.J.; Torres, D.M.; Harrison, S.A. Association of coffee and caffeine consumption with fatty liver disease, nonalcoholic steatohepatitis, and degree of hepatic fibrosis. Hepatology 2012, 55, 429–436. [Google Scholar]
- Malaguarnera, M.; Vacante, M.; Antic, T.; Giordano, M.; Chisari, G.; Acquaviva, R.; Mastrojeni, S.; Malaguarnera, G.; Mistretta, A.; Li Volti, G.; et al. Bifidobacterium longum with fructo-oligosaccharides in patients with non alcoholic steatohepatitis. Dig. Dis. Sci 2012, 57, 545–553. [Google Scholar]
- Malaguarnera, M.; Gargante, M.P.; Russo, C.; Antic, T.; Vacante, M.; Malaguarnera, M.; Avitabile, T.; Li Volti, G.; Galvano, F. l-carnitine supplementation to diet: A new tool in treatment of nonalcoholic steatohepatitis—A randomized and controlled clinical trial. Am. J. Gastroenterol 2010, 105, 1338–1345. [Google Scholar]
- Aigner, E.; Theurl, I.; Theurl, M.; Lederer, D.; Haufe, H.; Dietze, O.; Strasser, M.; Datz, C.; Weiss, G. Pathways underlying iron accumulation in human nonalcoholic fatty liver disease. Am. J. Clin. Nutr 2008, 87, 1374–1383. [Google Scholar]
- Equitani, F.; Fernandez-Real, J.M.; Menichella, G.; Koch, M.; Calvani, M.; Nobili, V.; Mingrone, G.; Manco, M. Bloodletting ameliorates insulin sensitivity and secretion in parallel to reducing liver iron in carriers of HFE gene mutations. Diabetes Care 2008, 31, 3–8. [Google Scholar]
- Fernandez-Real, J.M.; Penarroja, G.; Castro, A.; Garcia-Bragado, F.; Hernandez-Aguado, I.; Ricart, W. Blood letting in high-ferritin type 2 diabetes: Effects on insulin sensitivity and beta-cell function. Diabetes 2002, 51, 1000–1004. [Google Scholar]
- Adams, L.; House, M.J.; St Pierre, T.G.; Crawford, D.H.; Stuart, K.A.; Ching, H.; Kava, J.; Webb, M.; Olynyk, J.K. The impact of phlebotomy in non-alcoholic fatty liver disease: Interim results of a randomized controlled trial. Hepatology 2013, 58, 498A–499A. [Google Scholar]
- Zein, C.O.; Yerian, L.M.; Gogate, P.; Lopez, R.; Kirwan, J.P.; Feldstein, A.E.; McCullough, A.J. Pentoxifylline improves nonalcoholic steatohepatitis: A randomized placebo-controlled trial. Hepatology 2011, 54, 1610–1619. [Google Scholar]
- Zein, C.O.; Lopez, R.; Fu, X.; Kirwan, J.P.; Yerian, L.M.; McCullough, A.J.; Hazen, S.L.; Feldstein, A.E. Pentoxifylline decreases oxidized lipid products in nonalcoholic steatohepatitis: New evidence on the potential therapeutic mechanism. Hepatology 2012, 56, 1291–1299. [Google Scholar]
- Koca, S.S.; Bahcecioglu, I.H.; Poyrazoglu, O.K.; Ozercan, I.H.; Sahin, K.; Ustundag, B. The treatment with antibody of TNF-alpha reduces the inflammation, necrosis and fibrosis in the non-alcoholic steatohepatitis induced by methionine- and choline-deficient diet. Inflammation 2008, 31, 91–98. [Google Scholar]
- Aghazadeh, S.; Yazdanparast, R. Inhibition of JNK along with activation of ERK1/2 MAPK pathways improve steatohepatitis among the rats. Clin. Nutr 2010, 29, 381–385. [Google Scholar]
- Zhang, X.; Xu, A.; Chung, S.K.; Cresser, J.H.; Sweeney, G.; Wong, R.L.; Lin, A.; Lam, K.S. Selective inactivation of c-Jun NH2-terminal kinase in adipose tissue protects against diet-induced obesity and improves insulin sensitivity in both liver and skeletal muscle in mice. Diabetes 2011, 60, 486–495. [Google Scholar]
- Deng, Y.R.; Ma, H.D.; Tsuneyama, K.; Yang, W.; Wang, Y.H.; Lu, F.T.; Liu, C.H.; Liu, P.; He, X.S.; Diehl, A.M.; et al. STAT3-mediated attenuation of CCl4-induced mouse liver fibrosis by the protein kinase inhibitor sorafenib. J. Autoimmun 2013, 46, 25–34. [Google Scholar]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med 2009, 15, 921–929. [Google Scholar]
- Ilan, Y.; Maron, R.; Tukpah, A.M.; Maioli, T.U.; Murugaiyan, G.; Yang, K.; Wu, H.Y.; Weiner, H.L. Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9765–9770. [Google Scholar]
- Ilan, Y.; Zigmond, E.; Lalazar, G.; Dembinsky, A.; Ben Ya’acov, A.; Hemed, N.; Kasis, I.; Axelrod, E.; Zolotarov, L.; Klein, A.; et al. Oral administration of OKT3 monoclonal antibody to human subjects induces a dose-dependent immunologic effect in T cells and dendritic cells. J. Clin. Immunol 2010, 30, 167–177. [Google Scholar]
- da Cunha, A.P.; Weiner, H.L. Induction of immunological tolerance by oral anti-CD3. Clin. Dev. Immunol 2012, 2012, 425021. [Google Scholar]
- NasVax. NasVax Announces the Success of a Phase 2a Clinical Trial of a New Oral Immunotherapy for Fatty Liver Disease. 2011. Available online: http://www.businesswire.com/news/home/20110321005934/en/NasVax-Announces-Success-Phase-2a-Clinical-Trial accessed on 15 January 2014.
- Mizrahi, M.; Shabat, Y.; Ben Ya’acov, A.; Lalazar, G.; Adar, T.; Wong, V.; Muller, B.; Rawlin, G.; Ilan, Y. Alleviation of insulin resistance and liver damage by oral administration of Imm124-E is mediated by increased Tregs and associated with increased serum GLP-1 and adiponectin: Results of a phase I/II clinical trial in NASH. J. Inflamm. Res 2012, 5, 141–150. [Google Scholar]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig 2012, 122, 3476–3489. [Google Scholar]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther 2011, 34, 274–285. [Google Scholar]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann-Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.M.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar]
- Aithal, G.P.; Thomas, J.A.; Kaye, P.V.; Lawson, A.; Ryder, S.D.; Spendlove, I.; Austin, A.S.; Freeman, J.G.; Morgan, L.; Webber, J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology 2008, 135, 1176–1184. [Google Scholar]
- Promrat, K.; Lutchman, G.; Uwaifo, G.I.; Freedman, R.J.; Soza, A.; Heller, T.; Doo, E.; Ghany, M.; Premkumar, A.; Park, Y.; et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology 2004, 39, 188–196. [Google Scholar]
- Cusi, K.; Orsak, B.; Lomonaco, R.; Bril, F.; Ortiz-Lopez, C.; Hecht, J.; Webb, A.; Tio, F.; Darland, C.M.; Hardies, J. Extended treatment with pioglitazone improves liver histology in patients with prediabetes or type 2 diabetes mellitus and NASH. Hepatology 2013, 58, 248A. [Google Scholar]
- Nissen, S.E.; Wolski, K. Rosiglitazone revisited: An updated meta-analysis of risk for myocardial infarction and cardiovascular mortality. Arch. Intern. Med 2010, 170, 1191–1201. [Google Scholar]
- Argo, C.K.; Iezzoni, J.C.; Al-Osaimi, A.M.; Caldwell, S.H. Thiazolidinediones for the treatment in NASH: Sustained benefit after drug discontinuation? J. Clin. Gastroenterol 2009, 43, 565–568. [Google Scholar]
- Hyogo, H.; Tazuma, S.; Arihiro, K.; Iwamoto, K.; Nabeshima, Y.; Inoue, M.; Ishitobi, T.; Nonaka, M.; Chayama, K. Efficacy of atorvastatin for the treatment of nonalcoholic steatohepatitis with dyslipidemia. Metabolism 2008, 57, 1711–1718. [Google Scholar]
- Rallidis, L.S.; Drakoulis, C.K.; Parasi, A.S. Pravastatin in patients with nonalcoholic steatohepatitis: Results of a pilot study. Atherosclerosis 2004, 174, 193–196. [Google Scholar]
- Ekstedt, M.; Franzen, L.E.; Mathiesen, U.L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Statins in non-alcoholic fatty liver disease and chronically elevated liver enzymes: A histopathological follow-up study. J. Hepatol 2007, 47, 135–141. [Google Scholar]
- Athyros, V.G.; Mikhailidis, D.P.; Didangelos, T.P.; Giouleme, O.I.; Liberopoulos, E.N.; Karagiannis, A.; Kakafika, A.I.; Tziomalos, K.; Burroughs, A.K.; Elisaf, M.S. Effect of multifactorial treatment on non-alcoholic fatty liver disease in metabolic syndrome: A randomised study. Curr. Med. Res. Opin 2006, 22, 873–883. [Google Scholar]
- Chalasani, N.; Aljadhey, H.; Kesterson, J.; Murray, M.D.; Hall, S.D. Patients with elevated liver enzymes are not at higher risk for statin hepatotoxicity. Gastroenterology 2004, 126, 1287–1292. [Google Scholar]
- Georgescu, E.F.; Ionescu, R.; Niculescu, M.; Mogoanta, L.; Vancica, L. Angiotensin-receptor blockers as therapy for mild-to-moderate hypertension-associated non-alcoholic steatohepatitis. World J. Gastroenterol 2009, 15, 942–954. [Google Scholar]
- Yokohama, S.; Yoneda, M.; Haneda, M.; Okamoto, S.; Okada, M.; Aso, K.; Hasegawa, T.; Tokusashi, Y.; Miyokawa, N.; Nakamura, K. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology 2004, 40, 1222–1225. [Google Scholar]
- Yokohama, S.; Tokusashi, Y.; Nakamura, K.; Tamaki, Y.; Okamoto, S.; Okada, M.; Aso, K.; Hasegawa, T.; Aoshima, M.; Miyokawa, N.; et al. Inhibitory effect of angiotensin II receptor antagonist on hepatic stellate cell activation in non-alcoholic steatohepatitis. World J. Gastroenterol 2006, 12, 322–326. [Google Scholar]
- Day, C.P. Clinical spectrum and therapy of non-alcoholic steatohepatitis. Dig. Dis 2012, 30, 69–73. [Google Scholar]
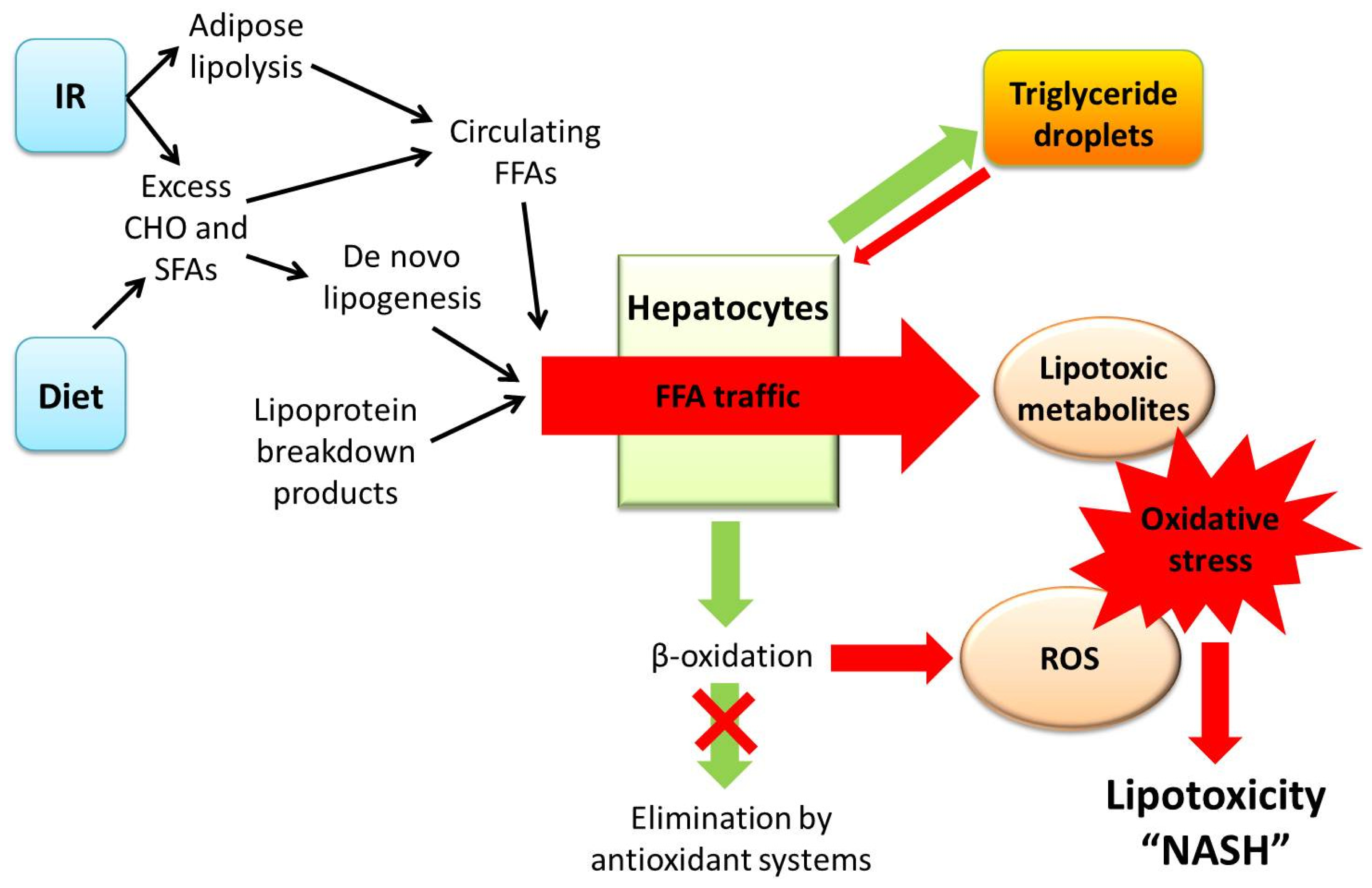
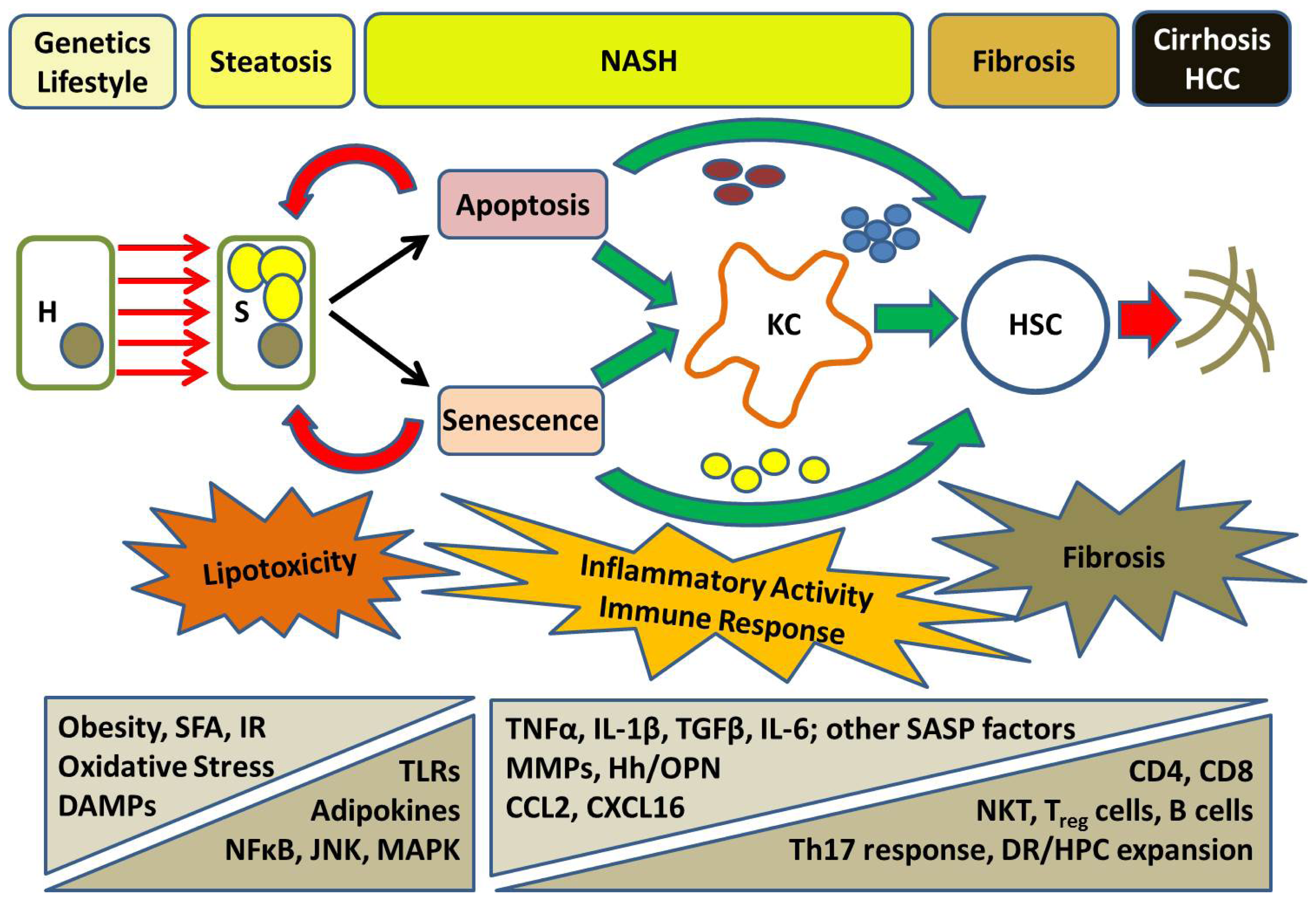

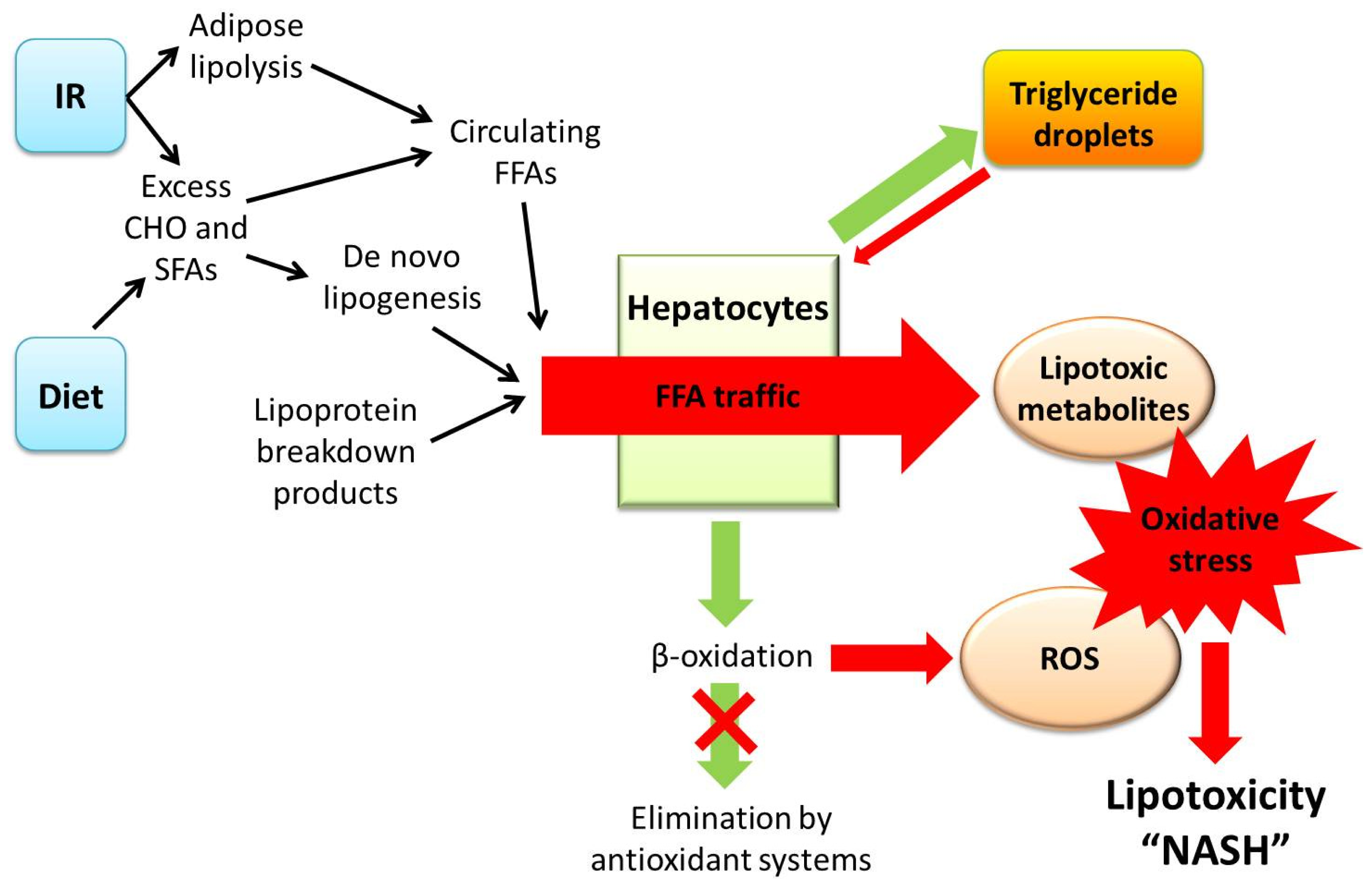{kind=link}
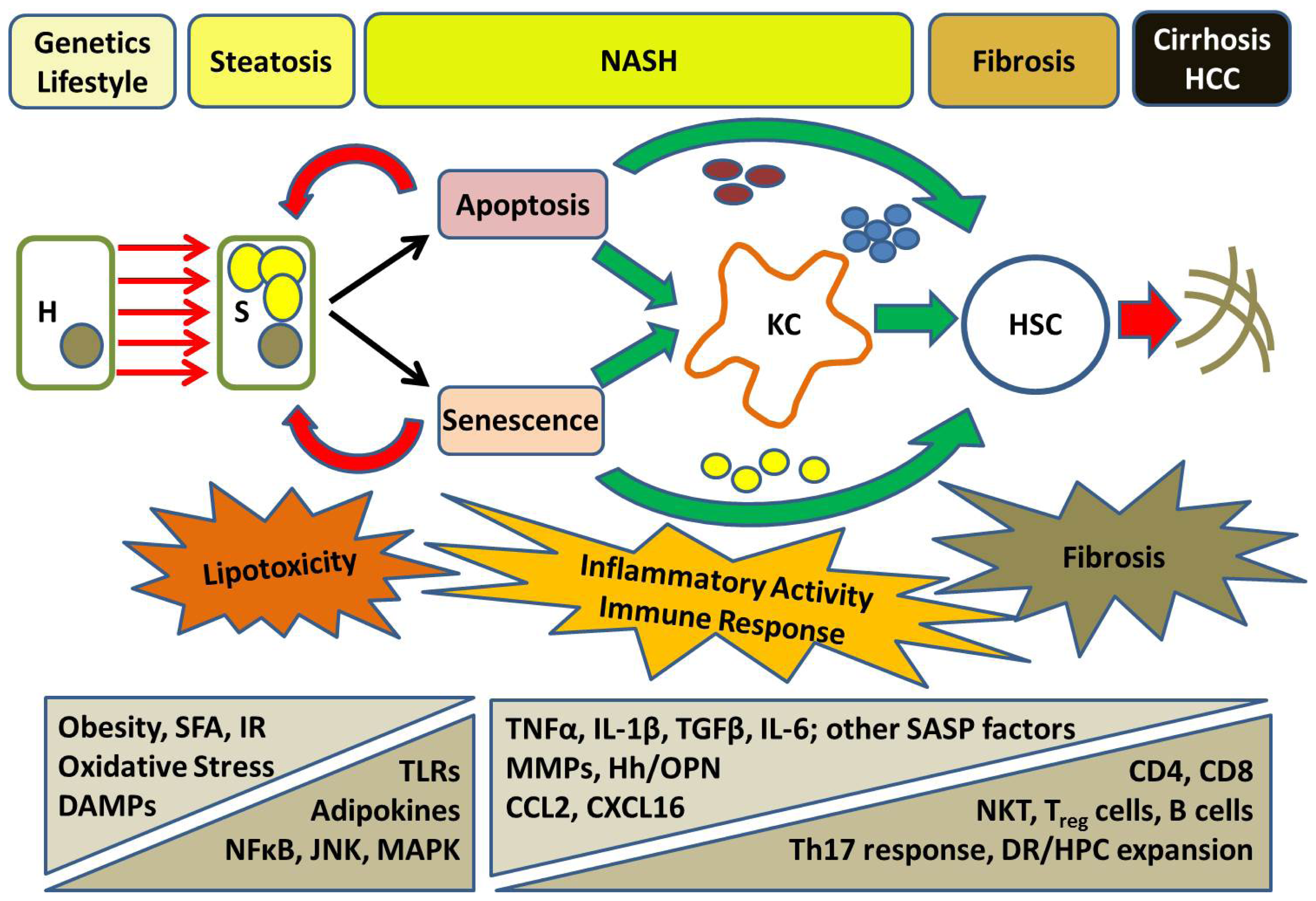{kind=link}
| Cell Parameter | Features of Senescent Cells |
|---|---|
| Cell cycling | Permanent, irreversible cell cycle arrest |
| Mechanisms of senescence | Critical telomere loss inducing a DDR-mediated growth arrest |
| Genomic damage (especially DNA double-strand breaks) | |
| Oncogene-induced senescence | |
| Stress-induced senescence (e.g., oxidative stress, serum-depletion in vitro) | |
| Cell morphology | Irregular shape and increased size |
| Markers of senescence | Increased beta-galactosidase activity and expression |
| Increased p21WAF1, p16INK4A, p15INK4B, p53 and RB expression | |
| Decreased expression of Ki-67, cyclin A and CDK2 | |
| Formation of SAHF and SDF (e.g., HP1β, γ-H2A.X) | |
| Formation of DNA-SCARS (reflecting DDR or telomere dysfunction) | |
| Cellular activity | Permanent growth arrest but metabolically active |
| Production of factors with autocrine/paracrine activity | |
| the senescence-associated secretory phenotype (SASP) | |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Peverill, W.; Powell, L.W.; Skoien, R. Evolving Concepts in the Pathogenesis of NASH: Beyond Steatosis and Inflammation. Int. J. Mol. Sci. 2014, 15, 8591-8638. https://doi.org/10.3390/ijms15058591
Peverill W, Powell LW, Skoien R. Evolving Concepts in the Pathogenesis of NASH: Beyond Steatosis and Inflammation. International Journal of Molecular Sciences. 2014; 15(5):8591-8638. https://doi.org/10.3390/ijms15058591
Chicago/Turabian StylePeverill, William, Lawrie W. Powell, and Richard Skoien. 2014. "Evolving Concepts in the Pathogenesis of NASH: Beyond Steatosis and Inflammation" International Journal of Molecular Sciences 15, no. 5: 8591-8638. https://doi.org/10.3390/ijms15058591
APA StylePeverill, W., Powell, L. W., & Skoien, R. (2014). Evolving Concepts in the Pathogenesis of NASH: Beyond Steatosis and Inflammation. International Journal of Molecular Sciences, 15(5), 8591-8638. https://doi.org/10.3390/ijms15058591