Detection of Borreliae in Archived Sera from Patients with Clinically Suspect Lyme Disease

Abstract
:1. Introduction
2. Results and Discussion
2.1. Detection of “Lyme Borreliae” in Archived Post-Treatment Sera from Lyme Disease Patients
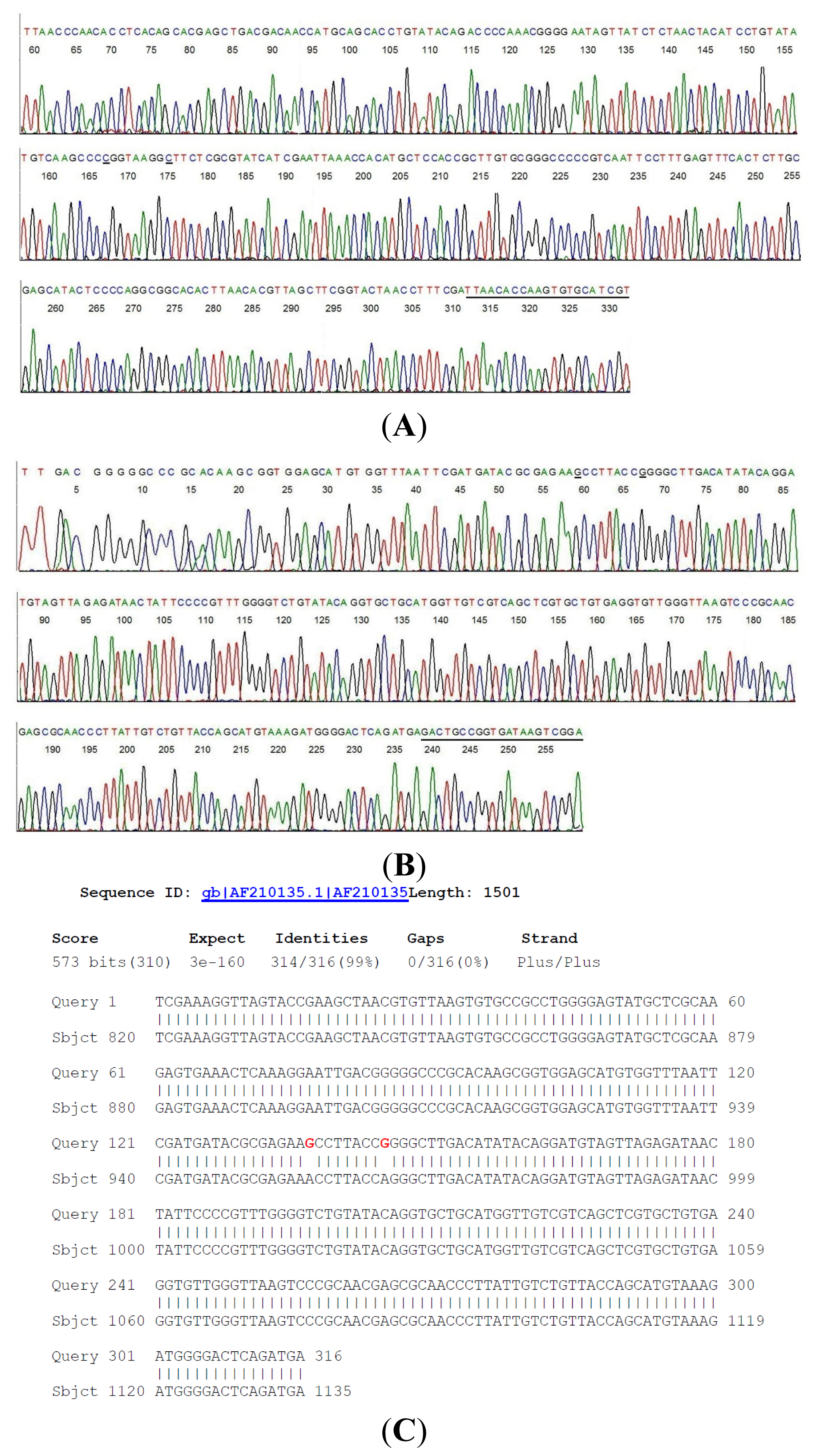
2.2. Sequencing of a Highly Conserved Segment of 16S rDNA of a Novel Borrelia
2.3. Detection of “Lyme Borreliae” in Archived Pre-Treatment Sera from Lyme Disease Patients
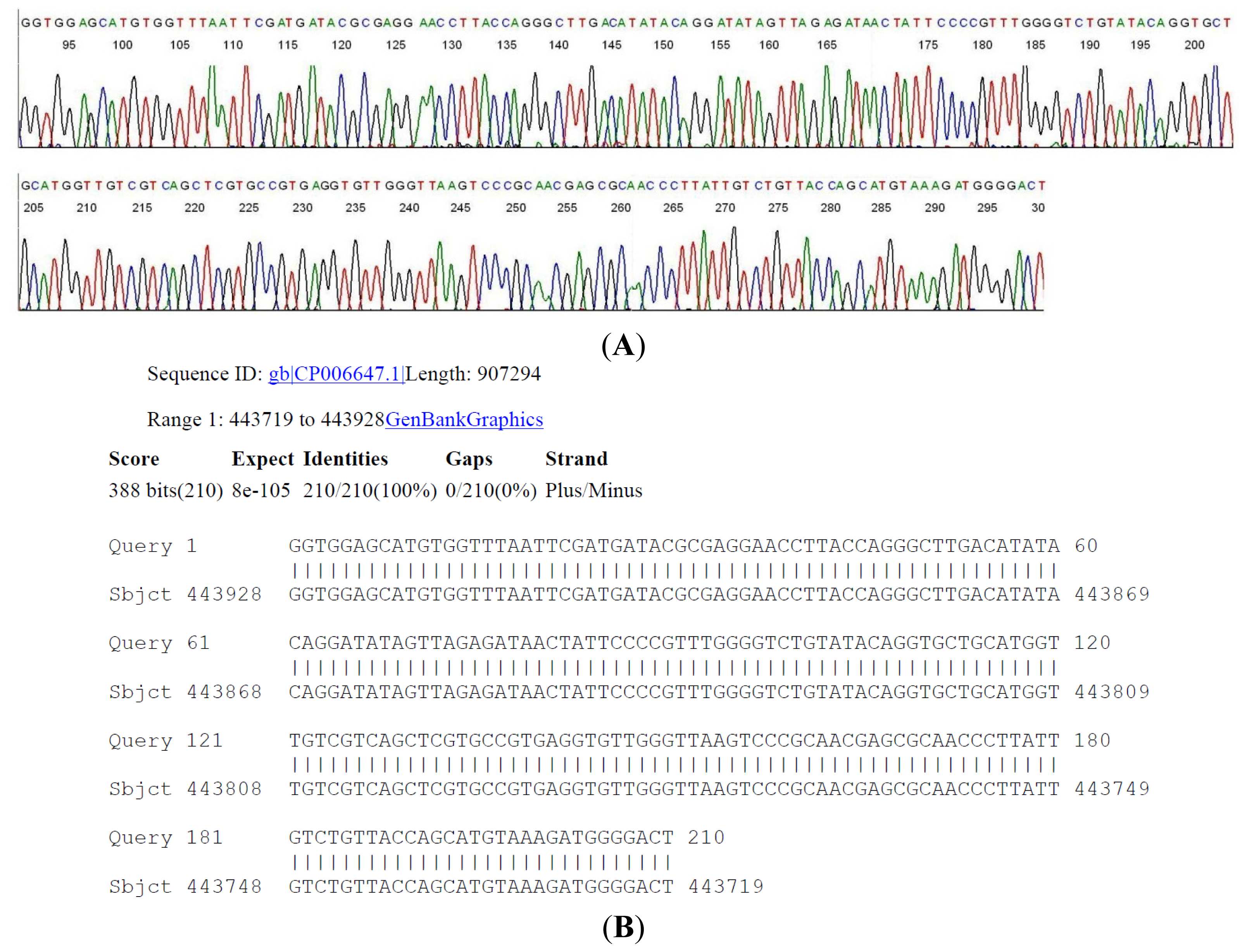
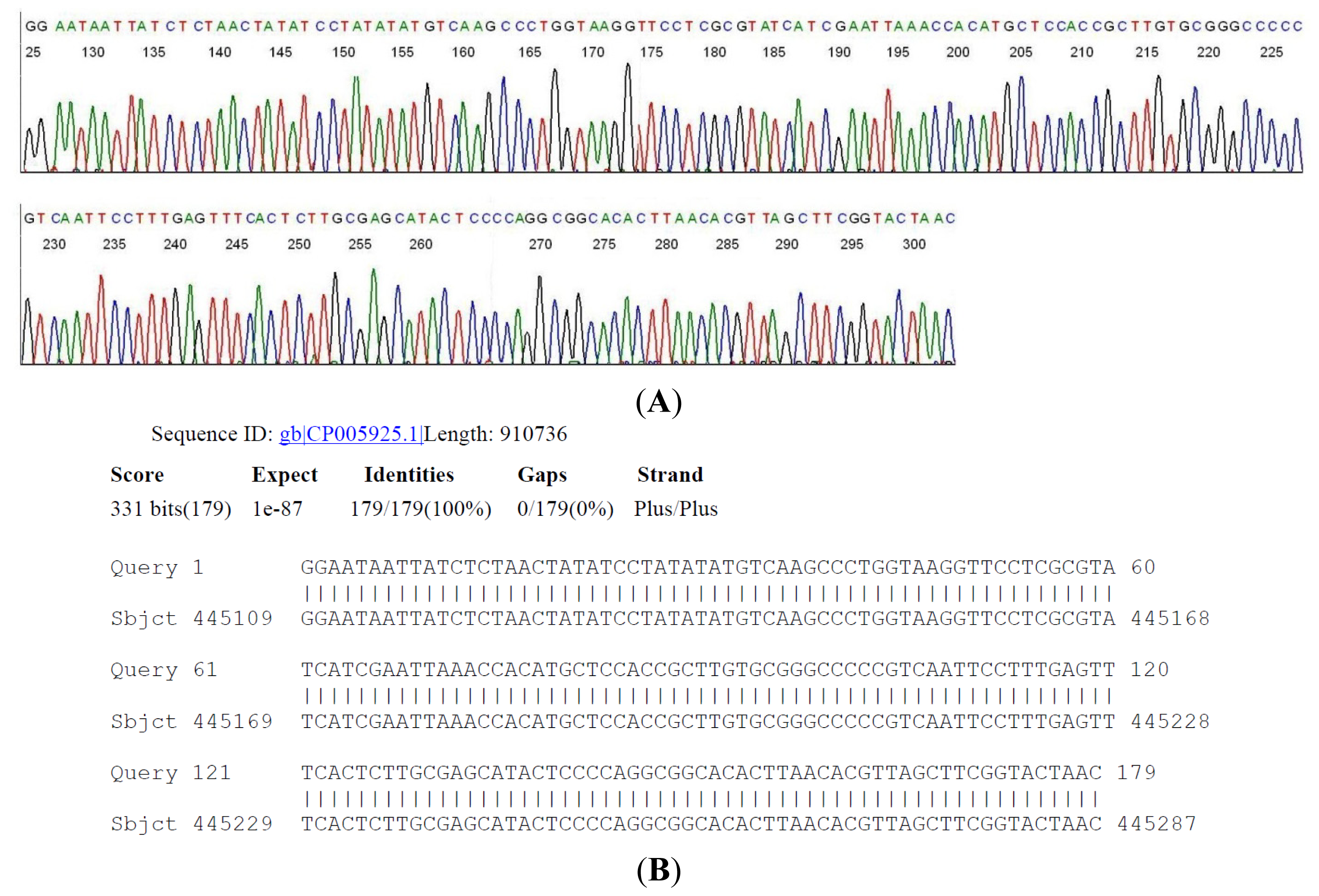
2.4. DNA Sequencing-Based Diagnosis of B. miyamotoi and B. burgdorferi
3. Experimental Section
3.1. Preparation of Borrelial DNA
3.2. Same-Nested PCR Amplification
3.3. Direct DNA Sequencing
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Feder, H.M., Jr; Johnson, B.J.; O’Connell, S.; Shapiro, E.D.; Steere, A.C.; Wormser, G.P. Ad Hoc International Lyme Disease Group. A critical appraisal of “chronic Lyme disease”. N. Engl. J. Med 2007, 357, 1422–1430. [Google Scholar]
- Centers for Disease Control and Prevention: Lyme Disease. Available online: http://www.cdc.gov/lyme/ (accessed on 25 January 2014).
- Johnston, B.; Conly, J. Lyme disease: Is it or is it not? Can. J. Infect. Dis. Med. Microbiol. 2005, 16, 325–328. [Google Scholar]
- Centers for Disease Control and Prevention. Atypical Erythema Migrans in Patients with PCR-Positive Lyme Disease. Available online: http://wwwnc.cdc.gov/eid/article/19/5/12-0796_article.htm (accessed on 20 February 2014).
- Steere, A.C.; Malawista, S.E.; Snydman, D.R.; Shope, R.E.; Andiman, W.A.; Ross, M.R.; Steele, F.M. Lyme arthritis: An epidemic of oligoarticular arthritis in children and adults in three connecticut communities. Arthritis Rheum 1977, 20, 7–17. [Google Scholar]
- Wormser, G.P. Hematogenous dissemination in early Lyme disease. Wien Klin. Wochenschr. 2006, 118, 634–637. [Google Scholar]
- Aucott, J.; Morrison, C.; Munoz, B.; Rowe, P.C.; Schwarzwalder, A.; West, S.K. Diagnostic challenges of early Lyme disease: Lessons from a community case series. BMC Infect. Dis. 2009, 9, 79. [Google Scholar]
- Morrison, C.; Seifter, A.; Aucott, J.N. Unusual presentation of Lyme disease: Horner syndrome with negative serology. J. Am. Board Fam. Med. 2009, 22, 219–222. [Google Scholar]
- Lee, S.H.; Vigliotti, V.S.; Vigliotti, J.S.; Jones, W.; Williams, J.; Walshon, J. Early lyme disease with spirochetemia—Diagnosed by DNA sequencing. BMC Res. Notes 2010, 3, 273–280. [Google Scholar]
- Recommendations for Test Performance and Interpretation from the Second National Conference on Serologic Diagnosis of Lyme Disease. Available online: http://www.cdc.gov/mmwr/preview/mmwrhtml/00038469.htm (accessed on 25 January 2014).
- Steere, A.C.; Taylor, E.; McHugh, G.L.; Logigian, E.L. The overdiagnosis of Lyme disease. JAMA 1993, 269, 1812–1816. [Google Scholar]
- Stricker, R.B.; Johnson, L. Lyme wars: Let’s tackle the testing. BMJ 2007, 335, 1008. [Google Scholar]
- Platonov, A.E.; Karan, L.S.; Kolyasnikova, N.M.; Makhneva, N.A.; Toporkova, M.G.; Maleev, V.V.; Fish, D.; Krause, P.J. Humans infected with relapsing fever spirochete Borrelia miyamotoi Russia. Emerg. Infect. Dis. 2011, 17, 1816–1823. [Google Scholar]
- Gugliotta, J.L.; Goethert, H.K.; Berardi, V.P.; Telford, S.R., III. Meningoencephalitis from Borrelia miyamotoi in an Immunocompromised Patient. N. Engl. J. Med 2013, 368, 240–245. [Google Scholar]
- Chowdri, H.R.; Gugliotta, J.L.; Berardi, V.P.; Goethert, H.K.; Molloy, P.J.; Sterling, S.L.; Telford, S.R. Borrelia miyamotoi infection presenting as human granulocytic anaplasmosis: A case report. Ann. Intern. Med. 2013, 159, 21–27. [Google Scholar]
- Centers for Disease Control and Prevention. Ticks. B. miyamotoi. Available online: http://www.cdc.gov/ticks/miyamotoi.html (accessed on 25 January 2014).
- Hong, G.; Lee, S.H.; Ge, S.; Zhou, S. A Novel Low Temperature PCR Assured High-Fidelity DNA Amplification. Int. J. Mol. Sci. 2013, 14, 12853–12862. [Google Scholar]
- Lee, S.H.; Vigliotti, V.S.; Vigliotti, J.S.; Pappu, S. Routine human papillomavirus genotyping by DNA sequencing in community hospital laboratories. Infect. Agent. Cancer 2007, 2, 11–21. [Google Scholar]
- Lee, S.H.; Vigliotti, V.S.; Vigliotti, J.S.; Jones, W.; Pappu, S. Increased sensitivity and specificity of Borrelia burgdorferi 16S ribosomal DNA detection. Am. J. Clin. Path. 2010, 133, 569–576. [Google Scholar]
- Lee, S.H. Guidelines for the use of molecular tests for the detection and genotyping of human papilloma virus from clinical specimens. Methods Mol. Biol. 2012, 903, 65–101. [Google Scholar]
- Wormser, G.P.; Bittker, S.; Cooper, D.; Nowakowski, J.; Nadelman, R.B.; Pavia, C. Comparison of the yields of blood cultures using serum or plasma from patients with early Lyme disease. J. Clin. Microbiol 2000, 38, 1648–1650. [Google Scholar]
- Niścigorska, J.; Skotarczak, B.; Wodecka, B. Borrelia burgdorferi infection among forestry workers-assessed with an immunoenzymatic method (ELISA), PCR and correlated with the clinical state of the patients. Ann. Agric. Environ. Med. 2003, 10, 15–19. [Google Scholar]
- Barthold, S.W.; Hodzic, E.; Imai, D.M.; Feng, S.; Yang, X.; Luft, B.J. Ineffectiveness of tigecycline against persistent Borrelia burgdorferi. Antimicrob Agents Chemother 2010, 54, 643–651. [Google Scholar]
- Richter, D.; Schlee, D.B.; Matuschka, F.R. Relapsing fever-like spirochetes infecting European vector tick of Lyme disease agent. Emerg. Infect. Dis. 2003, 9, 697–701. [Google Scholar]
- Janda, J.M.; Abbott, S.L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses perils and pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar]
- Hendson, M.; Lane, R.S. Genetic characteristics of Borrelia coriaceae isolates from the soft tick Ornithodoros coriaceus (Acari: Argasidae). J. Clin. Microbiol 2000, 38, 2678–2682. [Google Scholar]
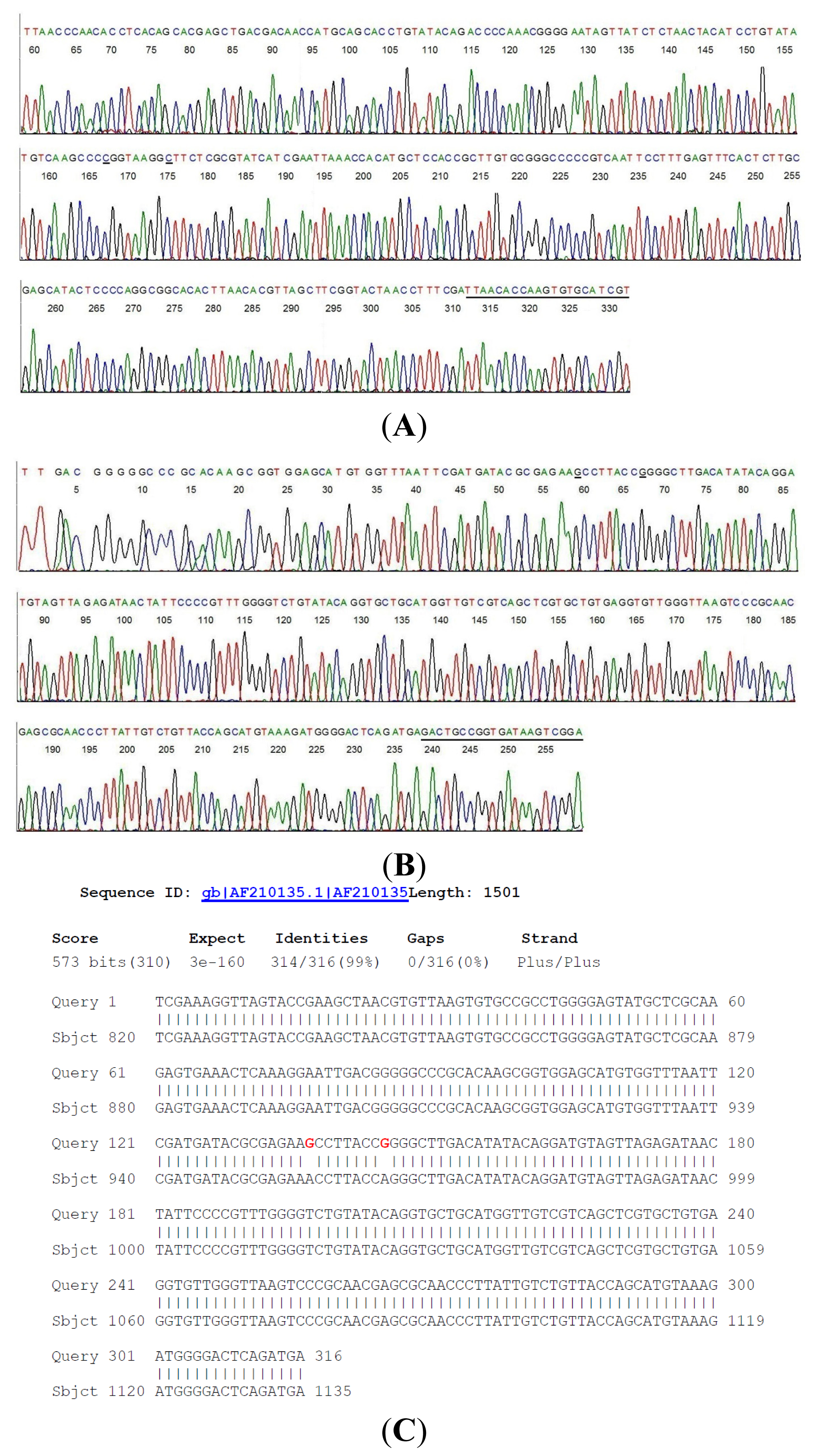
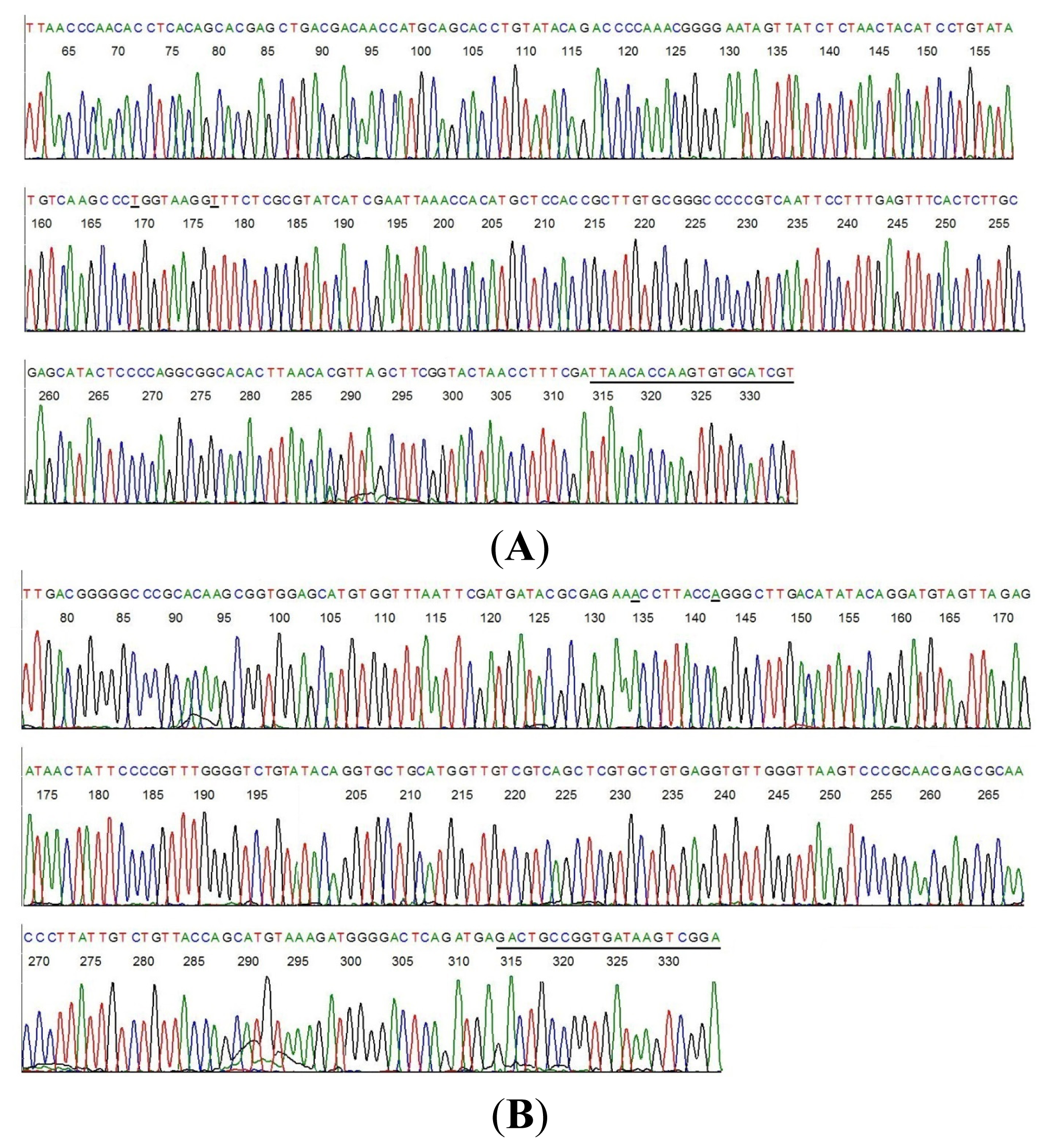
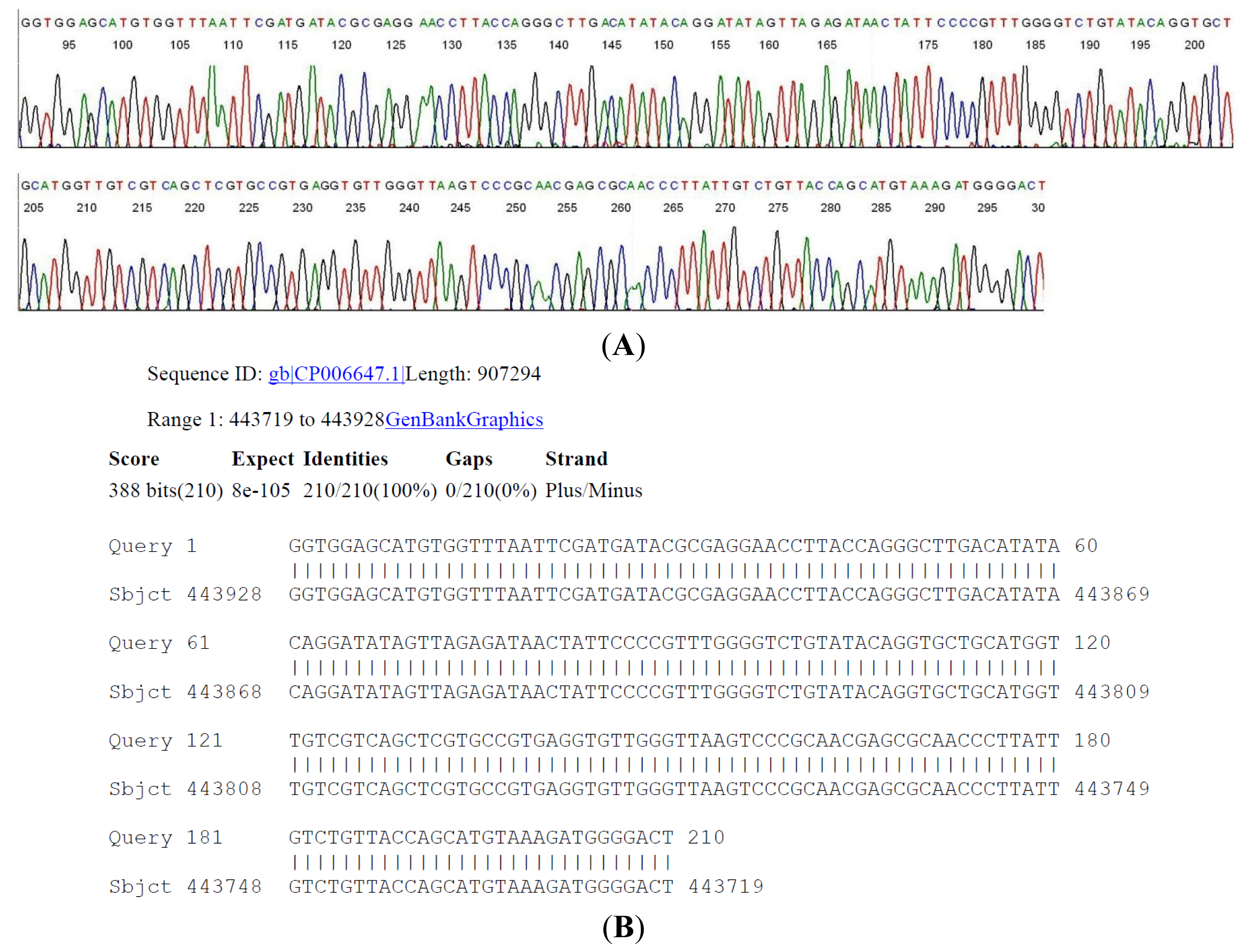
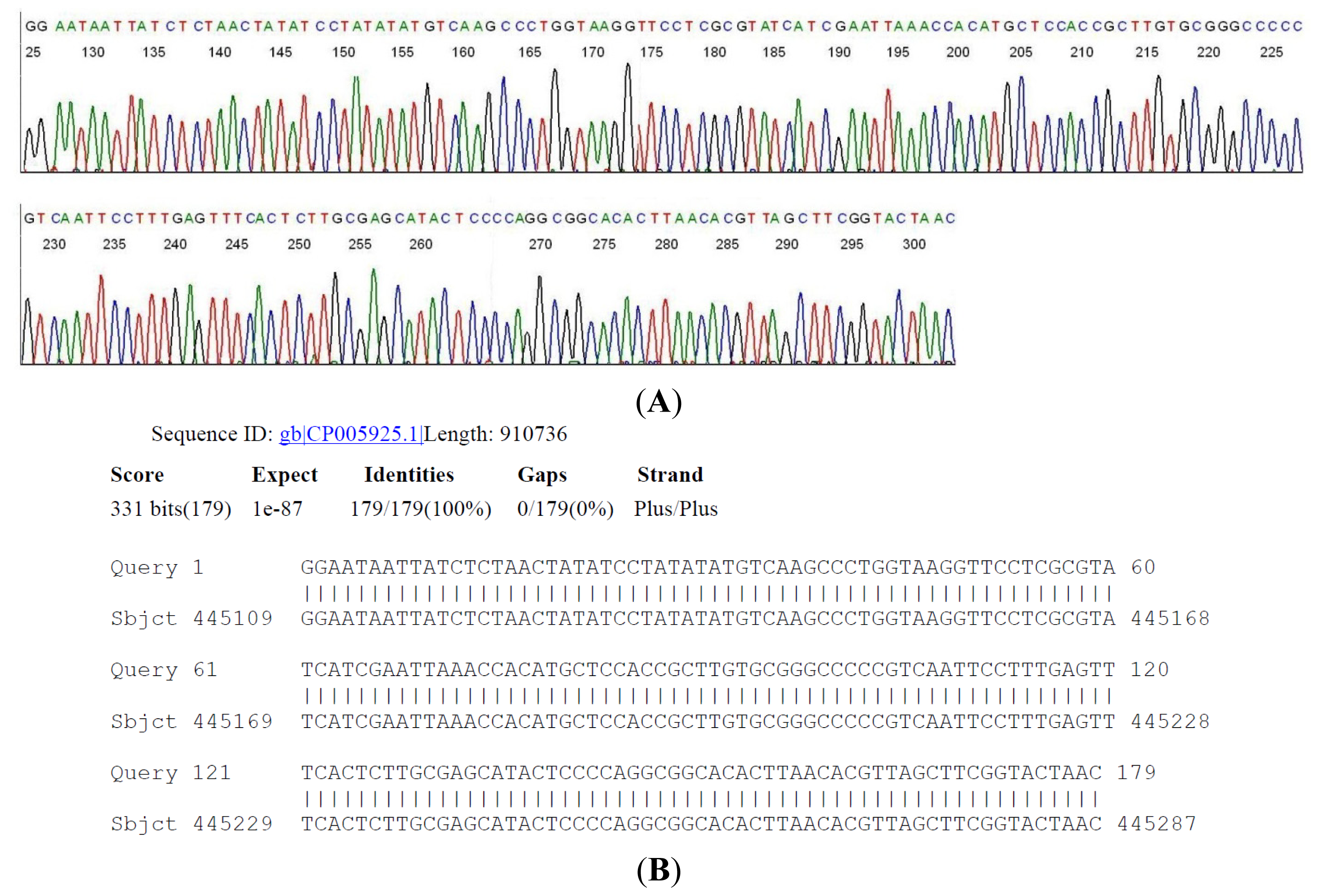

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case # | Clinical Diagnosis | Acute/Conv. | EIA Value | IgM WB Bands | IgG WB Bands | 2-Tier Interpretation | Borrelia by PCR |
|---|---|---|---|---|---|---|---|
| 1 | Early LD-EM | Acute | 0.07 | - | 30 | Neg. | Neg. |
| 2 | Early LD-EM | Acute | 0.39 | - | 18 | Neg. | Neg. |
| 3 | Early LD-EM | Acute | 0.11 | - | 45, 41 | Neg. | Neg. |
| 4 | Early LD-EM | Acute | 0.72 | 41, 23 | 66, 41 | Neg. | Neg. |
| 5 | Early LD-EM | Conv. | 2.05 | 41, 23 | 41, 23 | Pos. | Neg. |
| 6 | Early LD-EM | Conv. | 5.26 | 39, 23 | 45, 41, 23, 18 | Pos. | Neg. |
| 7 | Early LD-EM | Conv. | 3.08 | 41, 23 | 45, 41, 39, 30, 23 | Pos. | Neg. |
| 8 | Early LD-EM | Conv. | 5.01 | 41, 39, 23 | 66, 41, 23 | Pos. | Neg. |
| 9 | Neurologic Lyme | N | 4.24 | 41, 39, 23 | 93, 41, 39, 23, 18 | Pos. | Novel isolate |
| 10 | Neurologic Lyme | N | 5.04 | 41, 23 | 93, 66, 58, 45, 41, 39, 30, 28, 23, 18 | Pos. | Neg. |
| 11 | Lyme arthritis | N | 5.98 | 41 | 93, 66, 58, 45, 41, 39, 23, 18 | Pos. | Neg. |
| 12 | Lyme arthritis | N | 4.47 | 23 | 93, 66, 58, 45, 41, 39, 30, 28, 23, 18 | Pos. | Neg. |
| 13 | Fibromyalgia | N | 0.71 | - | 41 | Neg. | Neg. |
| 14 | Fibromyalgia | N | 0.20 | - | - | Neg. | Neg. |
| 15 | Rheumatoid arthritis | N | 0.07 | - | 66, 41 | Neg. | Neg. |
| 16 | Rheumatoid arthritis | N | 0.38 | - | - | Neg. | Neg. |
| 17 | Multiple sclerosis | N | 0.48 | - | 41 | Neg. | Neg. |
| 18 | Multiple sclerosis | N | 0.26 | - | 41 | Neg. | Neg. |
| 19 | Mononucleosis | N | 3.17 | - | - | Neg. | Neg. |
| 20 | Mononucleosis | N | 0.27 | 23 | - | Neg. | Neg. |
| 21 | Syphilis | N | 0.32 | - | 41 | Neg. | Neg. |
| 22 | Syphilis | N | 1.73 | 23 | - | Neg. | Neg. |
| 23 | Severe periodontitis | N | 0.24 | - | 41 | Neg. | Neg. |
| 24 | Severe periodontitis | N | 0.22 | - | - | Neg. | Neg. |
| 25 | Healthy endemic | N | 1.29 | - | - | Neg. | Neg. |
| 26 | Healthy endemic | N | 0.74 | - | 66 | Neg. | Neg. |
| 27 | Healthy endemic | N | 0.60 | - | - | Neg. | Neg. |
| 28 | Healthy endemic | N | 0.19 | 23 | 45, 41 | Neg. | Neg. |
| 29 | Healthy non-endemic | N | 0.72 | - | 66, 41, 39 | Neg. | Neg. |
| 30 | Healthy non-endemic | N | 0.42 | 23 | 45, 41 | Neg. | Neg. |
| 31 | Healthy non-endemic | N | 0.49 | 41 | 41 | Neg. | Neg. |
| 32 | Healthy non-endemic | N | 1.18 | - | - | Neg. | Neg. |
| Case # | Clinical Diagnosis | EIA Value | IgM WB Bands | IgG WB Bands | 2-Tier Interpretation | Borrelia by PCR |
|---|---|---|---|---|---|---|
| 33 | Early LD-EM | 0.82 | - | - | Neg. | Neg. |
| 34 | Early LD-EM | 4.41 | 41, 39, 23 | 41, 23, 18 | Pos. | Neg. |
| 35 | Early LD-EM | 1.29 | 41 | 30 | Neg. | Neg. |
| 36 | Early LD-EM | 0.11 | 23 | - | Neg. | Neg. |
| 37 | Early LD-EM | 0.25 | - | 23 | Neg. | Neg. |
| 38 | Early LD-EM | 0.49 | - | 66 | Neg. | Neg. |
| 39 | Early LD-EM | 0.72 | 41, 23 | 66, 41 | Neg. | B. miyamotoi |
| 40 | Early LD-EM | 0.07 | - | 30 | Neg. | Neg. |
| 41 | Early LD-EM | 0.25 | - | - | Neg. | Neg. |
| 42 | Early LD-EM | 4.52 | 41, 39, 23 | 66, 58, 45, 41, 39, 23, 18 | Pos. | Neg. |
| 43 | Early LD-EM | 5.10 | 41, 23 | 66, 58, 45, 41, 39, 23, 18 | Pos. | Neg. |
| 44 | Early LD-EM | 0.21 | 39, 23 | 23 | Neg. | Neg. |
| 45 | Early LD-EM | 0.65 | - | 41 | Neg. | B. burgdorferi |
| 46 | Early LD-EM | 0.11 | - | 45, 41 | Neg. | Neg. |
| 47 | Early LD-EM | 0.80 | 41, 23 | 41, 23 | Pos. | Neg. |
| 48 | Early LD-EM | 0.39 | - | 18 | Neg. | Neg. |
| 49 | Early LD-EM | 2.60 | 23 | 66, 41, 23 | Neg. | Neg. |
| 50 | Early LD-EM | 3.76 | 41, 23 | 41 | Pos. | B. burgdorferi |
| 51 | Early LD-EM | 4.25 | 41, 39, 23 | 41 | Pos. | Neg. |
| 52 | Early LD-EM | 1.95 | 41, 39, 23 | 58, 41 | Pos. | Neg. |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, S.H.; Vigliotti, J.S.; Vigliotti, V.S.; Jones, W.; Shearer, D.M. Detection of Borreliae in Archived Sera from Patients with Clinically Suspect Lyme Disease. Int. J. Mol. Sci. 2014, 15, 4284-4298. https://doi.org/10.3390/ijms15034284
Lee SH, Vigliotti JS, Vigliotti VS, Jones W, Shearer DM. Detection of Borreliae in Archived Sera from Patients with Clinically Suspect Lyme Disease. International Journal of Molecular Sciences. 2014; 15(3):4284-4298. https://doi.org/10.3390/ijms15034284
Chicago/Turabian StyleLee, Sin Hang, Jessica S. Vigliotti, Veronica S. Vigliotti, William Jones, and David M. Shearer. 2014. "Detection of Borreliae in Archived Sera from Patients with Clinically Suspect Lyme Disease" International Journal of Molecular Sciences 15, no. 3: 4284-4298. https://doi.org/10.3390/ijms15034284
APA StyleLee, S. H., Vigliotti, J. S., Vigliotti, V. S., Jones, W., & Shearer, D. M. (2014). Detection of Borreliae in Archived Sera from Patients with Clinically Suspect Lyme Disease. International Journal of Molecular Sciences, 15(3), 4284-4298. https://doi.org/10.3390/ijms15034284