Molecular Mechanisms Underlying the Effects of Statins in the Central Nervous System


Abstract
:1. Introduction
2. Pharmacology
2.1. Mechanism of Action
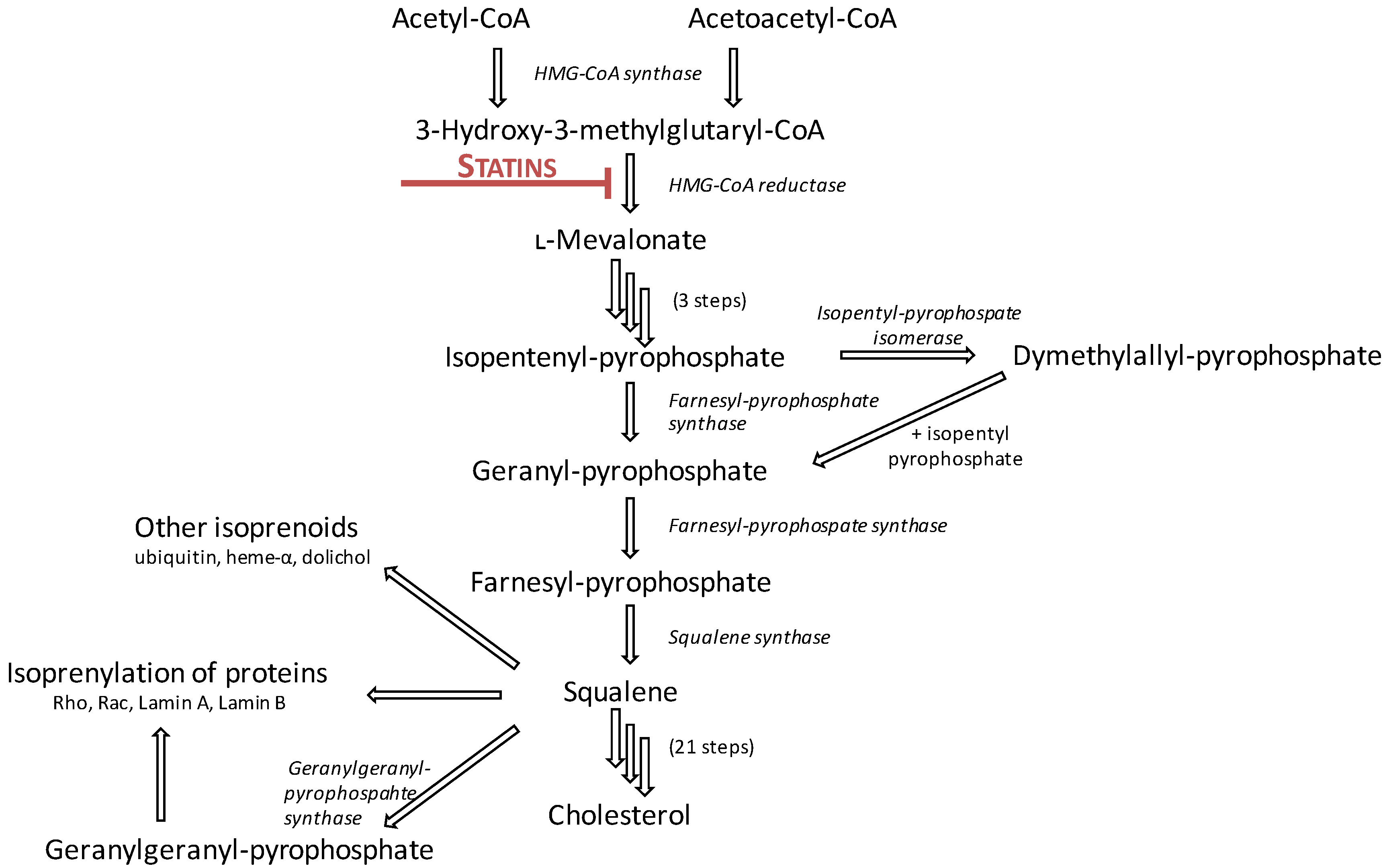
2.2. Pharmacokinetics

{kind=link}
| PK Parameter | Atorvastatin | Fluvastatin | Lovastatin | Pitavastatin | Pravastatin | Rosuvastatin | Simvastatin |
|---|---|---|---|---|---|---|---|
| Molecular Structure |  | 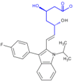 | 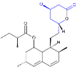 |  |  | 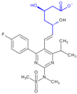 | 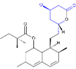 |
| Statin Type | II | II | I | II | I | II | I |
| Dosing Time | Any time of day | Bedtime | With food morning & night | Any time of day | Bedtime | Any time of day | Evening |
| Prodrug | No | No | Yes | No | No | No | Yes |
| Bioavailability | 12% | 9%–50% | 5% | 51% | 18% | 20% | <5% |
| Half-Life | 14 h | 2.3 h | 3 h | 12 h | 1.3–2.7 h | 19 h | 3 h |
| Volume of Distribution | 381 L | 330 L | (not available) | 148 L | 35 L | 134 L | (not available) |
| Log D * | 1.53 | 1.75 | 3.91 (lactone)/1.51 (acid) | 1.50 | −0.47 | −0.25 to −0.50 | 4.40 (lactone)/1.80 (acid) |
| Lipophilicity | Lipophilic | Lipophilic | Lipophilic | Lipophilic | Hydrophilic | Hydrophilic | Lipophilic |
| Active Metabolites | Yes | No | Yes | Yes (minimal) | Yes (minimal) | Yes (minimal) | Yes |
| CYP Substrate | 3A4 | 2C9 | 3A4 | 2C8, limited 2C9 mostly glucuronidation | Limited 3A4 mostly sulfation | Limited 2C9 mostly excreted unchanged | 3A4 |
| Effects on p-Glycoprotein | Substrate and inhibitor | No significant inhibition | Substrate and inhibitor | No significant inhibition | No significant inhibition | No significant inhibition | Substrate and inhibitor |
| OATP Transporters | 1B1, 2B1 | 1B1, 1B3, 2B1 | 1B1 | 1A2, 1B1, 1B3 | 1B1, 1B3, 2B1 | 1A2, 1B1, 1B3, 2B1 | 1B1 |
| Protein Binding | Very high (98%) | Very high (98%) | Very high (95%) | Very high (96%) | Moderate (50%) | High (90%) | Very high (95%) |
| Excretion (Renal) | <2% | 6% | 10% | 2% | 20% | 10% | 13% |
| Excretion (Faecal) | >98% | 93% | 83% | 79% | 70% | 90% | 60% |
3. Statins in the Central Nervous System (CNS)
3.1. Effects on Brain Cholesterol
3.2. CNS Entry
4. Statins and Cognition
| Disorder | Possible Statin-Induced Mechanisms | Strength of Evidence | Overall Consensus |
|---|---|---|---|
| General cognition | ↓ FPP and/or GGPP; modulation of adult neurogenesis; ↑ expression of neural growth factors. | Limited in vitro and in vivo studies. Conflicting evidence from epidemiological studies and randomised controlled trials. Case reports of negative effects on cognition. Recent meta-analyses suggest long term statin use may reduce incident dementia. | Long-term statin treatment appears to be beneficial for cognitive function. Whether statins can cause acute cognitive disruption as a rare adverse effect is unclear due to lack of causal evidence from case reports. Identification of underlying mechanisms in vitro or in vivo is difficult due to the subjective nature of acute cognition changes. |
| Alzheimer’s disease | ↓ FPP and/or GGPP; ↓ APP production; ↓ ROCK activity; ↓ amyloid-β production; ↑ amyloid-β degradation; ↓ neuroinflammation; ↓ ROS. | Numerous in vitro and in vivo studies, however some data appears model-dependent so requires careful interpretation. Several randomised controlled trials, and multiple systematic reviews and meta-analyses have been conducted. | Studies suggest statins, if started before old age and without cognitive dysfunction at baseline, may reduce incidence of AD. It is likely different statins have different capacities for inducing this effect. |
| Parkinson’s disease | ↓ ROS; ↓ nitric oxide; ↓ lipid peroxidation; ↓ neuroinflammation; ↓ NF-κB activity; ↓ neuronal loss. | Numerous in vitro and in vivo studies, however data from prospective studies or clinical trials is lacking. | Data from cell and animal models is encouraging, however further well-designed prospective studies are needed to evaluate statins’ clinical application in PD. |
| Multiple sclerosis | Altered Th1/Th2 ratio; ↓ neuroinflammation; ↑ remyelination-associated genes; ↑/↓ differentiation from OPC to OD; ↓ ROCK activity; modulation of NF-κB activity. | Numerous in vitro and in vivo studies, however results from these are highly conflicting. Simvastatin has recently completed phase II testing as a treatment for MS. | Vast discrepancies between models limits our understanding of the mechanisms of statins in MS. It appears likely that modulation of neuroinflammation and/or T cell immunity is involved. Further studies needed to determine if benefit is seen with statins other than simvastatin in MS. |
| Neurofibromatosis Type I | ↓ Ras activity; rescue long-term potentiation deficit. | Limited in vitro and in vivo data. Conflicting data from randomised controlled trials. | Further cell and animal studies are recommended to better understand possible clinical application in NF-1 before any further trials in children with the disorder are conducted. |
4.1. Cognitive Function
4.2. Alzheimer’s Disease
4.3. Parkinson’s Disease
4.4. Multiple Sclerosis
4.5. Neurofibromatosis Type I
5. Statins and Neurological Disease
| Disorder | Possible Statin-Induced Mechanisms | Strength of Evidence | Overall Consensus |
|---|---|---|---|
| Stroke | Modulation of eNOS; ↓ nitric oxide; ↓ ROS; ↓ MMPs. | Many in vitro and in vivo studies. Supported by meta-analyses and well-designed randomised controlled trials. | Statins reduce incidence of ischemic and haemorrhagic stroke, likely through antioxidant effects. |
| Epilepsy | Lipid raft disruption; ROCK inhibition; ↑ PI3K pathway activity. | Limited in vitro and in vivo studies. | Very different excitoprotective properties between statins. More studies are required. |
| Depression | Modulation of NMDA receptor activity; ↓ nitric oxide. | Mainly epidemiological studies. Recent meta-analysis suggested statins reduce risk of depression. Limited mechanism-based studies. | Whether the observed effects from qualitative studies are statin-induced, due to decreased cholesterol, or due to an improved quality of life, or a combination is unclear. |
| Psychiatric disorders | Unknown. | Limited observational studies. | Causality is unclear. If prevalence is affected by statins, it is thought to be rare and only in predisposed patients. |
| CNS cancers | ↑ caspase-3-mediated apoptosis; cell-cycle arrest; ↓ ERK1/2; ↓ Akt activity. | Limited in vitro and in vivo studies. Retrospective studies suggest no link between statin use and cancer incidence. | Further in vivo studies should be used to clarify statins’ effects. Directed epidemiological studies would also prove useful. |
| Brain and spinal cord injury | ↓ apoptosis; ↓ inflammation; ↓ RhoA/ROCK activity; ↓axonal degradation; ↓ myelin degradation. | Numerous in vivo studies. | Statins appear to exert beneficial effects in vivo if initiated immediately post-TBI/SCI. Due to some conflicting data, further well-designed studies are required before clinical application can be assessed. |
5.1. Stroke
5.2. Epilepsy
5.3. Depression
5.4. Psychiatric Disorders
5.5. CNS Cancers
5.6. Brain and Spinal Cord Injury
6. Conclusions
Acknowledgments
Author Contributions
Abbreviations
| HMG-CoA | 3-hydroxy-3-methylglutaryl coenzyme A |
| 6-OHDA | 6-hydroxydopamine |
| AD | Alzheimer’s disease |
| Aβ | amyloid beta peptide |
| APP | amyloid precursor protein |
| BBB | blood-brain barrier |
| BDNF | brain-derived neurotrophic factor |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| CYP | cytochrome P450 |
| eNOS | endothelial nitric oxide synthase |
| EAE | experimental autoimmune encephalomyelitis |
| ERK | extracellular-signal-regulated kinase |
| FPP | farnesylpyrophosphate |
| GGPP | geranylgeranylpyrophosphate |
| iNOS | inducible nitric oxide synthase |
| IFN | interferon |
| IL | interleukin |
| LPS | lipopolysaccharide |
| LDL | low-density lipoprotein |
| MMP | matrix metalloproteinase |
| MAPK | mitogen-activated protein kinase |
| MS | multiple sclerosis |
| NMDA | N-methyl-d-aspartate |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NF-1 | neurofibromatosis type 1 |
| OD | oligodendrocyte |
| OPC | oligodendrocyte progenitor cell |
| PD | Parkinson’s disease |
| PPAR | peroxisome proliferator-activated receptor |
| PK | pharmacokinetic |
| PI3K | phosphatidylinositol-3-kinase |
| ROS | reactive oxygen species |
| rht-PA | recombination human tissue plasminogen activator |
| ROCK | Rho-associated coiled-coil kinase1/2 |
| sAPP | soluble APP |
| SCI | spinal cord injury |
| Th | T helper |
| TBI | traumatic brain injury |
| TNF | tumour necrosis factor |
| VEGF | vascular endothelial growth factor |
Conflicts of Interest
References
- Parsaik, A.K.; Singh, B.; Hassan, M.M.; Singh, K.; Mascarenhas, S.S.; Williams, M.D.; Lapid, M.I.; Richardson, J.W.; West, C.P.; Rummans, T.A. Statins use and risk of depression: A systematic review and meta-analysis. J. Affect. Disord. 2014, 160, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Swiger, K.J.; Manalac, R.J.; Blumenthal, R.S.; Blaha, M.J.; Martin, S.S. Statins and cognition: A systematic review and meta-analysis of short- and long-term cognitive effects. Mayo Clin. Proc. 2013, 88, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Ni Chroinin, D.; Asplund, K.; Asberg, S.; Callaly, E.; Cuadrado-Godia, E.; Diez-Tejedor, E.; di Napoli, M.; Engelter, S.T.; Furie, K.L.; Giannopoulos, S.; et al. Statin therapy and outcome after ischemic stroke: Systematic review and meta-analysis of observational studies and randomized trials. Stroke 2013, 44, 448–456. [Google Scholar] [CrossRef] [PubMed]
- McKinney, J.S.; Kostis, W.J. Statin therapy and the risk of intracerebral hemorrhage: A meta-analysis of 31 randomized controlled trials. Stroke 2012, 43, 2149–2156. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: An update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, H.N.; Le, N.A.; Short, M.P.; Ramakrishnan, R.; Desnick, R.J. Suppression of apolipoprotein B production during treatment of cholesteryl ester storage disease with lovastatin. Implications for regulation of apolipoprotein B synthesis. J. Clin. Investig. 1987, 80, 1692–1697. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S. Consensus statement: Role of therapy with “statins” in patients with hypertriglyceridemia. Am. J. Cardiol. 1998, 81, 1B. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.; Steinman, L.; Zamvil, S.S. Statin therapy and autoimmune disease: From protein prenylation to immunomodulation. Nat. Rev. Immunol. 2006, 6, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Gotto, A.M., Jr.; Moon, J. Pitavastatin for the treatment of primary hyperlipidemia and mixed dyslipidemia. Expert Rev. Cardiovasc. Ther. 2010, 8, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.J.; Reinoso, R.F.; Sanchez Navarro, A.; Prous, J.R. Clinical pharmacokinetics of statins. Methods Find. Exp. Clin. Pharmacol. 2003, 25, 457–481. [Google Scholar] [CrossRef] [PubMed]
- Lennernas, H. Clinical pharmacokinetics of atorvastatin. Clin. Pharmacokinet. 2003, 42, 1141–1160. [Google Scholar] [CrossRef] [PubMed]
- Quion, J.A.V.; Jones, P.H. Clinical pharmacokinetics of pravastatin. Clin. Pharmacokinet. 1994, 27, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Davignon, J. Pleiotropic effects of pitavastatin. Br. J. Clin. Pharmacol. 2012, 73, 518–535. [Google Scholar] [CrossRef] [PubMed]
- Alagona, P., Jr. Pitavastatin: Evidence for its place in treatment of hypercholesterolemia. Core Evid. 2010, 5, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Beltowski, J.; Wojcicka, G.; Jamroz-Wisniewska, A. Adverse effects of statins—Mechanisms and consequences. Curr. Drug Saf. 2009, 4, 209–228. [Google Scholar] [CrossRef] [PubMed]
- Kowa Pharmaceuticals America Inc. Livalo (pitavastatin calcium) tablets prescribing information. Available online: http://www.kowapharma.com/documents/LIVALO_PI_CURRENT.pdf (accessed on 27 August 2014).
- Holtzman, C.W.; Wiggins, B.S.; Spinler, S.A. Role of P-glycoprotein in statin drug interactions. Pharmacotherapy 2006, 26, 1601–1607. [Google Scholar] [CrossRef] [PubMed]
- Goard, C.A.; Mather, R.G.; Vinepal, B.; Clendening, J.W.; Martirosyan, A.; Boutros, P.C.; Sharom, F.J.; Penn, L.Z. Differential interactions between statins and P-glycoprotein: Implications for exploiting statins as anticancer agents. Int. J. Cancer 2010, 127, 2936–2948. [Google Scholar] [CrossRef] [PubMed]
- Kajinami, K.; Takekoshi, N.; Saito, Y. Pitavastatin: Efficacy and safety profiles of a novel synthetic HMG-CoA reductase inhibitor. Cardiovasc. Drug Rev. 2003, 21, 199–215. [Google Scholar]
- Niemi, M. Transporter pharmacogenetics and statin toxicity. Clin. Pharmacol. Ther. 2010, 87, 130–133. [Google Scholar] [CrossRef] [PubMed]
- McKenney, J.M. Pharmacologic characteristics of statins. Clin. Cardiol. 2003, 26, 32–38. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D. Thematic review series: Brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M.; Turley, S.D. Cholesterol metabolism in the brain. Curr. Opin. Lipidol. 2001, 12, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Nieweg, K.; Schaller, H.; Pfrieger, F.W. Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J. Neurochem. 2009, 109, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Bellosta, S. Cholesterol: Its regulation and role in central nervous system disorders. Cholesterol 2012, 2012, 19. [Google Scholar] [CrossRef]
- Lutjohann, D.; Stroick, M.; Bertsch, T.; Kuhl, S.; Lindenthal, B.; Thelen, K.; Andersson, U.; Bjorkhem, I.; von Bergmann, K.; Fassbender, K. High doses of simvastatin, pravastatin and cholesterol reduce brain cholesterol synthesis in guinea pigs. Steroids 2004, 69, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Thelen, K.M.; Rentsch, K.M.; Gutteck, U.; Heverin, M.; Olin, M.; Andersson, U.; von Eckardstein, A.; Björkhem, I.; Lütjohann, D. Brain cholesterol synthesis in mice is affected by high dose of simvastatin but not of pravastatin. J. Pharmacol. Exp. Ther. 2006, 316, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Elmberger, P.O.; Edlund, C.; Kristensson, K.; Dallner, G. Rates of cholesterol, ubiquinone, dolichol and dolichyl-P biosynthesis in rat brain slices. FEBS Lett. 1990, 269, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Bjoand̈rkhem, I.; Heverin, M.; Leoni, V.; Meaney, S.; Diczfalusy, U. Oxysterols and Alzheimer’s disease. Acta Neurol. Scand. 2006, 114, 43–49. [Google Scholar] [CrossRef]
- Cibičková, L. Statins and their influence on brain cholesterol. J. Clin. Lipidol. 2011, 5, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Vega, G.L.; Weiner, M.F.; Lipton, A.M.; Von Bergmann, K.; Lutjohann, D.; Moore, C.; Svetlik, D. Reduction in levels of 24S-hydroxycholesterol by statin treatment in patients with Alzheimer disease. Arch. Neurol. 2003, 60, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Schwarzler, F.; Lutjohann, D.; von Bergmann, K.; Beyreuther, K.; Dichgans, J.; Wormstall, H.; Hartmann, T.; Schulz, J.B. Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann. Neurol. 2002, 52, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Stroick, M.; Bertsch, T.; Ragoschke, A.; Kuehl, S.; Walter, S.; Walter, J.; Brechtel, K.; Muehlhauser, F.; von Bergmann, K. Effects of statins on human cerebral cholesterol metabolism and secretion of Alzheimer amyloid peptide. Neurology 2002, 59, 1257–1258. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Vega, G.L.; Lutjohann, D.; Locascio, J.J.; Tennis, M.K.; Deng, A.; Atri, A.; Hyman, B.T.; Irizarry, M.C.; Growdon, J.H. Effects of simvastatin on cholesterol metabolism and Alzheimer disease biomarkers. Alzheimer Dis. Assoc. Disord. 2010, 24, 220–226. [Google Scholar] [PubMed]
- Locatelli, S.; Lutjohann, D.; Schmidt, H.H.; Otto, C.; Beisiegel, U.; von Bergmann, K. Reduction of plasma 24S-hydroxycholesterol (cerebrosterol) levels using high-dosage simvastatin in patients with hypercholesterolemia: Evidence that simvastatin affects cholesterol metabolism in the human brain. Arch. Neurol. 2002, 59, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Thelen, K.M.; Lutjohann, D.; Vesalainen, R.; Janatuinen, T.; Knuuti, J.; von Bergmann, K.; Lehtimaki, T.; Laaksonen, R. Effect of pravastatin on plasma sterols and oxysterols in men. Eur. J. Clin. Pharmacol. 2006, 62, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Thelen, K.M.; Laaksonen, R.; Paiva, H.; Lehtimaki, T.; Lutjohann, D. High-dose statin treatment does not alter plasma marker for brain cholesterol metabolism in patients with moderately elevated plasma cholesterol levels. J. Clin. Pharmacol. 2006, 46, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Anuna, L.N.; Eckert, G.P.; Keller, J.H.; Igbavboa, U.; Franke, C.; Fechner, T.; Schubert-Zsilavecz, M.; Karas, M.; Muller, W.E.; Wood, W.G. Chronic administration of statins alters multiple gene expression patterns in mouse cerebral cortex. J. Pharmacol. Exp. Ther. 2005, 312, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Botti, R.E.; Triscari, J.; Pan, H.Y.; Zayat, J. Concentrations of pravastatin and lovastatin in cerebrospinal fluid in healthy subjects. Clin. Neuropharmacol. 1991, 14, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M. Role of OATP transporters in the disposition of drugs. Pharmacogenomics 2007, 8, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Tamai, I.; Takanaga, H.; Maeda, H.; Ogihara, T.; Yoneda, M.; Tsuji, A. Proton-cotransport of pravastatin across intestinal brush-border membrane. Pharm. Res. 1995, 12, 1727–1732. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Fernandez, C.H.; Cameron, J.C. Is statin-associated cognitive impairment clinically relevant? A narrative review and clinical recommendations. Ann. Pharmacother. 2012, 46, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, L.R.; Mitton, M.W.; Arvik, B.M.; Doraiswamy, P.M. Statin-associated memory loss: Analysis of 60 case reports and review of the literature. Pharmacotherapy 2003, 23, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Mandas, A.; Congiu, M.G.; Abete, C.; Dessi, S.; Manconi, P.E.; Musio, M.; Columbu, S.; Racugno, W. Cognitive decline and depressive symptoms in late-life are associated with statin use: Evidence from a population-based study of Sardinian old people living in their own home. Neurol. Res. 2014, 36, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Sparks, D.L.; Kryscio, R.J.; Sabbagh, M.N.; Connor, D.J.; Sparks, L.M.; Liebsack, C. Reduced risk of incident AD with elective statin use in a clinical trial cohort. Curr. Alzheimer Res. 2008, 5, 416–421. [Google Scholar] [PubMed]
- Jick, H.; Zornberg, G.L.; Jick, S.S.; Seshadri, S.; Drachman, D.A. Statins and the risk of dementia. Lancet 2000, 356, 1627–1631. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Wang, S.; Li, N.-C.; Lee, A.; Lee, T.; Kazis, L. Simvastatin is associated with a reduced incidence of dementia and Parkinson’s disease. BMC Med. 2007, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bettermann, K.; Arnold, A.M.; Williamson, J.; Rapp, S.; Sink, K.; Toole, J.F.; Carlson, M.C.; Yasar, S.; DeKosky, S.; Burke, G.L. Statins, risk of dementia and cognitive function: Secondary analysis of the ginkgo evaluation of memory study. J. Stroke Cerebrovasc. Dis. 2012, 21, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; Simes, J.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [CrossRef] [PubMed]
- Anstey, K.J.; Lipnicki, D.M.; Low, L.-F. Cholesterol as a risk factor for dementia and cognitive decline: A systematic review of prospective studies with meta-analysis. Am. J. Geriatr. Psychiatry 2008, 16, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Dufouil, C.; Richard, F.; Fievet, N.; Dartigues, J.F.; Ritchie, K.; Tzourio, C.; Amouyel, P.; Alperovitch, A. APOE genotype, cholesterol level, lipid-lowering treatment and dementia: The three-city study. Neurology 2005, 64, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, K.; Barrett-Connor, E.; Lin, F.; Grady, D. Serum lipoprotein levels, statin use and cognitive function in older women. Arch. Neurol. 2002, 59, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Mans, R.A.; McMahon, L.L.; Li, L. Simvastatin-mediated enhancement of long-term potentiation is driven by farnesyl-pyrophosphate depletion and inhibition of farnesylation. Neuroscience 2012, 202, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kotti, T.J.; Ramirez, D.M.; Pfeiffer, B.E.; Huber, K.M.; Russell, D.W. Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3869–3874. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.G.; Bosel, J.; Stoeckel, M.; Megow, D.; Dirnagl, U.; Endres, M. HMG-CoA reductase inhibition causes neurite loss by interfering with geranylgeranylpyrophosphate synthesis. J. Neurochem. 2004, 89, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Ekdahl, C.T.; Claasen, J.H.; Bonde, S.; Kokaia, Z.; Lindvall, O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13632–13637. [Google Scholar] [CrossRef] [PubMed]
- Monje, M.L.; Toda, H.; Palmer, T.D. Inflammatory blockade restores adult hippocampal neurogenesis. Science 2003, 302, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Robin, N.C.; Agoston, Z.; Biechele, T.L.; James, R.G.; Berndt, J.D.; Moon, R.T. Simvastatin promotes adult hippocampal neurogenesis by enhancing Wnt/β-catenin signaling. Stem Cell Rep. 2014, 2, 9–17. [Google Scholar] [CrossRef]
- Lu, D.; Qu, C.; Goussev, A.; Jiang, H.; Lu, C.; Schallert, T.; Mahmood, A.; Chen, J.; Li, Y.; Chopp, M. Statins increase neurogenesis in the dentate gyrus, reduce delayed neuronal death in the hippocampal CA3 region, and improve spatial learning in rat after traumatic brain injury. J. Neurotrauma 2007, 24, 1132–1146. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lu, D.; Jiang, H.; Xiong, Y.; Qu, C.; Li, B.; Mahmood, A.; Zhou, D.; Chopp, M. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J. Neurotrauma 2008, 25, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.; Langui, D.; Ulrich, J. Alzheimer’s disease: A description of the structural lesions. Brain Pathol. 1991, 1, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Kellman, W.; Ruosseau, P.; Celesia, G.G.; Siegel, G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 2000, 57, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Haag, M.D.M.; Hofman, A.; Koudstaal, P.J.; Stricker, B.H.C.; Breteler, M.M.B. Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The Rotterdam study. J. Neurol. Neurosur. Psychiatry 2009, 80, 13–17. [Google Scholar] [CrossRef]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lutjohann, D.; Keller, P.; Runz, H.; Kuhl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease β-amyloid peptides Aβ42 and Aβ40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar] [CrossRef] [PubMed]
- Kurata, T.; Miyazaki, K.; Kozuki, M.; Panin, V.-L.; Morimoto, N.; Ohta, Y.; Nagai, M.; Ikeda, Y.; Matsuura, T.; Abe, K. Atorvastatin and pitavastatin improve cognitive function and reduce senile plaque and phosphorylated tau in aged APP mice. Brain Res. 2011, 1371, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Tramontina, A.C.; Wartchow, K.M.; Rodrigues, L.; Biasibetti, R.; Quincozes-Santos, A.; Bobermin, L.; Tramontina, F.; Goncalves, C.A. The neuroprotective effect of two statins: Simvastatin and pravastatin on a streptozotocin-induced model of Alzheimer’s disease in rats. J. Neural. Transm. 2011, 118, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Eckert, G.P.; Hooff, G.P.; Strandjord, D.M.; Igbavboa, U.; Volmer, D.A.; Muller, W.E.; Wood, W.G. Regulation of the brain isoprenoids farnesyl- and geranyl-geranylpyrophosphate is altered in male Alzheimer patients. Neurobiol. Dis. 2009, 35, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Pedrini, S.; Carter, T.L.; Prendergast, G.; Petanceska, S.; Ehrlich, M.E.; Gandy, S. Modulation of statin-activated shedding of Alzheimer APP ectodomain by ROCK. PLoS Med. 2005, 2, e18. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Sato, N.; Kurinami, H.; Takeuchi, D.; Takeda, S.; Shimamura, M.; Yamashita, T.; Uchiyama, Y.; Rakugi, H.; Morishita, R. Reduction of brain β-amyloid (Aβ) by fluvastatin, a hydroxymethylglutaryl-CoA reductase inhibitor, through increase in degradation of amyloid precursor protein C-terminal fragments (APP-CTFs) and Aβ clearance. J. Biol. Chem. 2010, 285, 22091–22102. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.M.; Wilkinson, B.L.; Golde, T.E.; Landreth, G. Statins reduce amyloid-β production through inhibition of protein isoprenylation. J. Biol. Chem. 2007, 282, 26832–26844. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, A.; Araki, W.; Oda, A.; Tomidokoro, Y.; Tamaoka, A. Statins reduce amyloid β-peptide production by modulating amyloid precursor protein maturation and phosphorylation through a cholesterol-independent mechanism in cultured neurons. Neurochem. Res. 2013, 38, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Pac-Soo, C.; Lloyd, D.G.; Vizcaychipi, M.P.; Ma, D. Statins: The role in the treatment and prevention of Alzheimer’s neurodegeneration. J. Alzheimers Dis. 2011, 27, 1–10. [Google Scholar] [PubMed]
- Clarke, R.M.; O’Connell, F.; Lyons, A.; Lynch, M.A. The HMG-CoA reductase inhibitor, atorvastatin, attenuates the effects of acute administration of amyloid-β1–42 in the rat hippocampus in vivo. Neuropharmacology 2007, 52, 136–145. [Google Scholar] [CrossRef]
- Cordle, A.; Landreth, G. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors attenuate β-amyloid-induced microglial inflammatory responses. J. Neurosci. 2005, 25, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Asahi, M.; Huang, Z.; Thomas, S.; Yoshimura, S.-I.; Sumii, T.; Mori, T.; Qiu, J.; Amin-Hanjani, S.; Huang, P.L.; Liao, J.K. Protective effects of statins involving both eNOS and tPA in focal cerebral ischemia. J. Cerebr. Blood Flow Metab. 2005, 25, 722–729. [Google Scholar] [CrossRef]
- Barone, E.; Cenini, G.; di Domenico, F.; Martin, S.; Sultana, R.; Mancuso, C.; Murphy, M.P.; Head, E.; Butterfield, D.A. Long-term high-dose atorvastatin decreases brain oxidative and nitrosative stress in a preclinical model of Alzheimer disease: A novel mechanism of action. Pharmacol. Res. 2011, 63, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Meske, V.; Albert, F.; Richter, D.; Schwarze, J.; Ohm, T.G. Blockade of HMG-CoA reductase activity causes changes in microtubule-stabilizing protein tau via suppression of geranylgeranylpyrophosphate formation: Implications for Alzheimer’s disease. Eur. J. Neurosci. 2003, 17, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Baudry, M.; Liu, J.; Yao, Y.; Fu, L.; Brucher, F.; Lynch, G. Inhibition of geranylgeranylation mediates the effects of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors on microglia. J. Biol. Chem. 2004, 279, 48238–48245. [Google Scholar] [CrossRef] [PubMed]
- Piermartiri, T.C.; Figueiredo, C.P.; Rial, D.; Duarte, F.S.; Bezerra, S.C.; Mancini, G.; de Bem, A.F.; Prediger, R.D.; Tasca, C.I. Atorvastatin prevents hippocampal cell death, neuroinflammation and oxidative stress following amyloid-β(1–40) administration in mice: Evidence for dissociation between cognitive deficits and neuronal damage. Exp. Neurol. 2010, 226, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.K.; Nicolakakis, N.; Fernandes, P.; Ongali, B.; Brouillette, J.; Quirion, R.; Hamel, E. Simvastatin improves cerebrovascular function and counters soluble amyloid-β, inflammation and oxidative stress in aged APP mice. Neurobiol. Dis. 2009, 35, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.K.; Lecrux, C.; Rosa-Neto, P.; Hamel, E. Age-dependent rescue by simvastatin of Alzheimer’s disease cerebrovascular and memory deficits. J. Neurosci. 2012, 32, 4705–4715. [Google Scholar] [CrossRef] [PubMed]
- Tamboli, I.Y.; Barth, E.; Christian, L.; Siepmann, M.; Kumar, S.; Singh, S.; Tolksdorf, K.; Heneka, M.T.; Lutjohann, D.; Wunderlich, P.; et al. Statins promote the degradation of extracellular amyloid β-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion. J. Biol. Chem. 2010, 285, 37405–37414. [Google Scholar] [CrossRef] [PubMed]
- Urano, Y.; Hayashi, I.; Isoo, N.; Reid, P.C.; Shibasaki, Y.; Noguchi, N.; Tomita, T.; Iwatsubo, T.; Hamakubo, T.; Kodama, T. Association of active γ-secretase complex with lipid rafts. J. Lipid Res. 2005, 46, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Phani, S.; Loike, J.D.; Przedborski, S. Neurodegeneration and inflammation in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18, S207–S209. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-C.; Lin, C.-H.; Wu, R.-M.; Lin, M.-S.; Lin, J.-W.; Chang, C.-H.; Lai, M.-S. Discontinuation of statin therapy associates with Parkinson disease. A population-based study. Neurology 2013, 81, 410–416. [Google Scholar] [PubMed]
- Tison, F.; Nègre-Pagès, L.; Meissner, W.G.; Dupouy, S.; Li, Q.; Thiolat, M.-L.; Thiollier, T.; Galitzky, M.; Ory-Magne, F.; Milhet, A.; et al. Simvastatin decreases levodopa-induced dyskinesia in monkeys, but not in a randomized, placebo-controlled, multiple cross-over (“n-of-1”) exploratory trial of simvastatin against levodopa-induced dyskinesia in Parkinson’s disease patients. Parkinsonism Relat. Disord. 2013, 19, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Q.; Long, L.; Yan, J.Q.; Wei, L.; Pan, M.Q.; Gao, H.M.; Zhou, P.; Liu, M.; Zhu, C.S.; Tang, B.S.; et al. Simvastatin induces neuroprotection in 6-OHDA-lesioned PC12 via the PI3K/AKT/caspase 3 pathway and anti-inflammatory responses. CNS Neurosci. Ther. 2013, 19, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Selley, M.L. Simvastatin prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced striatal dopamine depletion and protein tyrosine nitration in mice. Brain Res. 2005, 1037, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sharma, N.; Gupta, A.; Kalonia, H.; Mishra, J. Neuroprotective potential of atorvastatin and simvastatin (HMG-CoA reductase inhibitors) against 6-hydroxydopamine (6-OHDA) induced Parkinson-like symptoms. Brain Res. 2012, 1471, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Xu, Y.; Zhu, C.; Zhang, L.; Wu, A.; Yang, Y.; Xiong, Z.; Deng, C.; Huang, X.F.; Yenari, M.A.; et al. Simvastatin prevents dopaminergic neurodegeneration in experimental parkinsonian models: The association with anti-inflammatory responses. PLoS One 2011, 6, e20945. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Roy, A.; Matras, J.; Brahmachari, S.; Gendelman, H.E.; Pahan, K. Simvastatin inhibits the activation of p21Ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurosci. 2009, 29, 13543–13556. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Brück, W.; Lucchinetti, C. Heterogeneity of multiple sclerosis pathogenesis: Implications for diagnosis and therapy. Trends Mol. Med. 2001, 7, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Chataway, J.; Schuerer, N.; Alsanousi, A.; Chan, D.; MacManus, D.; Hunter, K.; Anderson, V.; Bangham, C.R.M.; Clegg, S.; Nielsen, C.; et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): A randomised, placebo-controlled, phase 2 trial. Lancet 2014, 383, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Paintlia, A.S.; Paintlia, M.K.; Singh, A.K.; Orak, J.K.; Singh, I. Activation of PPAR-γ and PTEN cascade participates in lovastatin-mediated accelerated differentiation of oligodendrocyte progenitor cells. Glia 2010, 58, 1669–1685. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Zehntner, S.P.; Kuhlmann, T.; Ludwin, S.K.; Owens, T.; Kennedy, T.E.; Bedell, B.J.; Antel, J.P. Statin therapy inhibits remyelination in the central nervous system. Am. J. Pathol. 2009, 174, 1880–1890. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.C.; Yang, D.Y.; Ou, Y.C.; Ho, S.P.; Cheng, F.C.; Chen, C.J. Neuroprotective effect of atorvastatin in an experimental model of nerve crush injury. Neurosurgery 2010, 67, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Rajasekharan, S.; Jarjour, A.A.; Zamvil, S.S.; Kennedy, T.E.; Antel, J.P. Simvastatin regulates oligodendroglial process dynamics and survival. Glia 2007, 55, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Klopfleisch, S.; Merkler, D.; Schmitz, M.; Klöppner, S.; Schedensack, M.; Jeserich, G.; Althaus, H.H.; Brück, W. Negative impact of statins on oligodendrocytes and myelin formation in vitro and in vivo. J. Neurosci. 2008, 28, 13609–13614. [Google Scholar] [CrossRef]
- Saher, G.; Brugger, B.; Lappe-Siefke, C.; Mobius, W.; Tozawa, R.; Wehr, M.C.; Wieland, F.; Ishibashi, S.; Nave, K.A. High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 2005, 8, 468–475. [Google Scholar] [PubMed]
- Stanislaus, R.; Pahan, K.; Singh, A.K.; Singh, I. Amelioration of experimental allergic encephalomyelitis in Lewis rats by lovastatin. Neurosci. Lett. 1999, 269, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Paintlia, A.S.; Paintlia, M.K.; Singh, A.K.; Singh, I. Inhibition of rho family functions by lovastatin promotes myelin repair in ameliorating experimental autoimmune encephalomyelitis. Mol. Pharmacol. 2008, 73, 1381–1393. [Google Scholar] [PubMed]
- Dunn, S.E.; Youssef, S.; Goldstein, M.J.; Prod’homme, T.; Weber, M.S.; Zamvil, S.S.; Steinman, L. Isoprenoids determine Th1/Th2 fate in pathogenic T cells, providing a mechanism of modulation of autoimmunity by atorvastatin. J. Exp. Med. 2006, 203, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Youssef, S.; Stuve, O.; Patarroyo, J.C.; Ruiz, P.J.; Radosevich, J.L.; Hur, E.M.; Bravo, M.; Mitchell, D.J.; Sobel, R.A.; Steinman, L.; et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature 2002, 420, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Aktas, O.; Waiczies, S.; Smorodchenko, A.; Dorr, J.; Seeger, B.; Prozorovski, T.; Sallach, S.; Endres, M.; Brocke, S.; Nitsch, R.; et al. Treatment of relapsing paralysis in experimental encephalomyelitis by targeting Th1 cells through atorvastatin. J. Exp. Med. 2003, 197, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Sim, F.J.; Lang, J.K.; Ali, T.A.; Roy, N.S.; Vates, G.E.; Pilcher, W.H.; Goldman, S.A. Statin treatment of adult human glial progenitors induces PPAR gamma-mediated oligodendrocytic differentiation. Glia 2008, 56, 954–962. [Google Scholar] [CrossRef] [PubMed]
- North, K.; Hyman, S.; Barton, B. Cognitive deficits in neurofibromatosis 1. J. Child. Neurol. 2002, 17, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cui, Y.; Kushner, S.A.; Brown, R.A.M.; Jentsch, J.D.; Frankland, P.W.; Cannon, T.D.; Silva, A.J. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr. Biol. 2005, 15, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Acosta, M.T.; Kardel, P.G.; Walsh, K.S.; Rosenbaum, K.N.; Gioia, G.A.; Packer, R.J. Lovastatin as treatment for neurocognitive deficits in neurofibromatosis type 1: Phase I study. Pediatr. Neurol. 2011, 45, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Van der Vaart, T.; Plasschaert, E.; Rietman, A.B.; Renard, M.; Oostenbrink, R.; Vogels, A.; de Wit, M.-C.Y.; Descheemaeker, M.-J.; Vergouwe, Y.; Catsman-Berrevoets, C.E.; et al. Simvastatin for cognitive deficits and behavioural problems in patients with neurofibromatosis type 1 (NF1-SIMCODA): A randomised, placebo-controlled trial. Lancet Neurol. 2013, 12, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Cappellari, M.; Bovi, P.; Moretto, G.; Zini, A.; Nencini, P.; Sessa, M.; Furlan, M.; Pezzini, A.; Orlandi, G.; Paciaroni, M. The THRombolysis and STatins (THRaST) study. Neurology 2013, 80, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Flint, A.C.; Kamel, H.; Navi, B.B.; Rao, V.A.; Faigeles, B.S.; Conell, C.; Klingman, J.G.; Hills, N.K.; Nguyen-Huynh, M.; Cullen, S.P.; et al. Inpatient statin use predicts improved ischemic stroke discharge disposition. Neurology 2012, 78, 1678–1683. [Google Scholar] [CrossRef] [PubMed]
- Flint, A.C.; Kamel, H.; Navi, B.B.; Rao, V.A.; Faigeles, B.S.; Conell, C.; Klingman, J.G.; Sidney, S.; Hills, N.K.; Sorel, M.; et al. Statin use during ischemic stroke hospitalization is strongly associated with improved poststroke survival. Stroke 2012, 43, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Cappellari, M.; Deluca, C.; Tinazzi, M.; Tomelleri, G.; Carletti, M.; Fiaschi, A.; Bovi, P.; Moretto, G. Does statin in the acute phase of ischemic stroke improve outcome after intravenous thrombolysis? A retrospective study. J. Neurol. Sci. 2011, 308, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Laufs, U.; Liao, J.K.; Moskowitz, M.A. Targeting eNOS for stroke protection. Trends Neurosci. 2004, 27, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Laufs, U.; Huang, Z.; Nakamura, T.; Huang, P.; Moskowitz, M.A.; Liao, J.K. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1998, 95, 8880–8885. [Google Scholar] [CrossRef] [PubMed]
- Sawada, N.; Liao, J.K. Targeting eNOS and beyond: Emerging heterogeneity of the role of endothelial Rho proteins in stroke protection. Expert Rev. Neurother. 2009, 9, 1171–1186. [Google Scholar] [CrossRef] [PubMed]
- Gasche, Y.; Copin, J.-C.; Sugawara, T.; Fujimura, M.; Chan, P.H. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood–brain barrier disruption after transient focal cerebral ischemia. J. Cerebr. Blood Flow Metab. 2001, 21, 1393–1400. [Google Scholar] [CrossRef]
- Woo, M.-S.; Park, J.-S.; Choi, I.-Y.; Kim, W.-K.; Kim, H.-S. Inhibition of MMP-3 or -9 suppresses lipopolysaccharide-induced expression of proinflammatory cytokines and iNOS in microglia. J. Neurochem. 2008, 106, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Zhang, Z.G.; Zhang, L.; Morris, D.C.; Kapke, A.; Lu, M.; Chopp, M. Atorvastatin downregulates tissue plasminogen activator-aggravated genes mediating coagulation and vascular permeability in single cerebral endothelial cells captured by laser microdissection. J. Cerebr. Blood Flow Metab. 2006, 26, 787–796. [Google Scholar] [CrossRef]
- Wang, S.; Lee, S.-R.; Guo, S.-Z.; Kim, W.J.; Montaner, J.; Wang, X.; Lo, E.H. Reduction of tissue plasminogen activator-induced matrix metalloproteinase-9 by simvastatin in astrocytes. Stroke 2006, 37, 1910–1912. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Zeng, J.-S.; Kreulen, D.L.; Kaufman, D.I.; Chen, A.F. Atorvastatin protects against cerebral infarction via inhibition of NADPH oxidase-derived superoxide in ischemic stroke. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2210–H2215. [Google Scholar] [CrossRef] [PubMed]
- Erdös, B.; Snipes, J.A.; Tulbert, C.D.; Katakam, P.; Miller, A.W.; Busija, D.W. Rosuvastatin improves cerebrovascular function in Zucker obese rats by inhibiting NAD(P)H oxidase-dependent superoxide production. Am. J. Physiol-Heart C 2006, 290, H1264–H1270. [Google Scholar]
- Ma, T.; Zhao, Y.; Kwak, Y.-D.; Yang, Z.; Thompson, R.; Luo, Z.; Xu, H.; Liao, F.-F. Statin’s excitoprotection is mediated by sAPP and the subsequent attenuation of calpain-induced truncation events, likely via Rho-ROCK signaling. J. Neurosci. 2009, 29, 11226–11236. [Google Scholar] [CrossRef] [PubMed]
- Etminan, M.; Samii, A.; Brophy, J.M. Statin use and risk of epilepsy: A nested case-control study. Neurology 2010, 75, 1496–1500. [Google Scholar] [CrossRef] [PubMed]
- Piermartiri, T.C.; Vandresen-Filho, S.; de Araujo Herculano, B.; Martins, W.C.; Dal’agnolo, D.; Stroeh, E.; Carqueja, C.L.; Boeck, C.R.; Tasca, C.I. Atorvastatin prevents hippocampal cell death due to quinolinic acid-induced seizures in mice by increasing Akt phosphorylation and glutamate uptake. Neurotox. Res. 2009, 16, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Won, J.S.; Singh, A.K.; Singh, I. Statin inhibits kainic acid-induced seizure and associated inflammation and hippocampal cell death. Neurosci. Lett. 2008, 440, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Ponce, J.; de la Ossa, N.P.; Hurtado, O.; Millan, M.; Arenillas, J.F.; Dávalos, A.; Gasull, T. Simvastatin reduces the association of NMDA receptors to lipid rafts: A cholesterol-mediated effect in neuroprotection. Stroke 2008, 39, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Zacco, A.; Togo, J.; Spence, K.; Ellis, A.; Lloyd, D.; Furlong, S.; Piser, T. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors protect cortical neurons from excitotoxicity. J. Neurosci. 2003, 23, 11104–11111. [Google Scholar] [PubMed]
- Ramirez, C.; Tercero, I.; Pineda, A.; Burgos, J.S. Simvastatin is the statin that most efficiently protects against kainate-induced excitotoxicity and memory impairment. J. Alzheimers Dis. 2011, 24, 161–174. [Google Scholar]
- Citraro, R.; Chimirri, S.; Aiello, R.; Gallelli, L.; Trimboli, F.; Britti, D.; de Sarro, G.; Russo, E. Protective effects of some statins on epileptogenesis and depressive-like behavior in WAG/Rij rats, a genetic animal model of absence epilepsy. Epilepsia 2014, 55, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Jick, S.S.; Jick, H. Lipid-lowering drugs and the risk of depression and suicidal behavior. Arch. Intern. Med. 2003, 163, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Young-Xu, Y.; Chan, K.A.; Liao, J.K.; Ravid, S.; Blatt, C.M. Long-term statin use and psychological well-being. J. Am. Coll. Cardiol. 2003, 42, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Tan, C.H.; Merchant, R.A.; Ng, T.P. Association between depressive symptoms and use of HMG-CoA reductase inhibitors (statins), corticosteroids and histamine H2 receptor antagonists in community-dwelling older persons: Cross-sectional analysis of a population-based cohort. Drugs Aging 2008, 25, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Stewart, R.; Kang, H.J.; Bae, K.Y.; Kim, S.W.; Shin, I.S.; Kim, J.T.; Park, M.S.; Cho, K.H.; Yoon, J.S. A prospective study of statin use and poststroke depression. J. Clin. Psychopharmacol. 2014, 34, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Otte, C.; Zhao, S.; Whooley, M.A. Statin use and risk of depression in patients with coronary heart disease: Longitudinal data from the Heart and Soul Study. J. Clin. Psychiatry 2012, 73, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Stafford, L.; Berk, M. The use of statins after a cardiac intervention is associated with reduced risk of subsequent depression: Proof of concept for the inflammatory and oxidative hypotheses of depression? J. Clin. Psychiatry 2011, 72, 1229–1235. [Google Scholar] [CrossRef]
- Chuang, C.S.; Yang, T.Y.; Muo, C.H.; Su, H.L.; Sung, F.C.; Kao, C.H. Hyperlipidemia, statin use and the risk of developing depression: A nationwide retrospective cohort study. Gen. Hosp. Psychiatry 2014, 36, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Agostini, J.V.; Tinetti, M.E.; Han, L.; McAvay, G.; Foody, J.M.; Concato, J. Effects of statin use on muscle strength, cognition and depressive symptoms in older adults. J. Am. Geriatr. Soc. 2007, 55, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Yap, K.B.; Kua, E.H.; Ng, T.P. Statin use and depressive symptoms in a prospective study of community-living older persons. Pharmacoepidemiol. Drug Saf. 2010, 19, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Al Badarin, F.J.; Spertus, J.A.; Gosch, K.L.; Buchanan, D.M.; Chan, P.S. Initiation of statin therapy after acute myocardial infarction is not associated with worsening depressive symptoms: Insights from the Prospective Registry Evaluating Outcomes After Myocardial Infarctions: Events and Recovery (PREMIER) and Translational Research Investigating Underlying Disparities in Acute Myocardial Infarction Patients’ Health Status (TRIUMPH) registries. Am. Heart J. 2013, 166, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.Y.; Chang, A.Y.; Lin, T.K. Simvastatin treatment exerts antidepressant-like effect in rats exposed to chronic mild stress. Pharmacol. Biochem. Behav. 2014, 124, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Ludka, F.K.; Zomkowski, A.D.E.; Cunha, M.P.; Dal-Cim, T.; Zeni, A.L.B.; Rodrigues, A.L.S.; Tasca, C.I. Acute atorvastatin treatment exerts antidepressant-like effect in mice via the l-arginine–nitric oxide–cyclic guanosine monophosphate pathway and increases BDNF levels. Eur. Neuropsychopharm. 2013, 23, 400–412. [Google Scholar] [CrossRef]
- Mansi, I.; Frei, C.R.; Pugh, M.J.; Mortensen, E.M. Psychologic disorders and statin use: A propensity score-matched analysis. Pharmacotherapy 2013, 33, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, J.; Blauw, G.J.; Murphy, M.B.; Bollen, E.L.; Buckley, B.M.; Cobbe, S.M.; Ford, I.; Gaw, A.; Hyland, M.; Jukema, J.W. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): A randomised controlled trial. Lancet 2002, 360, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Dale, K.M.; Coleman, C.I.; Henyan, N.N.; Kluger, J.; White, C. Statins and cancer risk: A meta-analysis. JAMA 2006, 295, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.H.; Voysey, M.; Gray, A.; Collins, R.; Baigent, C. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: Meta-analysis of individual data from 27 randomised trials. Lancet 2012, 380, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Thibault, A.; Samid, D.; Tompkins, A.C.; Figg, W.D.; Cooper, M.R.; Hohl, R.J.; Trepel, J.; Liang, B.; Patronas, N.; Venzon, D.J. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin. Cancer Res. 1996, 2, 483–491. [Google Scholar] [PubMed]
- Larner, J.; Jane, J.; Laws, E.; Packer, R.; Myers, C.; Shaffrey, M. A phase I–II trial of lovastatin for anaplastic astrocytoma and glioblastoma multiforme. Am. J. Clin. Oncol. 1998, 21, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Goto, T.; Saito, Y.; Goto, S.; Kochi, M.; Ushio, Y. The inhibitory effect of simvastatin on growth in malignant gliomas—With special reference to its local application with fibrin glue spray in vivo. Int. J. Oncol. 2001, 19, 525–531. [Google Scholar] [PubMed]
- Maltese, W.A.; Defendini, R.; Green, R.A.; Sheridan, K.M.; Donley, D.K. Suppression of murine neuroblastoma growth in vivo by mevinolin, a competitive inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Clin. Investig. 1985, 76, 1748–1754. [Google Scholar] [CrossRef] [PubMed]
- Dimitroulakos, J.; Ye, L.Y.; Benzaquen, M.; Moore, M.J.; Kamel-Reid, S.; Freedman, M.H.; Yeger, H.; Penn, L.Z. Differential sensitivity of various pediatric cancers and squamous cell carcinomas to lovastatin-induced apoptosis: Therapeutic implications. Clin. Cancer Res. 2001, 7, 158–167. [Google Scholar]
- Prasanna, P.; Thibault, A.; Liu, L.; Samid, D. Lipid metabolism as a target for brain cancer therapy: Synergistic activity of lovastatin and sodium phenylacetate against human glioma cells. J. Neurochem. 1996, 66, 710–716. [Google Scholar] [CrossRef]
- Yanae, M.; Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Yamazoe, Y.; Nishida, S. Statin-induced apoptosis via the suppression of ERK1/2 and Akt activation by inhibition of the geranylgeranyl-pyrophosphate biosynthesis in glioblastoma. J. Exp. Clin. Cancer Res. 2011, 30, 74. [Google Scholar] [CrossRef] [PubMed]
- Crick, D.C.; Andres, D.A.; Danesi, R.; Macchia, M.; Waechter, C.J. Geranylgeraniol overcomes the block of cell proliferation by lovastatin in C6 glioma cells. J. Neurochem. 1998, 70, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, A.; Zanin, V.; Piscianz, E.; Tricarico, P.M.; Vuch, J.; Girardelli, M.; Monasta, L.; Bianco, A.M.; Crovella, S. Lovastatin-induced apoptosis is modulated by geranylgeraniol in a neuroblastoma cell line. Int. J. Dev. Neurosci. 2012, 30, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Déry, M.A.; Rousseau, G.; Benderdour, M.; Beaumont, E. Atorvastatin prevents early apoptosis after thoracic spinal cord contusion injury and promotes locomotion recovery. Neurosci. Lett. 2009, 453, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Pannu, R.; Christie, D.K.; Barbosa, E.; Singh, I.; Singh, A.K. Post-trauma lipitor treatment prevents endothelial dysfunction, facilitates neuroprotection and promotes locomotor recovery following spinal cord injury. J. Neurochem. 2007, 101, 182–200. [Google Scholar] [CrossRef] [PubMed]
- Pannu, R.; Barbosa, E.; Singh, A.K.; Singh, I. Attenuation of acute inflammatory response by atorvastatin after spinal cord injury in rats. J. Neurosci. Res. 2005, 79, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, N.; Cui, Y.; Xu, Y.; Dang, G.; Song, C. Simvastatin mobilizes bone marrow stromal cells migrating to injured areas and promotes functional recovery after spinal cord injury in the rat. Neurosci. Lett. 2012, 521, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, N.; Xu, Y.; Zhu, J.; Chen, Z.; Liu, Z.; Dang, G.; Song, C. Simvastatin treatment improves functional recovery after experimental spinal cord injury by upregulating the expression of BDNF and GDNF. Neurosci. Lett. 2011, 487, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Kahveci, R.; Gökçe, E.C.; Gürer, B.; Gökçe, A.; Kisa, U.; Cemil, D.B.; Sargon, M.F.; Kahveci, F.O.; Aksoy, N.; Erdoğan, B. Neuroprotective effects of rosuvastatin against traumatic spinal cord injury in rats. Eur. J. Pharmacol. 2014, 741, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Mann, C.M.; Lee, J.H.T.; Hillyer, J.; Stammers, A.M.T.; Tetzlaff, W.; Kwon, B.K. Lack of robust neurologic benefits with simvastatin or atorvastatin treatment after acute thoracic spinal cord contusion injury. Exp. Neurol. 2010, 221, 285–295. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McFarland, A.J.; Anoopkumar-Dukie, S.; Arora, D.S.; Grant, G.D.; McDermott, C.M.; Perkins, A.V.; Davey, A.K. Molecular Mechanisms Underlying the Effects of Statins in the Central Nervous System. Int. J. Mol. Sci. 2014, 15, 20607-20637. https://doi.org/10.3390/ijms151120607
McFarland AJ, Anoopkumar-Dukie S, Arora DS, Grant GD, McDermott CM, Perkins AV, Davey AK. Molecular Mechanisms Underlying the Effects of Statins in the Central Nervous System. International Journal of Molecular Sciences. 2014; 15(11):20607-20637. https://doi.org/10.3390/ijms151120607
Chicago/Turabian StyleMcFarland, Amelia J., Shailendra Anoopkumar-Dukie, Devinder S. Arora, Gary D. Grant, Catherine M. McDermott, Anthony V. Perkins, and Andrew K. Davey. 2014. "Molecular Mechanisms Underlying the Effects of Statins in the Central Nervous System" International Journal of Molecular Sciences 15, no. 11: 20607-20637. https://doi.org/10.3390/ijms151120607
APA StyleMcFarland, A. J., Anoopkumar-Dukie, S., Arora, D. S., Grant, G. D., McDermott, C. M., Perkins, A. V., & Davey, A. K. (2014). Molecular Mechanisms Underlying the Effects of Statins in the Central Nervous System. International Journal of Molecular Sciences, 15(11), 20607-20637. https://doi.org/10.3390/ijms151120607