Alpha-Bulges in G Protein-Coupled Receptors

Abstract
:1. Introduction
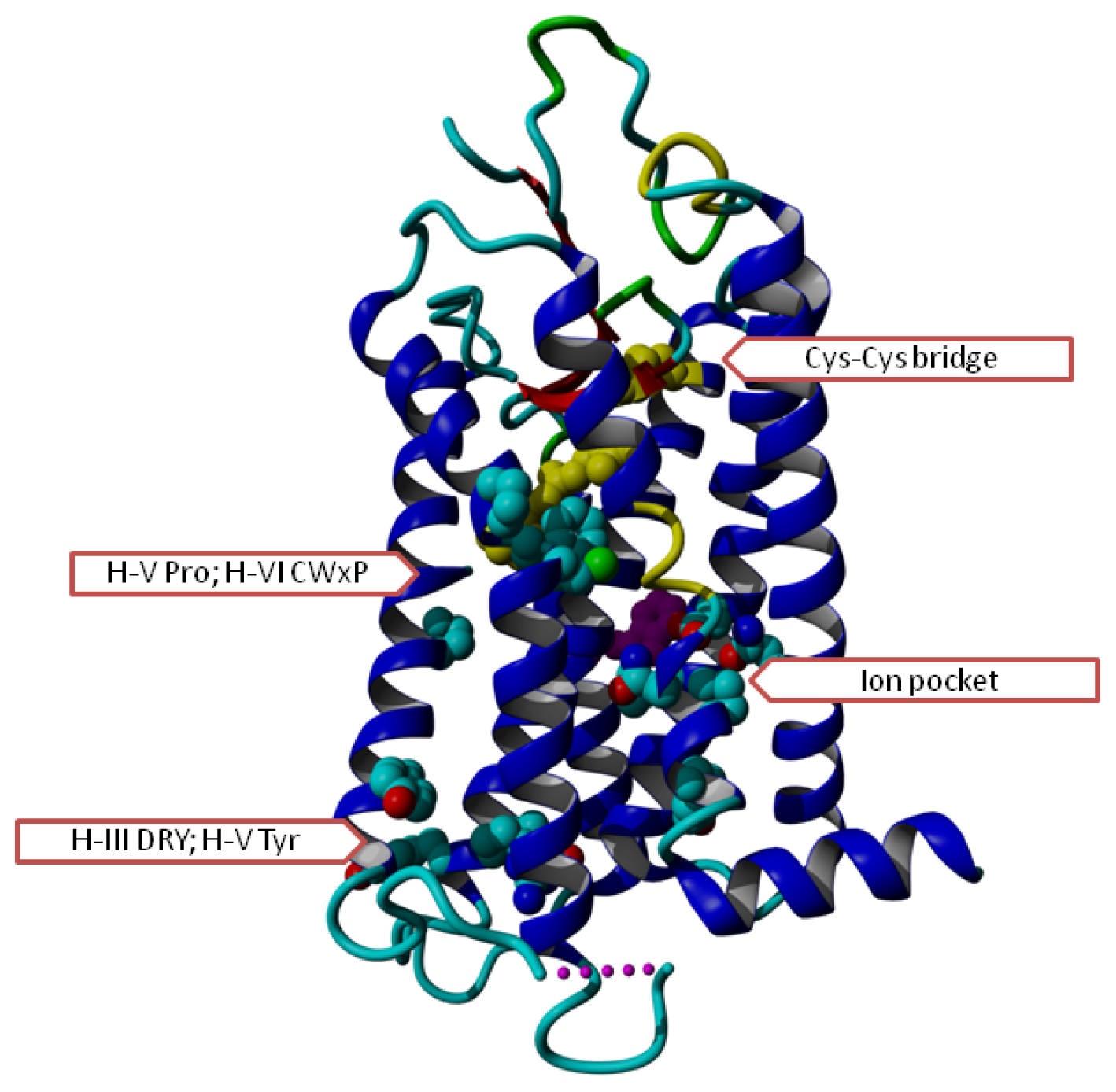
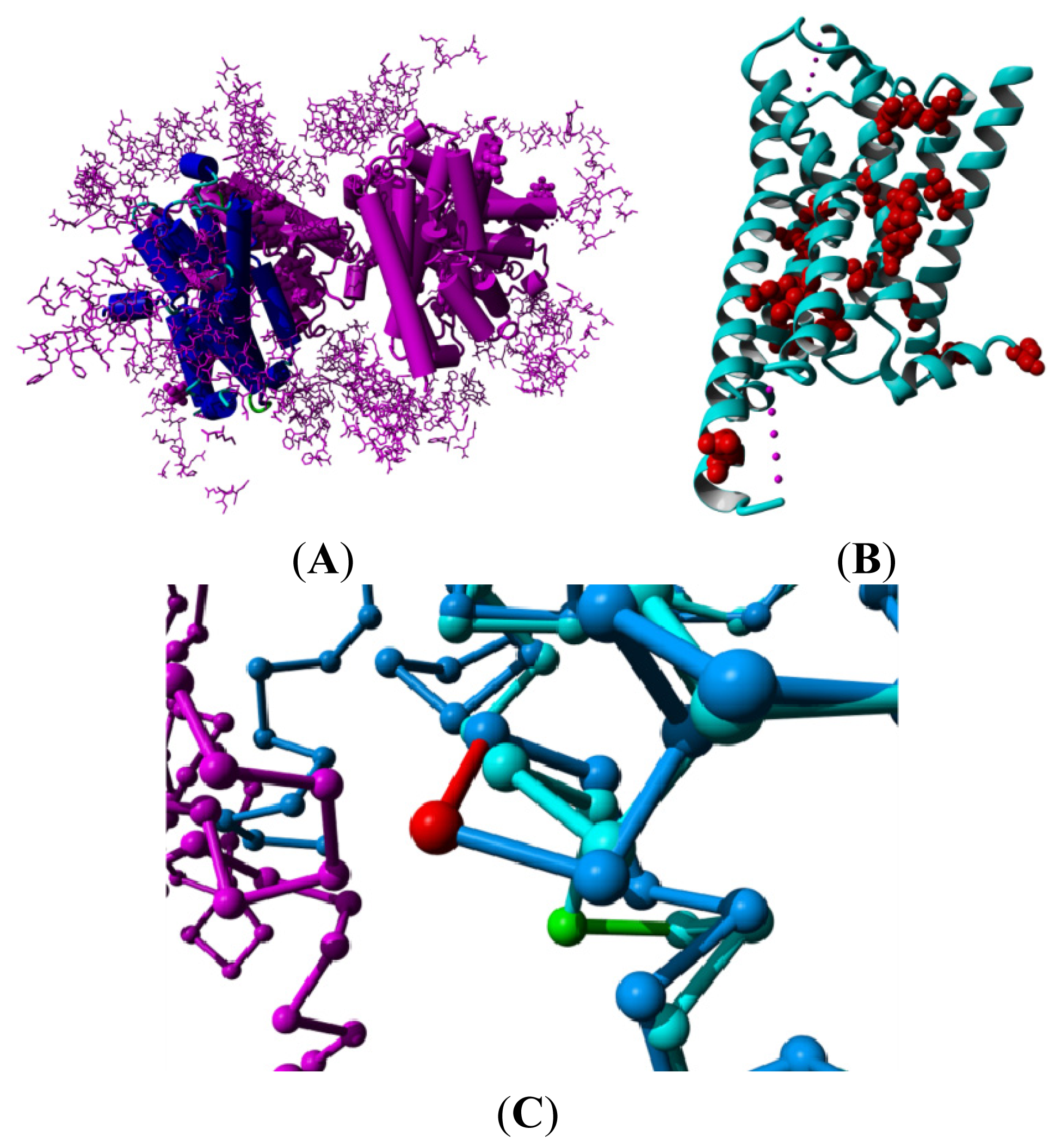
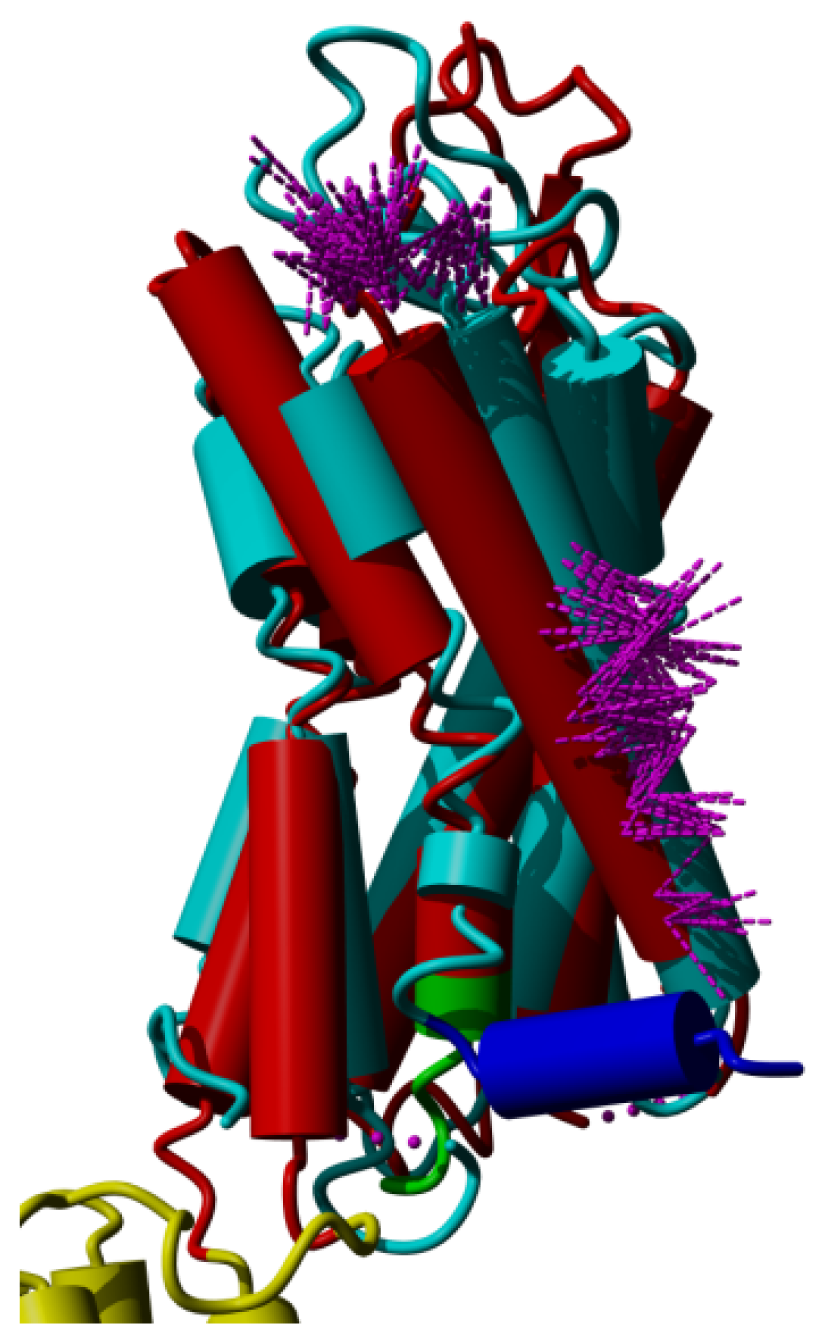
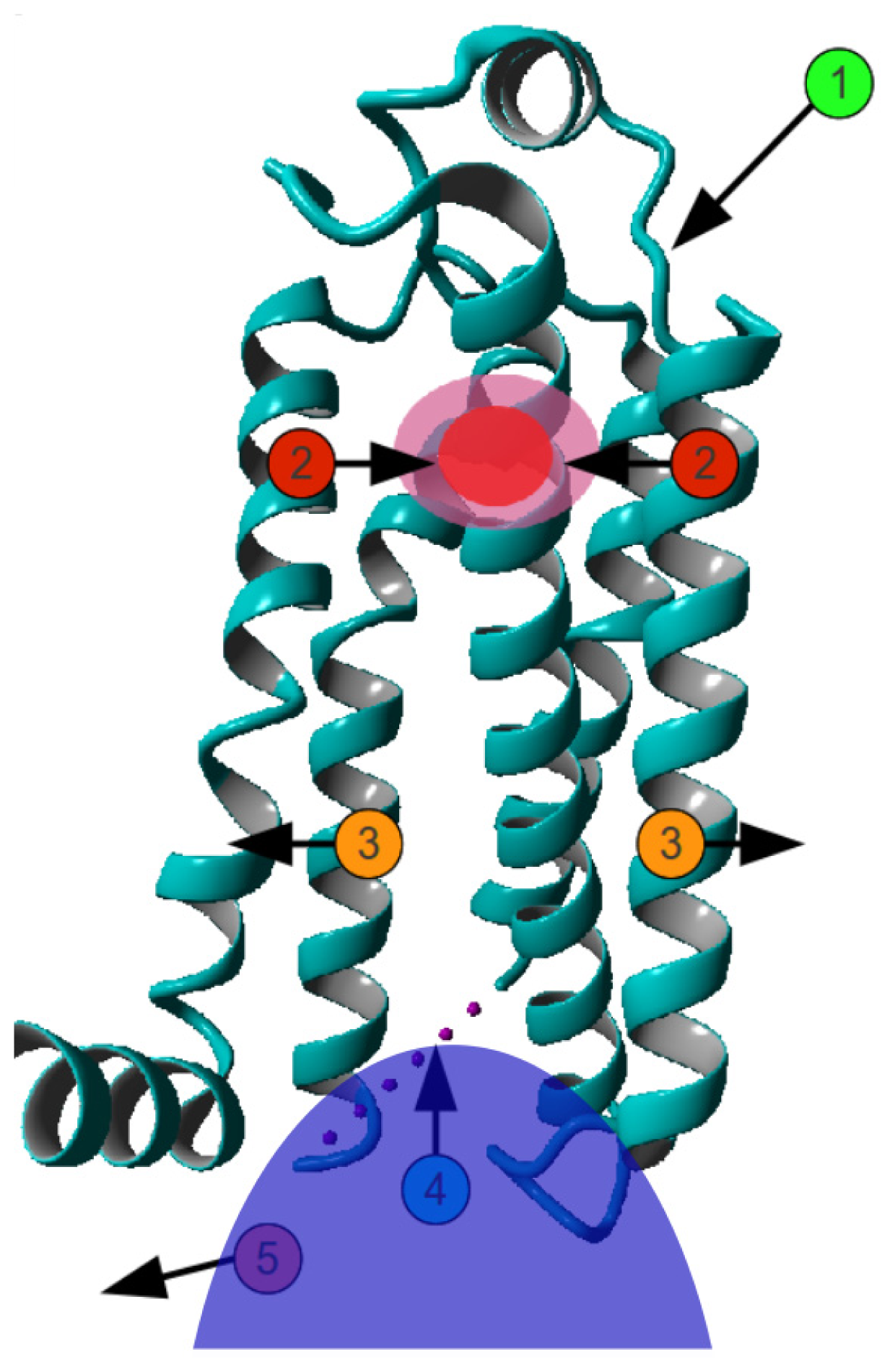
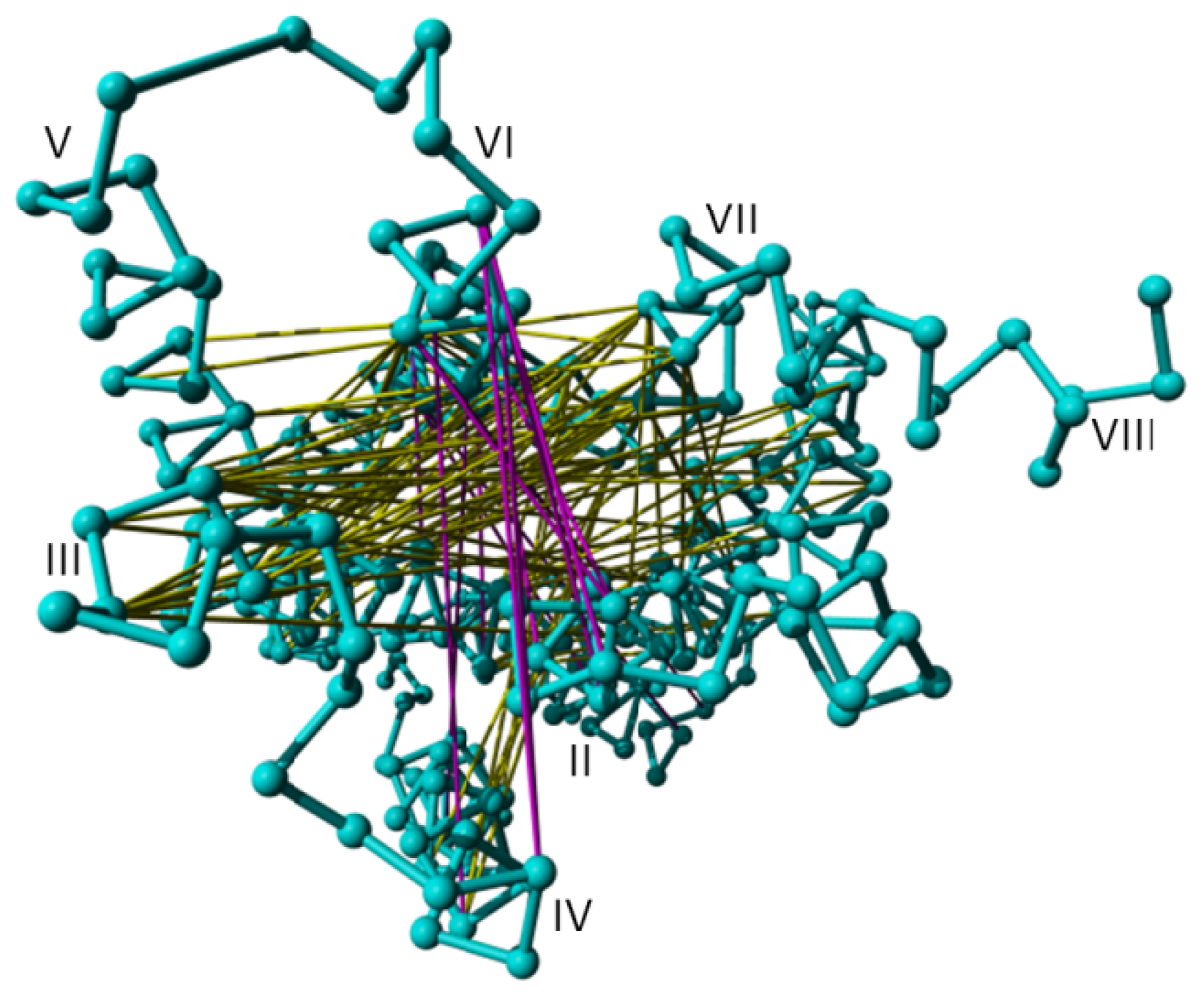
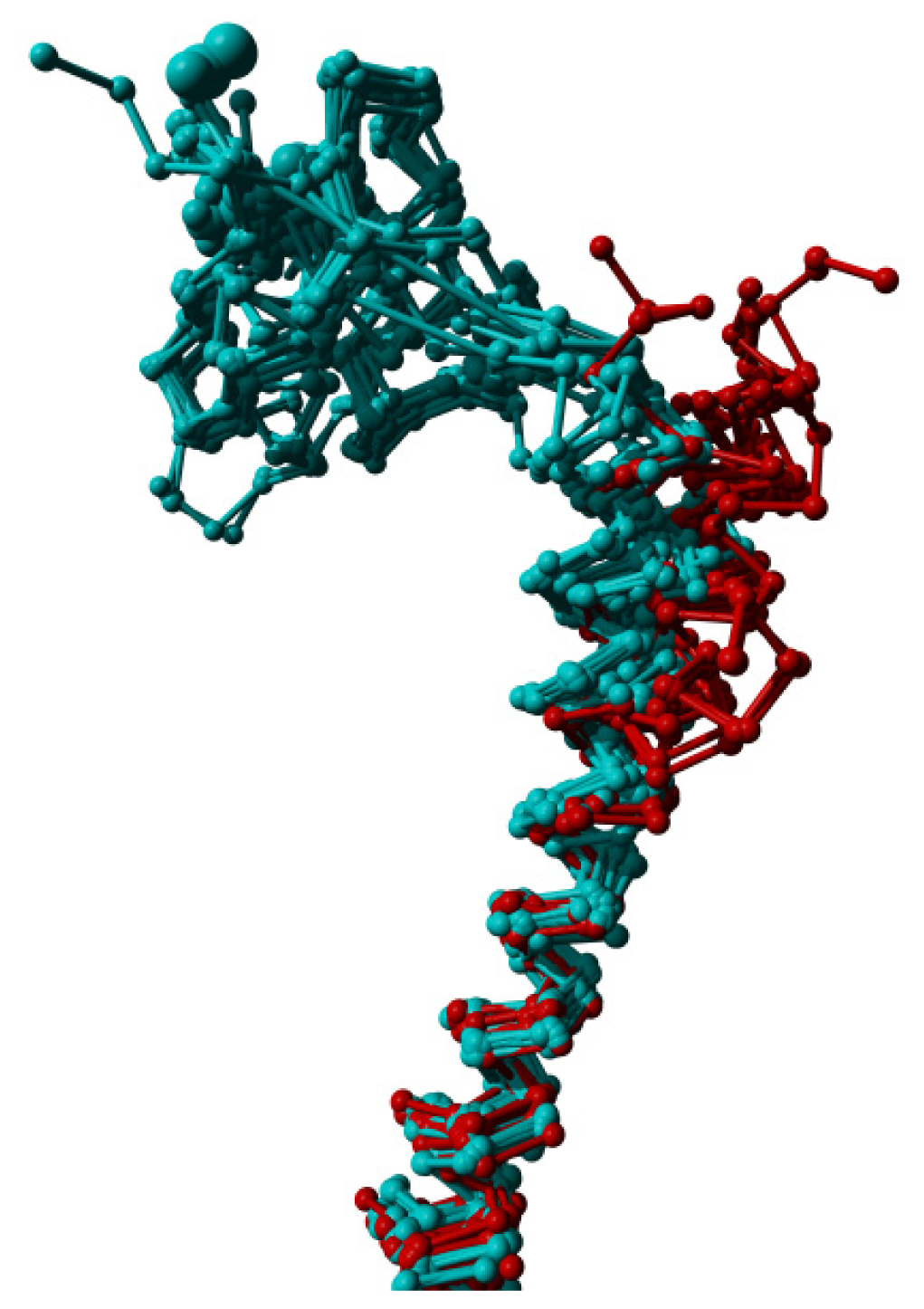
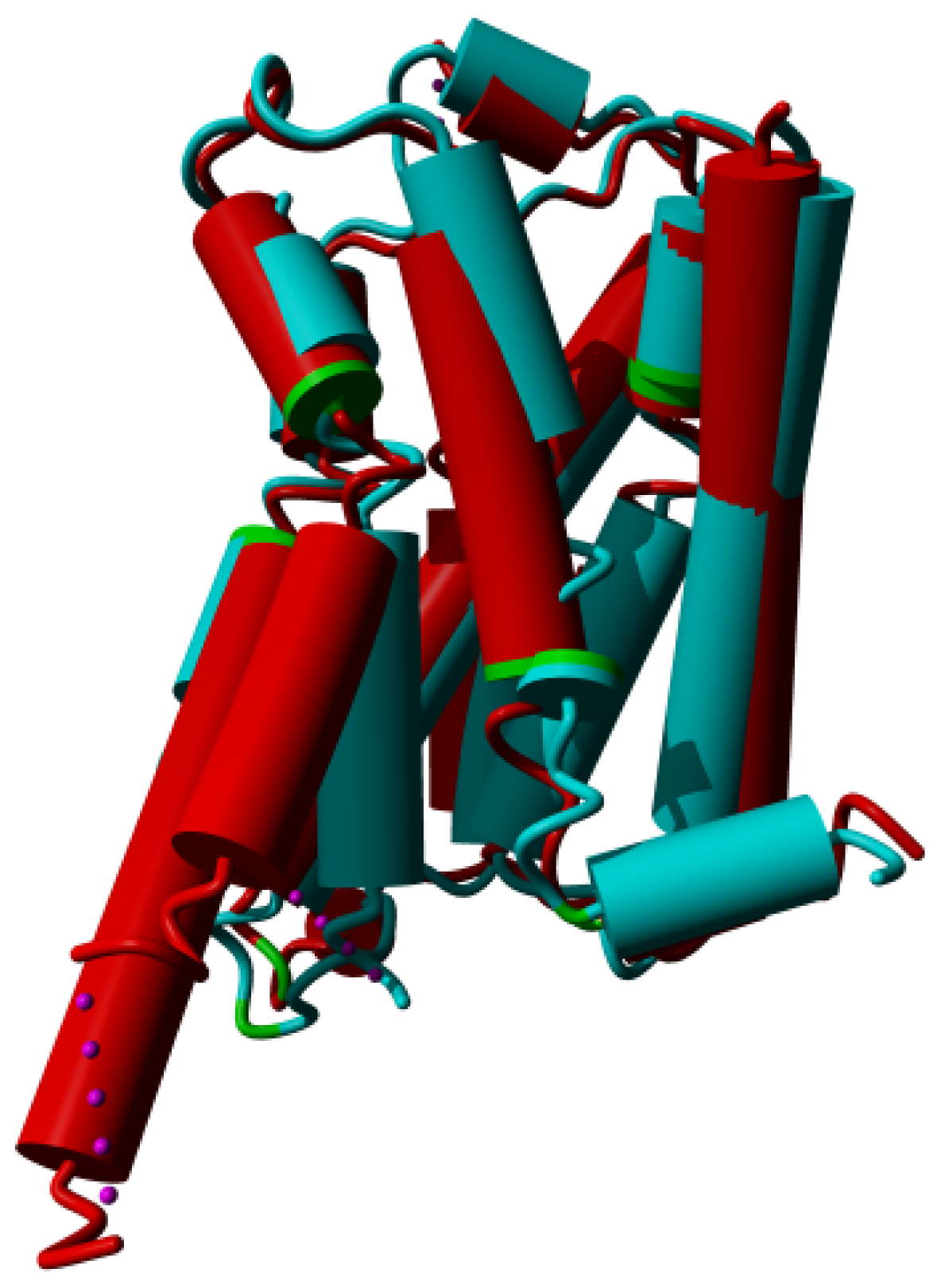
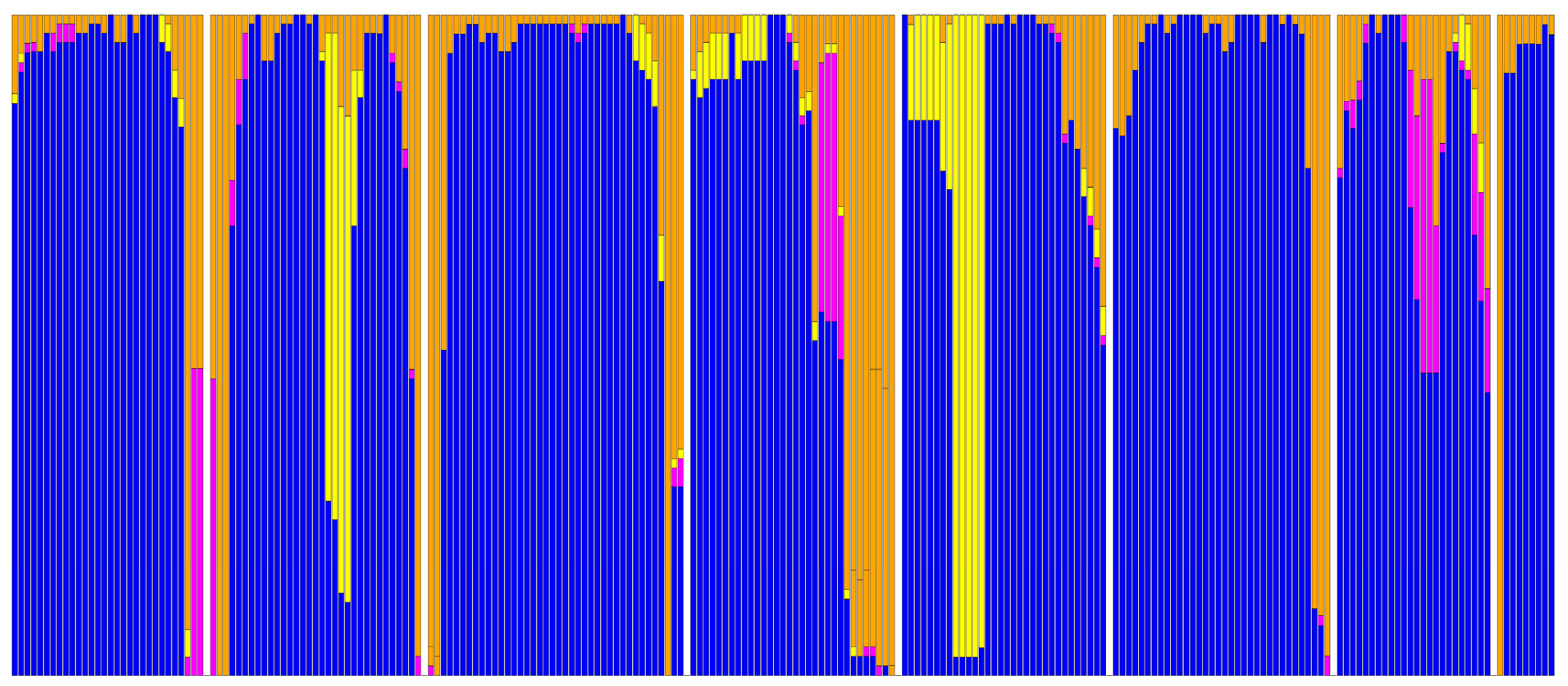
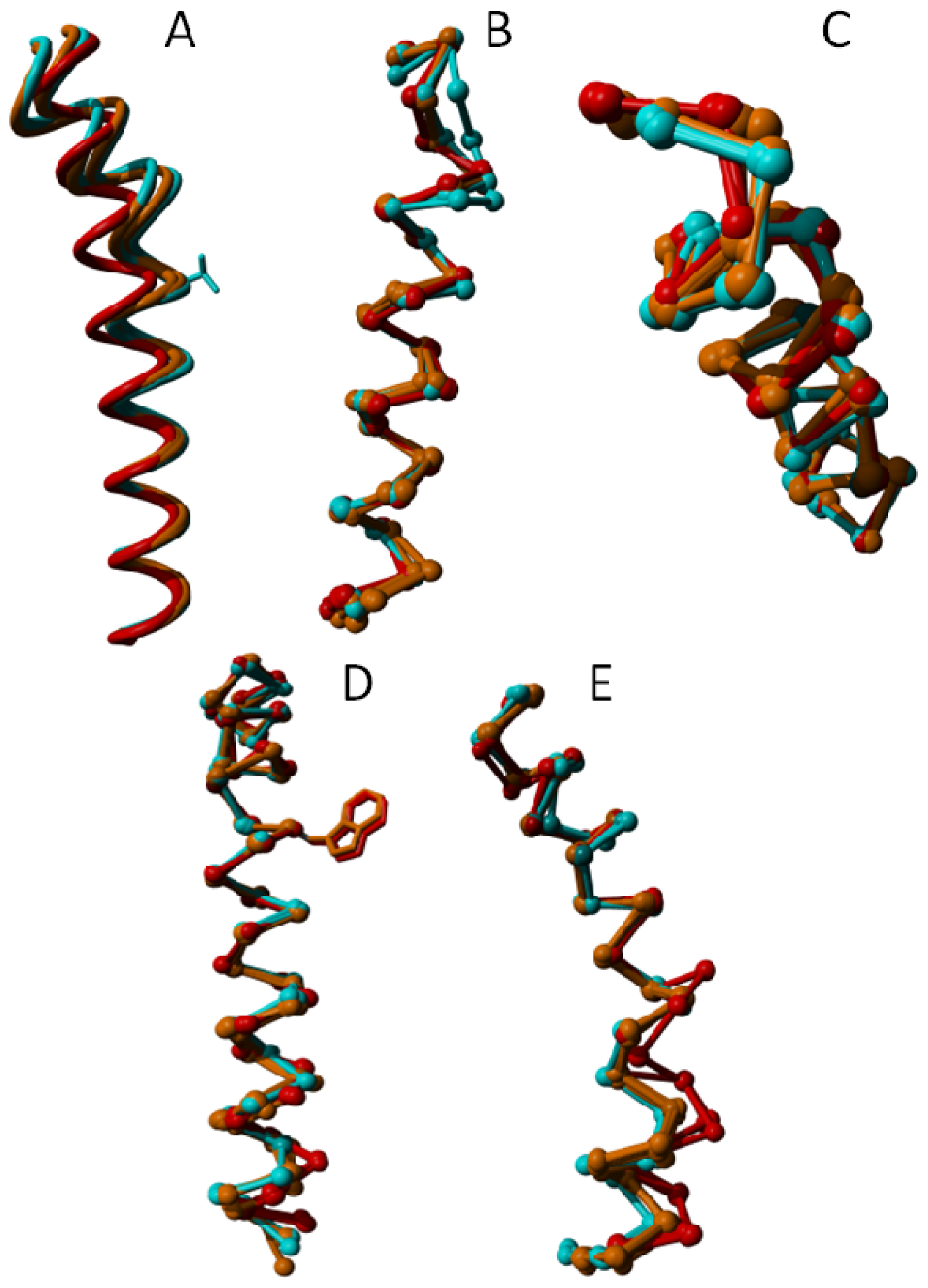
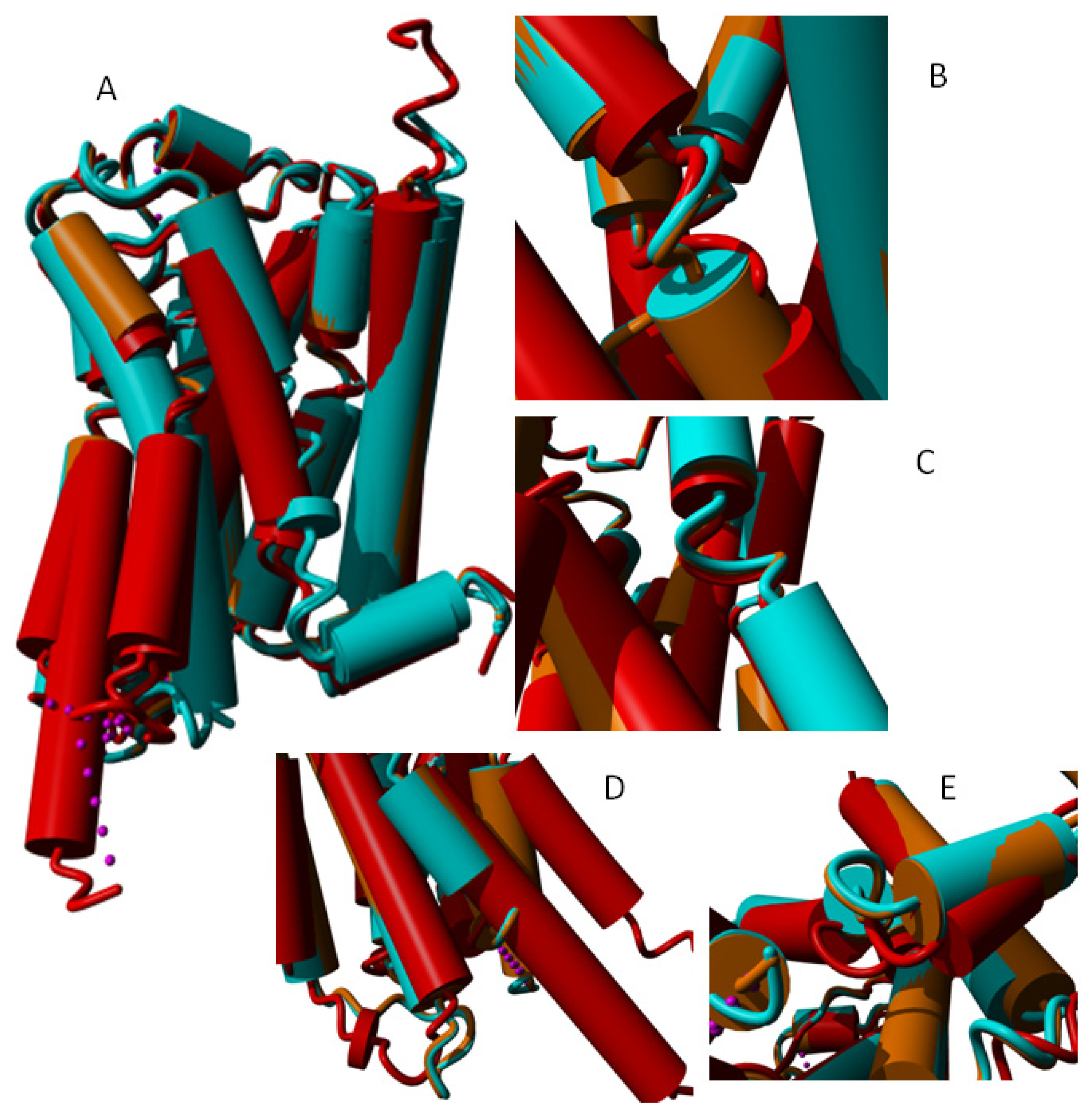
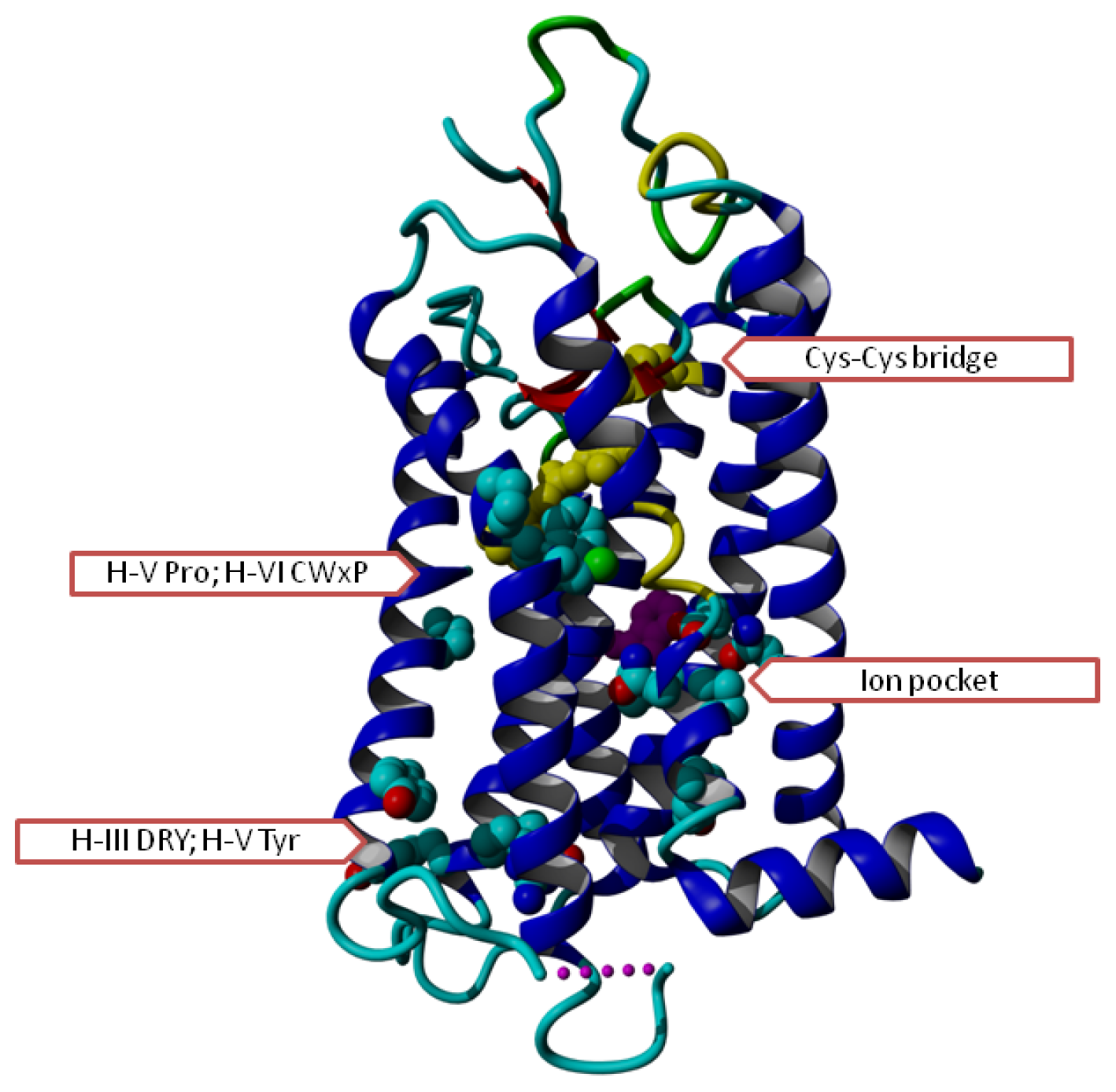
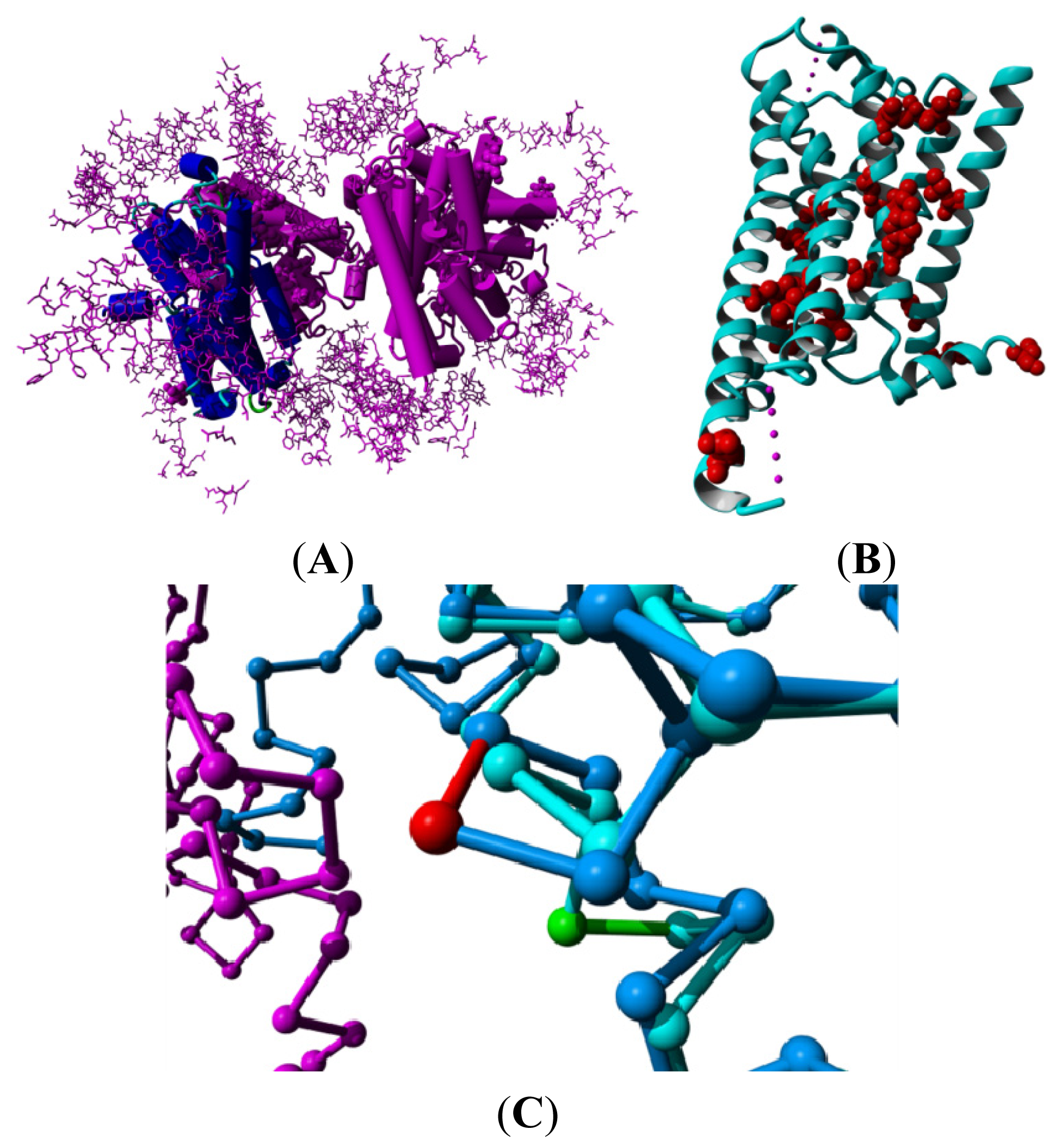
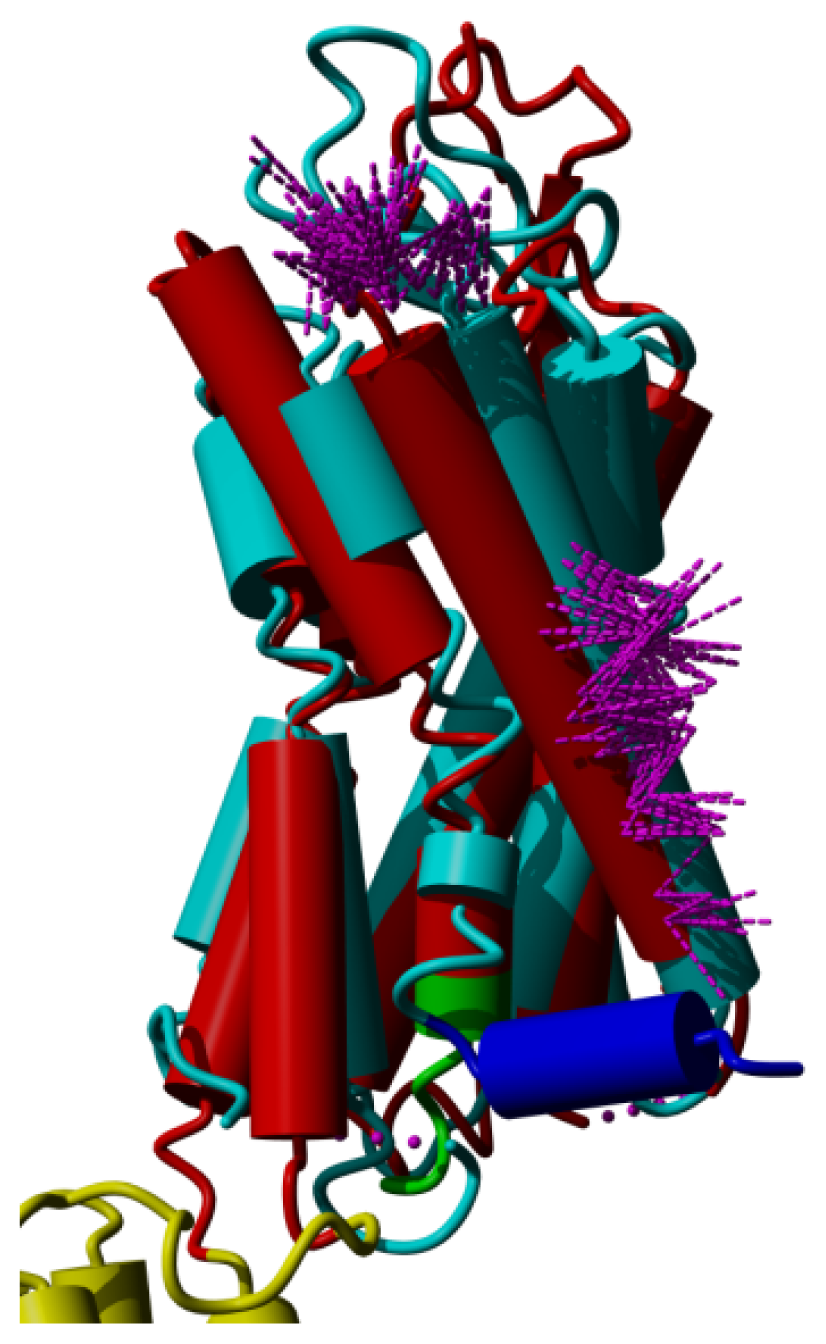
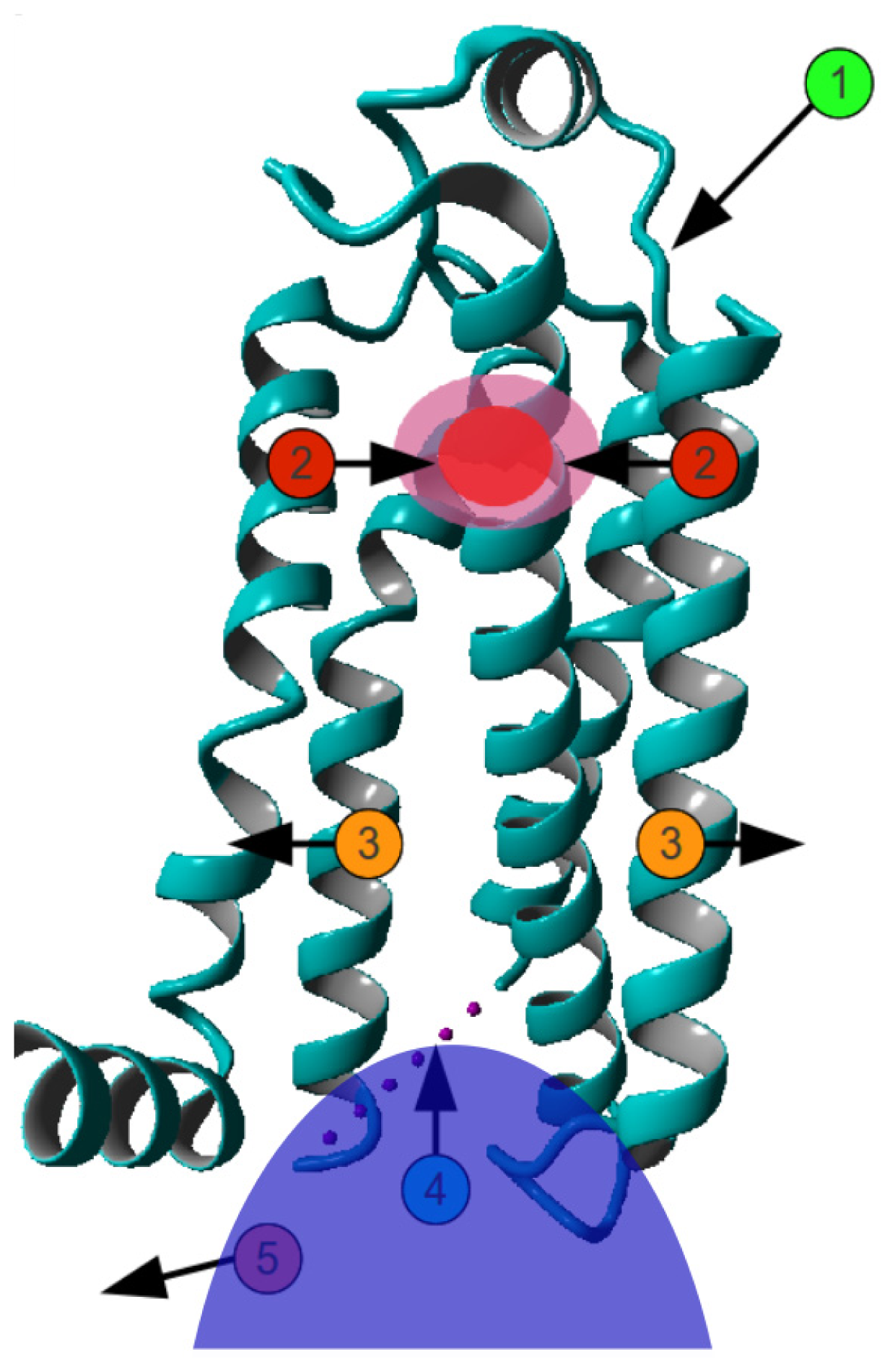
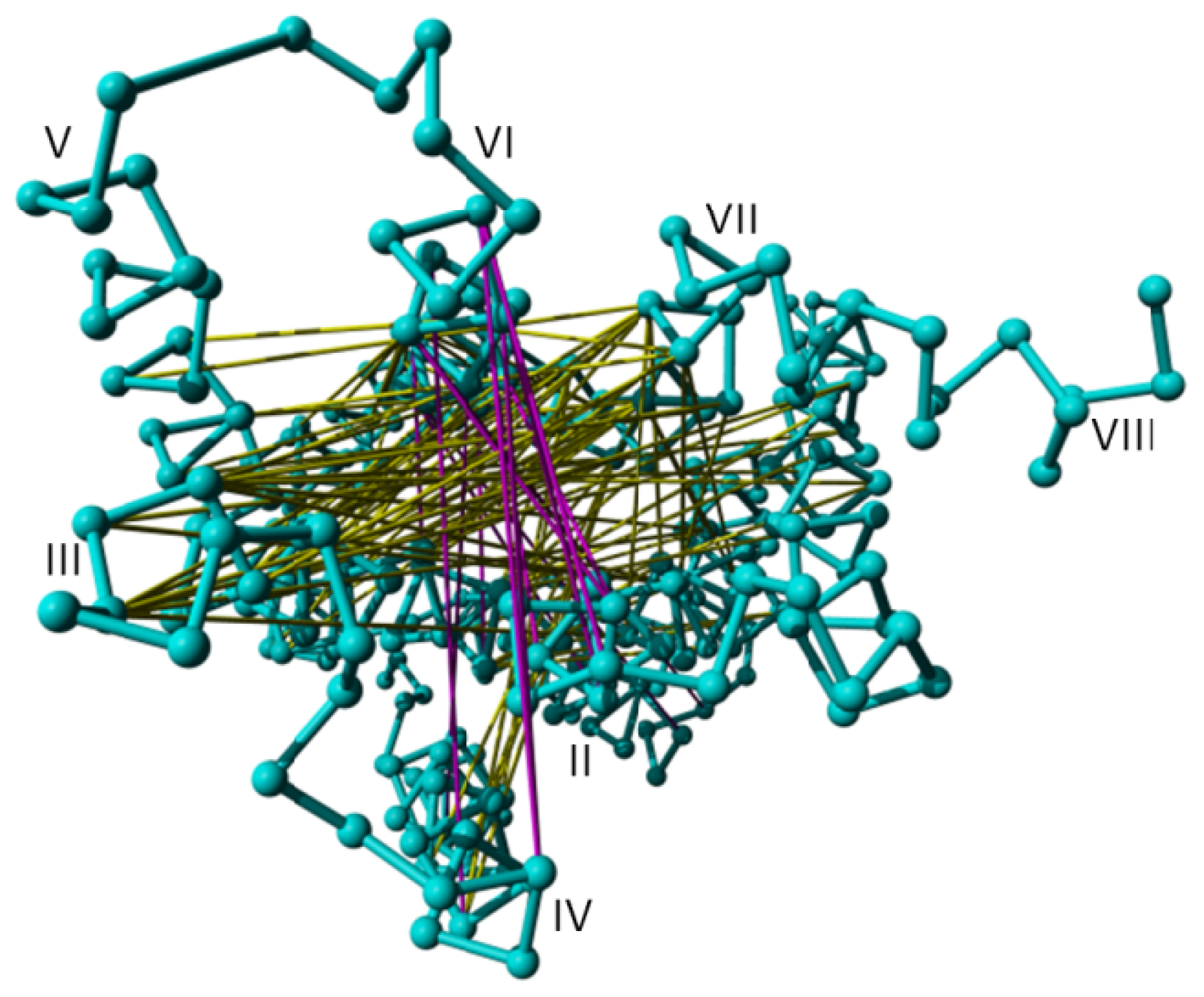
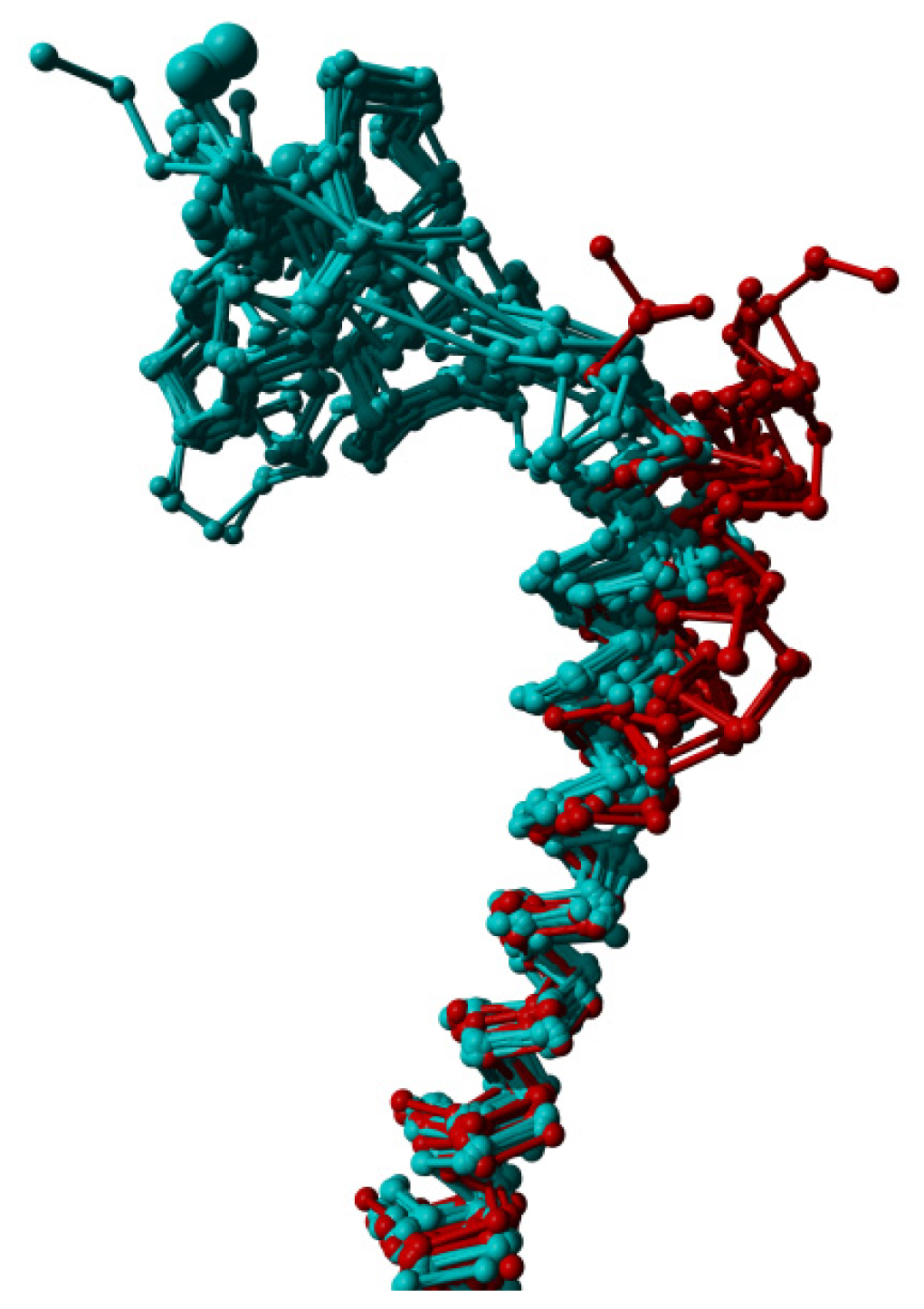
2. Results
3. Discussion
4. Methods
5. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsGV designed the project and supervised RvdK during the execution while RvdK was a master student in GV’s group at the CMBI. RvdK and GV jointly wrote the article.
References
- PubMed. Available online: http://www.ncbi.nlm.nih.gov/pubmed accessed on 19 April 2014.
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar]
- GPCR Network. Available online: http://gpcr.scripps.edu/ accessed on 19 April 2014.
- Kouranov, A.; Xie, L.; de la Cruz, J.; Chen, L.; Westbrook, J.; Bourne, P.E.; Berman, H.M. The RCSB PDB information portal for structural genomics. Nucleic Acids Res 2006, 34, D302–D305. [Google Scholar]
- Vroling, B.; Sanders, M.; Baakman, C.; Borrmann, A.; Verhoeven, S.; Klomp, J.; Oliveira, L.; de Vlieg, J.; Vriend, G. GPCRDB: Information system for G protein-coupled receptors. Nucleic Acids Res 2011, 39, D309–D319. [Google Scholar]
- Horn, F.; Bettler, E.; Oliveira, L.; Campagne, F.; Cohen, F.E.; Vriend, G. GPCRDB information system for G protein-coupled receptors. Nucleic Acids Res 2003, 31, 294–297. [Google Scholar]
- Van Durme, J.; Horn, F.; Costagliola, S.; Vriend, G.; Vassart, G. GRIS: Glycoprotein-hormone receptor information system. Mol. Endocrinol 2006, 20, 2247–2255. [Google Scholar]
- Oliveira, L.; Paiva, A.C.; Vriend, G. A low resolution model for the interaction of G proteins with G protein-coupled receptors. Protein Eng 1999, 12, 1087–1095. [Google Scholar]
- Oliveira, L.; Hulsen, T.; Lutje Hulsik, D.; Paiva, A.C.; Vriend, G. Heavier-than-air flying machines are impossible. FEBS Lett 2004, 564, 269–273. [Google Scholar]
- Oliveira, L.; Paiva, A.C.; Sander, C.; Vriend, G. A common step for signal transduction in G protein-coupled receptors. Trends Pharmacol. Sci 1994, 15, 170–172. [Google Scholar]
- Rasmussen, S.G.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar]
- Wacker, D.; Fenalti, G.; Brown, M.A.; Katritch, V.; Abagyan, R.; Cherezov, V.; Stevens, R.C. Conserved binding mode of human β2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J. Am. Chem. Soc 2010, 132, 11443–11445. [Google Scholar]
- Chun, E.; Thompson, A.A.; Liu, W.; Roth, C.B.; Griffith, M.T.; Katritch, V.; Kunken, J.; Xu, F.; Cherezov, V.; Hanson, M.A.; et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 2012, 20, 967–976. [Google Scholar]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar]
- Rasmussen, S.G.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar]
- Warne, T.; Serrano-Vega, M.J.; Baker, J.G.; Moukhametzianov, R.; Edwards, P.C.; Henderson, R.; Leslie, A.G.; Tate, C.G.; Schertler, G.F. Structure of a β1-adrenergic G protein-coupled receptor. Nature 2008, 454, 486–491. [Google Scholar]
- Doré, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 2011, 19, 1283–1293. [Google Scholar]
- GPCR Activation: What Moves Where? Van der Kant RWA. Available online: http://swift.cmbi.ru.nl/gv/GPCR/ accessed on 19 April 2014.
- White, J.F.; Noinaj, N.; Shibata, Y.; Love, J.; Kloss, B.; Xu, F.; Gvozdenovic-Jeremic, J.; Shah, P.; Shiloach, J.; Tate, C.G.; et al. Structure of the agonist-bound neurotensin receptor. Nature 2012, 490, 508–513. [Google Scholar]
- Mason, J.S.; Bortolato, A.; Congreve, M.; Marshall, F.H. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol. Sci 2012, 33, 249–260. [Google Scholar]
- Hino, T.; Arakawa, T.; Iwanari, H.; Yurugi-Kobayashi, T.; Ikeda-Suno, C.; Nakada-Nakura, Y.; Kusano-Arai, O.; Weyand, S.; Shimamura, T.; Nomura, N.; et al. G protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature 2012, 482, 237–240. [Google Scholar]
- Katritch, V.; Reynolds, K.A.; Cherezov, V.; Hanson, M.A.; Roth, C.B.; Yeager, M.; Abagyan, R. Analysis of full and partial agonists binding to β2-adrenergic receptor suggests a role of transmembrane helix V in agonist-specific conformational changes. J. Mol. Recognit 2009, 22, 307–318. [Google Scholar]
- Rasmussen, S.G.; Choi, H.J.; Rosenbaum, D.M.; Kobilka, T.S.; Thian, F.S.; Edwards, P.C.; Burghammer, M.; Ratnala, V.R.; Sanishvili, R.; Fischetti, R.F.; et al. Crystal structure of the human β2 adrenergic G protein-coupled receptor. Nature 2007, 450, 383–387. [Google Scholar]
- Rey, J.; Deville, J.; Chabbert, M. Structural determinants stabilizing helical distortions related to proline. J. Struct. Biol 2010, 171, 266–276. [Google Scholar]
- Van Arnam, E.B.; Lester, H.A.; Dougherty, D.A. Dissecting the functions of conserved prolines within transmembrane helices of the D2 dopamine receptor. ACS Chem. Biol 2011, 6, 1063–1068. [Google Scholar]
- Cartailler, J.P.; Luecke, H. Structural and functional characterization of pi bulges and other short intrahelical deformations. Structure 2004, 12, 133–144. [Google Scholar]
- Worth, C.L.; Kreuchwig, A.; Kleinau, G.; Krause, G. GPCR-SSFE: A comprehensive database of G protein-coupled receptor template predictions and homology models. BMC Bioinform 2011, 12, 185. [Google Scholar]
- Deupi, X. Quantification of structural distortions in the transmembrane helices of GPCRs. Methods Mol. Biol 2012, 914, 219–235. [Google Scholar]
- Devillé, J.; Rey, J.; Chabbert, M. An indel in transmembrane helix 2 helps to Trace the molecular evolution of class A G protein-coupled receptors. J. Mol. Evol 2009, 68, 475–489. [Google Scholar]
- Gonzalez, A.; Cordomí, A.; Caltabiano, G.; Pardo, L. Impact of helix irregularities on sequence alignment and homology modeling of G protein-coupled receptors. Chembiochem 2012, 13, 1393–1399. [Google Scholar]
- Isberg, V.; Vroling, B.; van der Kant, R.; Li, K.; Vriend, G.; Gloriam, D. GPCRDB: An information system for G protein-coupled receptors. Nucleic Acids Res 2014, 42, D422–D425. [Google Scholar]
- Strobl, C.; Boulesteix, A.L.; Kneib, T.; Augustin, T.; Zeileis, A. Conditional variable importance for random forests. BMC Bioinform 2008, 9, 307:1–307:11. [Google Scholar]
- Toloşi, L.; Lengauer, T. Classification with correlated features: Unreliability of feature ranking and solutions. Bioinformatics 2011, 27, 1986–1994. [Google Scholar]
- Hofmann, K.P.; Scheerer, P.; Hildebrand, P.W.; Choe, H.W.; Park, J.H.; Heck, M.; Ernst, O.P. A G protein-coupled receptor at work: The rhodopsin model. Trends Biochem. Sci 2009, 34, 540–552. [Google Scholar]
- Vogel, R.; Sakmar, T.P.; Sheves, M.; Siebert, F. Coupling of protonation switches during rhodopsin activation. Photochem. Photobiol 2007, 83, 286–292. [Google Scholar]
- Fritze, O.; Filipek, S.; Kuksa, V.; Palczewski, K.; Hofmann, K.P.; Ernst, O.P. Role of the conserved NPxxY(x)5,6F motif in the rhodopsin ground state and during activation. Proc. Natl. Acad. Sci. USA 2003, 100, 2290–2295. [Google Scholar]
- Yohannan, S.; Faham, S.; Yang, D.; Whitelegge, J.P.; Bowie, J.U. The evolution of transmembrane helix kinks and the structural diversity of G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 959–963. [Google Scholar]
- Ceruso, M.A.; Weinstein, H. Structural mimicry of proline kinks: Tertiary packing interactions support local structural distortions. J. Mol. Biol 2002, 318, 1237–1249. [Google Scholar]
- Riek, R.P.; Rigoutsos, I.; Novotny, J.; Graham, R.M. Non-α-helical elements modulate polytopic membrane protein architecture. J. Mol. Biol 2001, 306, 349–362. [Google Scholar]
- Hong, S.; Ryu, K.S.; Oh, M.S.; Ji, I.; Ji, T.H. Roles of transmembrane prolines and proline-induced kinks of the lutropin/choriogonadotropin receptor. J. Biol. Chem 1997, 272, 4166–4171. [Google Scholar]
- Geetha, V. Distortions in protein helices. Int. J. Biol. Macromol 1996, 19, 81–89. [Google Scholar]
- Von Heijne, G. Proline kinks in transmembrane α-helices. J. Mol. Biol 1991, 218, 499–503. [Google Scholar]
- Conner, A.C.; Hay, D.L.; Simms, J.; Howitt, S.G.; Schindler, M.; Smith, D.M.; Wheatley, M.; Poyner, D.R. A key role for transmembrane prolines in calcitonin receptor-like receptor agonist binding and signalling: Implications for family B G protein-coupled receptors. Mol. Pharmacol 2005, 67, 20–31. [Google Scholar]
- Krieger, E.; Vriend, G. Models@Home: Distributed computing in bioinformatics using a screensaver based approach. Bioinformatics 2002, 18, 315–318. [Google Scholar]
- Hanson, M.A.; Roth, C.B.; Jo, E.; Griffith, M.T.; Scott, F.L.; Reinhart, G.; Desale, H.; Clemons, B.; Cahalan, S.M.; Schuerer, S.C.; et al. Crystal structure of a lipid G protein-coupled receptor. Science 2012, 335, 851–855. [Google Scholar]
- Jaakola, V.P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.; Lane, J.R.; Ijzerman, A.P.; Stevens, R.C. The 2.6 Ångstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [Google Scholar]
- Murakami, M.; Kouyama, T. Crystal structure of squid rhodopsin. Nature 2008, 453, 363–367. [Google Scholar]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar]
- Haga, K.; Kruse, A.C.; Asada, H.; Yurugi-Kobayashi, T.; Shiroishi, M.; Zhang, C.; Weis, W.I.; Okada, T.; Kobilka, B.K.; Haga, T.; et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 2012, 482, 547–551. [Google Scholar]
- Park, J.H.; Scheerer, P.; Hofmann, K.P.; Choe, H.W.; Ernst, O.P. Crystal structure of the ligand-free G protein-coupled receptor opsin. Nature 2008, 454, 183–187. [Google Scholar]
- Granier, S.; Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Weis, W.I.; Kobilka, B.K. Structure of the δ-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. [Google Scholar]
- Chien, E.Y.; Liu, W.; Zhao, Q.; Katritch, V.; Han, G.W.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [Google Scholar]
- Schwartz, T.W.; Frimurer, T.M.; Holst, B.; Rosenkilde, M.M.; Elling, C.E. Molecular mechanism of 7TM receptor activation—A global toggle switch model. Annu. Rev. Pharmacol. Toxicol 2006, 46, 481–519. [Google Scholar]
- Congreve, M.; Andrews, S.P.; Doré, A.S.; Hollenstein, K.; Hurrell, E.; Langmead, C.J.; Mason, J.S.; Ng, I.W.; Tehan, B.; Zhukov, A.; et al. Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J. Med. Chem 2012, 55, 1898–1903. [Google Scholar]
- Xu, F.; Wu, H.; Katritch, V.; Han, G.W.; Jacobson, K.A.; Gao, Z.G.; Cherezov, V.; Stevens, R.C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [Google Scholar]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.P.; Chien, E.Y.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A specific cholesterol binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure 2008, 16, 897–905. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res 2000, 28, 235–242. [Google Scholar]
- Oliveira, L.; Paiva, A.C.M.; Vriend, G. A common motif in G protein-coupled seven transmembrane helix receptors. J. Comput-Aided Mol. Des 1993, 7, 649–658. [Google Scholar]
- Vroling, B.; Thorne, D.; McDermott, P.; Attwood, T.K.; Vriend, G.; Pettifer, S. Integrating GPCR-specific information with full text articles. BMC Bioinform 2011, 12, 362:1–362:10. [Google Scholar]
- GPCRDB. Available online: http://www.gpcr.org/7tm/ accessed on 19 April 2014.
- Konagurthu, A.S.; Whisstock, J.C.; Stuckey, P.J.; Lesk, A.M. MUSTANG: A multiple structural alignment algorithm. Proteins Struct. Funct. Bioinform 2006, 64, 559–574. [Google Scholar]
- Vriend, G. WHAT IF A molecular modelling and drug design program. J. Mol. Graph 1990, 8, 52–56. [Google Scholar]
- Vriend, G.; Sander, C. Detection of common three-dimensional substructures in proteins. Proteins 1991, 11, 52–58. [Google Scholar]
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008. Available online: http://www.R-project.org accessed on 19 April 2014.
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar]
- Joosten, R.P.; te Beek, T.A.; Krieger, E.; Hekkelman, M.L.; Hooft, R.W.; Schneider, R.; Sander, C.; Vriend, G. A series of PDB related databases for everyday needs. Nucleic Acids Res 2011, 39, D411–D419. [Google Scholar]
- CMBI Protein Structure Bioinformatics Facilities. Available online: http://swift.cmbi.ru.nl/gv/facilities/ accessed on 19 April 2014.
- Index of Software DSSP. Available online: ftp://ftp.cmbi.ru.nl/pub/software/dssp/ accessed on 19 April 2014.

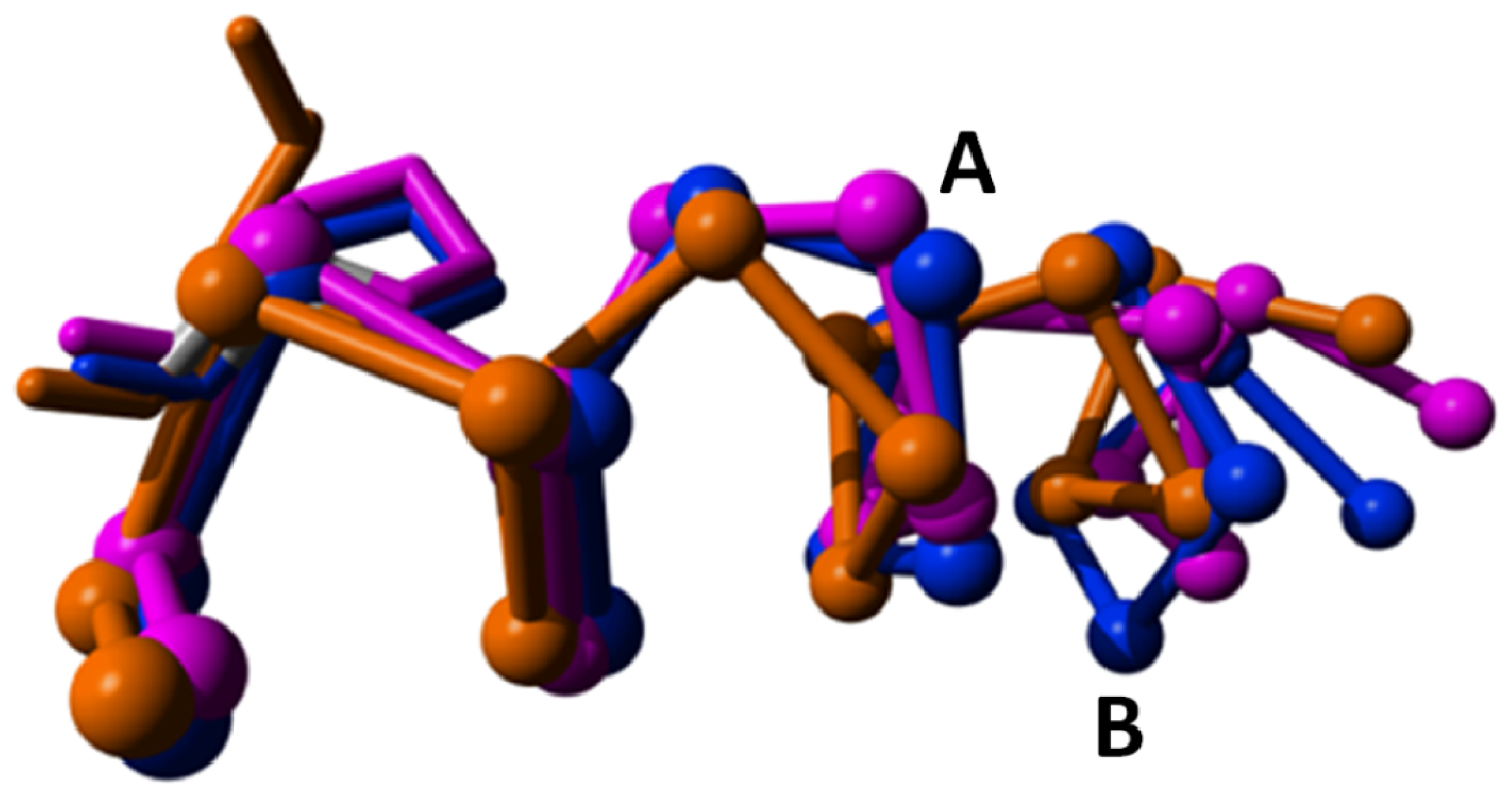
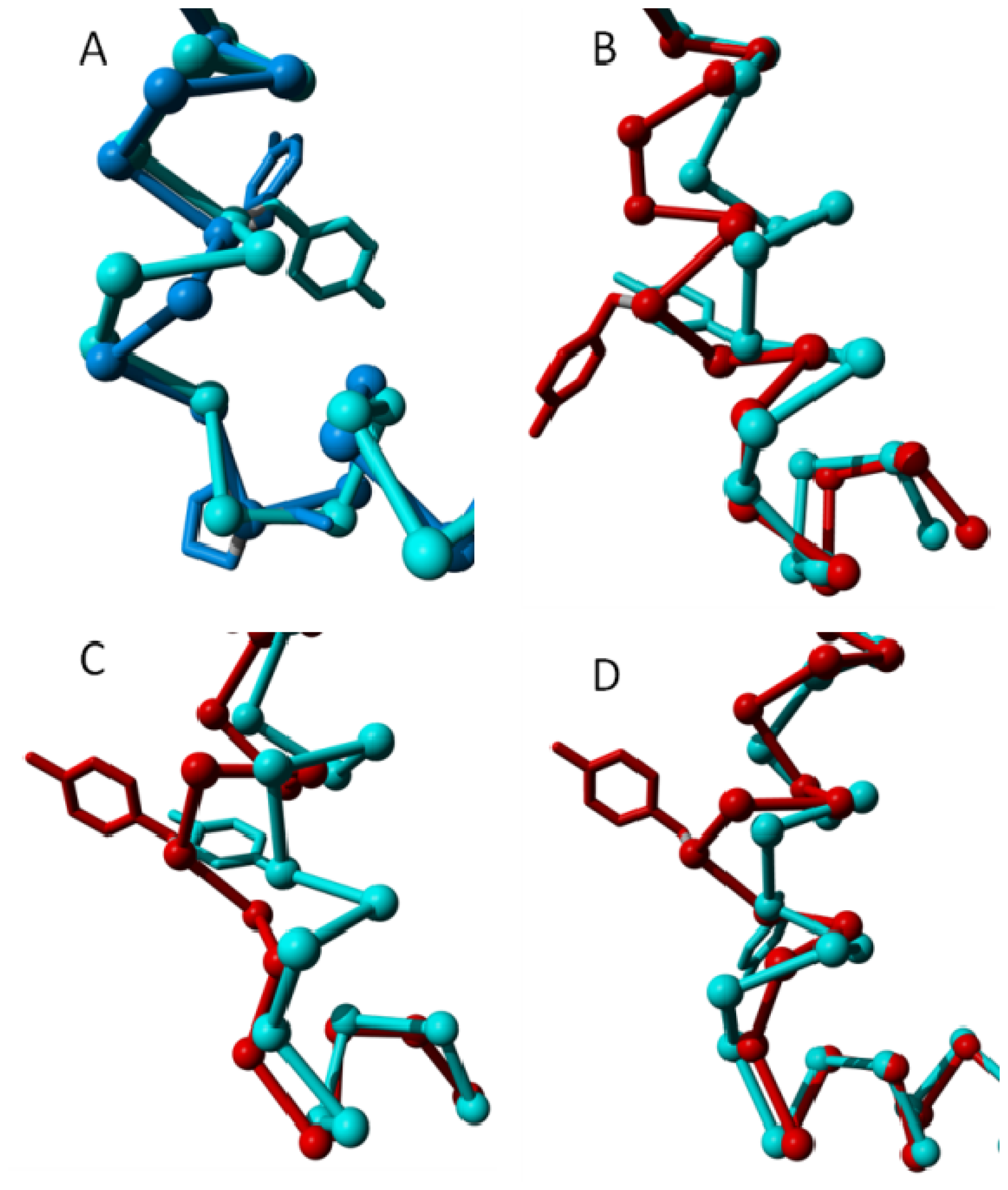
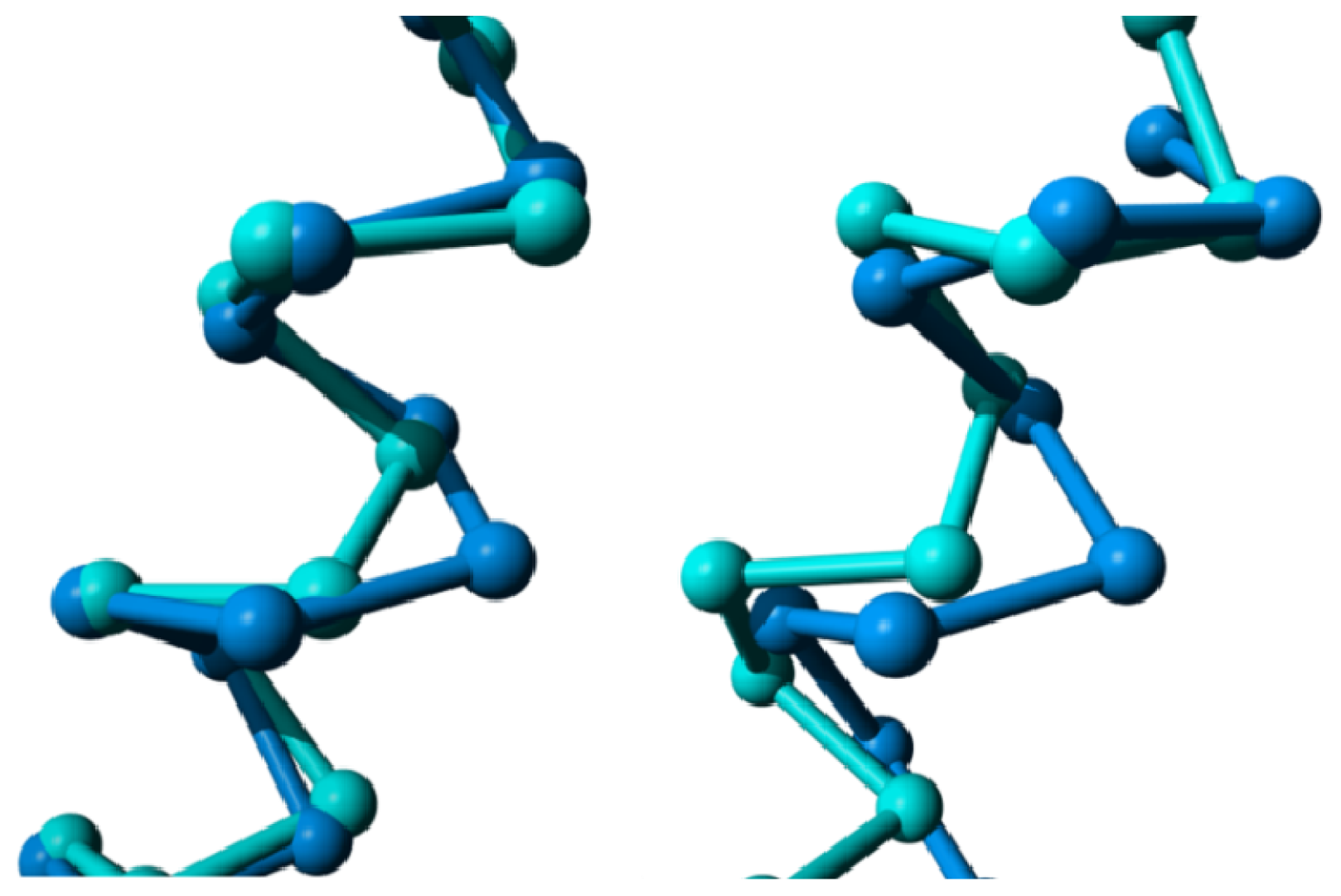
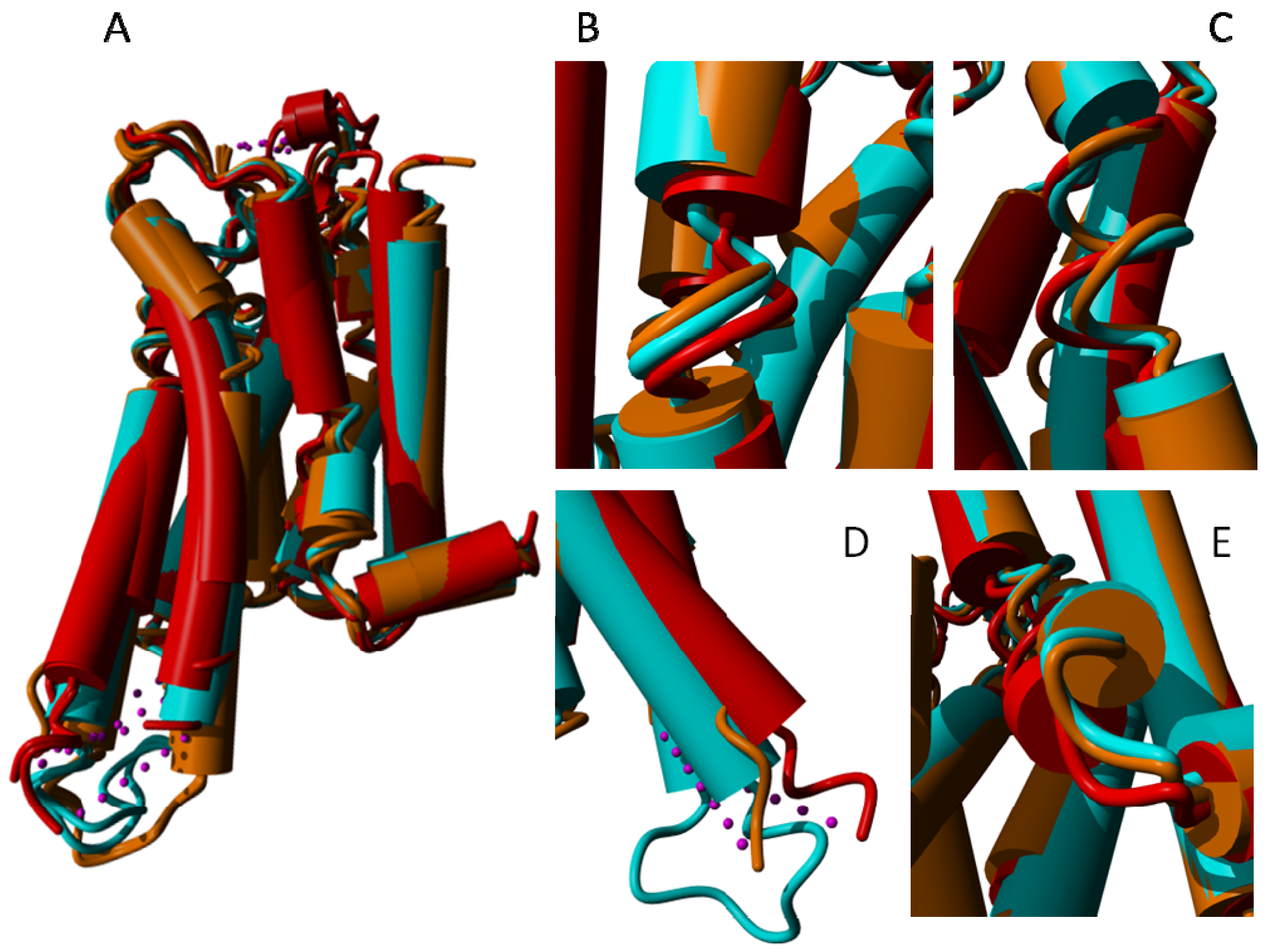

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | ID | TM1 | TM2 | TM3 | TM4 | TM5 | TM6 | TM7 |
|---|---|---|---|---|---|---|---|---|
| Rhodopsin | 1f88, 1gzm, 1hzx, 1l9h, 1u19, 2g87, 2hzy, 2i35, 2i36, 2i37, 2j4y, 2ped, 3c9l, 3c9m, 3oax | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| Opsin | 2x72, 3cap, 3dqb, 3pqr, 3pxo, 4a4m | 0 | 1 | 0 | 0 | 1 | 0 | 1 |
| Rhod squid | 2z73, 2ziy, 3aym, 3ayn | 0 | 2 | 0 | 0 | 1 | 0 | 0 |
| β1 AR | 2vt4, 2y00, 2y01, 2y02, 2y03, 2y04, 2ycw, 2ycx, 2ycy, 2ycz, 4ami, 4amj, 4gpo | 0 | 1 | 0 | 0 | 1 | 0 | −1 |
| β2 AR inact | 2rh1, 3d4s, 3ny8, 3ny9, 3nya | 0 | 1 | 0 | 0 | 1 | 0 | −1 |
| β2 AR act | 3p0g, 3pds, 3sn6, 4lde, 4ldl, 4ldo | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| A2A inact | 3eml, 3pwh, 3rey, 3rfm, 3uza, 3uac, 3vg9, 3vga, 4eiy | 0 | 1 | 0 | 0 | 2 * | 0 | 0 |
| A2A act | 2ydo, 2ydv, 3qak | 0 | 1 | 0 | 0 | 2 * | 0 | 1 |
| CXCR4 | 3odu, 3oe0, 3oe6, 3oe8, 3oe9 | 0 | 0 | 0 | 1 | 1 | 0 | 0 |
| Opioid | 4djh, 4dkl, 4ea3, 4ej4 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Lipid | 3v2w, 3v2y | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Serotonin 1B | 4iaq, 4iar | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| Serotonin 2B | 4ib4 | 1 | 1 | 0 | 0 | 1 | 0 | 0 |
| CCR5 | 4mbs | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| PAR1 | 3vw7 | 0 | 0 | 0 | 0 | 1 | 1 | 0 |
| NTSR1 | 4grv | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| Muscarinic | 3uon, 4daj | 0 | 1 | 0 | 1 | 1 | 0 | 0 |
| Histamine H1 | 3rze | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| Dopamine D3 | 3pbl | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Van der Kant, R.; Vriend, G. Alpha-Bulges in G Protein-Coupled Receptors. Int. J. Mol. Sci. 2014, 15, 7841-7864. https://doi.org/10.3390/ijms15057841
Van der Kant R, Vriend G. Alpha-Bulges in G Protein-Coupled Receptors. International Journal of Molecular Sciences. 2014; 15(5):7841-7864. https://doi.org/10.3390/ijms15057841
Chicago/Turabian StyleVan der Kant, Rob, and Gert Vriend. 2014. "Alpha-Bulges in G Protein-Coupled Receptors" International Journal of Molecular Sciences 15, no. 5: 7841-7864. https://doi.org/10.3390/ijms15057841
APA StyleVan der Kant, R., & Vriend, G. (2014). Alpha-Bulges in G Protein-Coupled Receptors. International Journal of Molecular Sciences, 15(5), 7841-7864. https://doi.org/10.3390/ijms15057841