The Inhibition of RANKL-Induced Osteoclastogenesis through the Suppression of p38 Signaling Pathway by Naringenin and Attenuation of Titanium-Particle-Induced Osteolysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
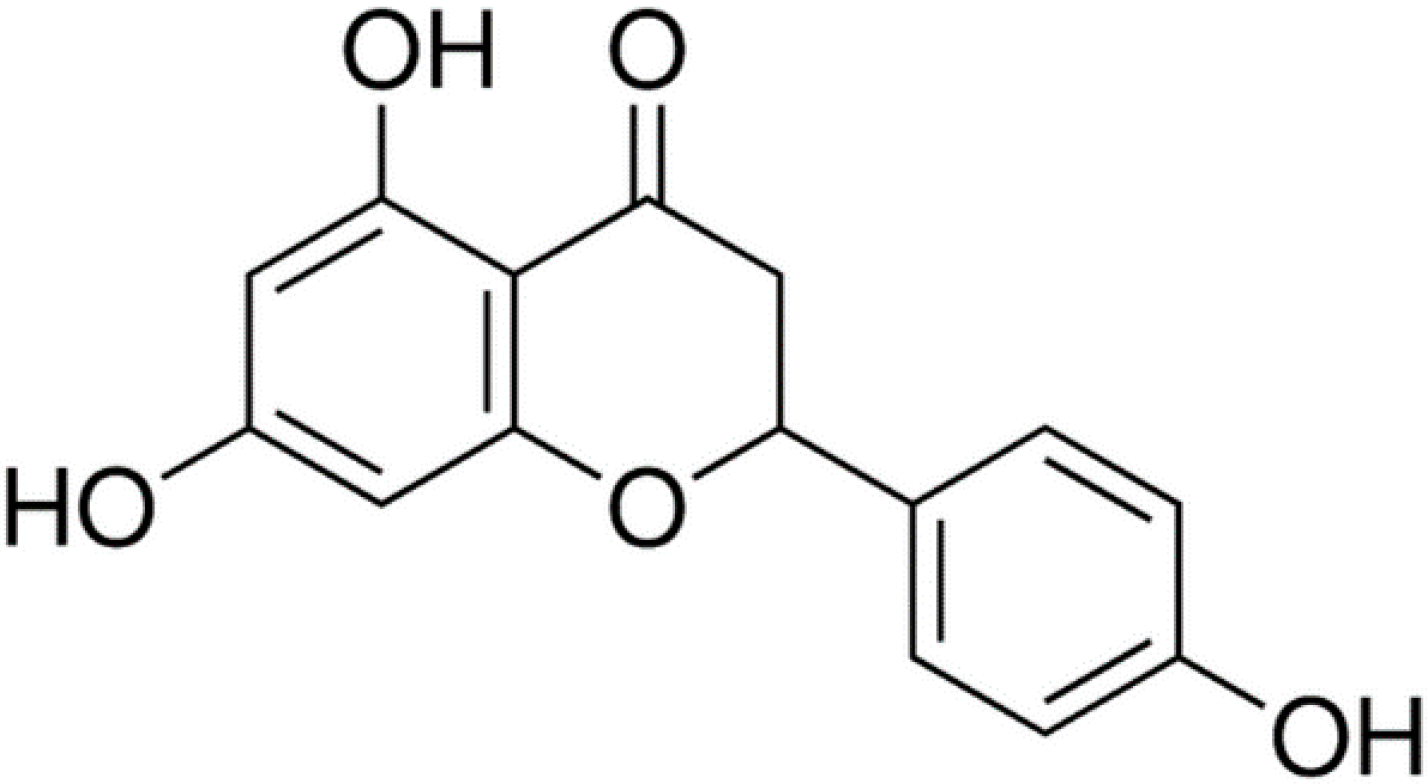
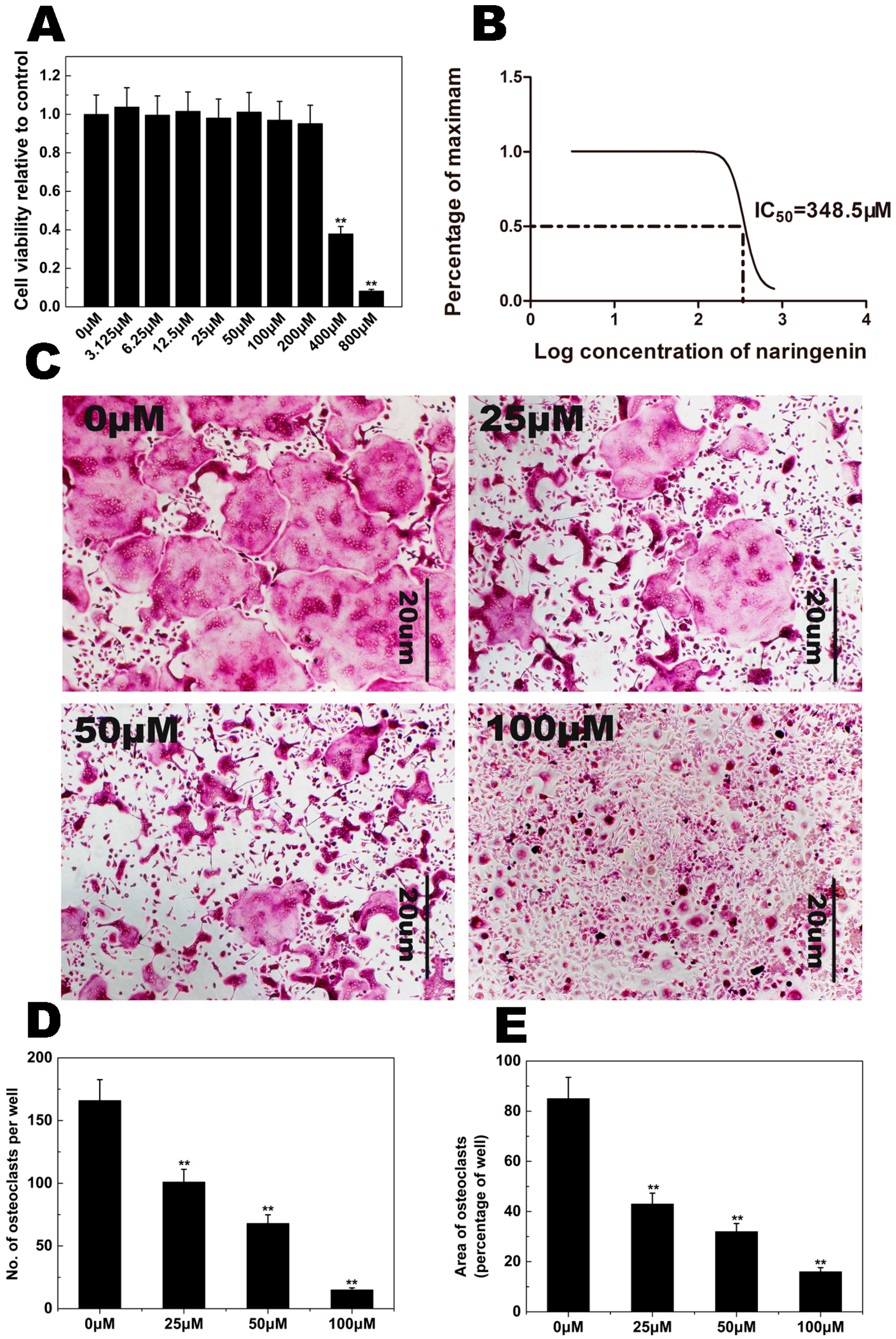
2.1. NAR Cytotoxicity and Effect of NAR on Osteoclastogenesis
2.2. F-Actin Ring Formation

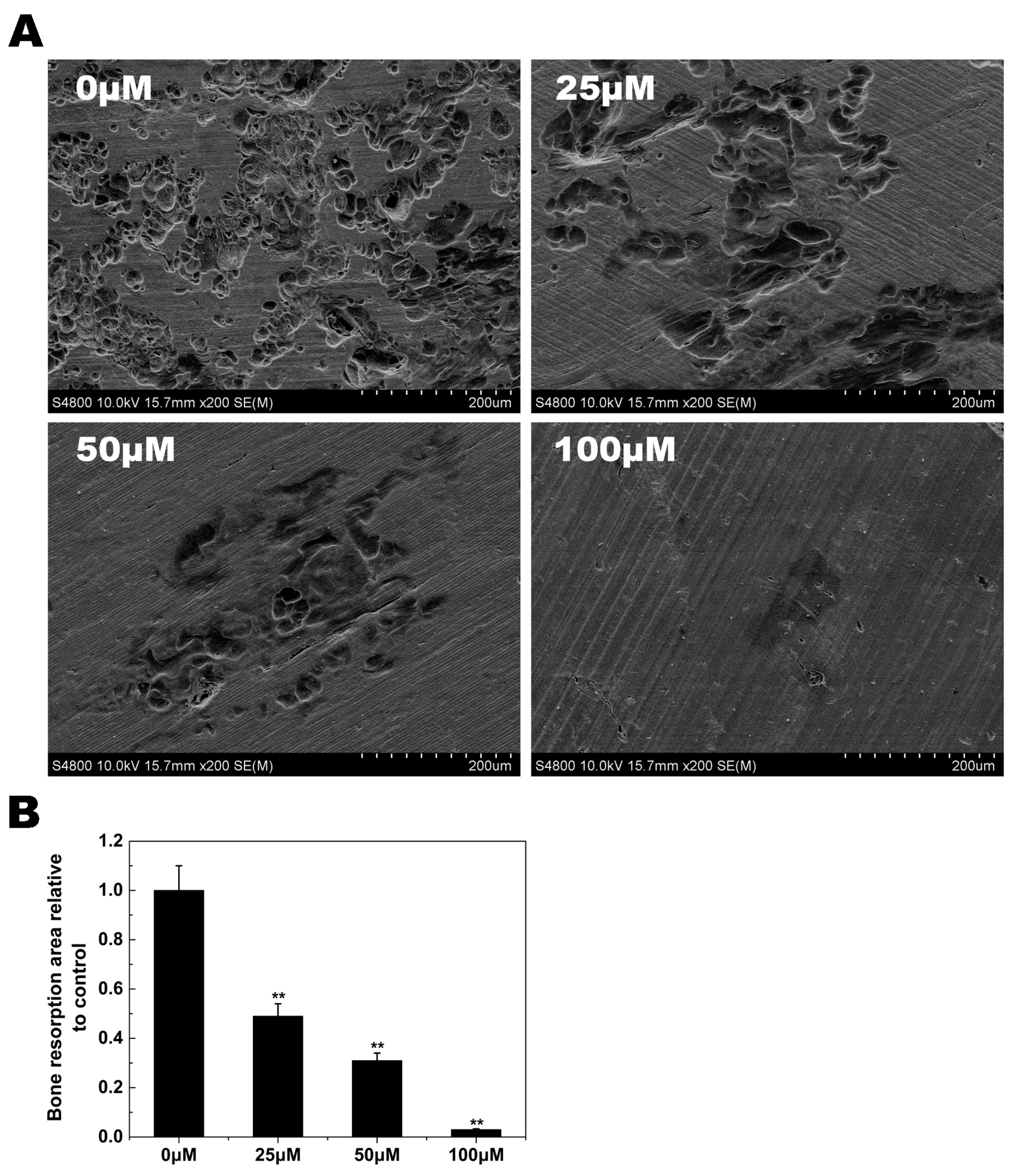
2.3. Osteoclast Bone Resorption
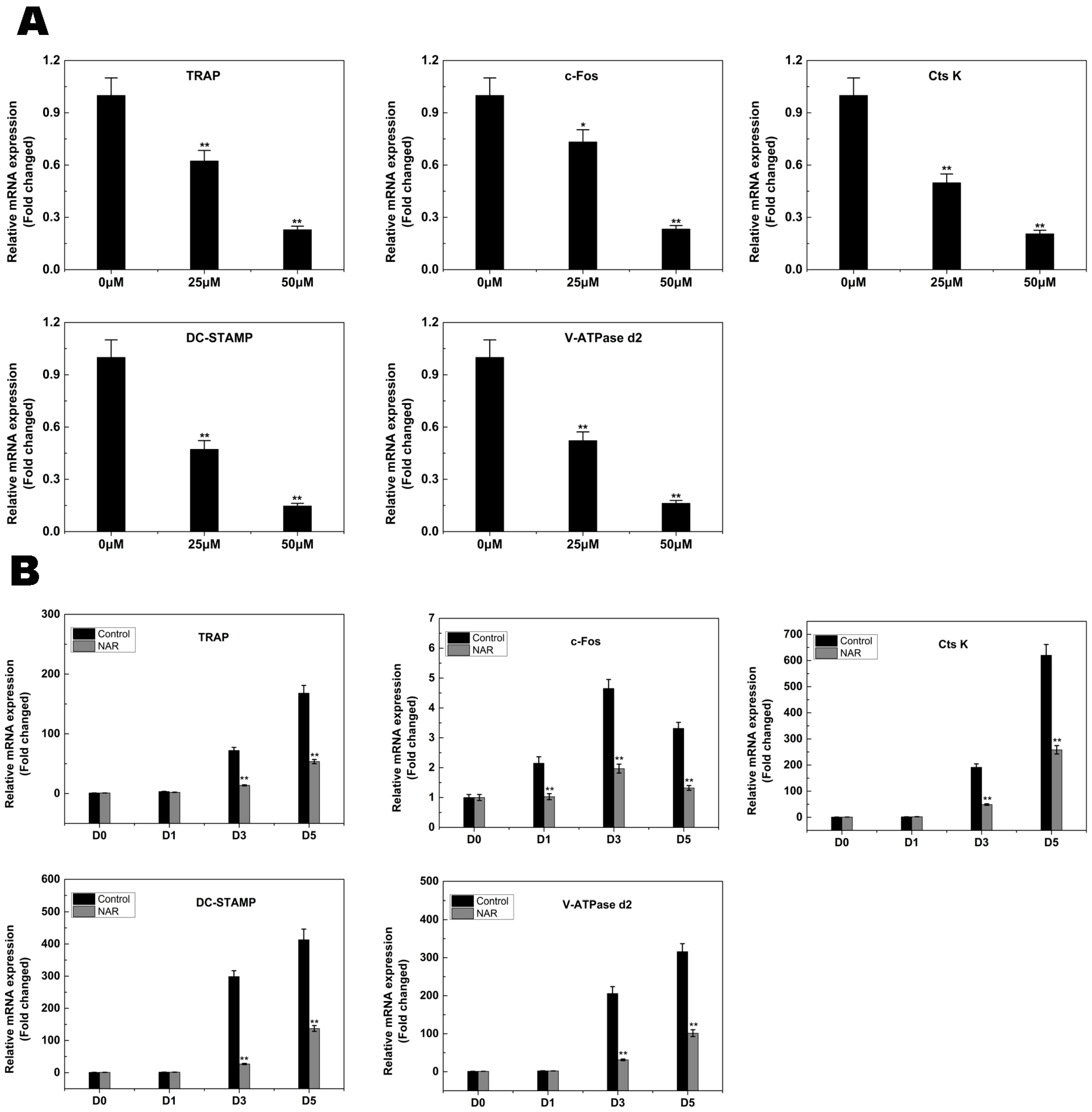
2.4. RANKL-Induced Gene Expression
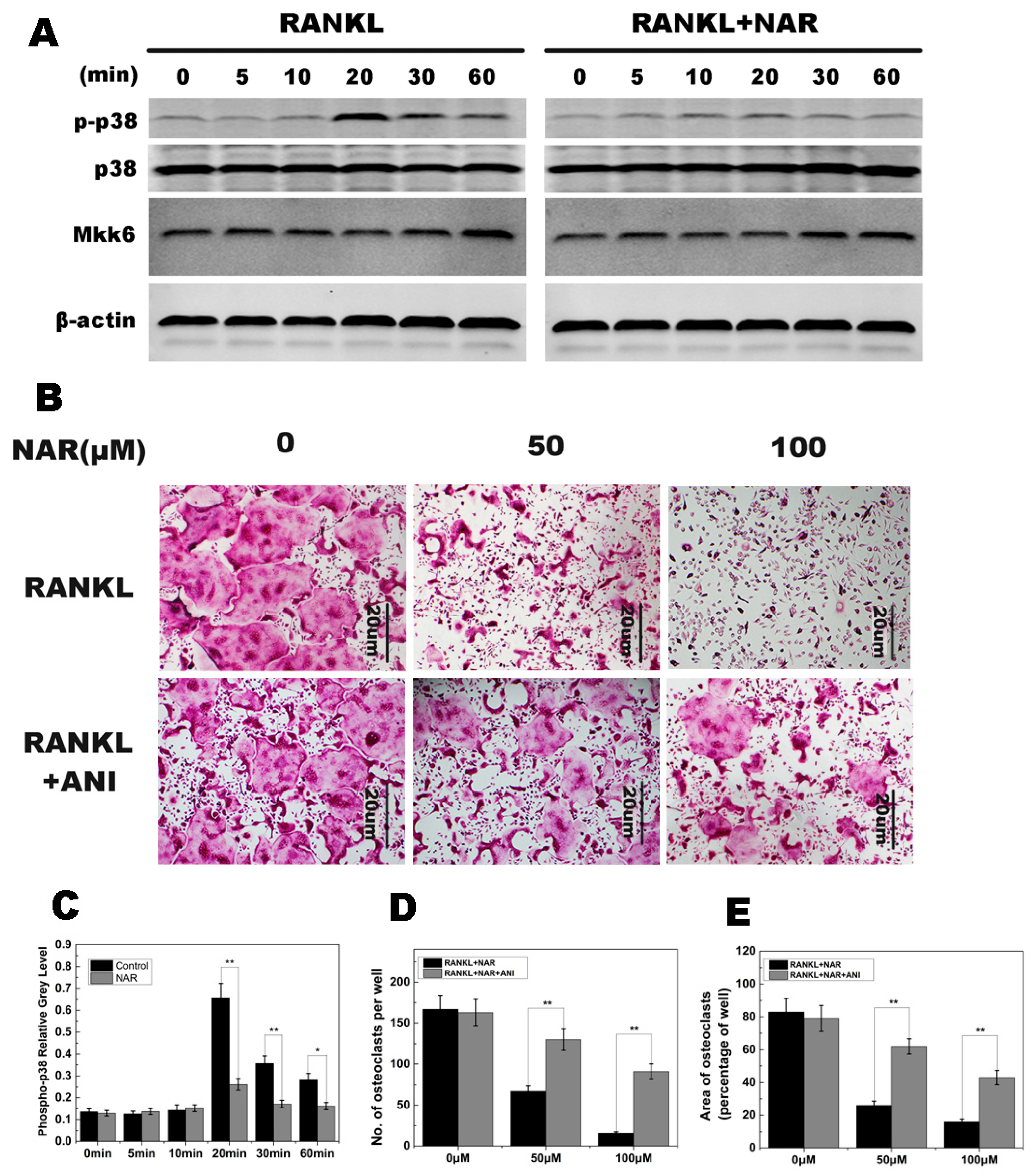
2.5. RANKL-Induced p38 Signaling and Anisomycin on NAR-Treated Osteoclastogenesis
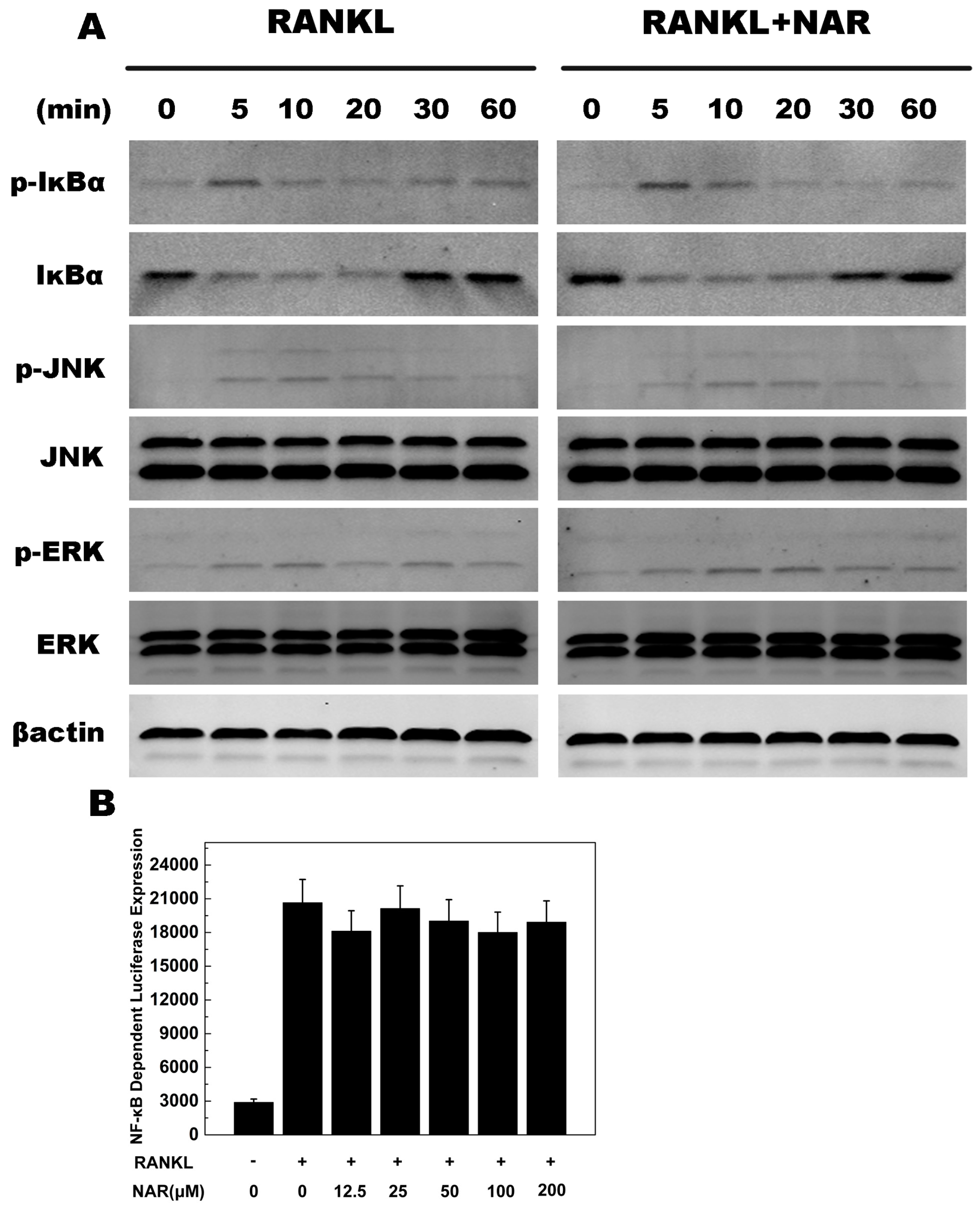
2.6. RANKL-Induced NF-κB, ERK, JNK Signaling
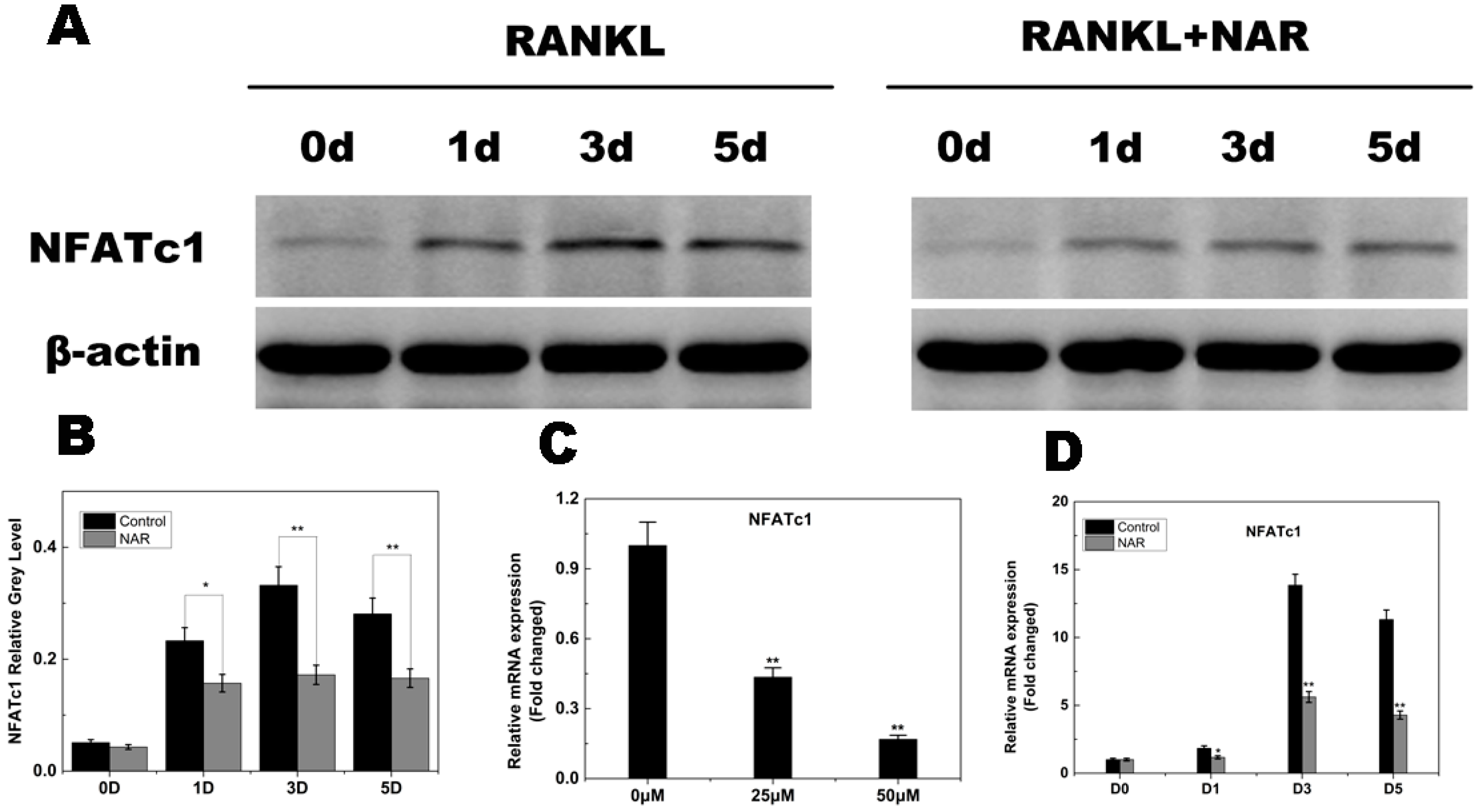
2.7. RANKL-Induced NFATc1 Expression
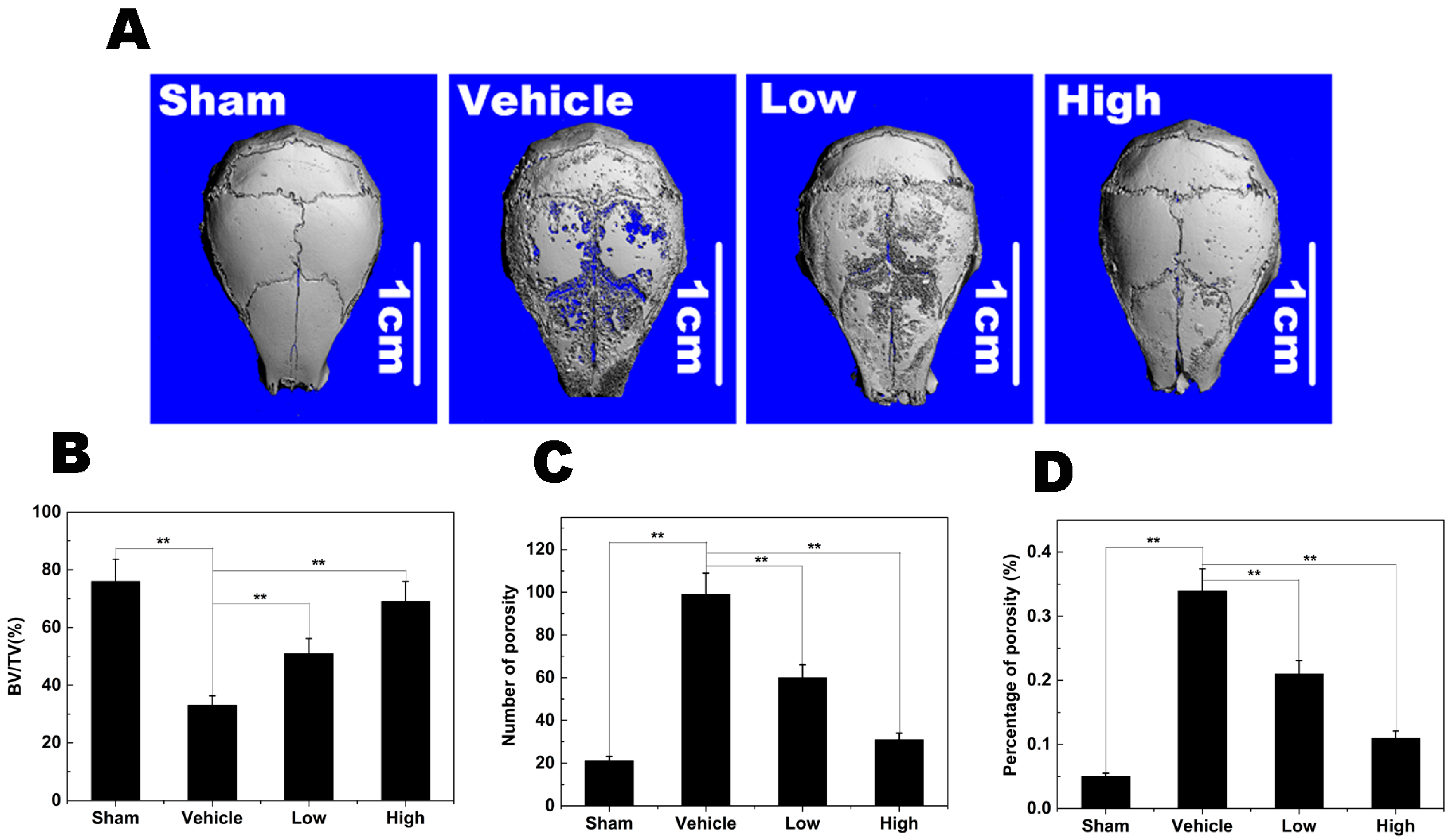
2.8. Titanium Particle-Induced Osteolysis
2.9. Schematic Diagram Describing the Mechanism by Which Naringenin (NAR) Inhibits Osteoclast Differentiation and Function
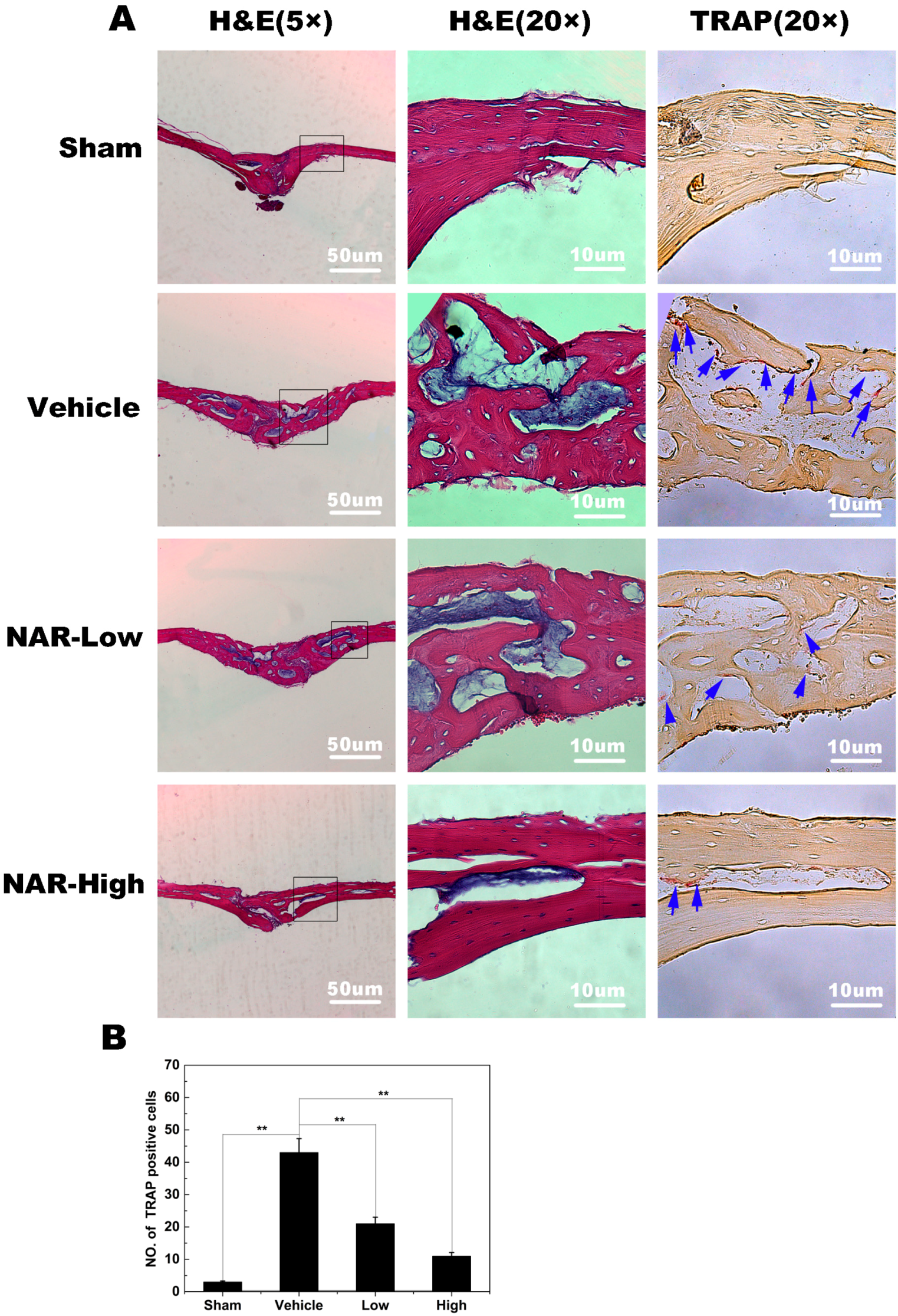
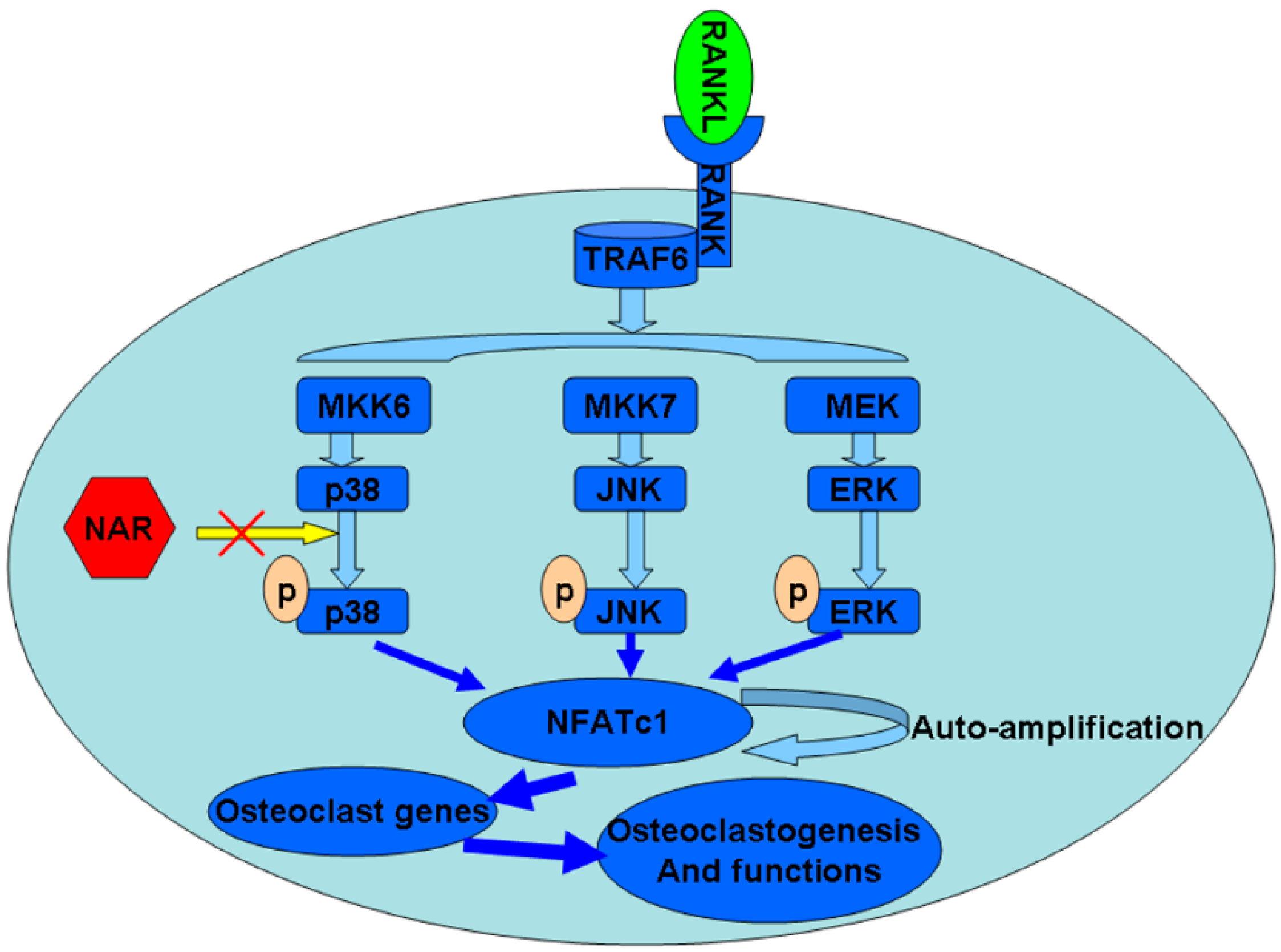
3. Discussion
4. Experimental Section
4.1. Media, Reagents, and Cells
4.2. Cytotoxicity Assay
4.3. BMMs Isolation and Osteoclasts Culture
4.4. F-Actin Ring Immunofluorescence
4.5. Bone Resorption Pit Assay
4.6. RNA Extraction and Quantitative PCR Assay
4.7. NF-κB Luciferase Reporter Gene Assay
4.8. Osteoclastogenesis Rescue Assay
4.9. Western Blot Analysis
4.10. Titanium Particle-Induced Calvarial Osteolysis Model
4.11. Micro-Computed Tomography (micro-CT) Scanning
4.12. Histological and Histomorphometric Analysis
4.13. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gallo, J.; Vaculova, J.; Goodman, S.B.; Konttinen, Y.T.; Thyssen, J.P. Contributions of human tissue analysis to understanding the mechanisms of loosening and osteolysis in total hip replacement. Acta Biomater. 2014, 10, 2354–2366. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Ong, K.; Lau, E.; Mowat, F.; Halpern, M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J. Bone Jt. Surg. Am. Vol. 2007, 89, 780–785. [Google Scholar] [CrossRef]
- Abu-Amer, Y.; Darwech, I.; Clohisy, J.C. Aseptic loosening of total joint replacements: mechanisms underlying osteolysis and potential therapies. Arthritis Res. Ther. 2007, 9, S6. [Google Scholar] [CrossRef] [PubMed]
- Purdue, P.E.; Koulouvaris, P.; Potter, H.G.; Nestor, B.J.; Sculco, T.P. The cellular and molecular biology of periprosthetic osteolysis. Clin. Orthop. Relat. Res. 2007, 454, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Wu, B.; Peng, X.; Hua, J.; Hao, H.N.; Wooley, P.H. Implant wear induces inflammation, but not osteoclastic bone resorption, in RANK(−/−) mice. J. Orthop. Res. 2006, 24, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Mercatali, L.; Ricci, M.; Scarpi, E.; Serra, P.; Fabbri, F.; Ricci, R.; Liverani, C.; Zanoni, M.; Zoli, W.; Maltoni, R.; et al. RANK/RANK-L/OPG in patients with bone metastases treated with anticancer agents and zoledronic acid: A prospective study. Int. J. Mol. Sci. 2013, 14, 10683–10693. [Google Scholar] [CrossRef] [PubMed]
- Mansky, K.C.; Sankar, U.; Han, J.; Ostrowski, M.C. Microphthalmia transcription factor is a target of the p38 MAPK pathway in response to receptor activator of NF-kappa B ligand signaling. J. Biol. Chem. 2002, 277, 11077–11083. [Google Scholar] [CrossRef] [PubMed]
- Gingery, A.; Bradley, E.; Shaw, A.; Oursler, M.J. Phosphatidylinositol 3-kinase coordinately activates the MEK/ERK and AKT/NFkappaB pathways to maintain osteoclast survival. J. Cell. Biochem. 2003, 89, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; He, J.; Wang, J.; Cao, Y.; Ling, J.; Qian, J.; Lu, Y.; Li, H.; Zheng, Y.; Lan, Y.; et al. Constitutive activation of p38 MAPK in tumor cells contributes to osteolytic bone lesions in multiple myeloma. Leukemia 2012, 26, 2114–2123. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Udagawa, N.; Itoh, K.; Suda, K.; Murase, Y.; Nishihara, T.; Suda, T.; Takahashi, N. p38 MAPK-mediated signals are required for inducing osteoclast differentiation but not for osteoclast function. Endocrinology 2002, 143, 3105–3113. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.B.; Ma, T.; Spanogle, J.; Chiu, R.; Miyanishi, K.; Oh, K.; Plouhar, P.; Wadsworth, S.; Smith, R.L. Effects of a p38 MAP kinase inhibitor on bone ingrowth and tissue differentiation in rabbit chambers. J. Biomed. Mater. Res. Part A 2007, 81, 310–316. [Google Scholar] [CrossRef]
- Chen, M.; Chen, P.M.; Dong, Q.R.; Huang, Q.; She, C.; Xu, W. p38 signaling in titanium particle-induced MMP-2 secretion and activation in differentiating MC3T3-E1 cells. J. Biomed. Mater. Res. Part A 2014, 102, 2824–2832. [Google Scholar] [CrossRef]
- Zwerina, J.; Hayer, S.; Redlich, K.; Bobacz, K.; Kollias, G.; Smolen, J. S.; Schett, G. Activation of p38 MAPK is a key step in tumor necrosis factor-mediated inflammatory bone destruction. Arthritis Rheumatol. 2006, 54, 463–472. [Google Scholar] [CrossRef]
- Mihara, K.; Almansa, C.; Smeets, R.L.; Loomans, E.E.; Dulos, J.; Vink, P.M.; Rooseboom, M.; Kreutzer, H.; Cavalcanti, F.; Boots, A.M.; et al. A potent and selective p38 inhibitor protects against bone damage in murine collagen-induced arthritis: A comparison with neutralization of mouse TNFalpha. Br. J. Pharmacol. 2008, 154, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Saklatvala, J. The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr. Opin. Pharmacol. 2004, 4, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Zhang, Z.H.; Sun, E.; Jia, X.B. Effect of beta-cyclodextrin complexation on solubility and enzymatic conversion of naringin. Int. J. Mol. Sci. 2012, 13, 14251–14261. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Inbaraj, B.S.; Chen, B.H. Determination of phenolic acids and flavonoids in Taraxacum formosanum Kitam by liquid chromatography-tandem mass spectrometry coupled with a post-column derivatization technique. Int. J. Mol. Sci. 2012, 13, 260–285. [Google Scholar] [CrossRef] [PubMed]
- Renugadevi, J.; Prabu, S.M. Naringenin protects against cadmium-induced oxidative renal dysfunction in rats. Toxicology 2009, 256, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Raso, G.M.; Meli, R.; di Carlo, G.; Pacilio, M.; di Carlo, R. Inhibition of inducible nitric oxide synthase and cyclooxygenase-2 expression by flavonoids in macrophage J774A.1. Life Sci. 2001, 68, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, N.; Carroll, K.K. Inhibition of mammary cancer by citrus flavonoids. Adv. Exp. Med. Biol. 1998, 439, 227–236. [Google Scholar] [PubMed]
- Ming, L.G.; Lv, X.; Ma, X.N.; Ge, B.F.; Zhen, P.; Song, P.; Zhou, J.; Ma, H.P.; Xian, C.J.; Chen, K.M. The prenyl group contributes to activities of phytoestrogen 8-prenynaringenin in enhancing bone formation and inhibiting bone resorption in vitro. Endocrinology 2013, 154, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- La, V.D.; Tanabe, S.; Grenier, D. Naringenin inhibits human osteoclastogenesis and osteoclastic bone resorption. J. Periodontal Res. 2009, 44, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Swarnkar, G.; Sharan, K.; Siddiqui, J.A.; Mishra, J.S.; Khan, K.; Khan, M.P.; Gupta, V.; Rawat, P.; Maurya, R.; Dwivedi, A.K.; et al. A naturally occurring naringenin derivative exerts potent bone anabolic effects by mimicking oestrogen action on osteoblasts. Br. J. Pharmacol. 2012, 165, 1526–1542. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhen, L.; Zhang, G.; Wong, M.S.; Qin, L.; Yao, X. Osteogenic effects of flavonoid aglycones from an osteoprotective fraction of Drynaria fortunei—An in vitro efficacy study. Phytomedicine 2011, 18, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Ming, L.G.; Ge, B.F.; Wang, M.G.; Chen, K.M. Comparison between 8-prenylnarigenin and narigenin concerning their activities on promotion of rat bone marrow stromal cellsʼ osteogenic differentiation in vitro. Cell Prolif. 2012, 45, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Chen, J.; Zhang, J.; Hu, X.; Zhou, X.; Lu, Z.; Jiang, H. Naringenin inhibits angiotensin II-induced vascular smooth muscle cells proliferation and migration and decreases neointimal hyperplasia in balloon injured rat carotid arteries through suppressing oxidative stress. Biol. Pharm. Bull. 2013, 36, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Yilma, A.N.; Singh, S.R.; Morici, L.; Dennis, V.A. Flavonoid naringenin: A potential immunomodulator for Chlamydia trachomatis inflammation. Mediat. Inflamm. 2013, 2013, 102457. [Google Scholar] [CrossRef]
- Nie, Y.C.; Wu, H.; Li, P.B.; Xie, L.M.; Luo, Y.L.; Shen, J.G.; Su, W.W. Naringin attenuates EGF-induced MUC5AC secretion in A549 cells by suppressing the cooperative activities of MAPKs-AP-1 and IKKs-IkappaB-NF-kappaB signaling pathways. Eur. J. Pharmacol. 2012, 690, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Vafeiadou, K.; Vauzour, D.; Lee, H.Y.; Rodriguez-Mateos, A.; Williams, R.J.; Spencer, J.P. The citrus flavanone naringenin inhibits inflammatory signalling in glial cells and protects against neuroinflammatory injury. Arch. Biochem. Biophys. 2009, 484, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Yagura, T.; Motomiya, T.; Ito, M.; Honda, G.; Iida, A.; Kiuchi, F.; Tokuda, H.; Nishino, H. Anticarcinogenic compounds in the Uzbek medicinal plant, Helichrysum maracandicum. J. Nat. Med. 2008, 62, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Inacio, M.C.; Ake, C.F.; Paxton, E.W.; Khatod, M.; Wang, C.; Gross, T.P.; Kaczmarek, R.G.; Marinac-Dabic, D.; Sedrakyan, A. Sex and risk of hip implant failure: Assessing total hip arthroplasty outcomes in the United States. JAMA Intern. Med. 2013, 173, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Rodan, G.A.; Martin, T.J. Therapeutic approaches to bone diseases. Science 2000, 289, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, R. A new treatment for post-menopausal osteoporosis: Strontium ranelate. J. Endocrinol. Investig. 2005, 28, 50–57. [Google Scholar]
- Weitzmann, M.N.; Pacifici, R. Estrogen regulation of immune cell bone interactions. Ann. N. Y. Acad. Sci. 2006, 1068, 256–274. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, E.M.; Bi, Y.; Ragab, A.A.; Goldberg, V.M.; van de Motter, R.R. The role of osteoclast differentiation in aseptic loosening. J. Orthop. Res. 2002, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lacey, D.L.; Boyle, W.J.; Simonet, W.S.; Kostenuik, P.J.; Dougall, W.C.; Sullivan, J.K.; San Martin, J.; Dansey, R. Bench to bedside: Elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat. Rev. Drug Discov. 2012, 11, 401–419. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Strait, K.; Li, Y.; Dillehay, D.L.; Weitzmann, M.N. Suppression of NF-kappaB activation blocks osteoclastic bone resorption during estrogen deficiency. Int. J. Mol. Med. 2008, 21, 521–525. [Google Scholar] [PubMed]
- Meissner, J.D.; Freund, R.; Krone, D.; Umeda, P.K.; Chang, K.C.; Gros, G.; Scheibe, R.J. Extracellular signal-regulated kinase 1/2-mediated phosphorylation of p300 enhances myosin heavy chain I/beta gene expression via acetylation of nuclear factor of activated T cells c1. Nucleic Acids Res. 2011, 39, 5907–5925. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.; Qu, X.; Li, H.; Yang, K.; Wan, P.; Tan, L.; Ouyang, Z.; Liu, X.; Tian, B.; Xiao, F.; et al. The effect of metallic magnesium degradation products on osteoclast-induced osteolysis and attenuation of NF-kappaB and NFATc1 signaling. Biomaterials 2014, 35, 6299–6310. [Google Scholar] [CrossRef]
- Liu, F.; Zhu, Z.; Mao, Y.; Liu, M.; Tang, T.; Qiu, S. Inhibition of titanium particle-induced osteoclastogenesis through inactivation of NFATc1 by VIVIT peptide. Biomaterials 2009, 30, 1756–1762. [Google Scholar] [CrossRef] [PubMed]
- Pivetta, E.; Scapolan, M.; Wassermann, B.; Steffan, A.; Colombatti, A.; Spessotto, P. Blood-derived human osteoclast resorption activity is impaired by Hyaluronan-CD44 engagement via a p38-dependent mechanism. J. Cell. Physiol. 2011, 226, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Choi, S.Y.; Lee, S.Y.; Lee, J.Y.; Kim, H.S.; Lee, N.K. Caffeine enhances osteoclast differentiation and maturation through p38 MAP kinase/Mitf and DC-STAMP/CtsK and TRAP pathway. Cell Signal. 2013, 25, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Cano, E.; Hazzalin, C.A.; Mahadevan, L.C. Anisomycin-activated protein kinases p45 and p55 but not mitogen-activated protein kinases ERK-1 and -2 are implicated in the induction of c-fos and c-jun. Mol. Cell. Biol. 1994, 14, 7352–7362. [Google Scholar] [PubMed]
- Hazzalin, C.A.; Le Panse, R.; Cano, E.; Mahadevan, L.C. Anisomycin selectively desensitizes signalling components involved in stress kinase activation and fos and jun induction. Mol. Cell. Biol. 1998, 18, 1844–1854. [Google Scholar] [PubMed]
- Okazaki, Y.; Gotoh, E. Comparison of metal release from various metallic biomaterials in vitro. Biomaterials 2005, 26, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Savarino, L.; Granchi, D.; Ciapetti, G.; Cenni, E.; Nardi Pantoli, A.; Rotini, R.; Veronesi, C.A.; Baldini, N.; Giunti, A. Ion release in patients with metal-on-metal hip bearings in total joint replacement: A comparison with metal-on-polyethylene bearings. J. Biomed. Mater. Res. 2002, 63, 467–474. [Google Scholar] [CrossRef]
- Taki, N.; Tatro, J.M.; Lowe, R.; Goldberg, V.M.; Greenfield, E.M. Comparison of the roles of IL-1, IL-6, and TNFalpha in cell culture and murine models of aseptic loosening. Bone 2007, 40, 1276–1283. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, K.; Bauer, T.W.; Stulberg, B.N.; Wilde, A.H. Comparison and quantitation of wear debris of failed total hip and total knee arthroplasty. J. Biomed. Mater. Res. 1996, 31, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Baumann, B.; Seufert, J.; Jakob, F.; Noth, U.; Rolf, O.; Eulert, J.; Rader, C.P. Activation of NF-kappaB signalling and TNFalpha-expression in THP-1 macrophages by TiAlV- and polyethylene-wear particles. J. Orthop. Res. 2005, 23, 1241–1248. [Google Scholar] [PubMed]
- Von Knoch, M.; Jewison, D.E.; Sibonga, J.D.; Sprecher, C.; Morrey, B.F.; Loer, F.; Berry, D.J.; Scully, S.P. The effectiveness of polyethylene versus titanium particles in inducing osteolysis in vivo. J. Orthop. Res. 2004, 22, 237–243. [Google Scholar]
- Masui, T.; Sakano, S.; Hasegawa, Y.; Warashina, H.; Ishiguro, N. Expression of inflammatory cytokines, RANKL and OPG induced by titanium, cobalt-chromium and polyethylene particles. Biomaterials 2005, 26, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- Baumann, B.; Rader, C.P.; Seufert, J.; Noth, U.; Rolf, O.; Eulert, J.; Jakob, F. Effects of polyethylene and TiAlV wear particles on expression of RANK, RANKL and OPG mRNA. Acta Orthop. Scand. 2004, 75, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Qin, A.; Cheng, T.S.; Lin, Z.; Cao, L.; Chim, S.M.; Pavlos, N. J.; Xu, J.; Zheng, M.H.; Dai, K.R. Prevention of wear particle-induced osteolysis by a novel V-ATPase inhibitor saliphenylhalamide through inhibition of osteoclast bone resorption. PLoS One 2012, 7, e34132. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Wu, C.; Tian, B.; Liu, X.; Zhai, Z.; Qu, X.; Jiang, C.; Ouyang, Z.; Mao, Y.; Tang, T.; et al. The Inhibition of RANKL-Induced Osteoclastogenesis through the Suppression of p38 Signaling Pathway by Naringenin and Attenuation of Titanium-Particle-Induced Osteolysis. Int. J. Mol. Sci. 2014, 15, 21913-21934. https://doi.org/10.3390/ijms151221913
Wang W, Wu C, Tian B, Liu X, Zhai Z, Qu X, Jiang C, Ouyang Z, Mao Y, Tang T, et al. The Inhibition of RANKL-Induced Osteoclastogenesis through the Suppression of p38 Signaling Pathway by Naringenin and Attenuation of Titanium-Particle-Induced Osteolysis. International Journal of Molecular Sciences. 2014; 15(12):21913-21934. https://doi.org/10.3390/ijms151221913
Chicago/Turabian StyleWang, Wengang, Chuanlong Wu, Bo Tian, Xuqiang Liu, Zanjing Zhai, Xinhua Qu, Chuan Jiang, Zhengxiao Ouyang, Yuanqing Mao, Tingting Tang, and et al. 2014. "The Inhibition of RANKL-Induced Osteoclastogenesis through the Suppression of p38 Signaling Pathway by Naringenin and Attenuation of Titanium-Particle-Induced Osteolysis" International Journal of Molecular Sciences 15, no. 12: 21913-21934. https://doi.org/10.3390/ijms151221913
APA StyleWang, W., Wu, C., Tian, B., Liu, X., Zhai, Z., Qu, X., Jiang, C., Ouyang, Z., Mao, Y., Tang, T., Qin, A., & Zhu, Z. (2014). The Inhibition of RANKL-Induced Osteoclastogenesis through the Suppression of p38 Signaling Pathway by Naringenin and Attenuation of Titanium-Particle-Induced Osteolysis. International Journal of Molecular Sciences, 15(12), 21913-21934. https://doi.org/10.3390/ijms151221913