Comparative Proteomic Analysis of Labellum and Inner Lateral Petals in Cymbidium ensifolium Flowers

Abstract
:1. Introduction
2. Results
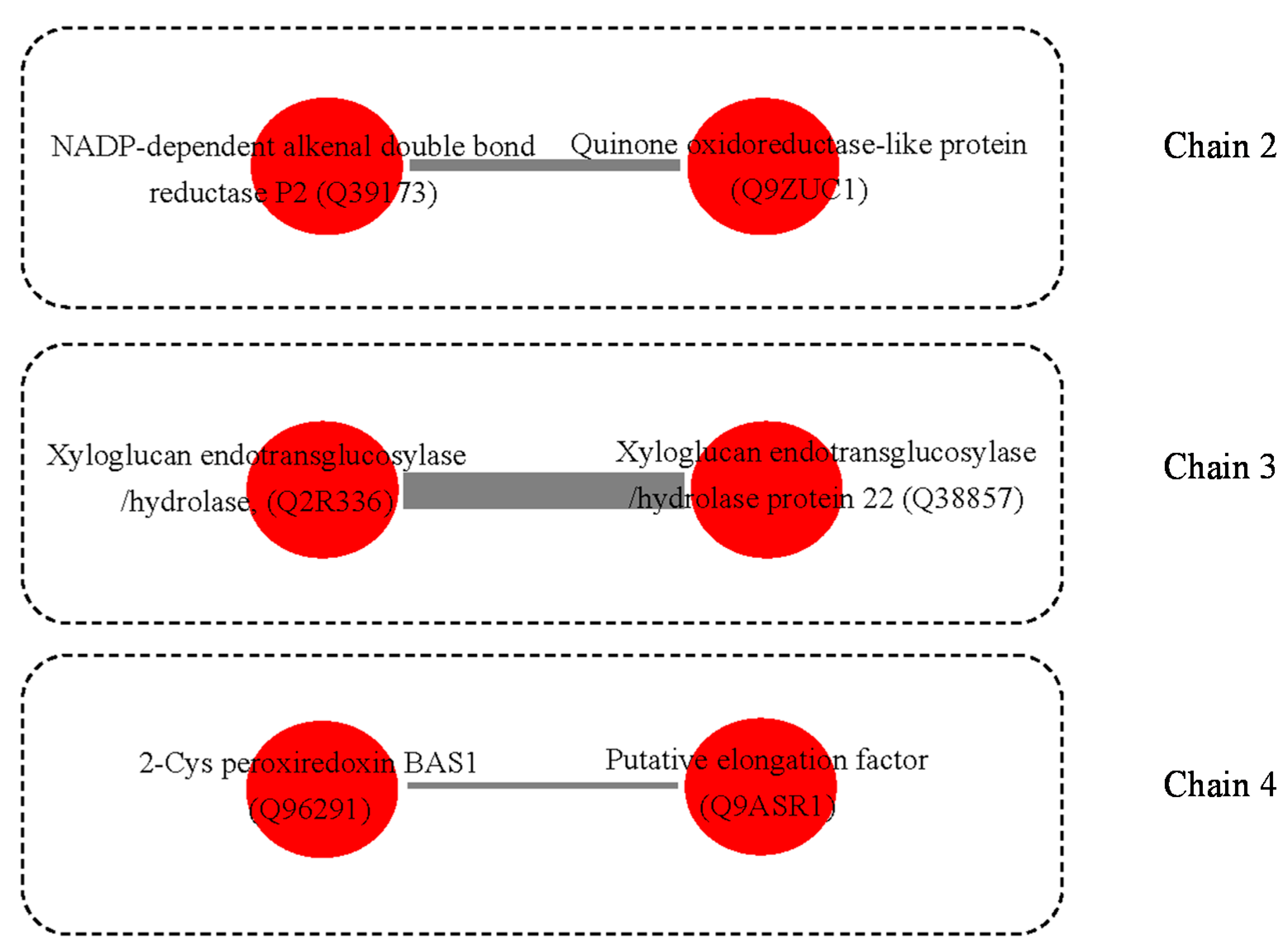
2.1. The Differentially Expressed Proteins
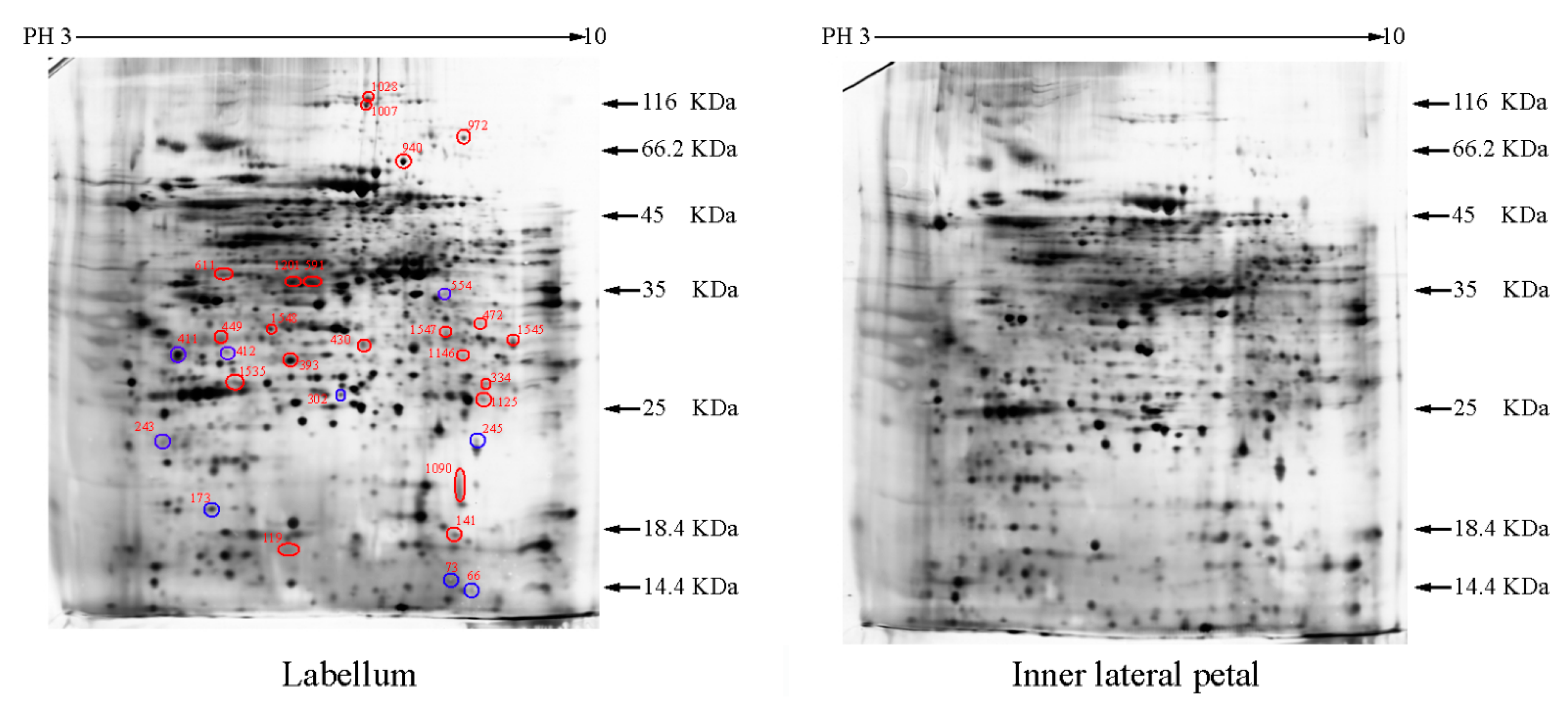

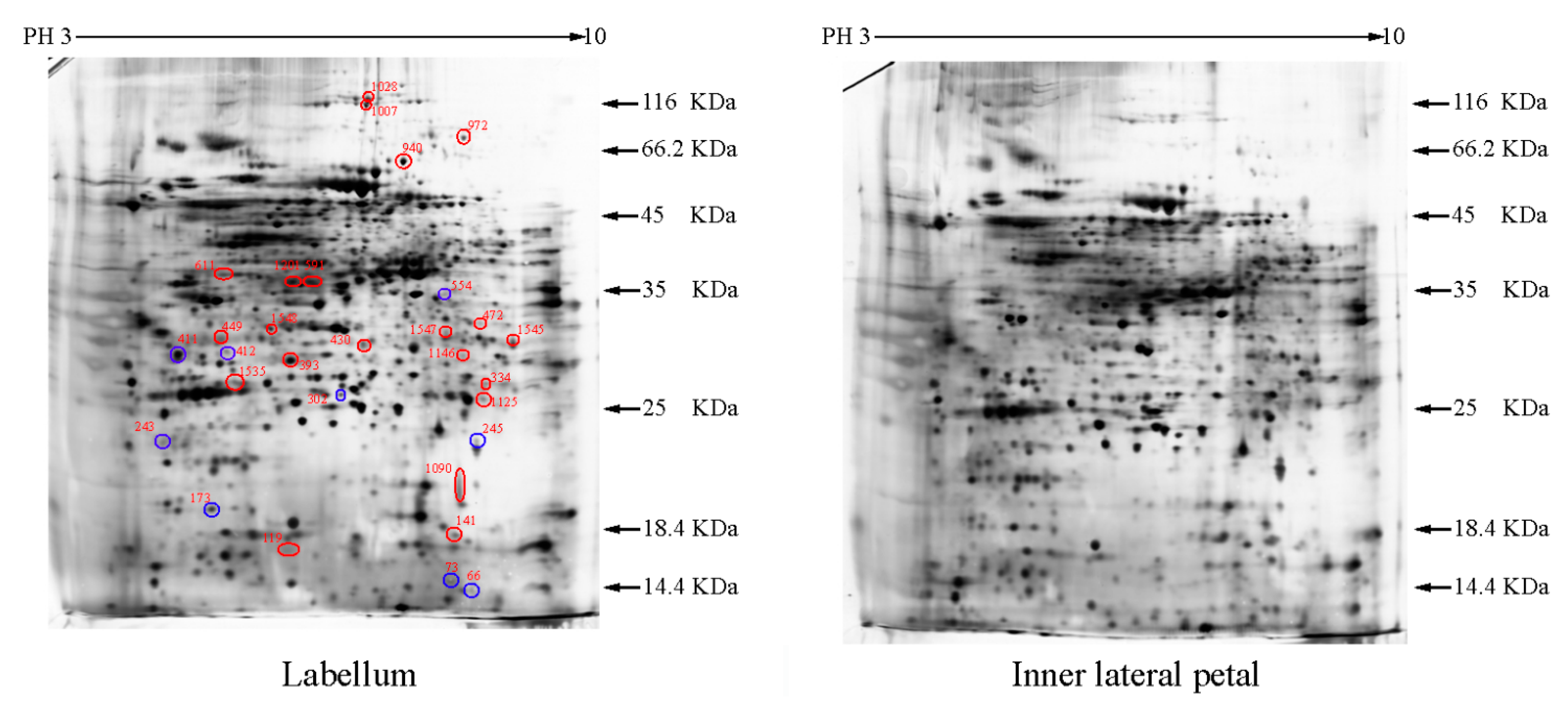{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group a | Spot Number b | Protein Name | Reference Organism | Accession c | Mascot Scores | Blast Score | Blast Expect | Spots % Volume Variations (p < 0.05) d |
|---|---|---|---|---|---|---|---|---|
| I | 173 | Superoxide dismutase [Cu-Zn], chloroplastic | Oryza sativa japonica group | P93407 | 214 | 306 | 9 × 10−103 | 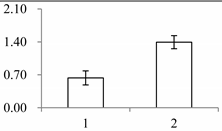 |
| I | 243 | 2-Cys peroxiredoxin BAS1, chloroplastic | Arabidopsis thaliana | Q96291 | 51 | 376 | 6 × 10−129 | 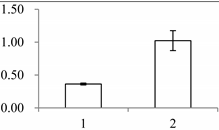 |
| I | 302 | Triosephosphate isomerase (TPI) | Gossypium mexicanum | D2D303 | 137 | 424 | 2 × 10−147 |  |
| I | 411 | Fibrillin-like protein (FIB) | Oncidium hybrid cultivar | B4F6G1 | 930 | 489 | 2 × 10−161 | 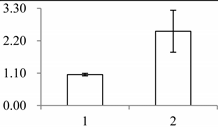 |
| I | 412 | Oxygen-evolving enhancer protein 1-2, chloroplastic | Oryza sativa subsp. japonica | Q9S841 | 131 | 485 | 3 × 10−169 | 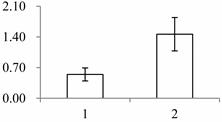 |
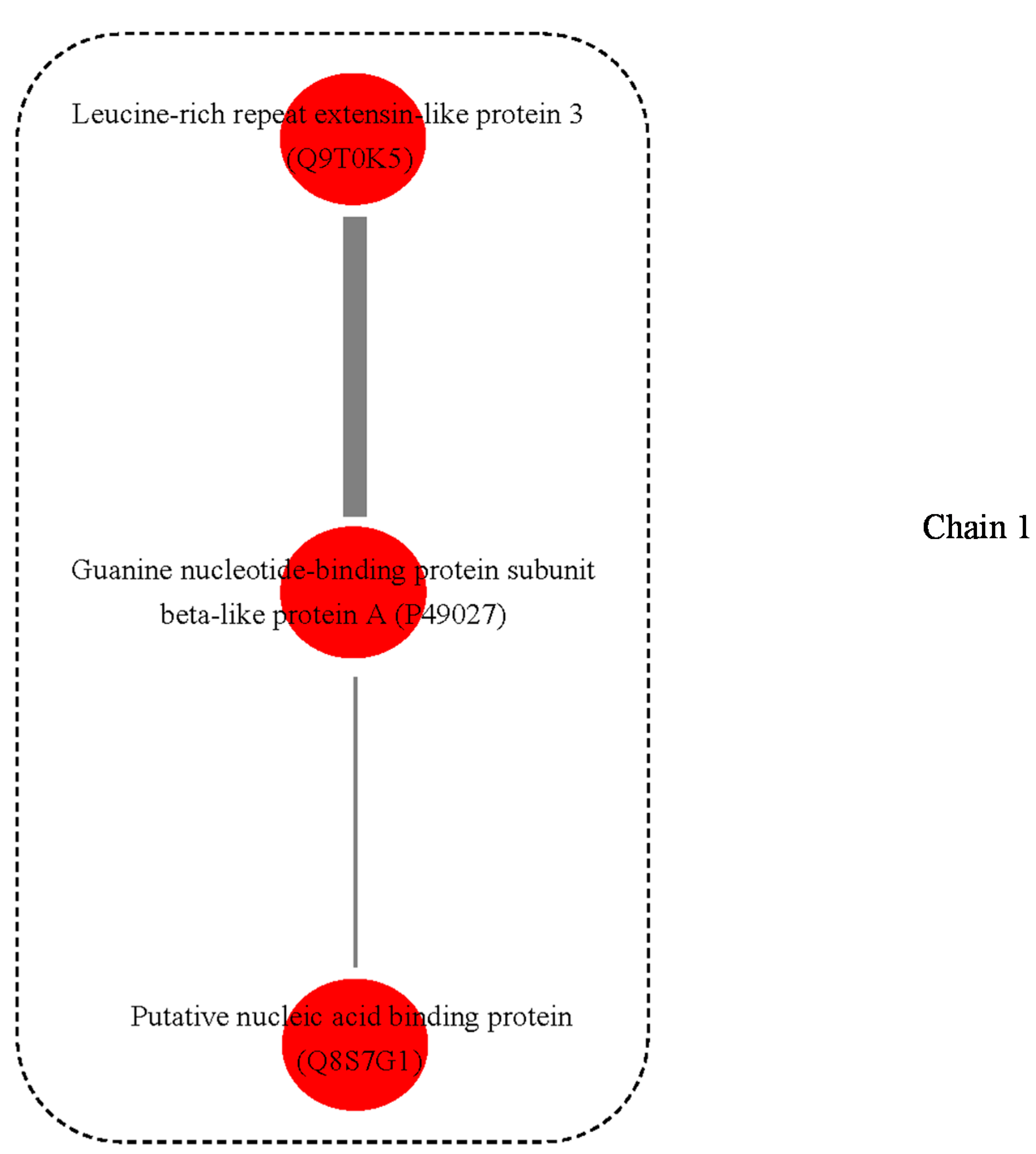
| I | 554 | Guanine nucleotide-binding protein subunit beta-like protein A | Oryza sativa subsp. japonica | P49027 | 93 | 521 | 0 | 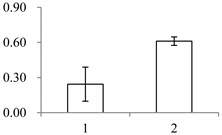 |
| II | 141 | SMALLER WITH VARIABLE BRANCHES (SVB) | Arabidopsis thaliana | Q9FXB0 | 274 | 164 | 6 × 10−48 | 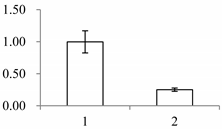 |
| II | 334 | NAD(P)-binding Rossmann-fold-containing protein | Arabidopsis thaliana | O80934 | 58 | 424 | 2 × 10−144 |  |
| II | 393 | Leucine-rich repeat extensin-like protein 3 | Arabidopsis thaliana | Q9T0K5 | 291 | 53.1 | 4 × 10−11 | 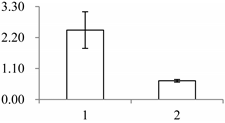 |
| II | 430 | Xyloglucan endotransglucosylase/hydrolase protein 22 | Arabidopsis thaliana | Q38857 | 262 | 420 | 1 × 10−144 | 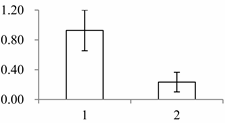 |
| II | 449 | Mannose-specific lectin | Allium sativum | P83886 | 239 | 63.2 | 3 × 10−11 | 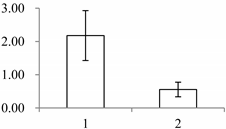 |
| II | 591 | NADP-dependent alkenal double bond reductase P2 | Arabidopsis thaliana | Q39173 | 65 | 479 | 2 × 10−166 | 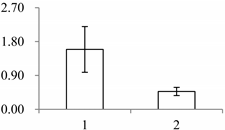 |
| II | 940 | NADP-dependent malic enzyme, chloroplastic (maeB) | Oryza sativa subsp. japonica | P43279 | 60 | 926 | 0 | 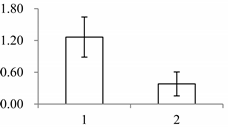 |
| II | 972 | Putative nucleic acid binding protein | Oryza sativa subsp. japonica | Q8S7G1 | 134 | 170 | 3 × 10−67 | 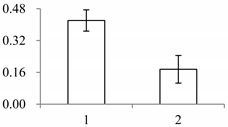 |
| II | 1028 | Putative elongation factor | Oryza sativa subsp. japonica | Q9ASR1 | 240 | 1524 | 0 |  |
| II | 1090 | serine carboxypeptidase (SCPL)-like 18 | Brachypodium distachyon | XP_003560245.1 | 48 | 440 | 2 × 10−144 | 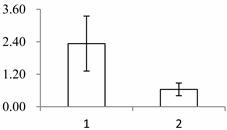 |
| II | 1146 | Proteasome subunit alpha type-7-B | Arabidopsis thaliana | O24616 | 181 | 443 | 3 × 10−156 |  |
| II | 1201 | Quinone oxidoreductase-like protein At1g23740, chloroplastic | Arabidopsis thaliana | Q9ZUC1 | 104 | 489 | 1 × 10−166 | 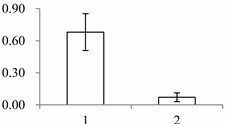 |
| III | 1535 | Cytosolic ascorbate peroxidase 1 | Gossypium mexicanum | A7KIX5 | 242 | 439 | 4 × 10−152 | 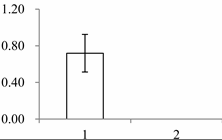 |
| III | 1545 | Xyloglucan endotransglucosylase/hydrolase, putative, expressed (XET/XTH) | Oryza sativa subsp. japonica | Q2R336 | 415 | 486 | 3 × 10−170 |  |
| III | 1547 | Beta-glucosidase 12 | Oryza sativa subsp. japonica | Q7XKV4 | 112 | 562 | 0 | 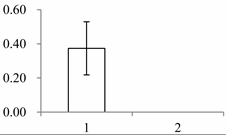 |
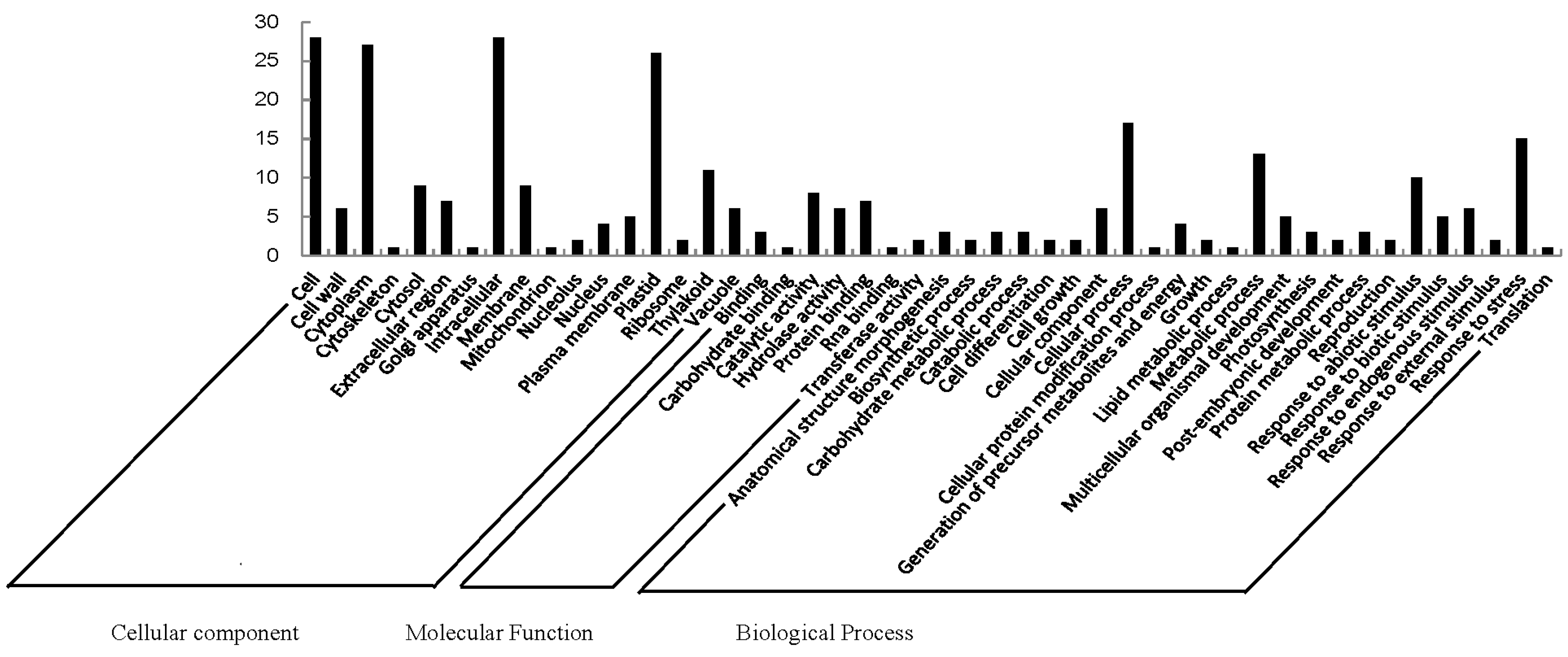
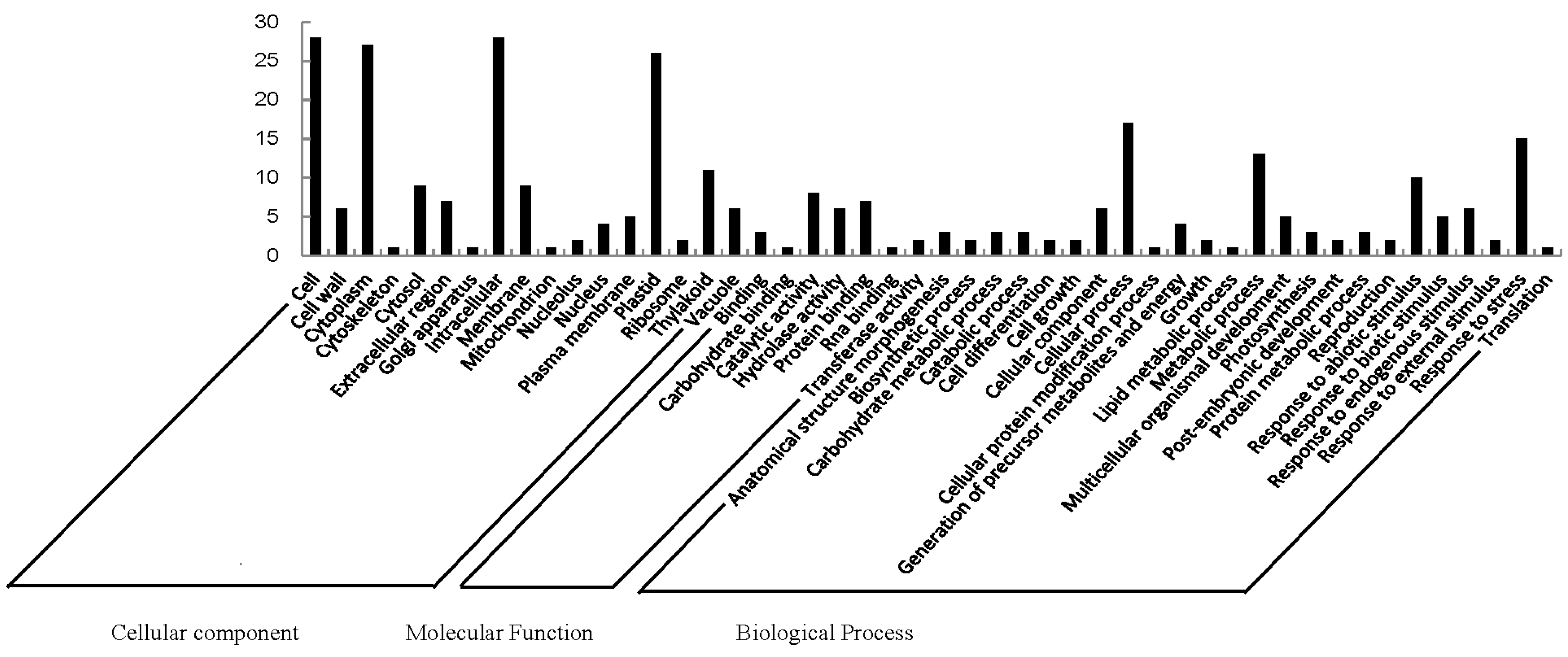
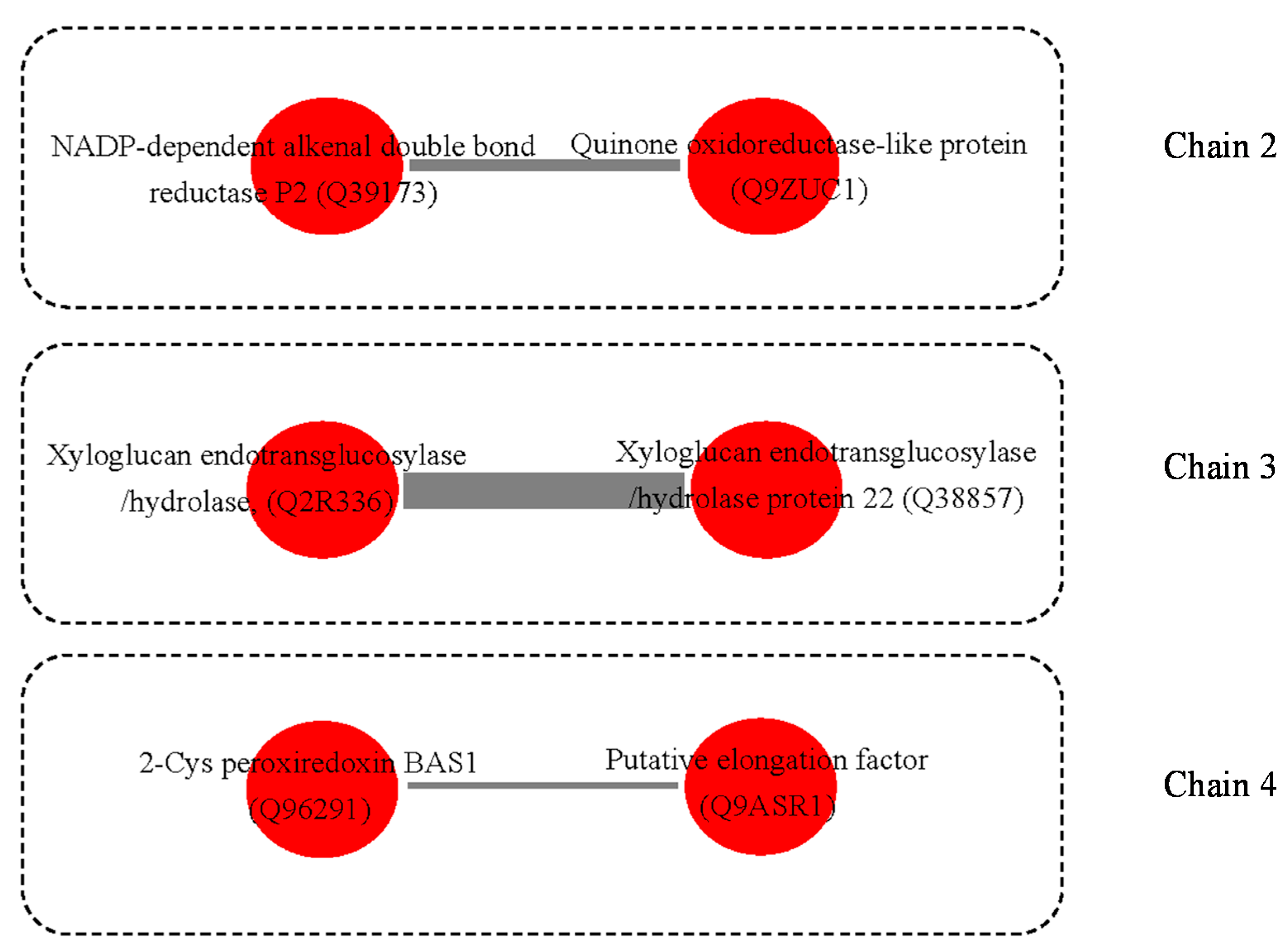
2.2. Functional Categorization and Protein-Protein Interactions (PPI) Prediction
| Pathway | Accession | Number | Differentially Expressed Proteins |
|---|---|---|---|
| Metabolic pathways | ath01100 | 4 | Malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+) (MaeB) [EC:1.1.1.40]; Beta-glucosidase [EC:3.2.1.21]; Triosephosphate isomerase (TPI) [EC:5.3.1.1]; Photosystem II oxygen-evolving enhancer protein 1 (psbO) |
| Biosynthesis of secondary metabolites | ath01110 | 2 | Beta-glucosidase [EC:3.2.1.21]; Triosephosphate isomerase (TPI) [EC:5.3.1.1] |
| Phenylpropanoid biosynthesis | ath00940 | 2 | Beta-glucosidase [EC:3.2.1.21]; Serine carboxypeptidase (SCPL)-like 19 [EC:3.4.16.-2.3.1.91] |
| Carbon fixation in photosynthetic organisms | ath00710 | 2 | Malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+) (MaeB) [EC:1.1.1.40]; Triosephosphate isomerase (TPI) [EC:5.3.1.1] |
| Carbon metabolism | ath01200 | 2 | Malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+) (MaeB) [EC:1.1.1.40]; Triosephosphate isomerase (TPI) [EC:5.3.1.1] |
| Starch and sucrose metabolism | ath00500 | 1 | Beta-glucosidase [EC:3.2.1.21] |
| Cyanoamino acid metabolism | ath00460 | 1 | Beta-glucosidase [EC:3.2.1.21] |
| Glutathione metabolism | ath00480 | 1 | l-ascorbate peroxidase [EC:1.11.1.11] |
| Ascorbate and aldarate metabolism | ath00053 | 1 | l-ascorbate peroxidase [EC:1.11.1.11] |
| Pyruvate metabolism | ath00620 | 1 | Malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+) (MaeB) [EC:1.1.1.40] |
| Peroxisome | ath04146 | 1 | Superoxide dismutase, Cu-Zn family (SOD1) [EC:1.15.1.1] |
| Inositol phosphate metabolism | ath00562 | 1 | Triosephosphate isomerase (TPI) [EC:5.3.1.1] |
| Fructose and mannose metabolism | ath00051 | 1 | Triosephosphate isomerase (TPI) [EC:5.3.1.1] |
| Proteasome | ath03050 | 1 | 20S proteasome subunit alpha 4 (PSMA7) [EC:3.4.25.1] |
| Photosynthesis | ath00195 | 1 | photosystem II oxygen-evolving enhancer protein 1 (psbO) |
| Glycolysis/Gluconeogenesis | ath00010 | 1 | Triosephosphate isomerase (TPI) [EC:5.3.1.1] |
| Biosynthesis of amino acids | ath01230 | 1 | Triosephosphate isomerase (TPI) [EC:5.3.1.1] |
| Plant hormone signal transduction | ath04075 | 1 | Xyloglucan:xyloglucosyl transferase (TCH4) [EC:2.4.1.207] |
3. Discussion
3.1. Protein Function
3.2. Possible Pathway
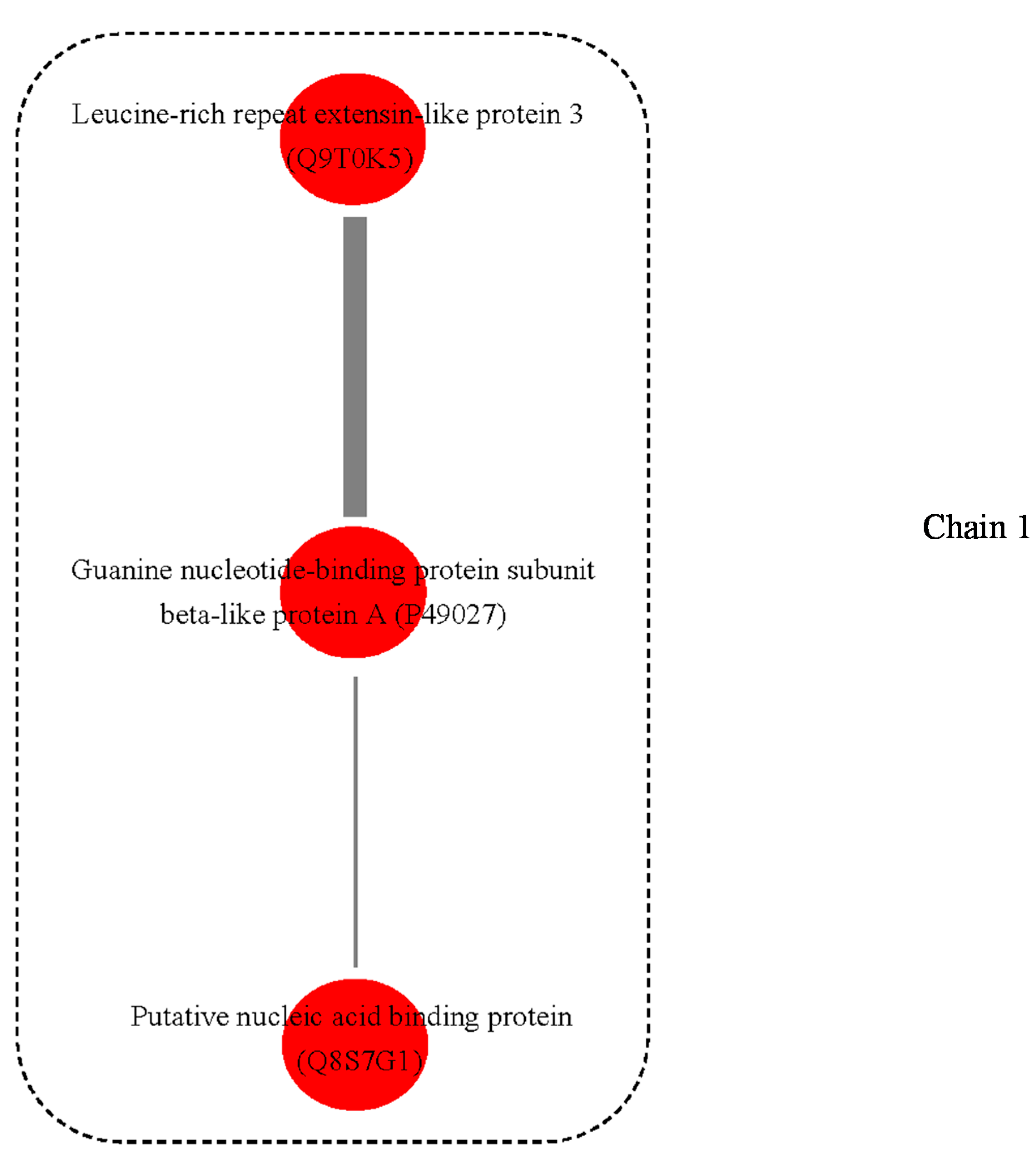
3.3. Possible Protein-Protein Interactions (PPI)
4. Methods
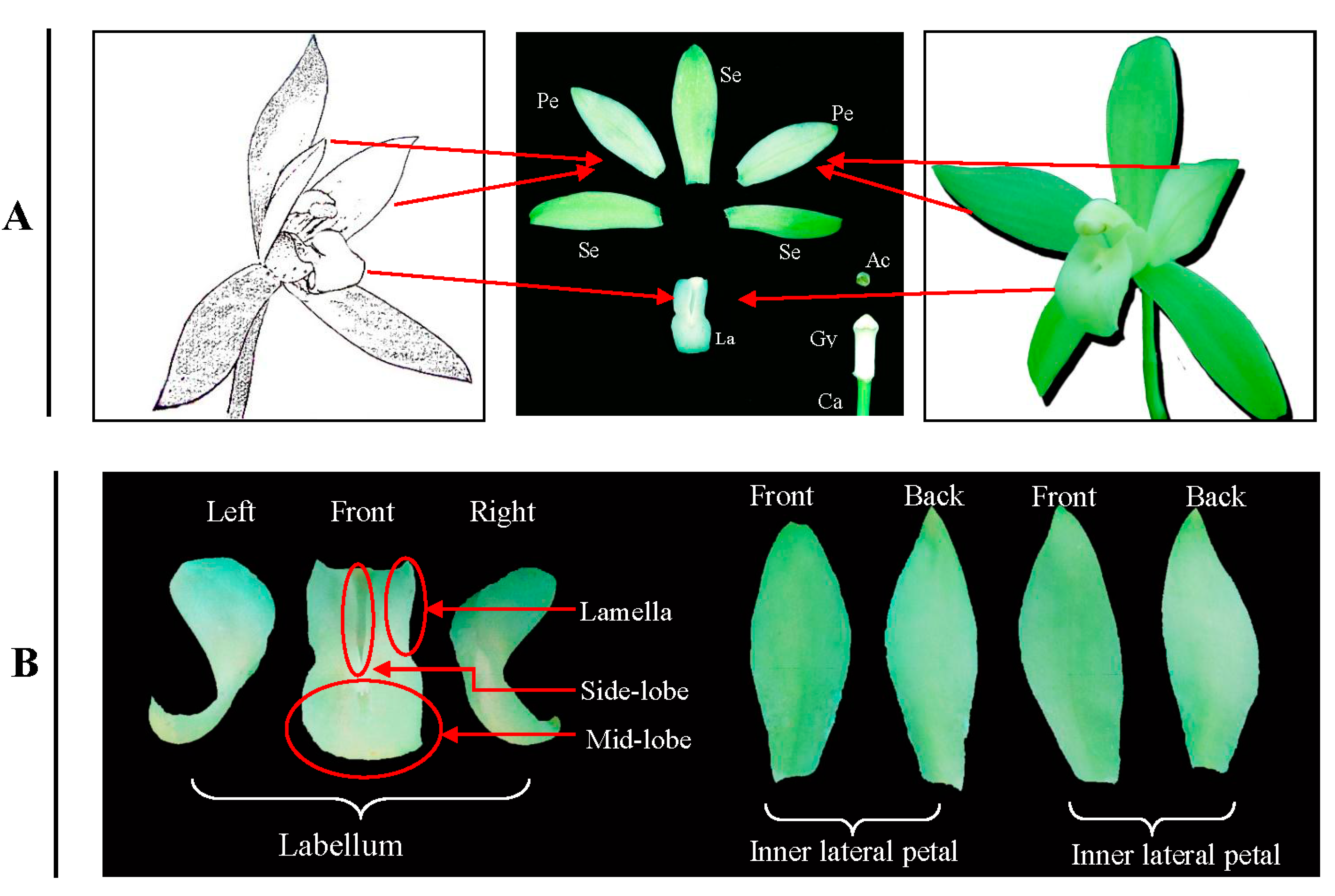
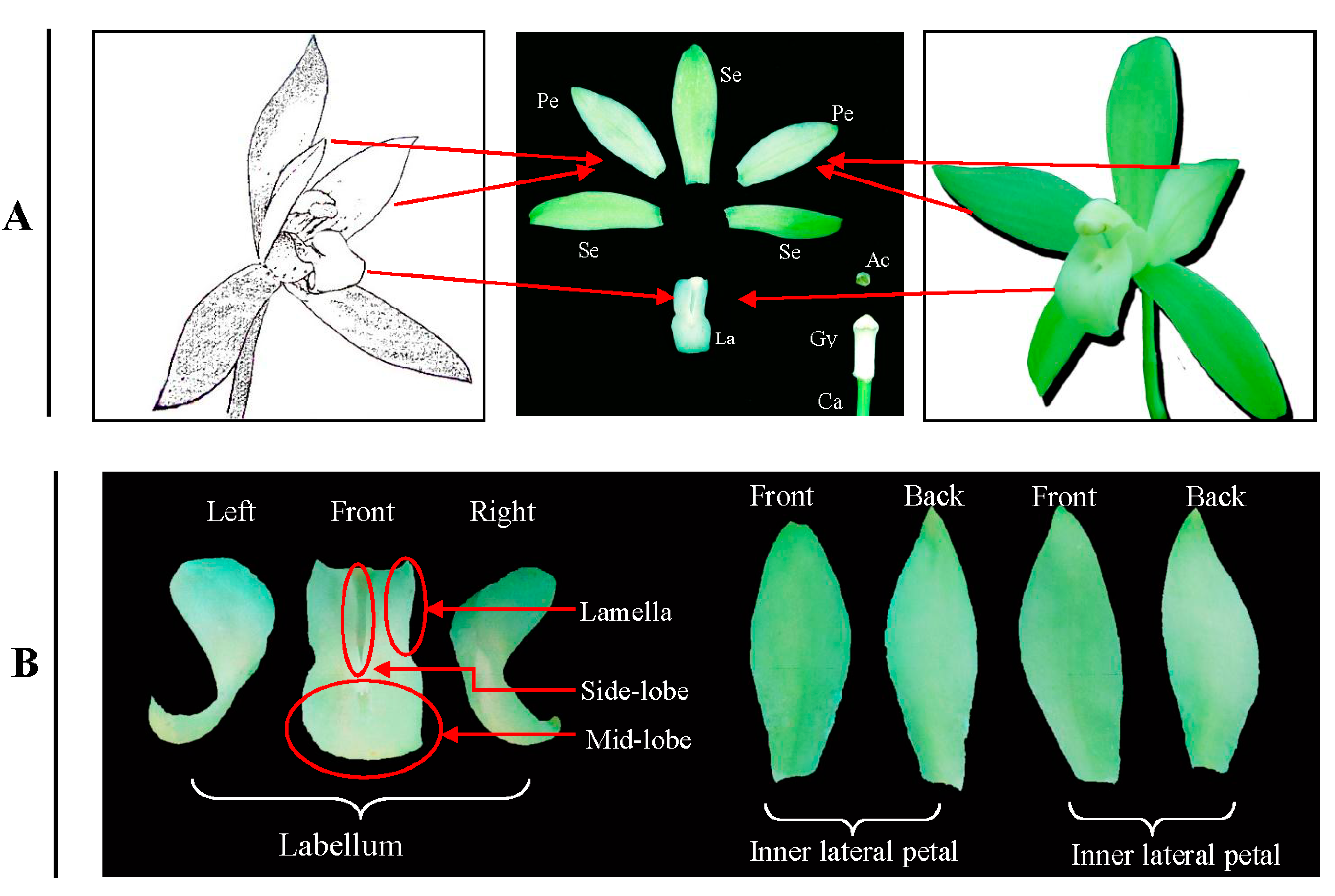
4.1. Plant Material
4.2. cDNA Library Construction and Sequencing
4.3. Protein Extraction and Quantification
4.4. Two-Dimensional Gel Electrophoresis
4.5. Image Acquisition and Analysis
4.6. In-Gel Protein Digestion and Mass Spectrometry
4.7. Protein Annotation and Classification
4.8. Protein and Protein Interactions (PPIs)
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aceto, S.; Gaudio, L. The mads and the beauty: Genes involved in the development of orchid flowers. Curr. Genomics 2011, 12, 342–356. [Google Scholar]
- Bateman, R.M.; Rudall, P.J. The good, the bad, and the ugly: Using naturally occurring terata to distinguish the possible from the impossible in orchid floral evolution. Aliso 2006, 22, 481–496. [Google Scholar]
- Rudall, P.J.; Bateman, R.M. Roles of synorganisation, zygomorphy and heterotopy in floral evolution: The gynostemium and labellum of orchids and other lilioid monocots. Biol. Rev. Camb. Philos. Soc. 2002, 77, 403–441. [Google Scholar]
- Krizek, B.A.; Fletcher, J.C. Molecular mechanisms of flower development: An armchair guide. Nat. Rev. Genet. 2005, 6, 688–698. [Google Scholar]
- Chen, S.; Harmon, A.C. Advances in plant proteomics. Proteomics 2006, 6, 5504–5516. [Google Scholar]
- Li, X.; Xiang, L.; Wang, Y.; Luo, J.; Wu, C.; Sun, C.; Xie, M. Genetic diversity, population structure, pollen morphology and cross-compatibility among Chinese Cymbidiums. Plant Breed. 2014, 133, 145–152. [Google Scholar]
- Li, X.; Luo, J.; Yan, T.; Xiang, L.; Jin, F.; Qin, D.; Sun, C.; Xie, M. Deep sequencing-based analysis of the cymbidium ensifolium floral transcriptome. PLoS One 2013, 8, e85480. [Google Scholar]
- Zhang, Y.; Wang, Z.; Zhang, L.; Cao, Y.; Huang, D.; Tang, K. Molecular cloning and stress-dependent regulation of potassium channel gene in Chinese cabbage (Brassica rapa ssp. Pekinensis). J. Plant Physiol. 2006, 163, 968–978. [Google Scholar]
- Hu, X.; Jiang, M.; Zhang, A.; Lu, J. Abscisic acid-induced apoplastic H2O2 accumulation up-regulates the activities of chloroplastic and cytosolic antioxidant enzymes in maize leaves. Planta 2005, 223, 57–68. [Google Scholar]
- Norman, C.; Howell, K.A.; Millar, A.H.; Whelan, J.M.; Day, D.A. Salicylic acid is an uncoupler and inhibitor of mitochondrial electron transport. Plant Physiol. 2004, 134, 492–501. [Google Scholar]
- Suhita, D.; Raghavendra, A.S.; Kwak, J.M.; Vavasseur, A. Cytoplasmic alkalization precedes reactive oxygen species production during methyl jasmonate- and abscisic acid-induced stomatal closure. Plant Physiol. 2004, 134, 1536–1545. [Google Scholar]
- Rogers, H.J. Is there an important role for reactive oxygen species and redox regulation during floral senescence? Plant Cell Environ. 2012, 35, 217–233. [Google Scholar]
- Albery, W.J.; Knowles, J.R. Free-energy profile for the reaction catalyzed by triosephosphate isomerase. Biochemistry 1976, 15, 5627–5631. [Google Scholar]
- Dorion, S.; Jeukens, J.; Matton, D.P.; Rivoal, J. Cloning and characterization of a cytosolic isoform of triosephosphate isomerase developmentally regulated in potato leaves. Plant Sci. 2005, 168, 183–194. [Google Scholar]
- Ben-Nissan, G.; Weiss, D. Developmental and hormonal regulation of a triosephosphate isomerase gene in petunia corollas. J. Plant Physiol. 1995, 147, 58–62. [Google Scholar]
- Ito, H.; Iwabuchi, M.; Ogawa, K. The sugar-metabolic enzymes aldolase and triose-phosphate isomerase are targets of glutathionylation in Arabidopsis thaliana: Detection using biotinylated glutathione. Plant Cell Physiol. 2003, 44, 655–660. [Google Scholar]
- Cox, M.; Lehninger, A.L.; Nelson, D.R. Lehninger Principles of Biochemistry; Worth Publishers: New York, NY, USA, 2000. [Google Scholar]
- Reuveni, M.; Sagi, Z.; Evnor, D.; Hetzroni, A. β-Glucosidase activity is involved in scent production in Narcissus flowers. Plant Sci. 1999, 147, 19–24. [Google Scholar]
- Leitner-Dagan, Y.; Ovadis, M.; Shklarman, E.; Elad, Y.; David, D.R.; Vainstein, A. Expression and functional analyses of the plastid lipid-associated protein CHRC suggest its role in chromoplastogenesis and stress. Plant Physiol. 2006, 142, 233–244. [Google Scholar]
- Ducreux, L.J.; Morris, W.L.; Hedley, P.E.; Shepherd, T.; Davies, H.V.; Millam, S.; Taylor, M.A. Metabolic engineering of high carotenoid potato tubers containing enhanced levels of β-carotene and lutein. J. Exp. Bot. 2005, 56, 81–89. [Google Scholar]
- Deruère, J.; Römer, S.; d’Harlingue, A.; Backhaus, R.A.; Kuntz, M.; Camara, B. Fibril assembly and carotenoid overaccumulation in chromoplasts: A model for supramolecular lipoprotein structures. Plant Cell Online 1994, 6, 119–133. [Google Scholar]
- Zhu, C.; Bai, C.; Sanahuja, G.; Yuan, D.; Farré, G.; Naqvi, S.; Shi, L.; Capell, T.; Christou, P. The regulation of carotenoid pigmentation in flowers. Arch. Biochem. Biophys. 2010, 504, 132–141. [Google Scholar]
- Tan, J.; Wang, H.; Yeh, K. Analysis of organ-specific, expressed genes in Oncidium orchid by subtractive expressed sequence tags library. Biotechnol. Lett. 2005, 27, 1517–1528. [Google Scholar]
- Loughrin, J.H.; Hamilton-Kemp, T.R.; Burton, H.R.; Andersen, R.A.; Hildebrand, D.F. Glycosidically bound volatile components of Nicotiana sylvestris and N. Suaveolens flowers. Phytochemistry 1992, 31, 1537–1540. [Google Scholar]
- Shirley, A.M.; McMichael, C.M.; Chapple, C. The sng2 mutant of Arabidopsis is defective in the gene encoding the serine carboxypeptidase—Like protein sinapoylglucose: Choline sinapoyltransferase. Plant J. 2001, 28, 83–94. [Google Scholar]
- Goldstein, G.; Sharifi, M.; Kohorn, L.; Lighton, J.; Shultz, L.; Rundel, P. Photosynthesis by inflated pods of a desert shrub, Isomeris arborea. Oecologia 1991, 85, 396–402. [Google Scholar]
- Galen, C.; Dawson, T.E.; Stanton, M.L. Carpels as leaves: Meeting the carbon cost of reproduction in an alpine buttercup. Oecologia 1993, 95, 187–193. [Google Scholar]
- Williams, K.; Koch, G.; Mooney, H. The carbon balance of flowers of Diplacus aurantiacus (Scrophulariaceae). Oecologia 1985, 66, 530–535. [Google Scholar]
- Christopher, J.T.; Holtum, J.A. Patterns of carbon partitioning in leaves of crassulacean acid metabolism species during deacidification. Plant Physiol. 1996, 112, 393–399. [Google Scholar]
- Kanai, R.; Edwards, G.E. The Biochemistry of C4 Photosynthesis; Academic Press: San Diego, CA, USA, 1999; pp. 49–87. [Google Scholar]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar]
- Youn, B.; Kim, S.-J.; Moinuddin, S.G.; Lee, C.; Bedgar, D.L.; Harper, A.R.; Davin, L.B.; Lewis, N.G.; Kang, C. Mechanistic and structural studies of apoform, binary, and ternary complexes of the Arabidopsis alkenal double bond reductase At5g1697. J. Biol. Chem. 2006, 281, 40076–40088. [Google Scholar]
- Gaikwad, A.; Long, D.J.; Stringer, J.L.; Jaiswal, A.K. In vivo role of NAD(P)H: Quinone oxidoreductase 1 (NQO1) in the regulation of intracellular redox state and accumulation of abdominal adipose tissue. J. Biol. Chem. 2001, 276, 22559–22564. [Google Scholar]
- Klein, D.; Fink, B.; Arold, B.; Eisenreich, W.; Schwab, W. Functional characterization of enone oxidoreductases from strawberry and tomato fruit. J. Agric. Food Chem. 2007, 55, 6705–6711. [Google Scholar]
- Hyodo, H.; Yamakawa, S.; Takeda, Y.; Tsuduki, M.; Yokota, A.; Nishitani, K.; Kohchi, T. Active gene expression of a xyloglucan endotransglucosylase/hydrolase gene, XTH9, in inflorescence apices is related to cell elongation in Arabidopsis thaliana. Plant Mol. Biol. 2003, 52, 473–482. [Google Scholar]
- Hayashi, T.; Kaida, R. Functions of xyloglucan in plant cells. Mol. Plant 2011, 4, 17–24. [Google Scholar]
- Maris, A.; Kaewthai, N.; Eklöf, J.M.; Miller, J.G.; Brumer, H.; Fry, S.C.; Verbelen, J.-P.; Vissenberg, K. Differences in enzymic properties of five recombinant xyloglucan endotransglucosylase/hydrolase (XTH) proteins of Arabidopsis thaliana. J. Exp. Bot. 2011, 62, 261–271. [Google Scholar]
- Harada, T.; Torii, Y.; Morita, S.; Onodera, R.; Hara, Y.; Yokoyama, R.; Nishitani, K.; Satoh, S. Cloning, characterization, and expression of xyloglucan endotransglucosylase/hydrolase and expansin genes associated with petal growth and development during carnation flower opening. J. Exp. Bot. 2011, 62, 815–823. [Google Scholar]
- Fang, X.; Chen, W.; Xin, Y.; Zhang, H.; Yan, C.; Yu, H.; Liu, H.; Xiao, W.; Wang, S.; Zheng, G.; et al. Proteomic analysis of strawberry leaves infected with Colletotrichum fragariae. J. Proteomics 2012, 75, 4074–4090. [Google Scholar]
- McCarthy, F.M.; Wang, N.; Magee, G.B.; Nanduri, B.; Lawrence, M.L.; Camon, E.B.; Barrell, D.G.; Hill, D.P.; Dolan, M.E.; Williams, W.P. AgBase: A functional genomics resource for agriculture. BMC Genomics 2006, 7, 229. [Google Scholar]
- Huang, C.; Morcos, F.; Kanaan, S.P.; Wuchty, S.; Chen, D.Z.; Izaguirre, J.A. Predicting protein-protein interactions from protein domains using a set cover approach. IEEE/ACM Trans. Comput. Biol. Bioinform. (TCBB) 2007, 4, 78–87. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Xu, W.; Chowdhury, M.R.; Jin, F. Comparative Proteomic Analysis of Labellum and Inner Lateral Petals in Cymbidium ensifolium Flowers. Int. J. Mol. Sci. 2014, 15, 19877-19897. https://doi.org/10.3390/ijms151119877
Li X, Xu W, Chowdhury MR, Jin F. Comparative Proteomic Analysis of Labellum and Inner Lateral Petals in Cymbidium ensifolium Flowers. International Journal of Molecular Sciences. 2014; 15(11):19877-19897. https://doi.org/10.3390/ijms151119877
Chicago/Turabian StyleLi, Xiaobai, Weiwei Xu, Moytri Roy Chowdhury, and Feng Jin. 2014. "Comparative Proteomic Analysis of Labellum and Inner Lateral Petals in Cymbidium ensifolium Flowers" International Journal of Molecular Sciences 15, no. 11: 19877-19897. https://doi.org/10.3390/ijms151119877
APA StyleLi, X., Xu, W., Chowdhury, M. R., & Jin, F. (2014). Comparative Proteomic Analysis of Labellum and Inner Lateral Petals in Cymbidium ensifolium Flowers. International Journal of Molecular Sciences, 15(11), 19877-19897. https://doi.org/10.3390/ijms151119877